Foamy Virus Vectors for HIV Gene Therapy

1
Department of Pharmaceutical Sciences, Washington State University, Pullman, WA 99164, USA
2
School of Molecular Biosciences, Washington State University, Pullman, WA 99164, USA
*
Author to whom correspondence should be addressed.
Viruses 2013, 5(10), 2585-2600; https://doi.org/10.3390/v5102585
Submission received: 1 September 2013
/
Revised: 10 October 2013
/
Accepted: 16 October 2013
/
Published: 22 October 2013
(This article belongs to the Special Issue Gene Therapy for Retroviral Infections)
Abstract
:Highly active antiretroviral therapy (HAART) has vastly improved outcomes for patients infected with HIV, yet it is a lifelong regimen that is expensive and has significant side effects. Retroviral gene therapy is a promising alternative treatment for HIV/AIDS; however, inefficient gene delivery to hematopoietic stem cells (HSCs) has so far limited the efficacy of this approach. Foamy virus (FV) vectors are derived from non-pathogenic viruses that are not endemic to the human population. FV vectors have been used to deliver HIV-inhibiting transgenes to human HSCs, and they have several advantages relative to other retroviral vectors. These include an attractive safety profile, broad tropism, a large transgene capacity, and the ability to persist in quiescent cells. In addition, the titers of FV vectors are not reduced by anti-HIV transgenes that affect the production of lentivirus (LV) vectors. Thus FV vectors are very promising for anti-HIV gene therapy. This review covers the advantages of FV vectors and describes their preclinical development for anti-HIV gene therapy.
1. HIV and HAART: Limitations of the Current Standard of Care
HIV infection remains a global health crisis. There were an estimated 34 million cases worldwide in 2010 [1], and there is still no effective vaccine. The current standard of care for HIV infection is highly active antiretroviral therapy (HAART), a treatment strategy that uses cocktails of antiretroviral drugs to control HIV replication and to delay disease progression to AIDS. HAART has greatly improved outcomes for individuals living with HIV [2,3]. It efficiently inhibits viral replication and is highly effective at suppressing viral loads.
However, there are several limitations to HAART that make the development of novel therapies including gene therapy a high priority. So far, HAART therapy has not been able to target latent proviruses residing in long-lived cellular reservoirs [4]. These reservoirs appear to be stable [5], and they are thought to be the source of the low level viremia that is typically observed over the lifetime of an HIV patient on HAART [4,5]. Lack of adherence to HAART regimens and the possibility of pharmacological sanctuary sites [6,7,8] are also major concerns as they may accelerate the generation of drug resistant HIV mutants [7,9,10]. Because infectious viral particles generally persist at a baseline level in the blood under a HAART regimen [11,12], interruption of treatment leads to viral rebound [13]. HAART is a lifelong treatment and is associated with toxicity that may have significant effects on longevity and quality of life [14,15,16]. In addition, HAART is expensive. The annual cost for HAART therapy in the U.S was $13,000 per person per year for antiretrovirals alone, based on 2006 data [17]. Although HAART has served as the cornerstone of anti-HIV therapeutics for nearly two decades, it has limitations that justify the exploration of alternative treatments.
2. HIV Gene Therapy
Retroviral gene therapy is a potential alternative to HAART. Under this strategy, hematopoietic cells are harvested from a patient and gene-modified ex vivo by transduction with a retroviral vector. The transduced cells are then reintroduced to the patient’s body. Both hematopoietic stem cells (HSCs) and CD4 T cells have been explored as cell targets. A major advantage of HSC gene therapy is that HSCs produce all the mature cells that are infected by HIV including CD4 cells, macrophages and dendritic cells. HSCs carrying anti-HIV transgenes would persist over the lifetime of the patient, continually producing differentiated daughter cells that are protected against HIV infection. A major advantage of this approach is that it would be a one-time procedure and, thus, eliminate the need for patients to comply with complicated and expensive HAART treatment regimens.
Early trials have demonstrated the efficacy of anti-HIV transgenes delivered by retroviral gene therapy. However, use of these therapies has been complicated with low levels of gene marking [18,19]. It is clear that the efficiency of gene transfer of anti-HIV transgenes must be improved. Another challenge for gene therapy is safety. Following the development of leukemia in SCID-X1 patients who received HSC gene therapy [20,21,22,23], major efforts have gone into better understanding the risks of different vector systems and into improving the safety of retroviral vectors. Safe vector systems will be an important consideration to move HIV gene therapy to a front line treatment for HIV/AIDS.
4. Limitations of LV Vectors
Much of the recent focus in HIV gene therapy has been directed towards efforts utilizing LV vectors derived from HIV-1. These vectors are widely used in part because of their ability to efficiently transduce non-dividing cells. However, the use of LV vectors is complicated by the fact that HIV-1 based vectors have nucleotide sequences and also some proteins of the HIV virus itself. The titers of LV vectors can be severely suppressed by the expression of anti-HIV transgenes that target functions that are shared by LV vectors and HIV [26,27,28,29,30] (Figure 1). For in vitro studies using transformed cell lines as models for protection, high titer vector preparations are not needed to efficiently deliver anti-HIV transgenes. However, vector titer is a critically important consideration for clinical studies where low anti-HIV vector titers can severely reduce gene transfer efficiency to quiescent HSCs. While some investigators have been able to compensate for inhibited vector production on a case-by-case basis [26,28,29,30], the use of LV vectors for anti-HIV gene therapy can complicate vector design. It may even preclude the use of some anti-HIV transgenes, or some transgene combinations if they synergize to reduce anti-HIV LV vector titers. Because even SIN LV vectors have residual transcriptional activity from their LTRs [31], another potential problem with using LV vectors is that integrated proviruses could recombine with and/or be mobilized by HIV. So far, however, the risk appears to be small for SIN LV vectors [29]. One approach to address the problems with HIV-1-based vectors is the use of LV vectors that are not based on HIV-1. HIV-2-based vectors have been used in anti-HIV gene therapy, but much of the work has been done on vectors that are mobilized by HIV-1 [32,33]. Other LV vectors such as feline immunodeficiency virus (FIV) [34] and equine infectious anemia virus (EIAV) [35] vectors have also been developed. However, EIAV vectors do not transduce human HSCs as efficiently as second generation HIV-1-based vectors [36], and inefficient transgene expression from FIV vectors in human hematopoietic cells has been reported [37].
