Is there a Role for Cyclophilin Inhibitors in the Management of Primary Biliary Cirrhosis?

{kind=link}
{kind=link}
{kind=link}
Abstract
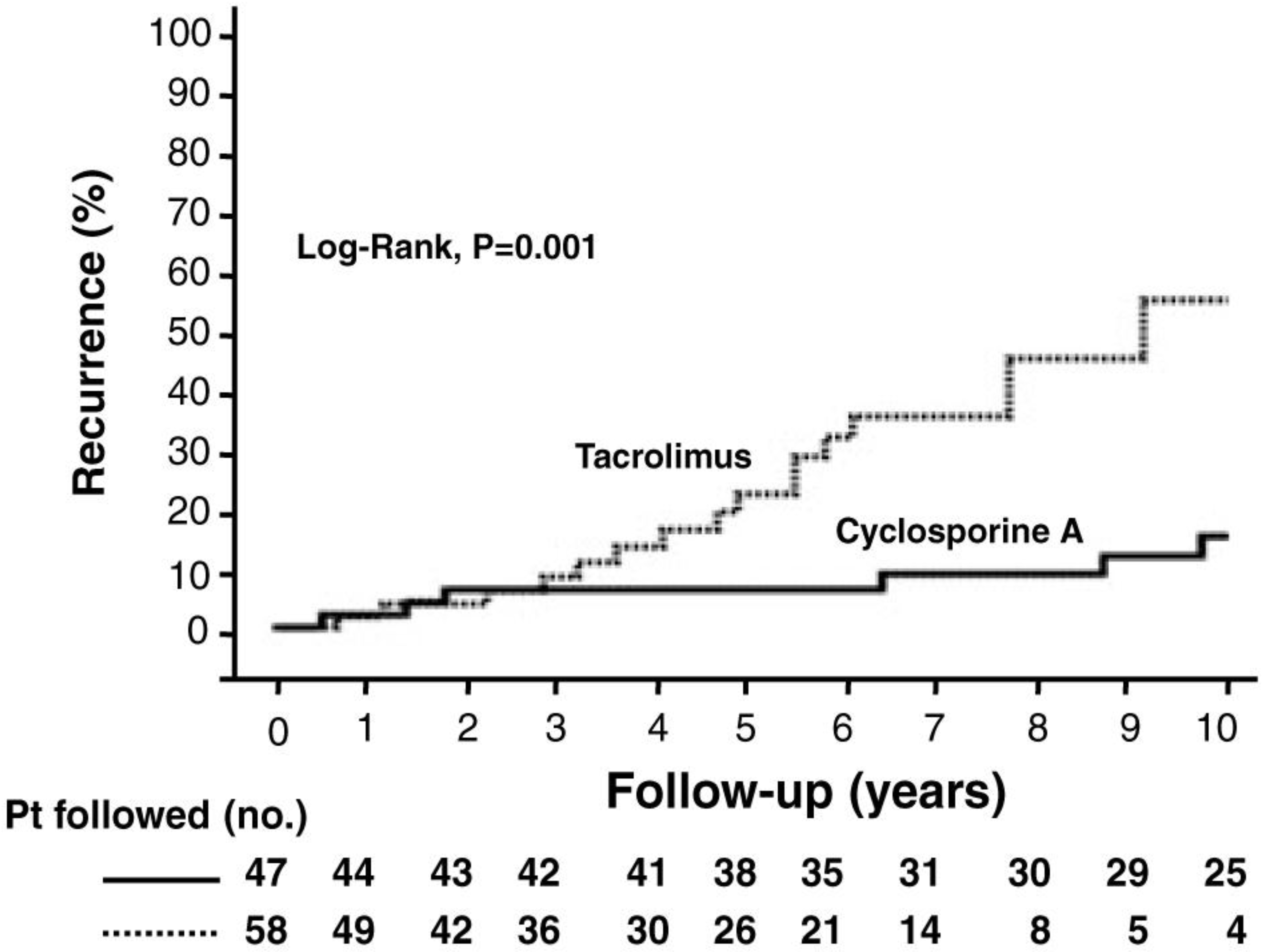
:1. Primary Biliary Cirrhosis

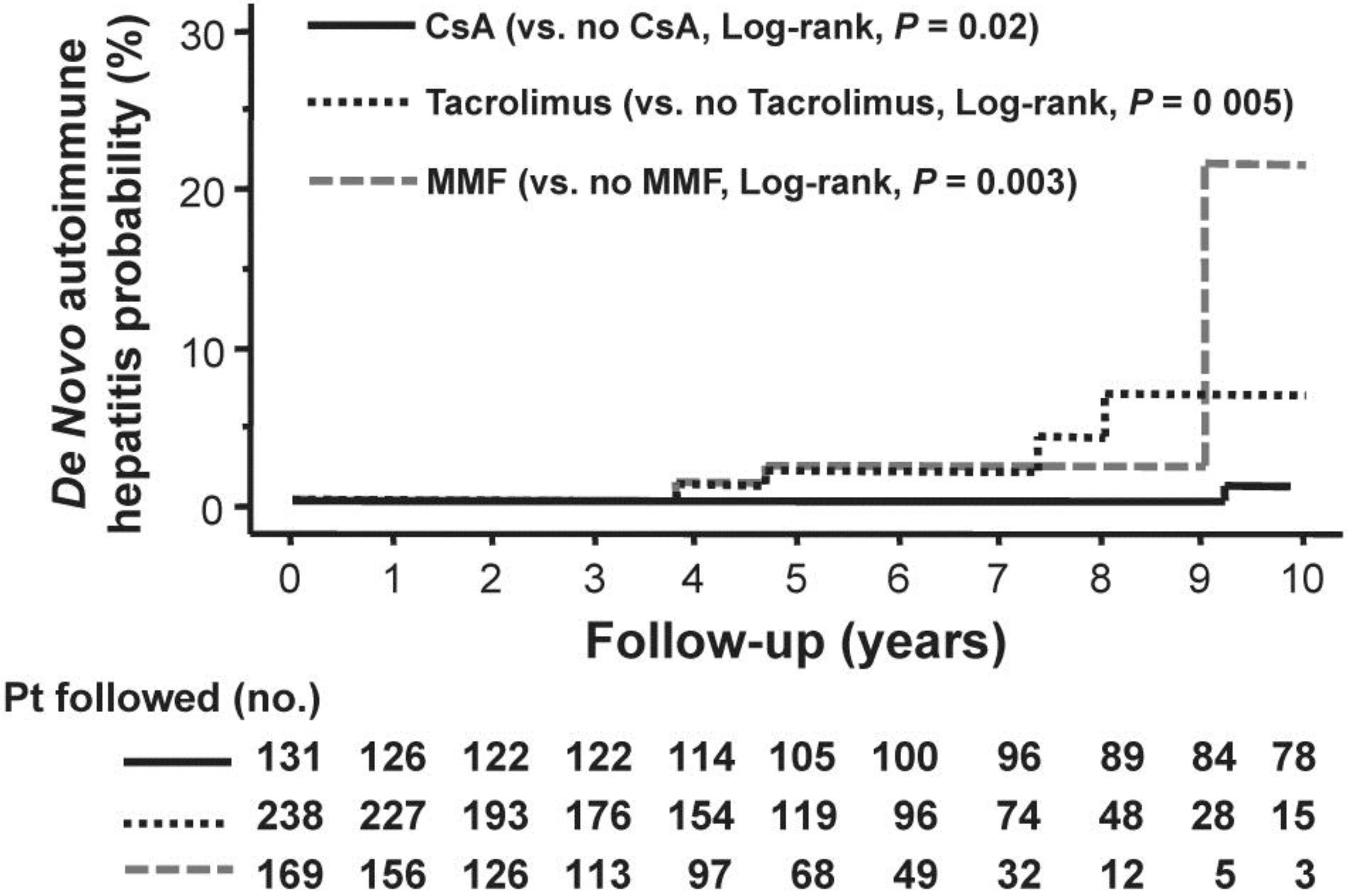
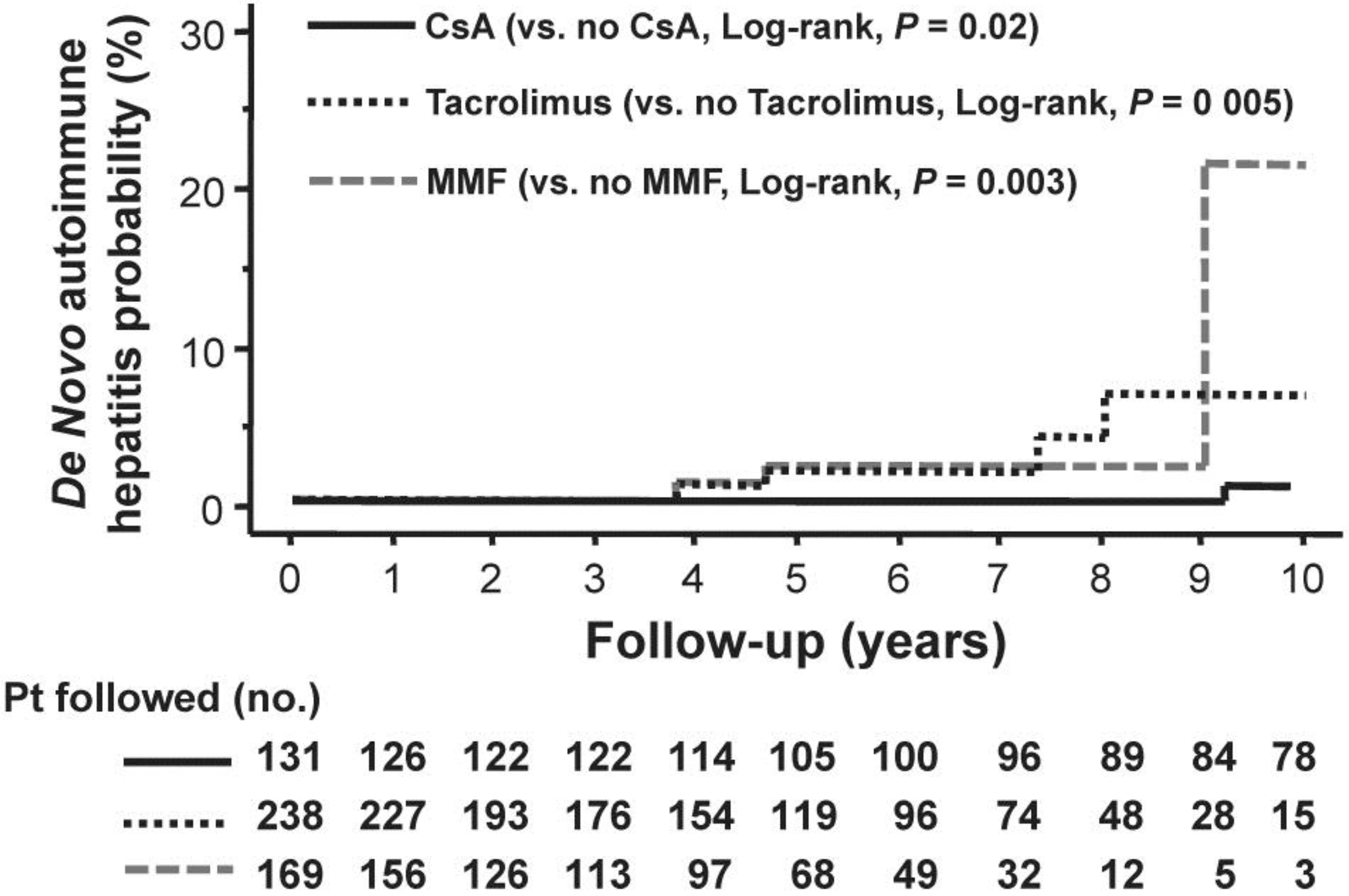
2. De Novo Autoimmune Hepatitis
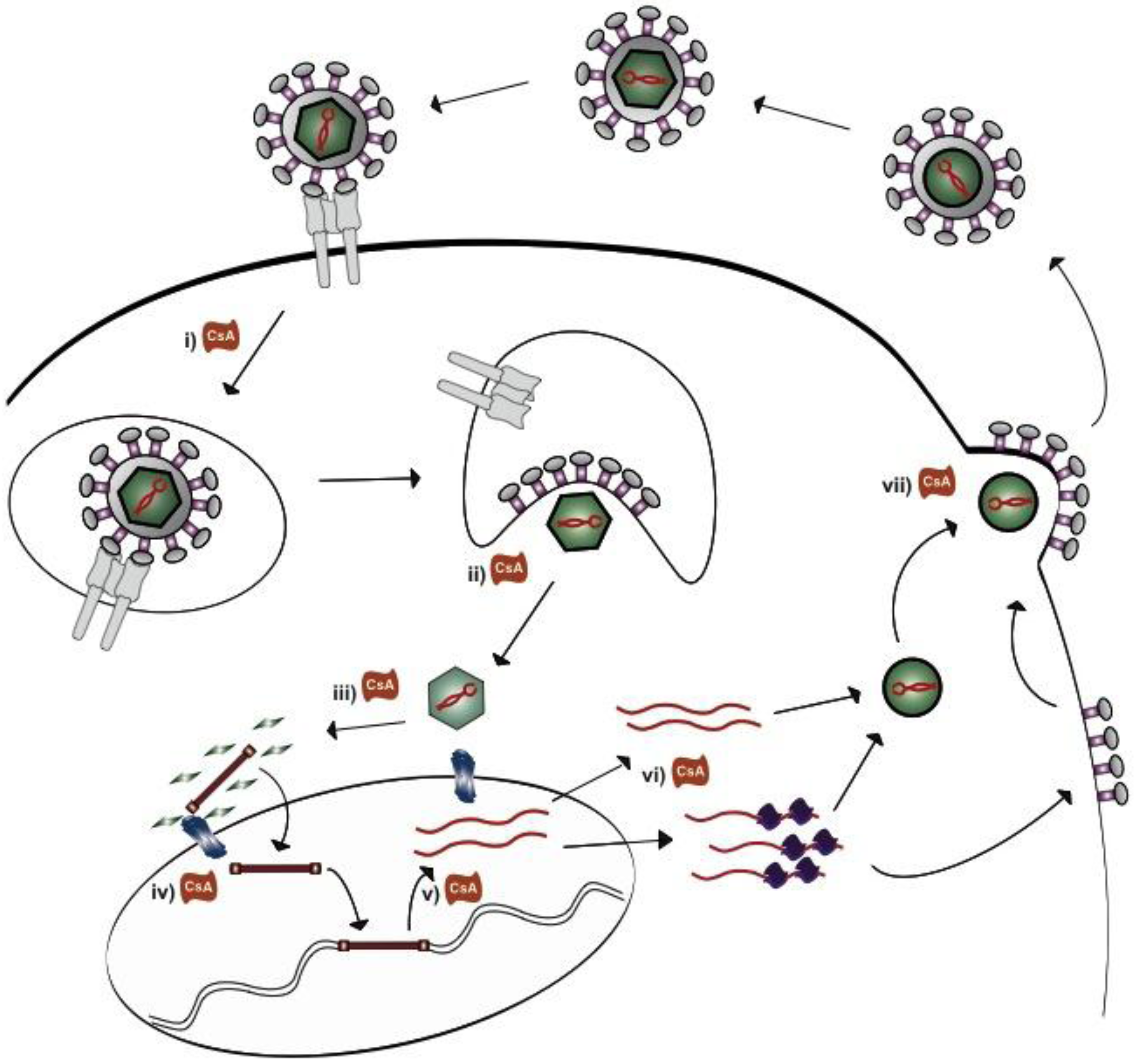
3. Pathophysiology of Recurrent PBC and de novo AIH
4. Is There a Potential Role of Cyclophilin Inhibitors in PBC?

5. How do Cyclosporine A and NIM811 Decrease Betaretrovirus Production
6. Prospectus
References
- Poupon, R. Primary biliary cirrhosis: A 2010 update. J. Hepatol. 2010, 52, 745–58. [Google Scholar] [CrossRef]
- Lindor, K.D.; Therneau, T.M.; Jorgensen, R.A.; Malinchoc, M.; Dickson, E.R. Effects of ursodeoxycholic acid on survival in patients with primary biliary cirrhosis. Gastroenterology 1996, 110, 1515–1518. [Google Scholar] [CrossRef]
- Poupon, R.E.; Balkau, B.; Eschwège, E.; Poupon, R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC study group. N. Eng. J. Med. 1991, 324, 1548–1554. [Google Scholar] [CrossRef]
- Neuberger, J. Liver transplantation for primary biliary cirrhosis. Autoimmun Rev. 2003, 2, 1–7. [Google Scholar] [CrossRef]
- Gish, R.G.; Mason, A. Autoimmune liver disease. Current standards, future directions. Clin. Liver Dis. 2001, 5, 287–314. [Google Scholar] [CrossRef]
- Mason, A.L.; Wasilenko, S.T. Other potential medical therapies: the use of antiviral agents to investigate and treat primary ciliary cirrhosis. Clin. Liver. Dis. 2008, 12, 445–460. [Google Scholar] [CrossRef]
- Lombard, M.; Portmann, B.; Neuberger, J.; Williams, R.; Tygstrup, N.; Ranek, L.; Ring-Larsen, H.; Rodes, J.; Navasa, M.; Trepo, C.; et al. Cyclosporin A treatment in primary biliary cirrhosis: Results of a long-term placebo controlled trial. Gastroenterology 1993, 104, 519–526. [Google Scholar]
- Perrillo, R.P.; Mason, A.L.; Jacob, S.; Gerber, M.A. Hepatitis and cholestasis in a middle-aged woman. Hepatology 1996, 24, 730–734. [Google Scholar] [CrossRef]
- Neuberger, J. Recurrent primary biliary cirrhosis. Liver. Transpl. 2003, 9, 539–546. [Google Scholar] [CrossRef]
- Neuberger, J.; Portmann, B.; Macdougall, B.R.; Calne, R.Y.; Williams, R. Recurrence of primary biliary cirrhosis after liver transplantation. N. Engl. J. Med. 1982, 306, 1–4. [Google Scholar] [CrossRef]
