Apoptosis in Pneumovirus Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
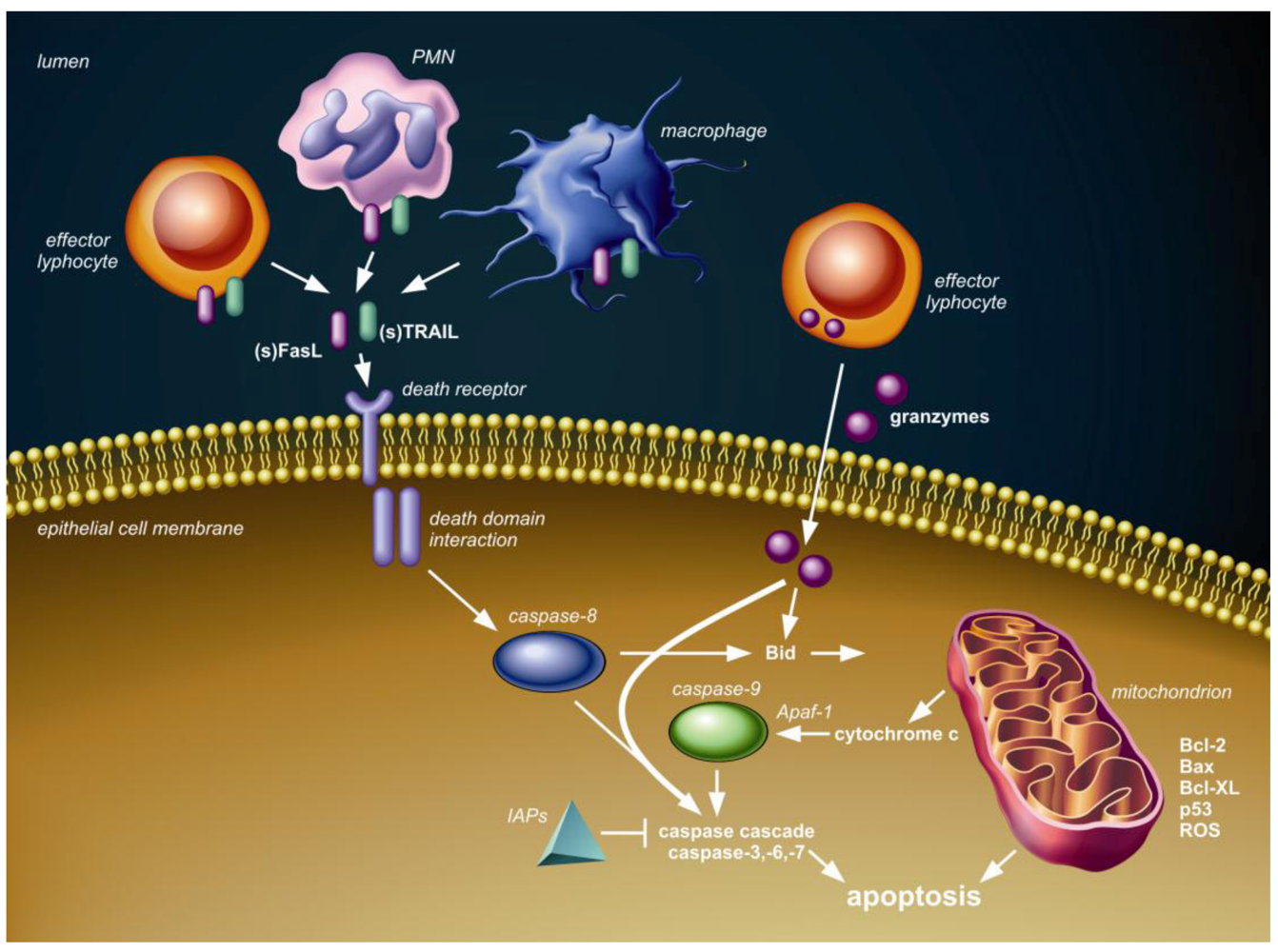
2. Apoptotic Signaling Pathways
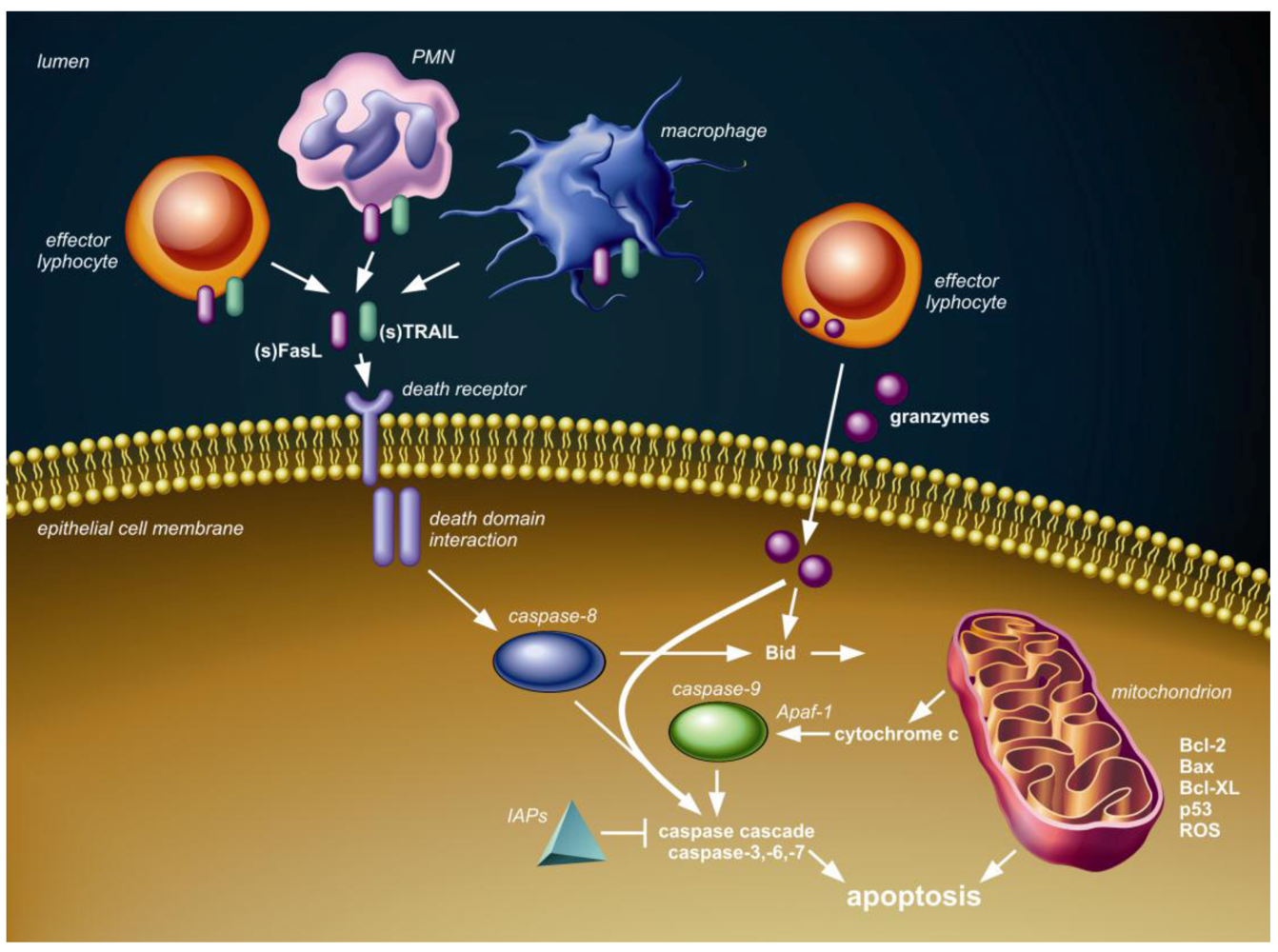
3. Lung (Airway and Alveolar) Epithelial Cell Apoptosis


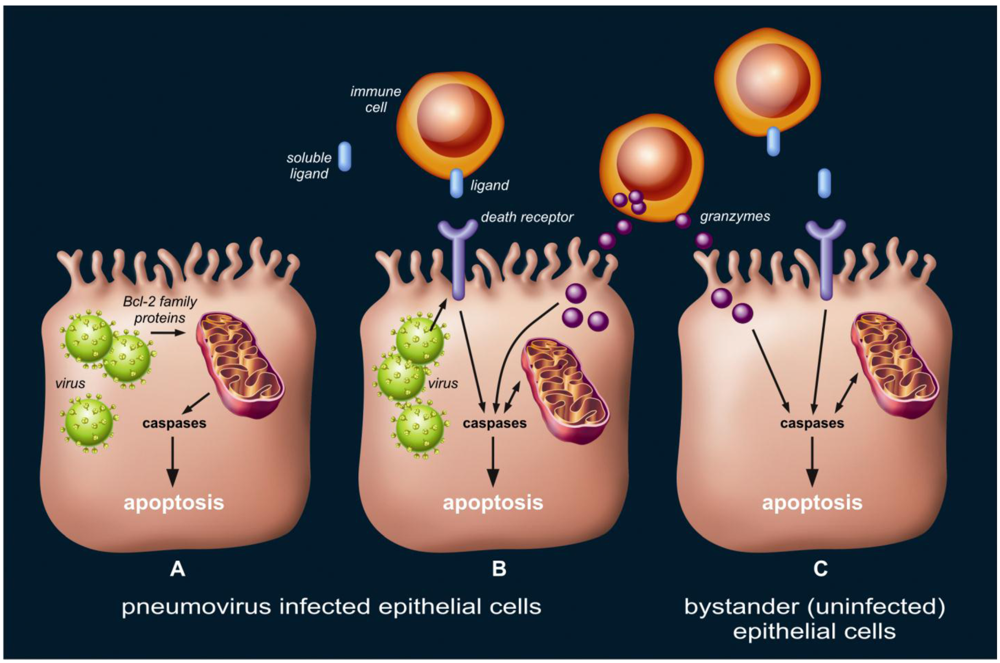
3.1. Pneumovirus Infected Cells May Undergo Apoptosis as a Result of Direct Activation of Intrinsic Pathways.
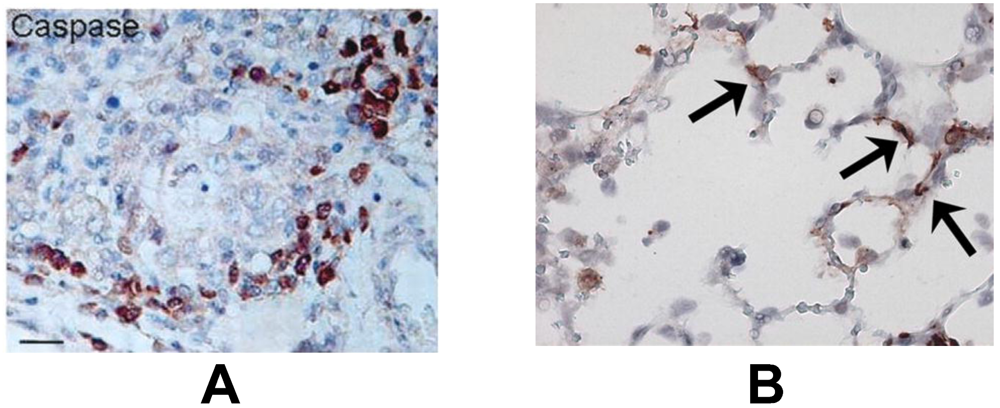
3.2. Pneumovirus Infected Cells May Undergo Apoptosis by Activation of Extrinsic and Granule-Mediated Pathways
3.3. Uninfected Bystander Epithelial Cells May Undergo Apoptosis as a Result of Extensive and Non-Specific Activation of Extrinsic and Granule-Mediated Pathways
4. Neutrophil Apoptosis
5. Macrophage Apoptosis
6. Apoptosis-Based Pharmacological Intervention
7. Conclusion
Conflict of Interest
Acknowledgments
References
- Easton, A.J.; Domachowske, J.B.; Rosenberg, H.F. Animal pneumoviruses: molecular genetics and pathogenesis. Clin. Microbiol. Rev. 2004, 17, 390–412. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O'Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Bem, R.A.; Domachowske, J.B.; Rosenberg, H.F. Animal models of human respiratory syncytial virus disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, 148–156. [Google Scholar] [CrossRef]
- ARDS definition task force Acute respiratory distress syndrome—The Berlin definition. JAMA 2012, 307, 256–2533.