5. FV Vectors
FV vectors have several important advantages for HIV gene therapy. The FVs, or spumaviruses, are ancient retroviruses that have undergone extensive co-evolution with their natural hosts [38,39]. They are endemic in non-human primates (NHPs) and other mammals, but have not been detected in humans except in cases of benign zoonosis. These zoonotic infections are usually acquired through hunting or occupational exposure to NHPs [40,41]. FVs have not been observed to be transmitted between humans, and unlike the LVs, FVs do not cause disease in their hosts. FV vectors have a broad cell tropism [42]. In addition, methods for pseudotyping FV vectors have been described [43]. In terms of genome size, FVs are among the largest of the retroviruses [44] and FV vectors are capable of packaging large transgenes. In one study, a vector was generated with a 9.2 kb insert, bringing the total vector length close to the parent virus size. This vector could be produced at approximately one third the titer of a vector with a 2.4 kb insert [45]. FV vectors also have an attractive safety profile relative to GV vectors and LV vectors (See below). FVs are unusual among retroviruses in that reverse transcription frequently takes place in the cell producing virions, rather than prior to integration in the infected cell. As a consequence, unlike other retroviruses, many of the infectious particles of FVs contain dsDNA genomes [46]. While mitosis is required for FV vector transduction, FV vectors form a highly stable transduction intermediate in quiescent cells [47] and this may explain their efficient transduction of HSCs. Transduction efficiencies of FV vectors in HSCs are comparable to those of LV vectors [48]. A stable FV vector transduction intermediate may also explain why very short ex vivo transduction protocols can be used for gene delivery to HSCs in a large animal model [49]. This is important because protocols with extended ex vivo culture times reduce engraftment [50]. Advanced, third generation SIN vectors [45] (Figure 2) based on the prototypic FV and other FVs have shown great promise in preclinical studies.
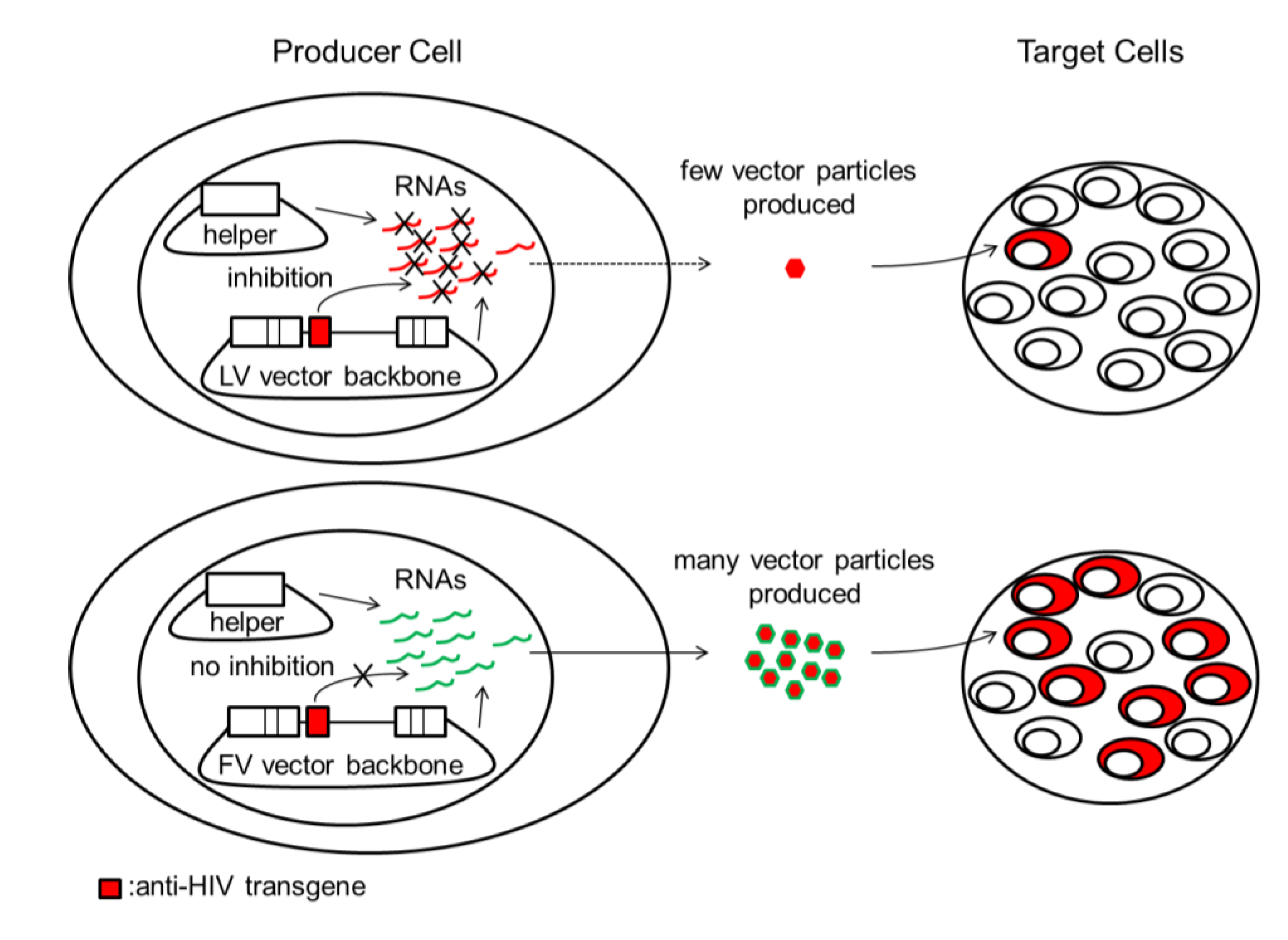
Figure 1.
Inhibition of LV vector production by anti-HIV transgenes can lead to low titers and poor transduction efficiency in target cells. HIV-based LV vectors and HIV share identical nucleotide sequences and proteins. During LV vector production, LV vector backbones and LV helper plasmids are cotransfected into producer cells to produce LV vector virions for infecting target cells. LV vector plasmids and/or LV helper plasmids and their respective RNAs can be targeted by some anti-HIV transgenes such as short hairpin (sh)RNAs (red box). This can result in a reduction in the number of vector particles produced, leading to inefficient transduction of target cells by low titer LV vector. FV vector plasmids, FV helper plasmids, and their respective RNAs are not affected because FV vectors do not share significant sequence identity with HIV. HIV/LV vector components and anti-HIV transgenes are indicated in red. FV vector components are indicated in green. LV, lentivirus; FV, foamy virus.
Figure 1.