- Charatcharoenwitthaya, P.; Pimentel, S.; Talwalkar, J.A.; Enders, F.T.; Lindor, K.D.; Krom, R.A.; Wiesner, R.H. Long-term survival and impact of ursodeoxycholic acid treatment for recurrent primary biliary cirrhosis after liver transplantation. Liver. Transpl. 2007, 13, 1236–1245. [Google Scholar] [CrossRef]
- Guy, J.E.; Qian, P.; Lowell, J.A.; Peters, M.G. Recurrent primary biliary cirrhosis: Peritransplant factors and ursodeoxycholic acid treatment post-liver transplant. Liver. Transpl. 2005, 11, 1252–1257. [Google Scholar] [CrossRef]
- Montano-Loza, A.J.; Wasilenko, S.; Bintner, J.; Mason, A.L. Cyclosporine A protects against primary biliary cirrhosis recurrence after liver transplantation. Am. J. Transplant. 2010, 10, 852–858. [Google Scholar] [CrossRef]
- Jacob, D.A.; Neumann, U.P.; Bahra, M.; Klupp, J.; Puhl, G.; Neuhaus, R.; Langrehr, J.M. Long-term follow-up after recurrence of primary biliary cirrhosis after liver transplantation in 100 patients. Clin. Transplant. 2006, 20, 211–220. [Google Scholar] [CrossRef]
- Levitsky, J.; Hart, J.; Cohen, S.M.; Te, H.S. The effect of immunosuppressive regimens on the recurrence of primary biliary cirrhosis after liver transplantation. Liver. Transpl. 2003, 9, 733–736. [Google Scholar] [CrossRef]
- Liermann Garcia, R.F.; Evangelista Garcia, C.; McMaster, P.; Neuberger, J. Transplantation for primary biliary cirrhosis: retrospective analysis of 400 patients in a single center. Hepatology 2001, 33, 22–27. [Google Scholar] [CrossRef]
- Neuberger, J.; Gunson, B.; Hubscher, S.; Nightingale, P. Immunosuppression affects the rate of recurrent primary biliary cirrhosis after liver transplantation. Liver. Transpl. 2004, 10, 488–491. [Google Scholar] [CrossRef]
- Sanchez, E.Q.; Levy, M.F.; Goldstein, R.M.; Fasola, C.G.; Tillery, G.W.; Netto, G.J.; Watkins, D.L.; Weinstein, J.S.; Murray, N.G.; Byers, D.; et al. The changing clinical presentation of recurrent primary biliary cirrhosis after liver transplantation. Transplantation 2003, 76, 1583–1588. [Google Scholar]
- Montano-Loza, A.J.; Wasilenko, S.; Bintner, J.; Mason, A.L. Cyclosporine A inhibits in vitro replication of betaretrovirus associated with primary biliary cirrhosis. Liver. Int. 2010, 30, 871–877. [Google Scholar]
- Montano-Loza, A.J.; Vargas-Vorackova, F.; Ma, M.; Bain, V.G.; Burak, K.; Kumar, T.; Mason, A.L. Incidence and risk factors associated with de novo autoimmune hepatitis after liver transplantation. Liver. Int. 2012, 32, 1426–33. [Google Scholar] [CrossRef]