- Dahlem, P.; van Aalderen, W.M.; Hamaker, M.E.; Dijkgraaf, M.G.; Bos, A.P. Incidence and short-term outcome of acute lung injury in mechanically ventilated children. Eur. Respir. J. 2003, 22, 980–985. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death. Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef]
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To kill or be killed: Viral evasion of apoptosis. Nat. Immunol. 2002, 3, 1013–1018. [Google Scholar] [CrossRef]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog 2008, 4, e1000018. [Google Scholar] [CrossRef]
- Herold, S.; Ludwig, S.; Pleschka, S.; Wolff, T. Apoptosis signaling in influenza virus propagation, innate host defense and lung injury. J. Leukoc. Biol. 2012, 92, 75–82. [Google Scholar] [CrossRef]
- Martin, T.R.; Hagimoto, N.; Nakamura, M.; Matute-Bello, G. Apoptosis and epithelial injury in the lungs. Proc. Am. Thorac. Soc. 2005, 2, 214–220. [Google Scholar] [CrossRef]
- Denecker, G.; Vercammen, D.; Declercq, W.; Vandenabeele, P. Apoptotic and necrotic cell death induced by death domain receptors. Cell Mol. Life Sci. 2001, 58, 356–370. [Google Scholar] [CrossRef]
- Leist, M.; Jaattela, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2001, 2, 589–598. [Google Scholar] [CrossRef]
- Wang, X.; Ryter, S.W.; Dai, C.; Tang, Z.L.; Watkins, S.C.; Yin, X.M.; Song, R.; Choi, A.M. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J. Biol. Chem. 2003, 278, 29184–29191. [Google Scholar]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar] [CrossRef]
- Bonville, C.A.; Bennett, N.J.; Koehnlein, M.; Haines, D.M.; Ellis, J.A.; DelVecchio, A.M.; Rosenberg, H.F.; Domachowske, J.B. Respiratory dysfunction and proinflammatory chemokines in the pneumonia virus of mice (PVM) model of viral bronchiolitis. Virology 2006, 349, 87–95. [Google Scholar] [CrossRef]
- Johnson, J.E.; Gonzales, R.A.; Olson, S.J.; Wright, P.F.; Graham, B.S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2007, 20, 108–119. [Google Scholar] [CrossRef]
- Viuff, B.; Tjornehoj, K.; Larsen, L.E.; Rontved, C.M.; Uttenthal, A.; Ronsholt, L.; Alexandersen, S. Replication and clearance of respiratory syncytial virus: Apoptosis is an important pathway of virus clearance after experimental infection with bovine respiratory syncytial virus. Am. J. Pathol. 2002, 161, 2195–2207. [Google Scholar] [CrossRef]
- Welliver, T.P.; Reed, J.L.; Welliver, R.C., Sr. Respiratory syncytial virus and influenza virus infections: observations from tissues of fatal infant cases. Pediatr. Infect. Dis. J. 2008, 27, 92–96. [Google Scholar] [CrossRef]
- Cook, P.M.; Eglin, R.P.; Easton, A.J. Pathogenesis of pneumovirus infections in mice: Detection of pneumonia virus of mice and human respiratory syncytial virus mRNA in lungs of infected mice by in situ hybridization. J. Gen. Virol. 1998, 79, 2411–2417. [Google Scholar]
- Aherne, W.; Bird, T.; Court, S.D.; Gardner, P.S.; McQuillin, J. Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 1970, 23, 7–18. [Google Scholar] [CrossRef]
- Reed, J.L.; Brewah, Y.A.; Delaney, T.; Welliver, T.; Burwell, T.; Benjamin, E.; Kuta, E.; Kozhich, A.; McKinney, L.; Suzich, J.; et al. Macrophage impairment underlies airway occlusion in primary respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2008, 198, 1783–1793. [Google Scholar] [CrossRef]
- Welliver, T.P.; Garofalo, R.P.; Hosakote, Y.; Hintz, K.H.; Avendano, L.; Sanchez, K.; Velozo, L.; Jafri, H.; Chavez-Bueno, S.; Ogra, P.L.; et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 2007, 195, 1126–1136. [Google Scholar] [CrossRef]