Inhibition of LV vector production by anti-HIV transgenes can lead to low titers and poor transduction efficiency in target cells. HIV-based LV vectors and HIV share identical nucleotide sequences and proteins. During LV vector production, LV vector backbones and LV helper plasmids are cotransfected into producer cells to produce LV vector virions for infecting target cells. LV vector plasmids and/or LV helper plasmids and their respective RNAs can be targeted by some anti-HIV transgenes such as short hairpin (sh)RNAs (red box). This can result in a reduction in the number of vector particles produced, leading to inefficient transduction of target cells by low titer LV vector. FV vector plasmids, FV helper plasmids, and their respective RNAs are not affected because FV vectors do not share significant sequence identity with HIV. HIV/LV vector components and anti-HIV transgenes are indicated in red. FV vector components are indicated in green. LV, lentivirus; FV, foamy virus.
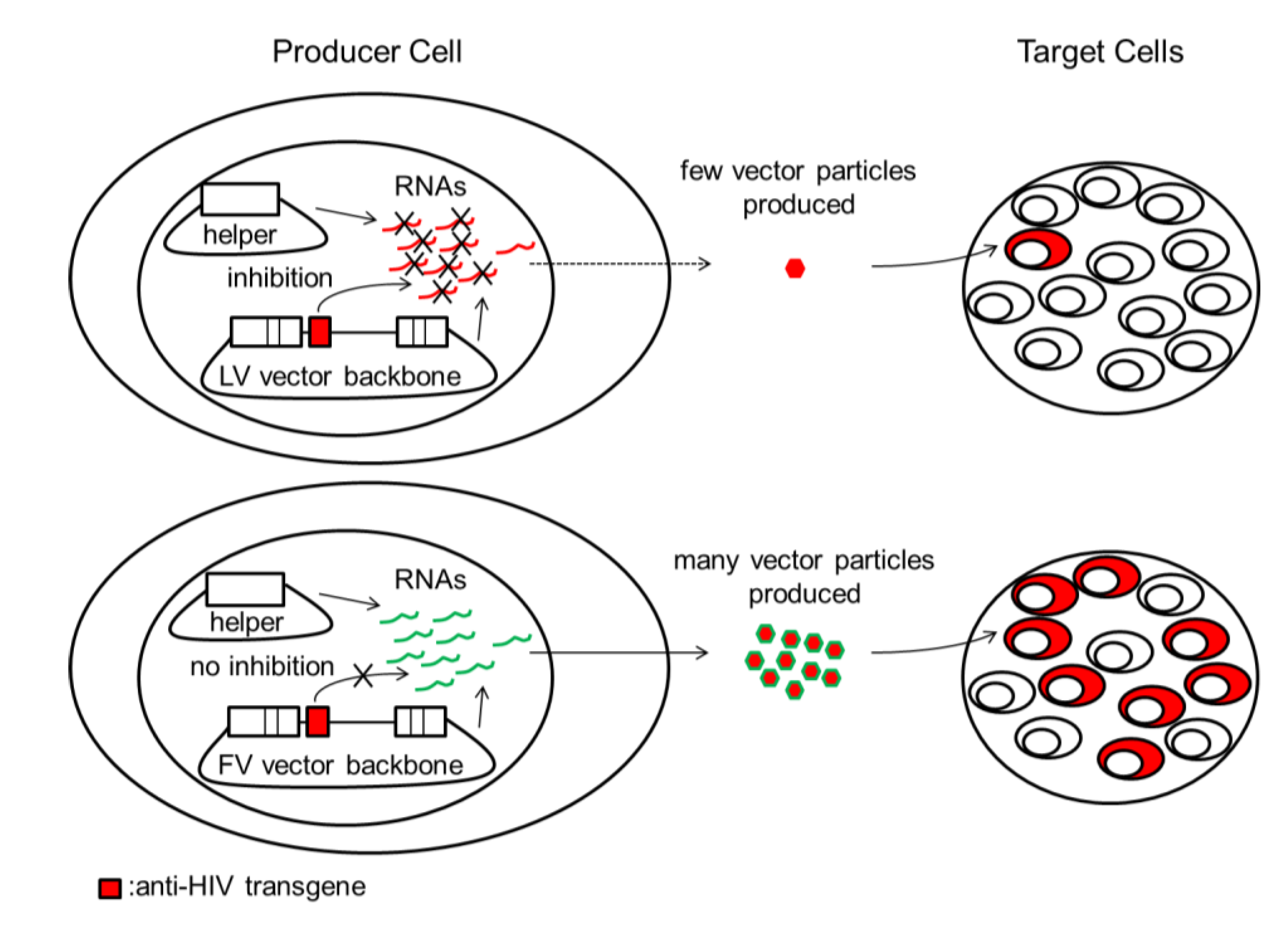
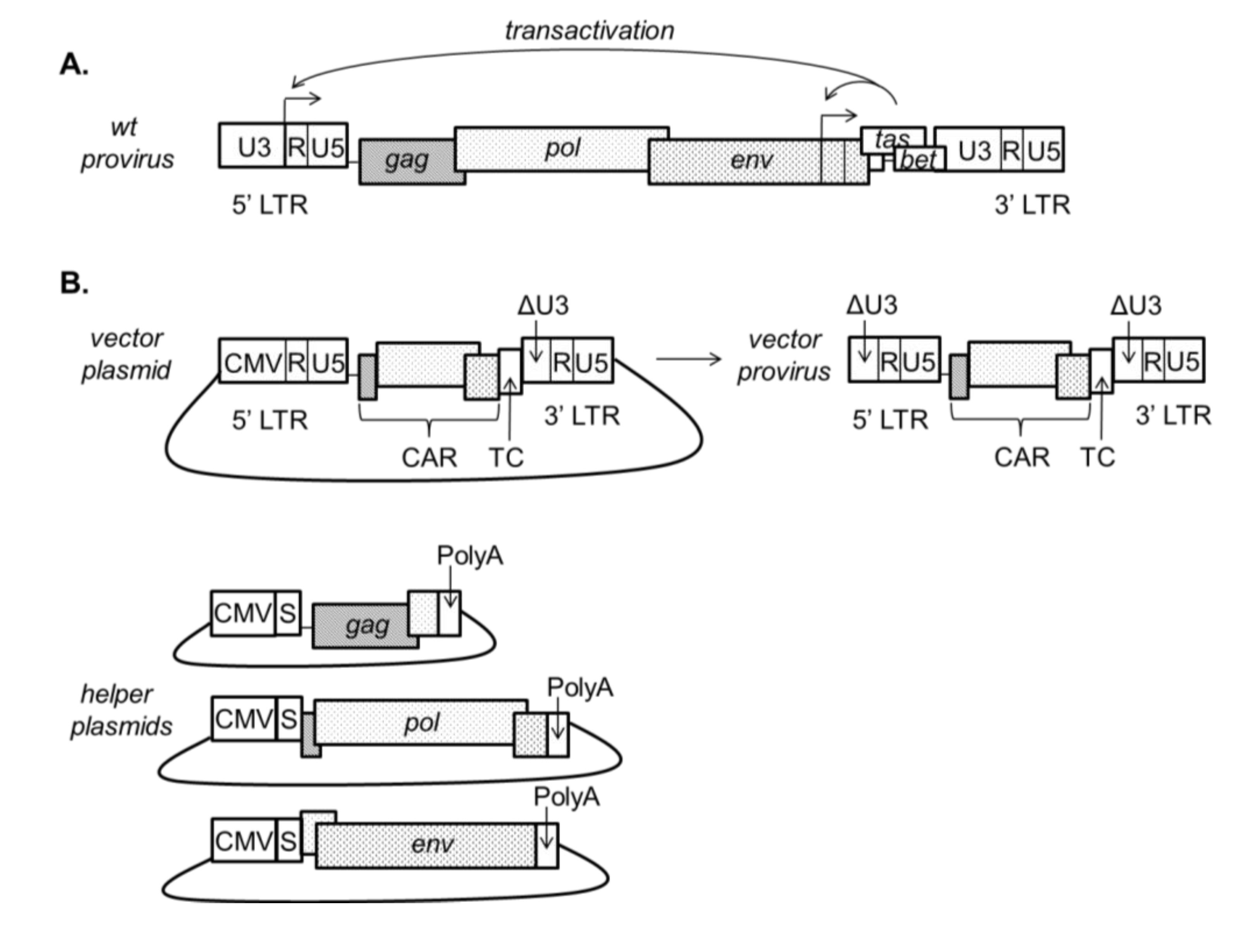
Figure 2.
