Innate and Adaptive Immune Response to Pneumonia Virus of Mice in a Resistant and a Susceptible Mouse Strain

Abstract
:1. Introduction
2. Results and Discussion
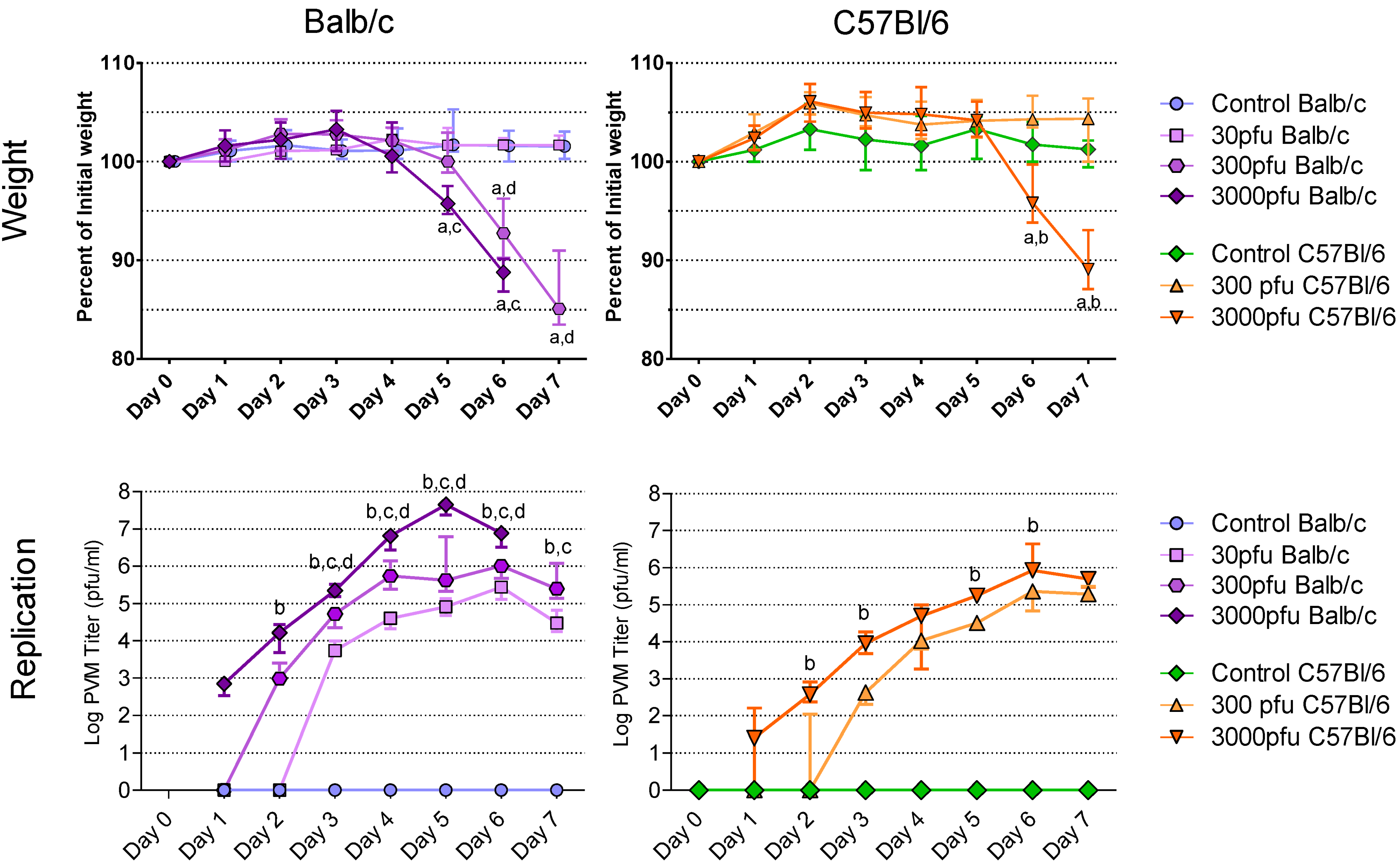