- Bem, R.A.; van Woensel, J.B.; Lutter, R.; Domachowske, J.B.; Medema, J.P.; Rosenberg, H.F.; Bos, A.P. Granzyme A- and B-cluster deficiency delays acute lung injury in pneumovirus-infected mice. J. Immunol. 2010, 184, 931–938. [Google Scholar] [CrossRef]
- Laham, F.R.; Trott, A.A.; Bennett, B.L.; Kozinetz, C.A.; Jewell, A.M.; Garofalo, R.P.; Piedra, P.A. LDH concentration in nasal-wash fluid as a biochemical predictor of bronchiolitis severity. Pediatrics 2010, 125, 225–233. [Google Scholar] [CrossRef]
- Herold, S.; Steinmueller, M.; von, W.W.; Cakarova, L.; Pinto, R.; Pleschka, S.; Mack, M.; Kuziel, W.A.; Corazza, N.; Brunner, T.; et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008, 205, 3065–3077. [Google Scholar] [CrossRef]
- Bitko, V.; Barik, S. An endoplasmic reticulum-specific stress-activated caspase (caspase-12) is implicated in the apoptosis of A549 epithelial cells by respiratory syncytial virus. J. Cell Biochem. 2001, 80, 441–454. [Google Scholar] [CrossRef]
- Bitko, V.; Shulyayeva, O.; Mazumder, B.; Musiyenko, A.; Ramaswamy, M.; Look, D.C.; Barik, S. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 2007, 81, 1786–1795. [Google Scholar] [CrossRef]
- Eckardt-Michel, J.; Lorek, M.; Baxmann, D.; Grunwald, T.; Keil, G.M.; Zimmer, G. The fusion protein of respiratory syncytial virus triggers p53-dependent apoptosis. J. Virol. 2008, 82, 3236–3249. [Google Scholar] [CrossRef]
- Fuentes, S.; Tran, K.C.; Luthra, P.; Teng, M.N.; He, B. Function of the respiratory syncytial virus small hydrophobic protein. J. Virol. 2007, 81, 8361–8366. [Google Scholar]
- Groskreutz, D.J.; Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Quelle, D.E.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J. Immunol. 2007, 179, 2741–2747. [Google Scholar]
- Groskreutz, D.J.; Monick, M.M.; Babor, E.C.; Nyunoya, T.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Cigarette smoke alters respiratory syncytial virus-induced apoptosis and replication. Am. J. Respir. Cell Mol. Biol. 2009, 41, 189–198. [Google Scholar] [CrossRef]
- Kotelkin, A.; Prikhod'ko, E.A.; Cohen, J.I.; Collins, P.L.; Bukreyev, A. Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J. Virol. 2003, 77, 9156–9172. [Google Scholar] [CrossRef]
- Monick, M.M.; Cameron, K.; Staber, J.; Powers, L.S.; Yarovinsky, T.O.; Koland, J.G.; Hunninghake, G.W. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J. Biol. Chem. 2005, 280, 2147–2158. [Google Scholar]
- Othumpangat, S.; Gibson, L.F.; Samsell, L.; Piedimonte, G. NGF is an essential survival factor for bronchial epithelial cells during respiratory syncytial virus infection. PLoS One 2009, 4, e6444. [Google Scholar]
- Takeuchi, R.; Tsutsumi, H.; Osaki, M.; Haseyama, K.; Mizue, N.; Chiba, S. Respiratory syncytial virus infection of human alveolar epithelial cells enhances interferon regulatory factor 1 and interleukin-1beta-converting enzyme gene expression, but does not cause apoptosis. J. Virol. 1998, 72, 4498–4502. [Google Scholar]
- Thomas, K.W.; Monick, M.M.; Staber, J.M.; Yarovinsky, T.; Carter, A.B.; Hunninghake, G.W. Respiratory syncytial virus inhibits apoptosis and induces NF-kappa B activity through a phosphatidylinositol 3-kinase-dependent pathway. J. Biol. Chem. 2002, 277, 492–501. [Google Scholar]
- Villenave, R.; Thavagnanam, S.; Sarlang, S.; Parker, J.; Douglas, I.; Skibinski, G.; Heaney, L.G.; McKaigue, J.P.; Coyle, P.V.; Shields, M.D.; et al. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 5040–5045. [Google Scholar]