(A) Wild type FV provirus; (B) A third generation minimal FV vector backbone and helper plasmids. The vector is shown as it would appear in a plasmid for vector production and as an integrated provirus. Third generation FV vectors include a deletion of the transcriptional transactivator, tas (Previously known as bel-1), which acts at the viral LTR in wild type FV. These vectors are SIN due to the removal of the transactivator tas and have a deletion encompassing the TATA box and enhancers in the U3 region of the 3' LTR of the vector plasmid (ΔU3 in figure). This deletion is copied to the 5' end of the viral genome during reverse transcription, resulting in the silencing of both LTRs in the integrated provirus. In contrast to LV vector systems, gag and pol in FV vector systems are translated from separate mRNAs, and gag, pol and env genes are provided in trans on three separate helper plasmids. Cis-acting regions (CAR) remain on the vector backbone. Abbreviations: CAR, cis-acting region; CMV, cytomegalovirus promoter; LTR, long terminal repeat; S, SV40 intron; TC, transgene cassette; Poly A, poly adenylation site.
Figure 2.
(A) Wild type FV provirus; (B) A third generation minimal FV vector backbone and helper plasmids. The vector is shown as it would appear in a plasmid for vector production and as an integrated provirus. Third generation FV vectors include a deletion of the transcriptional transactivator, tas (Previously known as bel-1), which acts at the viral LTR in wild type FV. These vectors are SIN due to the removal of the transactivator tas and have a deletion encompassing the TATA box and enhancers in the U3 region of the 3' LTR of the vector plasmid (ΔU3 in figure). This deletion is copied to the 5' end of the viral genome during reverse transcription, resulting in the silencing of both LTRs in the integrated provirus. In contrast to LV vector systems, gag and pol in FV vector systems are translated from separate mRNAs, and gag, pol and env genes are provided in trans on three separate helper plasmids. Cis-acting regions (CAR) remain on the vector backbone. Abbreviations: CAR, cis-acting region; CMV, cytomegalovirus promoter; LTR, long terminal repeat; S, SV40 intron; TC, transgene cassette; Poly A, poly adenylation site.
6. Vector Genotoxicity
Integrating retroviral vectors are insertional mutagens that modify the genome. Hence, they pose a risk of oncogenesis when used for gene therapy. Clonal expansion and leukemia have occurred in gene therapy clinical trials as a result of vector-mediated dysregulation of nearby genes [20,21,22,23,51,52]. As a result, improving vector safety is a major priority for the field of gene therapy. Integrating vectors differ in their preferences for insertion sites within the host genome [53]. They also differ in their likelihood to dysregulate nearby genes [54]. Together, these factors influence vector genotoxicity.
6.2. Dysregulation of Neighboring Genes
Vector-mediated dysregulation of neighboring genes occurs by several different mechanisms including enhancer-mediated activation, truncation of cellular transcripts, and read through transcription. For a review see [60]. While SIN LV vectors appear less likely than GV vectors to dysregulate nearby genes through enhancer-mediated activation, there is evidence that LV vector proviruses allow significant read-through transcription and generate chimeric transcripts [54,61]. This can contribute to clonal expansion and oncogenic potential [62]. FV vectors are more resistant to read-through transcription than LV and GV vectors, presumably in part due to efficient polyadenylation although other factors may be involved [54]. This can reduce the potential to activate nearby genes and is an important safety advantage for FV vectors.
6.3. Vector Design to Reduce Genotoxicity
The use of insulator elements [63] and elimination of potential splice sites can improve safety. In addition, efficient polyadenylation signals in vector LTRs [64] can reduce the potential for genotoxicity. The use of weaker housekeeping promoters, such as the elongation factor 1α promoter, rather than strong viral promoters such as the spleen focus forming virus (SFFV) promoter to drive the transgene cassette can decrease the probability of vector-mediated dysregulation of neighboring genes [65]. However, some vector modifications may reduce efficacy. For example, using weaker promoters can result in reduced transgene expression. Thus, efforts to improve safety must be balanced with the need for clinical efficacy.