- Verdonk, R.C.; Dijkstra, G.; Haagsma, E.B.; Shostrom, V.K.; Van den Berg, A.P.; Kleibeuker, J.H.; Langnas, A.N.; Sudan, D.L. Inflammatory bowel disease after liver transplantation: risk factors for recurrence and de novo disease. Am. J. Transplant. 2006, 6, 1422–1429. [Google Scholar] [CrossRef]
- Kerkar, N.; Hadzic, N.; Davies, E.T.; Portmann, B.; Donaldson, P.T.; Rela, M.; Heaton, N.D.; Vergani, D.; Mieli-Vergani, G. De-novo autoimmune hepatitis after liver transplantation. Lancet 1998, 351, 409–413. [Google Scholar]
- Muratori, L.; Muratori, P.; Granito, A.; Pappas, G.; Cassani, F.; Lenzi, M. Current topics in autoimmune hepatitis. Dig. Liver. Dis. 2010, 42, 757–764. [Google Scholar] [CrossRef]
- Rodriguez-Diaz, Y.; Reyes-Rodriguez, R.; Dorta-Francisco, M.C.; Aguilera, I.; Perera-Molinero, A.; Moneva-Arce, E.; Aviles-Ruiz, J.F. De novo autoimmune hepatitis following liver transplantation for primary biliary cirrhosis. Transplant. Proc. 2006, 38, 1467–1470. [Google Scholar] [CrossRef]
- Gibelli, N.E.; Tannuri, U.; Mello, E.S.; Cancado, E.R.; Santos, M.M.; Ayoub, A.A.; Maksoud-Filho, J.G.; Velhote, M.C.; Silva, M.M.; Pinho-Apezzato, M.L.; et al. Successful treatment of de novo autoimmune hepatitis and cirrhosis after pediatric liver transplantation. Pediatr. Transplant. 2006, 10, 371–376. [Google Scholar] [CrossRef]
- Andries, S.; Casamayou, L.; Sempoux, C.; Burlet, M.; Reding, R.; Bernard Otte, J.; Buts, J.P.; Sokal, E. Posttransplant immune hepatitis in pediatric liver transplant recipients: Incidence and maintenance therapy with azathioprine. Transplantation 2001, 72, 267–272. [Google Scholar] [CrossRef]
- Gupta, P.; Hart, J.; Millis, J.M.; Cronin, D.; Brady, L. De novo hepatitis with autoimmune antibodies and atypical histology: A rare cause of late graft dysfunction after pediatric liver transplantation. Transplantation 2001, 71, 664–668. [Google Scholar] [CrossRef]
- D'Antiga, L.; Dhawan, A.; Portmann, B.; Francavilla, R.; Rela, M.; Heaton, N.; Mieli-Vergani, G. Late cellular rejection in paediatric liver transplantation: aetiology and outcome. Transplantation 2002, 73, 80–84. [Google Scholar] [CrossRef]