- Othumpangat, S.; Walton, C.; Piedimonte, G. MicroRNA-221 modulates RSV replication in human bronchial epithelium by targeting NGF expression. PLoS One 2012, 7, e30030. [Google Scholar]
- Spann, K.M.; Tran, K.C.; Chi, B.; Rabin, R.L.; Collins, P.L. Suppression of the induction of alpha, beta and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J. Virol. 2004, 78, 4363–4369. [Google Scholar] [CrossRef]
- Bem, R.A.; Bos, A.P.; Wosten-van Asperen, R.M.; Bruijn, M.; Lutter, R.; Sprick, M.R.; van Woensel, J.B. Potential Role of Soluble TRAIL in Epithelial Injury in Children with Severe RSV Infection. Am. J. Respir. Cell Mol. Biol. 2009, 42, 697–705. [Google Scholar]
- Aung, S.; Rutigliano, J.A.; Graham, B.S. Alternative mechanisms of respiratory syncytial virus clearance in perforin knockout mice lead to enhanced disease. J. Virol. 2001, 75, 9918–9924. [Google Scholar] [CrossRef]
- Domachowske, J.B.; Bonville, C.A.; Mortelliti, A.J.; Colella, C.B.; Kim, U.; Rosenberg, H.F. Respiratory syncytial virus infection induces expression of the anti-apoptosis gene IEX-1L in human respiratory epithelial cells. J. Infect. Dis. 2000, 181, 824–830. [Google Scholar] [CrossRef]
- O'donnell, D.R.; Milligan, L.; Stark, J.M. Induction of CD95 (Fas) and apoptosis in respiratory epithelial cell cultures following respiratory syncytial virus infection. Virology 1999, 257, 198–207. [Google Scholar] [CrossRef]
- Rutigliano, J.A.; Graham, B.S. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J. Immunol. 2004, 173, 3408–3417. [Google Scholar]
- van den Berg, E.; van Woensel, J.B.; Bos, A.P.; Bem, R.A.; Altemeier, W.A.; Gill, S.E.; Martin, T.R.; Matute-Bello, G. Role of the Fas/FasL system in a model of RSV infection in mechanically ventilated mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, 451–460. [Google Scholar]
- Bem, R.A.; Bos, A.P.; Bots, M.; Wolbink, A.M.; van Ham, S.M.; Medema, J.P.; Lutter, R.; van Woensel, J.B. Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr. Res. 2008, 63, 650–655. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Liles, W.C.; Steinberg, K.P.; Kiener, P.A.; Mongovin, S.; Chi, E.Y.; Jonas, M.; Martin, T.R. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J. Immunol. 1999, 163, 2217–2225. [Google Scholar]
- Serrao, K.L.; Fortenberry, J.D.; Owens, M.L.; Harris, F.L.; Brown, L.A. Neutrophils induce apoptosis of lung epithelial cells via release of soluble Fas ligand. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, 298–305. [Google Scholar]
- Powell, W.C.; Fingleton, B.; Wilson, C.L.; Boothby, M.; Matrisian, L.M. The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr. Biol. 1999, 9, 1441–1447. [Google Scholar] [CrossRef]
- Everard, M.L.; Swarbrick, A.; Wrightham, M.; McIntyre, J.; Dunkley, C.; James, P.D.; Sewell, H.F.; Milner, A.D. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch. Dis. Child 1994, 71, 428–432. [Google Scholar] [CrossRef]
- van Woensel, J.B.; Lutter, R.; Biezeveld, M.H.; Dekker, T.; Nijhuis, M.; van Aalderen, W.M.; Kuijpers, T.W. Effect of dexamethasone on tracheal viral load and interleukin-8 tracheal concentration in children with respiratory syncytial virus infection. Pediatr. Infect. Dis. J. 2003, 22, 721–726. [Google Scholar] [CrossRef]
- McNamara, P.S.; Ritson, P.; Selby, A.; Hart, C.A.; Smyth, R.L. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 2003, 88, 922–926. [Google Scholar] [CrossRef]