7. FV Vector HSC Gene Therapy Models
Encouraging data obtained from preclinical studies with FV vectors have increased interest in FV vectors for HSC gene therapy. FV vectors have been investigated in several animal models, notably the non-obese diabetic/severe combined immunodeficiency (NOD-SCID) and NOD-SCID IL2Rγnull (NSG) mouse, and also the dog large animal model. Xenotransplantation of FV transduced human CD34 cells has been demonstrated in immunodeficient mouse strains such as the NOD-SCID and NSG models [27,66,67]. Gene marking has been observed in multiple hematopoietic lineages, indicating the potential of FV anti-HIV vectors to protect mature cells in the myeloid and lymphoid lineages from HIV infection. While mouse models have provided important preclinical data for the use of FV vectors for HSC gene therapy, the short lifespans of mice and differences in HSC characteristics in mice and primates impose some limitations [68,69]. Large animal models have better predicted clinical efficacy of HSC gene therapies and also allow for studies of long term repopulating cells. The dog large animal model has several advantages. Dogs are easily cared for, they reproduce quickly, their HSC physiology is similar to that of humans [70], and they can be used to model several human hematopoietic diseases [71,72,73]. FV vectors efficiently transduce canine long-term repopulating HSCs [49]. In a direct comparison with LV vectors, FV vectors transduced canine long term repopulating HSCs at similar efficiencies to LV vectors [48]. Canine leukocyte adhesion deficiency (CLAD) [74] and pyruvate kinase deficiency [75] have been corrected in dogs using FV vector HSC gene therapy. CLAD dogs receiving FV vector HSC gene therapy did not develop leukemia as a result of vector mediated oncogenesis in the years following infusion [76]. This large animal data strongly supports the safety of FV vector gene therapy.
8. FV Vector Anti-HIV Studies
A number of anti-HIV transgenes and transgene combinations have been explored for use in FV vector-mediated anti-HIV gene therapy [27,77,78,79]. These are summarized in Table 1.
8.1. In Vitro Studies
An FV vector with a single shRNA targeting viral rev/env was used to inhibit simian immunodeficiency virus, a close relative of HIV, in in-vitro challenge assays. Results were encouraging, with inhibition of viral replication reaching 68%–80% [77]. However, single RNAi therapies against HIV are of limited use due to the ability of HIV to escape inhibition through mutation [80]. RNAi therapies are less vulnerable to viral escape when two or more sequences are expressed in combination, or when RNAi is expressed together with other classes of anti-HIV transgenes. Park et al. reported [78] using FV vectors to deliver anti-HIV transgene cassettes under the control of either a cytomegalovirus (CMV) promoter or a minimal heat shock promoter (Hsp), which is activated in the presence of HIV Tat. Transgenes under the control of the Hsp were expressed conditionally in the presence of HIV Tat through interaction of Hsp with TAR recruited proteins at an upstream partial HIV LTR, while transgenes under the control of the CMV promoter were constitutively expressed. An anti-HIV miRNA cassette targeting HIV Rev (R) and the HIV LTR (L2) was highly effective under the control of either promoter, inhibiting HIV replication by >98% in a challenge assay. The investigators also tested the effectiveness of an anti-rev miRNA cassette under the control of Hsp, as well as the antiviral activity of the TAR expressed without any additional transgenes. The anti-rev miRNA cassette under the control of Hsp was found to inhibit HIV replication by >98% when challenged with HIV. The LTR expressing only TAR inhibited HIV replication to the same degree. The authors speculated that a TAR miRNA processed from the vector might have been responsible for the inhibitory effect.

{kind=link}
{kind=link}
Table 1.
Anti-HIV Transgenes in FV Vectors. C46: membrane associated HIV fusion inhibitor; CMV: cytomegalovirus immediate early promoter; LTR: long terminal repeat; H1: human H1 RNA promoter; Hsp: heat shock promoter; L2R: LTR + rev miRNA; MSCV: murine stem cell virus promoter; PGK: phosphoglycerate kinase promoter; R2: SIV rev shRNA; R5: CCR5 shRNA; RevM10: dominant negative Rev; SFFV: spleen focus forming virus promoter; Sh1: anti tat/rev shRNA; SHIV: simian-human immunodeficiency virus; SI: tat/rev shRNA; SII: tat/rev shRNA; SIV: simian immunodeficiency virus; TAR: HIV trans-activation response element; U6: human U6 small nuclear RNA Pol III promoter.
| Transgene | Description | Efficacy | Promoter | Assay | Publication |
|---|---|---|---|---|---|
| R2 | SIV rev + env shRNA | 68%–80% inhibition of viral replication | U6 | SIV challenge, CEMx174 cell line | Park et al. 2005 [77] |
| L2R | HIV LTR + rev miRNA cassette | >98% inhibition of viral replication | CMV | HIV challenge, U87.CD4.CXCR4 cell line | Park et al. 2009 [78] |
| TAR + L2R | Tat inducible HIV LTR + rev miRNA cassette + TAR | >98% inhibition of viral replication | Tat inducible LTR-Hsp fusion | ||
| TAR + R | Tat inducible rev miRNA cassette + TAR | >98% inhibition of viral replication | Tat-inducible LTR-Hsp fusion | ||
| TAR | TAR | >98% inhibition of viral replication | LTR | ||
| Sh1 | anti tat/rev shRNA | 4 log reduction of viral replication | U6 | HIV challenge, CD34-derived macrophages | Taylor et al. 2008 [79] |
| C46 | membrane associated fusion inhibitor | 4 log reduction of viral replication | MSCV | ||
| Sh1 + C46 + RevM10 | tat/rev shRNA + membrane-associated fusion inhibitor + dominant negative Rev | significantly increased relative to C46 alone | U6, MSCV, PGK | HIV challenge of protected and unprotected cells in CEMx174 cell line | |
| C46 | membrane associated fusion inhibitor | 5.2-fold increase in cell survival +3.1-fold decrease in HIV p24/cell | MSCV | ||
| 4 log reduction of viral replication | SFFV | SHIV challenge, CEM.NKR-CCR5 lymphocytes | Kiem et al. 2010 [27] | ||
| 15–20 fold reduction of viral replication | SFFV | SHIV or HIV single viral cycle challenge, MAGI-CCR5 cell line | |||
| SI + C46 | tat/rev shRNA + membrane associated fusion inhibitor | 5 fold reduction of viral replication | U6, SFFV | ||
| SII + SI + R5 + C46 | two tat/rev shRNAs + CCR5 shRNA + membrane associated fusion inhibitor | 23 fold reduction of viral replication | H1, SFFV | ||
| 4 log reduction of viral replication | SHIV challenge, CEM.NKR-CCR5 lymphocytes | ||||
| SI + C46 | tat/rev shRNA + C46 | 4 log reduction of viral replication | U6, SFFV | ||
| SII + SI + R5 | two tat/rev shRNAs + CCR5 shRNA | 180 fold reduction of viral replication | H1 |
Taylor et al. investigated FV vectors with an anti-tat/rev shRNA, a dominant negative mutant of HIV rev, and a membrane-associated HIV fusion inhibitor (C46) [79]. Both the anti-tat/rev shRNA and C46 potently inhibited viral replication in CD34-derived macrophages. When a challenge was performed on a mixture of gene-modified and also unprotected CEMx174 cells, improved survival and reduction of viral replication was observed. A vector expressing all three anti-HIV transgenes offered significantly better protection than C46 expressed alone. Finally, the relative effectiveness of each transgene and also a combination of all three transgenes was compared in a competitive challenge assay. Cells were transduced with FV vectors expressing each transgene individually, or all three. These FV-transduced cells were then combined in equal proportion and challenged with HIV. The ratio of cells expressing C46 or all three transgenes increased with time relative to cells expressing the rev mutant or the shRNA. This again suggested that C46 and the triple combination cassette were more effective at inhibiting HIV infection than the mutant rev or the shRNA expressed alone [79].