- Riva, S.; Sonzogni, A.; Bravi, M.; Bertani, A.; Alessio, M.G.; Candusso, M.; Stroppa, P.; Melzi, M.L.; Spada, M.; Gridelli, B.; et al. Late graft dysfunction and autoantibodies after liver transplantation in children: preliminary results of an Italian experience. Liver. Transpl. 2006, 12, 573–577. [Google Scholar] [CrossRef]
- Inui, A.; Sogo, T.; Komatsu, H.; Miyakawa, H.; Fujisawa, T. Antibodies against cytokeratin 8/18 in a patient with de novo autoimmune hepatitis after living-donor liver transplantation. Liver. Transpl. 2005, 11, 504–507. [Google Scholar] [CrossRef]
- Salcedo, M.; Vaquero, J.; Banares, R.; Rodriguez-Mahou, M.; Alvarez, E.; Vicario, J.L.; Hernandez-Albujar, A.; Tiscar, J.L.; Rincon, D.; Alonso, S.; et al. Response to steroids in de novo autoimmune hepatitis after liver transplantation. Hepatology 2002, 35, 349–356. [Google Scholar] [CrossRef]
- Di Cocco, P.; Barletta, A.; Clemente, K.; D'Angelo, M.; Greco, S.; Mazzotta, C.; Orlando, G.; Izza, V.; Famulari, A.; Grimaldi, A.; et al. De novo autoimmune hepatitis following liver transplantation: a case report. Transplant. Proc. 2008, 40, 2073–2074. [Google Scholar] [CrossRef]
- Manns, M.P.; Czaja, A.J.; Gorham, J.D.; Krawitt, E.L.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M. Diagnosis and management of autoimmune hepatitis. Hepatology 2010, 51, 2193–2213. [Google Scholar] [CrossRef]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; Gershwin, M.E. Primary Biliary Cirrhosis in Monozygotic and Dizygotic Twins: Genetics, Epigenetics, and Environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef]
- Selmi, C.; Invernizzi, P.; Zuin, M.; Podda, M.; Gershwin, M.E. Genetics and geoepidemiology of primary biliary cirrhosis: following the footprints to disease etiology. Semin. Liver. Dis. 2005, 25, 265–280. [Google Scholar] [CrossRef]
- Donaldson, P.; Agarwal, K.; Craggs, A.; Craig, W.; James, O.; Jones, D. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut 2001, 48, 397–402. [Google Scholar] [CrossRef]
- Hirschfield, G.; Chapman, R.; Karlsen, T.; Lammert, F.; Lazaridis, K.; Mason, A. The Genetics of Complex Cholestatic Disorders. in Submission 2012.
- Hirschfield, G.M.; Liu, X.; Han, Y.; Gorlov, I.P.; Lu, Y.; Xu, C.; Chen, W.; Juran, B.D.; Coltescu, C.; Mason, A.L.; et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 655–657. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; Mason, A.L.; et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Xie, G.; Lu, E.; Sun, Y.; Juran, B.D.; Chellappa, V.; Coltescu, C.; Mason, A.L.; Milkiewicz, P.; Myers, R.P.; et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes and Immunity 2012, 13, 328–335. [Google Scholar] [CrossRef]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; Selmi, C.; et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 658–660. [Google Scholar] [CrossRef]