2.1. Balb/c Mice Show Earlier and More Weight Loss, and Enhanced Virus Replication in the Lungs after PVM 15 Infection in Comparison with C57Bl/6 Mice

2.2. Comparison of Lung Pathology in PVM-Infected Balb/c and C57Bl/6 Mice

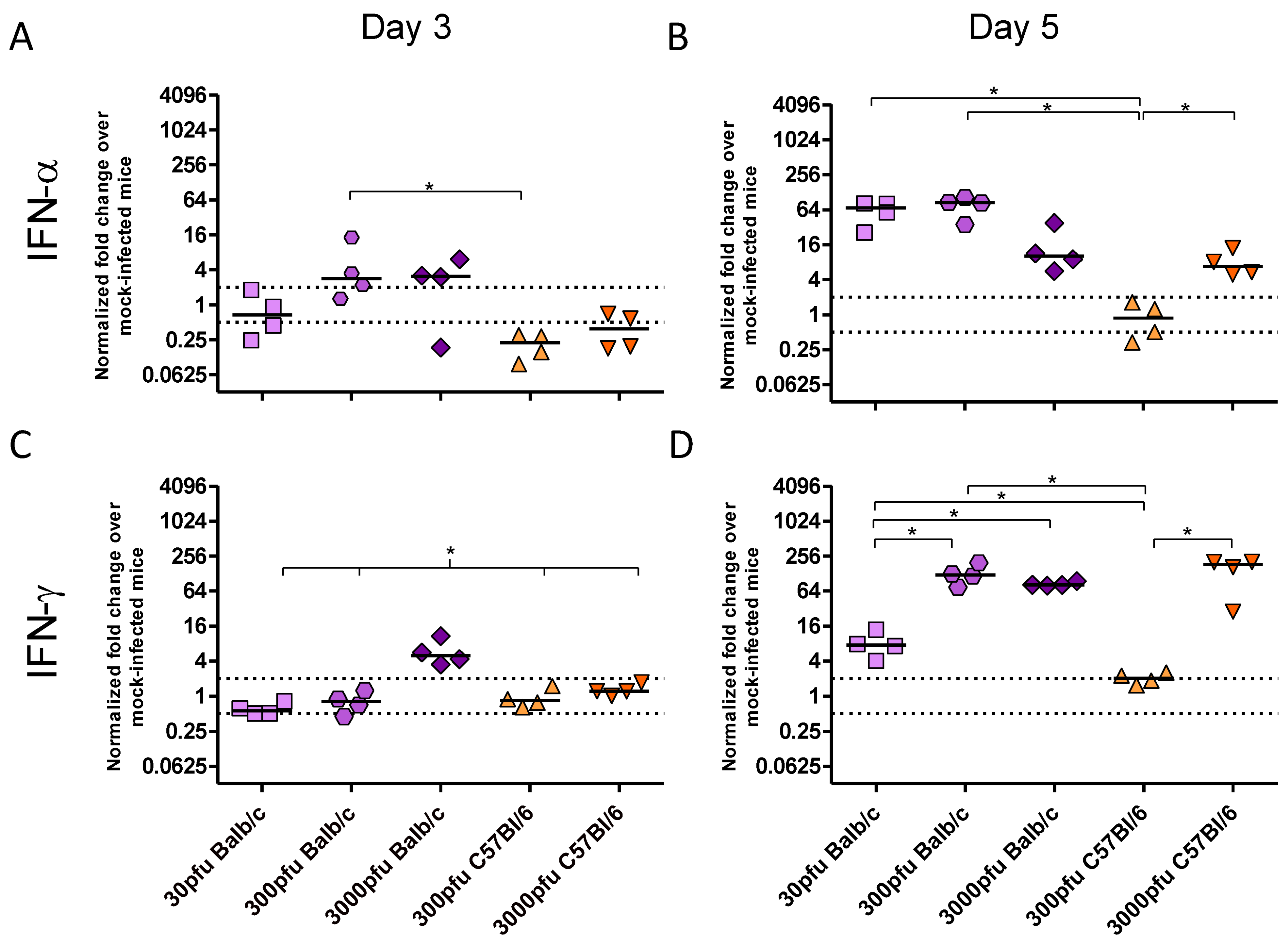