8.2. In Vivo Selection of Human SCID Repopulating Cells
The P140K mutant of the methylguanine methyltransferase gene (MGMTP140K) can be included in retroviral vectors to allow selection of transduced cells in vivo. The gene product of wild-type MGMT repairs alkylated guanine bases that are induced by chemotherapy drugs such as bis-chloroethyl nitrosourea (BCNU). Wild-type MGMT can be deactivated by guanine analogs such as O6-benzylguanine (O6BG) [81]; however, MGMTP140K is highly resistant to deactivation by this compound [82]. Administration of O6BG and BCNU to an animal engrafted with hematopoietic cells transduced with a vector expressing MGMTP140K kills unprotected (untransduced) cells. The result is the enrichment of cells that express MGMTP140K and the anti-HIV cassette. Importantly, this type of selection allows expansion of long term repopulating hematopoietic cells in vivo [83].
A study in the NSG mouse model demonstrated efficient engraftment and expansion of human CD34 cells transduced with an FV vector that incorporated an anti-HIV transgene cassette and MGMTP140K [27]. The anti-HIV cassette encoded a combination of C46, two shRNAs against HIV-1 rev and tat, and a shRNA against CCR5, a macrophage-tropic HIV-1 coreceptor. Of several transgene cassettes tested in a single cycle in vitro assay, this combination was found to most potently inhibit both HIV and simian-human immunodeficiency virus (SHIV), a chimeric human-simian immunodeficiency virus used to model HIV infection in primates. The inclusion of the MGMTP140K transgene allowed for in vivo selection of gene modified cells using O6BG and BCNU. This allowed for a significant increase in the percentage of marked cells in the bone marrow of NSG mice. Importantly, the vector used in this study could be produced at a titer of 3.8 × 107 transducing units∙mL−1, sufficient for clinical studies.
8.3. Anti-HIV shRNAs Inhibit LV but Not FV Vector Production
The titers of LV and FV vectors expressing the same anti-HIV transgene cassette that included a tat/rev shRNA have also been directly compared [27]. The FV vector titer was not affected compared to a control vector, however a LV vector was reduced in titer 150-fold. This is consistent with observations that transgenes targeting rev or its gene product reduce LV vector titers by inhibiting expression of Rev from LV helper plasmids or LV vector backbones during vector production [28,29].
9. Conclusions
FV vectors offer several important advantages over other retroviral vectors for HIV gene therapy. They can package large transgene cassettes and they have a desirable safety profile. Although FV vectors are potentially suited for broad use in HSC gene therapy, they may prove particularly useful in the treatment of HIV/AIDS. Lower sequence and functional homology between FV vectors and HIV reduces the probability of recombination with and/or mobilization by endogenous HIV, and FV vectors avoid the reduced titers observed in LV vectors carrying some anti-HIV transgenes. FV vector platforms may therefore better avoid complications in vector design and more efficiently produce safer, high titer anti-HIV-transgene containing vector than LV vector systems. FV anti-HIV vectors have been developed by several groups and have shown great promise in preclinical studies. Additional studies to improve FV vector safety by modifying vector components, and continued development of FV vectors with potent anti-HIV transgene combinations should lead to FV vectors with excellent clinical potential.
Acknowledgments
This work was supported in part by Grant Numbers AI097100, AI102672 from the National Institutes of Health, Bethesda, MD, USA.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO, Global HIV/AIDS Response. Epidemic Update and Health Sector Progress towards Universal Access Progress Report 2011; WHO Press: Geneva, Switzerland, 2011.
- Ray, M.; Logan, R.; Sterne, J.A.; Hernandez-Diaz, S.; Robins, J.M.; Sabin, C.; Bansi, L.; van Sighem, A.; de Wolf, F.; Costagliola, D.; et al. The effect of combined antiretroviral therapy on the overall mortality of hiv-infected individuals. AIDS 2010, 24, 123–137. [Google Scholar] [CrossRef]
- Altmann, M.; An der Heiden, M.; Scheufele, R.; Hartmann, K.; Houareau, C.; Bartmeyer, B.; Hamouda, O.; German, H.I.V.S.C. The risk of aids-defining events is decreasing over time in the german HIV-1 seroconverter cohort. BMC Infect. Dis. 2012, 12, e94. [Google Scholar]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef]
- Valentin, A.; Morrow, M.; Poirier, R.H.; Aleman, K.; Little, R.; Yarchoan, R.; Pavlakis, G.N. Identification of a potential pharmacological sanctuary for HIV Type 1 in a fraction of CD4(+) primary cells. AIDS Res. Hum. Retroviruses 2010, 26, 79–88. [Google Scholar] [CrossRef]
- Delobel, P.; Sandres-Saune, K.; Cazabat, M.; L’Faqihi, F.E.; Aquilina, C.; Obadia, M.; Pasquier, C.; Marchou, B.; Massip, P.; Izopet, J. Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. AIDS 2005, 19, 1739–1750. [Google Scholar] [CrossRef]
- Yukl, S.A.; Shergill, A.K.; McQuaid, K.; Gianella, S.; Lampiris, H.; Hare, C.B.; Pandori, M.; Sinclair, E.; Gunthard, H.F.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef]
- Lambotte, O.; Chaix, M.L.; Gubler, B.; Nasreddine, N.; Wallon, C.; Goujard, C.; Rouzioux, C.; Taoufik, Y.; Delfraissy, J.F. The lymphocyte HIV reservoir in patients on long-term HAART is a memory of virus evolution. AIDS 2004, 18, 1147–1158. [Google Scholar] [CrossRef]
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Richman, D.D. Update of the drug resistance mutations in HIV-1: March 2013. Top. Antivir. Med. 2013, 21, 6–14. [Google Scholar]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef]