- Manns, M.P.; Bremm, A.; Schneider, P.M.; Notghi, A.; Gerken, G.; Prager-Eerbele, M.; Stradmann-Bellinghausen, B.; Mayer-zum-Buschenfelde, K.H.; Ritner, C. HLA DRw8 and complement C4 deficiency as risk factors in primary biliary cirrhosis. Gastroenterology 1991, 101, 1367–1373. [Google Scholar]
- Sherlock, S. Primary Biliary Cirrhosis: Definition and Epidemiological Features; Kluwer Academic Publishers: Boston, MA, USA, 1993. [Google Scholar]
- Trigger, D.R. Primary biliary cirrhosis: an epidemiological study. Br. Med. J. 1980, 281, 772–775. [Google Scholar] [CrossRef]
- Prince, M.I.; James, O.F. The epidemiology of primary biliary cirrhosis. Clin. Liver. Dis. 2003, 7, 795–819. [Google Scholar] [CrossRef]
- Metcalf, J.; James, O. The geoepidemiology of primary biliary cirrhosis. Semin. Liver. Dis. 1997, 17, 13–22. [Google Scholar] [CrossRef]
- Neuberger, J. Primary Biliary Cirrhosis. Lancet 1997, 350, 875–879. [Google Scholar] [CrossRef]
- Mason, A.L.; Zhang, G. Linking human betaretrovirus infection with primary biliary cirrhosis. Gastroenterol Clin. Biol. 2010, 34, 359–366. [Google Scholar] [CrossRef]
- Xu, L.; Sakalian, M.; Shen, Z.; Loss, G.; Neuberger, J.; Mason, A. Cloning the human betaretrovirus proviral genome from patients with primary biliary cirrhosis. Hepatology 2004, 39, 151–156. [Google Scholar] [CrossRef]
- Xu, L.; Shen, Z.; Guo, L.; Fodera, B.; Keogh, A.; Joplin, R.; O'Donnell, B.; Aitken, J.; Carman, W.; Neuberger, J.; et al. Does a betaretrovirus infection trigger primary biliary cirrhosis? Proc. Natl. Acad. Sci.U.S.A. 2003, 100, 8454–8459. [Google Scholar]
- Selmi, C.; Ross, S.R.; Ansari, A.A.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Gershwin, M.E. Lack of immunological or molecular evidence for a role of mouse mammary tumor retrovirus in primary biliary cirrhosis. Gastroenterology 2004, 127, 493–501. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, M.; Graham, D.; Subsin, B.; McDougall, C.; Gilady, S.; Kneteman, M.; Swain, M.; Trauner, M.; Wrzesinski, S.; Flavell, R.; Wasilenko, S.; Mason, A. Mouse Mammary Tumor Virus in Anti-Mitochondrial Antibody Producing Mouse Models. J. Hepatology 2011, 55, 876–884. [Google Scholar] [CrossRef]