2.3. PVM 15 Induces Earlier Transcription of Chemokines and Cytokines in Balb/c Mice When Compared to C57Bl/6 Mice

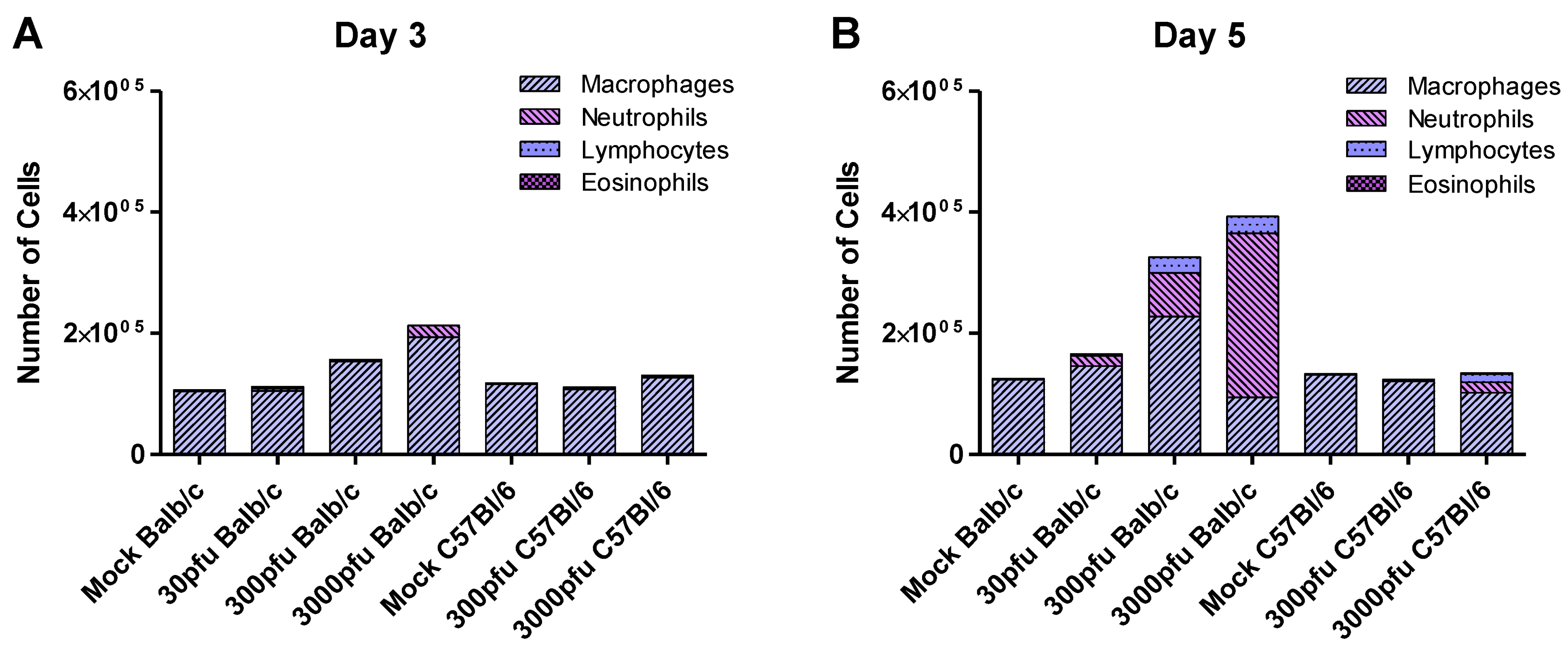
2.4. Balb/c Mice Experience Earlier and Enhanced Infiltration of Immune