- Maldarelli, F.; Palmer, S.; King, M.S.; Wiegand, A.; Polis, M.A.; Mican, J.; Kovacs, J.A.; Davey, R.T.; Rock-Kress, D.; Dewar, R.; et al. Art suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007, 3, e46. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875. [Google Scholar] [CrossRef]
- Smith, R.L.; de Boer, R.; Brul, S.; Budovskaya, Y.; van Spek, H. Premature and accelerated aging: HIV or HAART? Front. Genet. 2012, 3, e328. [Google Scholar]
- Pinti, M.; Salomoni, P.; Cossarizza, A. Anti-HIV drugs and the mitochondria. Biochim. Biophys. Acta 2006, 1757, 700–707. [Google Scholar]
- Fernandez-Fernandez, B.; Montoya-Ferrer, A.; Sanz, A.B.; Sanchez-Nino, M.D.; Izquierdo, M.C.; Poveda, J.; Sainz-Prestel, V.; Ortiz-Martin, N.; Parra-Rodriguez, A.; Selgas, R.; et al. Tenofovir nephrotoxicity: 2011 update. AIDS Res. Treat. 2011, 2011, e354908. [Google Scholar]
- Gebo, K.A.; Fleishman, J.A.; Conviser, R.; Hellinger, J.; Hellinger, F.J.; Josephs, J.S.; Keiser, P.; Gaist, P.; Moore, R.D.; Network, H.I.V.R. Contemporary costs of HIV healthcare in the HAART era. AIDS 2010, 24, 2705–2715. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Zack, J.A.; Macpherson, J.L.; Symonds, G.P. Phase I/II clinical trials using gene-modified adult hematopoietic stem cells for HIV: Lessons learnt. Stem Cells Int. 2011, 2011, e393698. [Google Scholar]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Yu, S.F.; von Ruden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Ruther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3194–3198. [Google Scholar] [CrossRef]
- Bokhoven, M.; Stephen, S.L.; Knight, S.; Gevers, E.F.; Robinson, I.C.; Takeuchi, Y.; Collins, M.K. Insertional gene activation by lentiviral and gammaretroviral vectors. J. Virol. 2009, 83, 283–294. [Google Scholar] [CrossRef]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-ccr5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef]
- Liu, Y.P.; Vink, M.A.; Westerink, J.T.; Ramirez de Arellano, E.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010, 16, 1328–1339. [Google Scholar] [CrossRef]
- Bahner, I.; Sumiyoshi, T.; Kagoda, M.; Swartout, R.; Peterson, D.; Pepper, K.; Dorey, F.; Reiser, J.; Kohn, D.B. Lentiviral vector transduction of a dominant-negative rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol. Ther. 2007, 15, 76–85. [Google Scholar] [CrossRef]
- Mautino, M.R.; Morgan, R.A. Potent inhibition of human immunodeficiency virus type 1 replication by conditionally replicating human immunodeficiency virus-based lentiviral vectors expressing envelope antisense mrna. Hum. Gene Ther. 2000, 11, 2025–2037. [Google Scholar] [CrossRef]
- Logan, A.C.; Haas, D.L.; Kafri, T.; Kohn, D.B. Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. J. Virol. 2004, 78, 8421–8436. [Google Scholar] [CrossRef]
- Morris, K.V.; Looney, D.J. Characterization of human immunodeficiency virus (HIV)-2 vector mobilization by HIV-1. Hum. Gene Ther. 2005, 16, 1463–1472. [Google Scholar] [CrossRef]
- Turner, A.M.; de La Cruz, J.; Morris, K.V. Mobilization-competent lentiviral vector-mediated sustained transcriptional modulation of HIV-1 expression. Mol. Ther. 2009, 17, 360–368. [Google Scholar] [CrossRef]
- Poeschla, E.M.; Wong-Staal, F.; Looney, D.J. Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nat. Med. 1998, 4, 354–357. [Google Scholar] [CrossRef]
- Mitrophanous, K.; Yoon, S.; Rohll, J.; Patil, D.; Wilkes, F.; Kim, V.; Kingsman, S.; Kingsman, A.; Mazarakis, N. Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther. 1999, 6, 1808–1818. [Google Scholar] [CrossRef]
- Siapati, E.K.; Bigger, B.W.; Miskin, J.; Chipchase, D.; Parsley, K.L.; Mitrophanous, K.; Themis, M.; Thrasher, A.J.; Bonnet, D. Comparison of HIV- and EIAV-based vectors on their efficiency in transducing murine and human hematopoietic repopulating cells. Mol. Ther. 2005, 12, 537–546. [Google Scholar] [CrossRef]
- Price, M.A.; Case, S.S.; Carbonaro, D.A.; Yu, X.J.; Petersen, D.; Sabo, K.M.; Curran, M.A.; Engel, B.C.; Margarian, H.; Abkowitz, J.L.; et al. Expression from second-generation feline immunodeficiency virus vectors is impaired in human hematopoietic cells. Mol. Ther. 2002, 6, 645–652. [Google Scholar] [CrossRef]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathog. 2012, 8, e1002790. [Google Scholar] [CrossRef]
- Betsem, E.; Rua, R.; Tortevoye, P.; Froment, A.; Gessain, A. Frequent and recent human acquisition of simian foamy viruses through apes’ bites in central Africa. PLoS Pathog. 2011, 7, e1002306. [Google Scholar] [CrossRef]
- Switzer, W.M.; Bhullar, V.; Shanmugam, V.; Cong, M.E.; Parekh, B.; Lerche, N.W.; Yee, J.L.; Ely, J.J.; Boneva, R.; Chapman, L.E.; et al. Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J. Virol. 2004, 78, 2780–2789. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar]
- Ho, Y.P.; Schnabel, V.; Swiersy, A.; Stirnnagel, K.; Lindemann, D. A small-molecule-controlled system for efficient pseudotyping of prototype foamy virus vectors. Mol. Ther. 2012, 20, 1167–1176. [Google Scholar] [CrossRef]
- Flugel, R.M. Spumaviruses: A group of complex retroviruses. J. Acquir. Immune Defic. Syndr. 1991, 4, 739–750. [Google Scholar]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Allen, J.; Peterson, L.; Ironside, C.; Russell, D.W.; Kiem, H.P. Foamy and lentiviral vectors transduce canine long-term repopulating cells at similar efficiency. Hum. Gene Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy-virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar] [CrossRef]