- Mason, A.L.; Farr, G.H.; Xu, L.; Hubscher, S.G.; Neuberger, J.M. Pilot studies of single and combination antiretroviral therapy in patients with primary biliary cirrhosis. Am. J. Gastroenterol. 2004, 99, 2348–2355. [Google Scholar] [CrossRef]
- Mason, A.L.; Lindor, K.D.; Bacon, B.R.; Vincent, C.; Neuberger, J.M.; Wasilenko, S.T. Clinical Trial: Randomized controlled trial of zidovudine and lamivudine for patients with primary biliary cirrhosis stabilized on ursodiol. Aliment. Pharmacol. Ther. 2008, 28, 886–894. [Google Scholar] [CrossRef]
- McDermid, J.; Chen, M.; Li, Y.; Wasilenko, S.; Bintner, J.; McDougall, C.; Pang, X.; Bain, V.G.; Mason, A.L. Reverse transcriptase activity in patients with primary biliary cirrhosis and other autoimmune liver disorders. Aliment. Pharmacol. Ther. 2007, 26, 587–595. [Google Scholar] [CrossRef]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 2009, 5, e1000524. [Google Scholar] [CrossRef]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins Facilitate Dissociation of the Human Papillomavirus Type 16 Capsid Protein L1 from the L2/DNA Complex following Virus Entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef]
- Watanabe, A.; Yoneda, M.; Ikeda, F.; Terao-Muto, Y.; Sato, H.; Kai, C. CD147/EMMPRIN acts as a functional entry receptor for measles virus on epithelial cells. J. Virol. 2010, 84, 4183–4193. [Google Scholar] [CrossRef]
- Pushkarsky, T.; Zybarth, G.; Dubrovsky, L.; Yurchenko, V.; Tang, H.; Guo, H.; Toole, B.; Sherry, B.; Bukrinsky, M. CD147 facilitates HIV-1 infection by interacting with virus-associated cyclophilin A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 6360–6365. [Google Scholar]
- Hatziioannou, T.; Perez-Caballero, D.; Cowan, S.; Bieniasz, P.D. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 2005, 79, 176–183. [Google Scholar] [CrossRef]
- Sokolskaja, E.; Sayah, D.M.; Luban, J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 2004, 78, 12800–12808. [Google Scholar] [CrossRef]
- Braaten, D.; Franke, E.K.; Luban, J. Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J. Virol. 1996, 70, 4220–4227. [Google Scholar]