- Tate, M.D.; Deng, Y.M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Garcia-Sastre, A.; Taubenberger, J.K.; Palese, P.; Swayne, D.E.; Pantin-Jackwood, M.J.; Schultz-Cherry, S.; Solorzano, A.; van, R.N.; Katz, J.M.; et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 2005, 79, 14933–14944. [Google Scholar]
- Halfhide, C.P.; Flanagan, B.F.; Brearey, S.P.; Hunt, J.A.; Fonceca, A.M.; McNamara, P.S.; Howarth, D.; Edwards, S.; Smyth, R.L. Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J. Infect. Dis. 2011, 204, 451–458. [Google Scholar] [CrossRef]
- Wang, S.Z.; Forsyth, K.D. The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology 2000, 5, 1–10. [Google Scholar] [CrossRef]
- Segel, G.B.; Halterman, M.W.; Lichtman, M.A. The paradox of the neutrophil's role in tissue injury. J. Leukoc. Biol. 2011, 89, 359–372. [Google Scholar] [CrossRef]
- Martin, T.R. Neutrophils and lung injury: Getting it right. J. Clin. Invest. 2002, 110, 1603–1605. [Google Scholar]
- Perl, M.; Lomas-Neira, J.; Chung, C.S.; Ayala, A. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury-a unifying hypothesis? What we have learned from small interfering RNAs. Mol. Med. 2008, 14, 465–475. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Coffer, P.J.; Schellens, I.M.; de Graaff, P.M.; Kimpen, J.L.; Koenderman, L. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J. Immunol. 2006, 176, 5529–5537. [Google Scholar]
- Coleman, C.M.; Plant, K.; Newton, S.; Hobson, L.; Whyte, M.K.; Everard, M.L. The Anti-Apoptotic Effect of Respiratory Syncytial Virus on Human Peripheral Blood Neutrophils is Mediated by a Monocyte Derived Soluble Factor. Open Virol. J. 2011, 5, 114–123. [Google Scholar] [CrossRef]
- Wang, S.Z.; Smith, P.K.; Lovejoy, M.; Bowden, J.J.; Alpers, J.H.; Forsyth, K.D. The apoptosis of neutrophils is accelerated in respiratory syncytial virus (RSV)-induced bronchiolitis. Clin. Exp. Immunol. 1998, 114, 49–54. [Google Scholar] [CrossRef]
- Pillay, J.; Ramakers, B.P.; Kamp, V.M.; Loi, A.L.; Lam, S.W.; Hietbrink, F.; Leenen, L.P.; Tool, A.T.; Pickkers, P.; Koenderman, L. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J. Leukoc. Biol. 2010, 88, 211–220. [Google Scholar] [CrossRef]
- Simon, H.U. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 2003, 193, 101–110. [Google Scholar] [CrossRef]
- Pribul, P.K.; Harker, J.; Wang, B.; Wang, H.; Tregoning, J.S.; Schwarze, J.; Openshaw, P.J. Alveolar macrophages are a major determinant of early responses to viral lung infection, but do not influence subsequent disease development. J. Virol. 2008, 82, 4441–4448. [Google Scholar] [CrossRef]
- Rigaux, P.; Killoran, K.E.; Qiu, Z.; Rosenberg, H.F. Depletion of alveolar macrophages prolongs survival in response to acute pneumovirus infection. Virology 2012, 422, 338–345. [Google Scholar] [CrossRef]
- Bem, R.A.; van Woensel, J.B.; Bos, A.P.; Koski, A.; Farnand, A.W.; Domachowske, J.B.; Rosenberg, H.F.; Martin, T.R.; Matute-Bello, G. Mechanical ventilation enhances lung inflammation and caspase activity in a model of mouse pneumovirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, 46–56. [Google Scholar]
- Midulla, F.; Villani, A.; Panuska, J.R.; Dab, I.; Kolls, J.K.; Merolla, R.; Ronchetti, R. Respiratory syncytial virus lung infection in infants: immunoregulatory role of infected alveolar macrophages. J. Infect. Dis. 1993, 168, 1515–1519. [Google Scholar] [CrossRef]
- Midulla, F.; Huang, Y.T.; Gilbert, I.A.; Cirino, N.M.; McFadden, E.R., Jr.; Panuska, J.R. Respiratory syncytial virus infection of human cord and adult blood monocytes and alveolar macrophages. Am. Rev. Respir. Dis. 1989, 140, 771–777. [Google Scholar]