- Tisdale, J.F.; Hanazono, Y.; Sellers, S.E.; Agricola, B.A.; Metzger, M.E.; Donahue, R.E.; Dunbar, C.E. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood 1998, 92, 1131–1141. [Google Scholar]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to evi1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. Bet proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Gijsbers, R.; Vets, S.; de Rijck, J.; Ocwieja, K.E.; Ronen, K.; Malani, N.; Bushman, F.D.; Debyser, Z. Role of the PWWP domain of lens epithelium-derived growth factor (LEDGF)/p75 cofactor in lentiviral integration targeting. J. Biol. Chem. 2011, 286, 41812–41825. [Google Scholar] [CrossRef]
- Vets, S.; de Rijck, J.; Brendel, C.; Grez, M.; Bushman, F.; Debyser, Z.; Gijsbers, R. Transient expression of an LEDGF/p75 chimera retargets lentivector integration and functionally rescues in a model for X-CGD. Mol. Ther. 2013, 2, e77. [Google Scholar]
- Rae, D.T.; Trobridge, G.D. Retroviral Genotoxicity. In Gene therapy—Tools and Potential Applications; Martin, F., Ed.; InTech: Rijeka, Croatia, 2013; pp. 399–427. [Google Scholar]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Invest. 2012, 122, 1667–1676. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Maroney, P.A.; Goodwin, R.G.; Rottman, F.M.; Crittenden, L.B.; Raines, M.A.; Kung, H.J. C-erbB activation in ALV-induced erythroblastosis: Novel RNA processing and promoter insertion result in expression of an amino-truncated EGF receptor. Cell 1985, 41, 719–726. [Google Scholar] [CrossRef]
- Li, C.L.; Xiong, D.; Stamatoyannopoulos, G.; Emery, D.W. Genomic and functional assays demonstrate reduced gammaretroviral vector genotoxicity associated with use of the CHS4 chromatin insulator. Mol. Ther. 2009, 17, 716–724. [Google Scholar] [CrossRef]
- Schambach, A.; Galla, M.; Maetzig, T.; Loew, R.; Baum, C. Improving transcriptional termination of self-inactivating gamma-retroviral and lentiviral vectors. Mol. Ther. 2007, 15, 1167–1173. [Google Scholar]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef]
- Josephson, N.C.; Vassilopoulos, G.; Trobridge, G.D.; Priestley, G.V.; Wood, B.L.; Papayannopoulou, T.; Russell, D.W. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc. Natl. Acad. Sci. USA 2002, 99, 8295–8300. [Google Scholar] [CrossRef]
- Zucali, J.R.; Ciccarone, T.; Kelley, V.; Park, J.; Johnson, C.M.; Mergia, A. Transduction of umbilical cord blood CD34+ NOD/SCID-repopulating cells by simian foamy virus type 1 (SFV-1) vector. Virology 2002, 302, 229–235. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Kiem, H.P. Large animal models of hematopoietic stem cell gene therapy. Gene Ther. 2010, 17, 939–948. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Horn, P.A.; Beard, B.C.; Kiem, H.P. Large animal models for foamy virus vector gene therapy. Viruses 2012, 4, 3572–3588. [Google Scholar] [CrossRef]
- Suter, S.E.; Gouthro, T.A.; McSweeney, P.A.; Nash, R.A.; Haskins, M.E.; Felsburg, P.J.; Henthorn, P.S. Isolation and characterization of pediatric canine bone marrow CD34+ cells. Vet. Immunol. Immunopathol. 2004, 101, 31–47. [Google Scholar] [CrossRef]
- Creevy, K.E.; Bauer, T.R., Jr.; Tuschong, L.M.; Embree, L.J.; Silverstone, A.M.; Bacher, J.D.; Romines, C.; Garnier, J.; Thomas, M.L. Mixed chimeric hematopoietic stem cell transplant reverses the disease phenotype in canine leukocyte adhesion deficiency. Vet. Immunol. Immunopathol. 2003, 95, 113–121. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Gu, Y.C.; Tuschong, L.M.; Burkholder, T.; Bacher, J.D.; Starost, M.F.; Donahue, R.E.; Sokolic, R.A.; Hickstein, D.D. Nonmyeloablative hematopoietic stem cell transplantation corrects the disease phenotype in the canine model of leukocyte adhesion deficiency. Exp. Hematol. 2005, 33, 706–712. [Google Scholar] [CrossRef]
- Zaucha, J.A.; Yu, C.; Lothrop, C.D., Jr.; Nash, R.A.; Sale, G.; Georges, G.; Kiem, H.P.; Niemeyer, G.P.; Dufresne, M.; Cao, Q.; et al. Severe canine hereditary hemolytic anemia treated by nonmyeloablative marrow transplantation. Biol. Blood Marrow Transplant. 2001, 7, 14–24. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PLoS One 2012, 7, e45173. [Google Scholar]
- Bauer, T.R., Jr.; Tuschong, L.M.; Calvo, K.R.; Shive, H.R.; Burkholder, T.H.; Karlsson, E.K.; West, R.R.; Russell, D.W.; Hickstein, D.D. Long-term follow-up of foamy viral vector-mediated gene therapy for canine leukocyte adhesion deficiency. Mol. Ther. 2013, 21, 964–972. [Google Scholar] [CrossRef]
- Park, J.; Nadeau, P.; Zucali, J.R.; Johnson, C.M.; Mergia, A. Inhibition of simian immunodeficiency virus by foamy virus vectors expressing sirnas. Virology 2005, 343, 275–282. [Google Scholar] [CrossRef]
- Park, J.; Nadeau, P.E.; Mergia, A. Activity of TAR in inducible inhibition of HIV replication by foamy virus vector expressing siRNAs under the control of HIV LTR. Virus Res. 2009, 140, 112–120. [Google Scholar] [CrossRef]
- Taylor, J.A.; Vojtech, L.; Bahner, I.; Kohn, D.B.; Laer, D.V.; Russell, D.W.; Richard, R.E. Foamy virus vectors expressing anti-HIV transgenes efficiently block HIV-1 replication. Mol. Ther. 2008, 16, 46–51. [Google Scholar] [CrossRef]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef]
- Xu-Welliver, M.; Kanugula, S.; Pegg, A.E. Isolation of human O6-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by O6-benzylguanine. Cancer Res. 1998, 58, 1936–1945. [Google Scholar]
- Beard, B.C.; Trobridge, G.D.; Ironside, C.; McCune, J.S.; Adair, J.E.; Kiem, H.P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Invest. 2010, 120, 2345–2354. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Olszko, M.E.; Trobridge, G.D. Foamy Virus Vectors for HIV Gene Therapy. Viruses 2013, 5, 2585-2600. https://doi.org/10.3390/v5102585
AMA Style
Olszko ME, Trobridge GD. Foamy Virus Vectors for HIV Gene Therapy. Viruses. 2013; 5(10):2585-2600. https://doi.org/10.3390/v5102585
Chicago/Turabian StyleOlszko, Miles E., and Grant D. Trobridge. 2013. "Foamy Virus Vectors for HIV Gene Therapy" Viruses 5, no. 10: 2585-2600. https://doi.org/10.3390/v5102585