- Schmidt, A.; Hennighausen, L.; Siebenlist, U. Inducible nuclear factor binding to the kappa B elements of the human immunodeficiency virus enhancer in T cells can be blocked by cyclosporin A in a signal-dependent manner. J. Virol. 1990, 64, 4037–4041. [Google Scholar]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef]
- Yang, F.; Robotham, J.M.; Grise, H.; Frausto, S.; Madan, V.; Zayas, M.; Bartenschlager, R.; Robinson, M.; Greenstein, A.E.; Nag, A.; et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog 2010, 6, e1001118. [Google Scholar] [CrossRef]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 2005, 19, 111–122. [Google Scholar] [CrossRef]
- Heck, J.A.; Meng, X.; Frick, D.N. Cyclophilin B stimulates RNA synthesis by the HCV RNA dependent RNA polymerase. Biochem. Pharmacol. 2009, 77, 1173–1180. [Google Scholar] [CrossRef]
- Fernandes, F.; Ansari, I.U.; Striker, R. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 2010, 5, e9815. [Google Scholar]
- Foster, T.L.; Gallay, P.; Stonehouse, N.J.; Harris, M. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 2011, 85, 7460–7464. [Google Scholar] [CrossRef]
- Puyang, X.; Poulin, D.L.; Mathy, J.E.; Anderson, L.J.; Ma, S.; Fang, Z.; Zhu, S.; Lin, K.; Fujimoto, R.; Compton, T.; et al. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob. Agents. Chemother. 2010, 54, 1981–1987. [Google Scholar]
- Qing, M.; Yang, F.; Zhang, B.; Zou, G.; Robida, J.M.; Yuan, Z.; Tang, H.; Shi, P.Y. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob. Agents. Chemother. 2009, 53, 3226–3235. [Google Scholar] [CrossRef]
- de Wilde, A.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Thiel, V.; Narayanan, K.; Makino, S.; Snijder, E.J.; van Hemert, M.J. Cyclosporin A inhibits the replication of diverse coronaviruses. J. Gen. Virol. 2011, 92, 2542–2548. [Google Scholar] [CrossRef]
- Renoir, J.M.; Mercier-Bodard, C.; Hoffmann, K.; Le Bihan, S.; Ning, Y.M.; Sanchez, E.R.; Handschumacher, R.E.; Baulieu, E.E. Cyclosporin A potentiates the dexamethasone-induced mouse mammary tumor virus-chloramphenicol acetyltransferase activity in LMCAT cells: A possible role for different heat shock protein-binding immunophilins in glucocorticosteroid receptor-mediated gene expression. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 4977–4981. [Google Scholar]
- Schiltknecht, E.; Ada, G.L. In vivo effects of cyclosporine on influenza A virus-infected mice. Cell Immunol. 1985, 91, 227–239. [Google Scholar] [CrossRef]