- Panuska, J.R.; Cirino, N.M.; Midulla, F.; Despot, J.E.; McFadden, E.R., Jr.; Huang, Y.T. Productive infection of isolated human alveolar macrophages by respiratory syncytial virus. J. Clin. Invest. 1990, 86, 113–119. [Google Scholar] [CrossRef]
- Krilov, L.R.; McCloskey, T.W.; Harkness, S.H.; Pontrelli, L.; Pahwa, S. Alterations in apoptosis of cord and adult peripheral blood mononuclear cells induced by in vitro infection with respiratory syncytial virus. J. Infect. Dis. 2000, 181, 349–353. [Google Scholar] [CrossRef]
- Nakamura-Lopez, Y.; Villegas-Sepulveda, N.; Sarmiento-Silva, R.E.; Gomez, B. Intrinsic apoptotic pathway is subverted in mouse macrophages persistently infected by RSV. Virus Res. 2011, 158, 98–107. [Google Scholar] [CrossRef]
- Fischer, U.; Schulze-Osthoff, K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005, 12, 942–961. [Google Scholar] [CrossRef]
- van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- Janssen, W.J.; Barthel, L.; Muldrow, A.; Oberley-Deegan, R.E.; Kearns, M.T.; Jakubzick, C.; Henson, P.M. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2011, 184, 547–560. [Google Scholar] [CrossRef]
- Nakamura, M.; Matute-Bello, G.; Liles, W.C.; Hayashi, S.; Kajikawa, O.; Lin, S.M.; Frevert, C.W.; Martin, T.R. Differential response of human lung epithelial cells to fas-induced apoptosis. Am. J. Pathol. 2004, 164, 1949–1958. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Liles, W.C.; Frevert, C.W.; Dhanireddy, S.; Ballman, K.; Wong, V.; Green, R.R.; Song, H.Y.; Witcher, D.R.; Jakubowski, J.A.; et al. Blockade of the Fas/FasL system improves pneumococcal clearance from the lungs without preventing dissemination of bacteria to the spleen. J. Infect. Dis. 2005, 191, 596–606. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Liles, W.C.; Frevert, C.W.; Nakamura, M.; Ballman, K.; Vathanaprida, C.; Kiener, P.A.; Martin, T.R. Recombinant human Fas ligand induces alveolar epithelial cell apoptosis and lung injury in rabbits. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, 328–335. [Google Scholar]
- Perl, M.; Chung, C.S.; Lomas-Neira, J.; Rachel, T.M.; Biffl, W.L.; Cioffi, W.G.; Ayala, A. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation and neutrophil influx after hemorrhagic shock and sepsis. Am. J. Pathol. 2005, 167, 1545–1559. [Google Scholar] [CrossRef]
- Kawasaki, M.; Kuwano, K.; Hagimoto, N.; Matsuba, T.; Kunitake, R.; Tanaka, T.; Maeyama, T.; Hara, N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am. J. Pathol. 2000, 157, 597–603. [Google Scholar] [CrossRef]
- Altemeier, W.A.; Sinclair, S.E. Hyperoxia in the intensive care unit: why more is not always better. Curr. Opin. Crit. Care 2007, 13, 73–78. [Google Scholar]
- Bem, R.A.; Bos, A.P.; Matute-Bello, G.; van, T.M.; van Woensel, J.B. Lung epithelial cell apoptosis during acute lung injury in infancy. Pediatr. Crit. Care Med. 2007, 8, 132–137. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Van den Berg, E.; Van Woensel, J.B.M.; Bem, R.A. Apoptosis in Pneumovirus Infection. Viruses 2013, 5, 406-422. https://doi.org/10.3390/v5010406
Van den Berg E, Van Woensel JBM, Bem RA. Apoptosis in Pneumovirus Infection. Viruses. 2013; 5(1):406-422. https://doi.org/10.3390/v5010406
Chicago/Turabian StyleVan den Berg, Elske, Job B.M. Van Woensel, and Reinout A. Bem. 2013. "Apoptosis in Pneumovirus Infection" Viruses 5, no. 1: 406-422. https://doi.org/10.3390/v5010406