- Welbourn, S.; Pause, A. The hepatitis C virus NS2/3 protease. Curr. Issues Mol. Biol. 2007, 9, 63–69. [Google Scholar]
- Liu, X.; Zhao, Z.; Li, Z.; Xu, C.; Sun, L.; Chen, J.; Liu, W. Cyclosporin A inhibits the influenza virus replication through cyclophilin A-dependent and -independent pathways. PLoS One 2012, 7, e37277. [Google Scholar]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 7408–7413. [Google Scholar]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 2009, 5, e1000546. [Google Scholar] [CrossRef]
- Ciesek, S.; Steinmann, E.; Wedemeyer, H.; Manns, M.P.; Neyts, J.; Tautz, N.; Madan, V.; Bartenschlager, R.; von Hahn, T.; Pietschmann, T. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 2009, 50, 1638–1645. [Google Scholar] [CrossRef]
- Zander, K.; Sherman, M.P.; Tessmer, U.; Bruns, K.; Wray, V.; Prechtel, A.T.; Schubert, E.; Henklein, P.; Luban, J.; Neidleman, J.; et al. Cyclophilin A interacts with HIV-1 Vpr and is required for its functional expression. J. Biol. Chem. 2003, 278, 43202–43213. [Google Scholar]
- Franke, E.K.; Yuan, H.E.; Luban, J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 1994, 372, 359–362. [Google Scholar] [CrossRef]
- Thali, M.; Bukovsky, A.; Kondo, E.; Rosenwirth, B.; Walsh, C.T.; Sodroski, J.; Gottlinger, H.G. Functional association of cyclophilin A with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar] [CrossRef]
- Schembri, G.; Schober, P. Killing two birds with one stone. Lancet 2011, 377, 96. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wasilenko, S.T.; Montano-Loza, A.J.; Mason, A.L. Is there a Role for Cyclophilin Inhibitors in the Management of Primary Biliary Cirrhosis? Viruses 2013, 5, 423-438. https://doi.org/10.3390/v5020423
Wasilenko ST, Montano-Loza AJ, Mason AL. Is there a Role for Cyclophilin Inhibitors in the Management of Primary Biliary Cirrhosis? Viruses. 2013; 5(2):423-438. https://doi.org/10.3390/v5020423
Chicago/Turabian StyleWasilenko, Shawn T., Aldo J. Montano-Loza, and Andrew L. Mason. 2013. "Is there a Role for Cyclophilin Inhibitors in the Management of Primary Biliary Cirrhosis?" Viruses 5, no. 2: 423-438. https://doi.org/10.3390/v5020423
APA StyleWasilenko, S. T., Montano-Loza, A. J., & Mason, A. L. (2013). Is there a Role for Cyclophilin Inhibitors in the Management of Primary Biliary Cirrhosis? Viruses, 5(2), 423-438. https://doi.org/10.3390/v5020423