Cells Compared to C57Bl/6 Mice in Response to PVM Infection

2.5. Balb/c Mice Show an Increase in IFN-γ Producing NK Cells in the Lung Compared to C57Bl/6 Mice in Response to PVM Infection

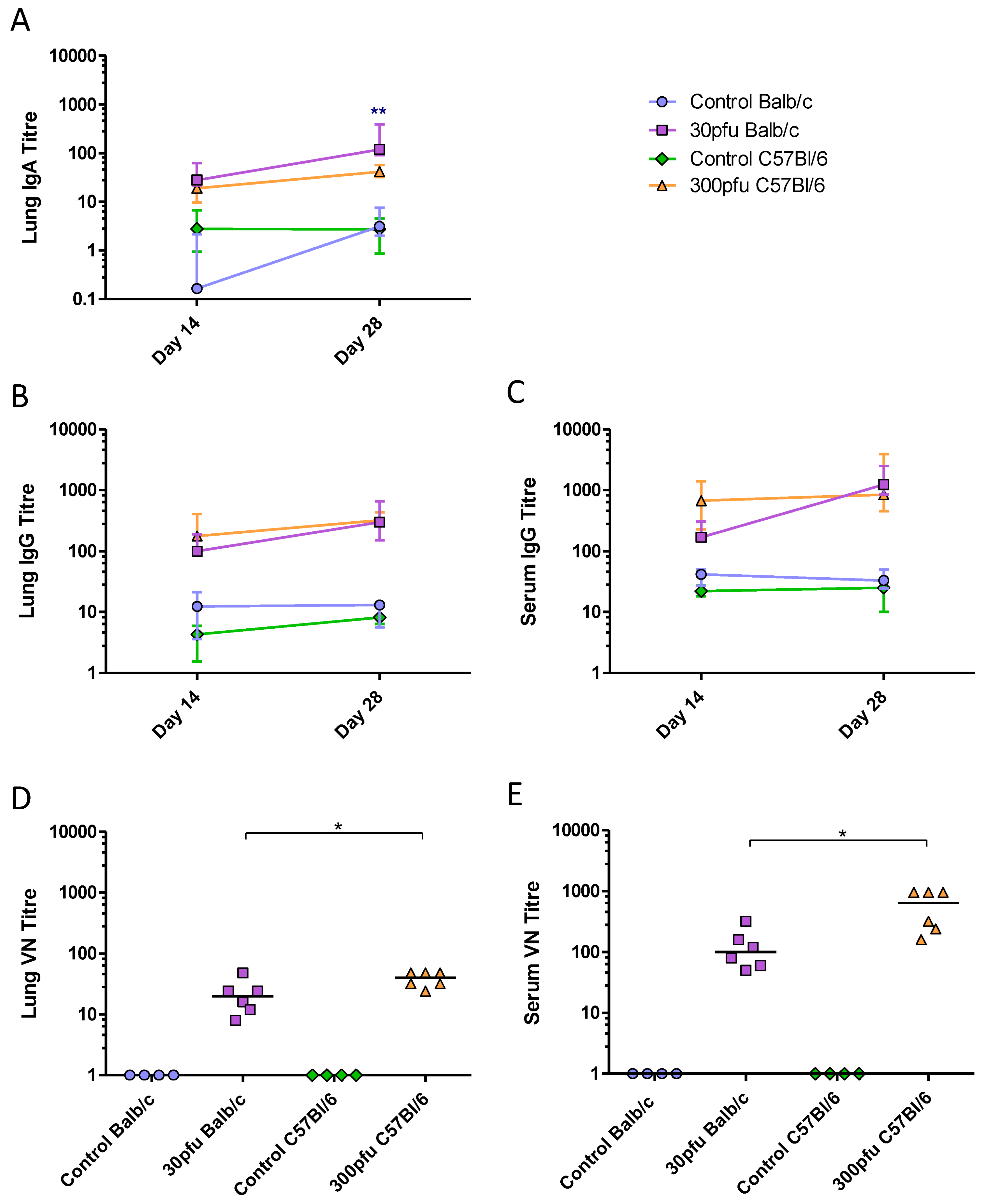
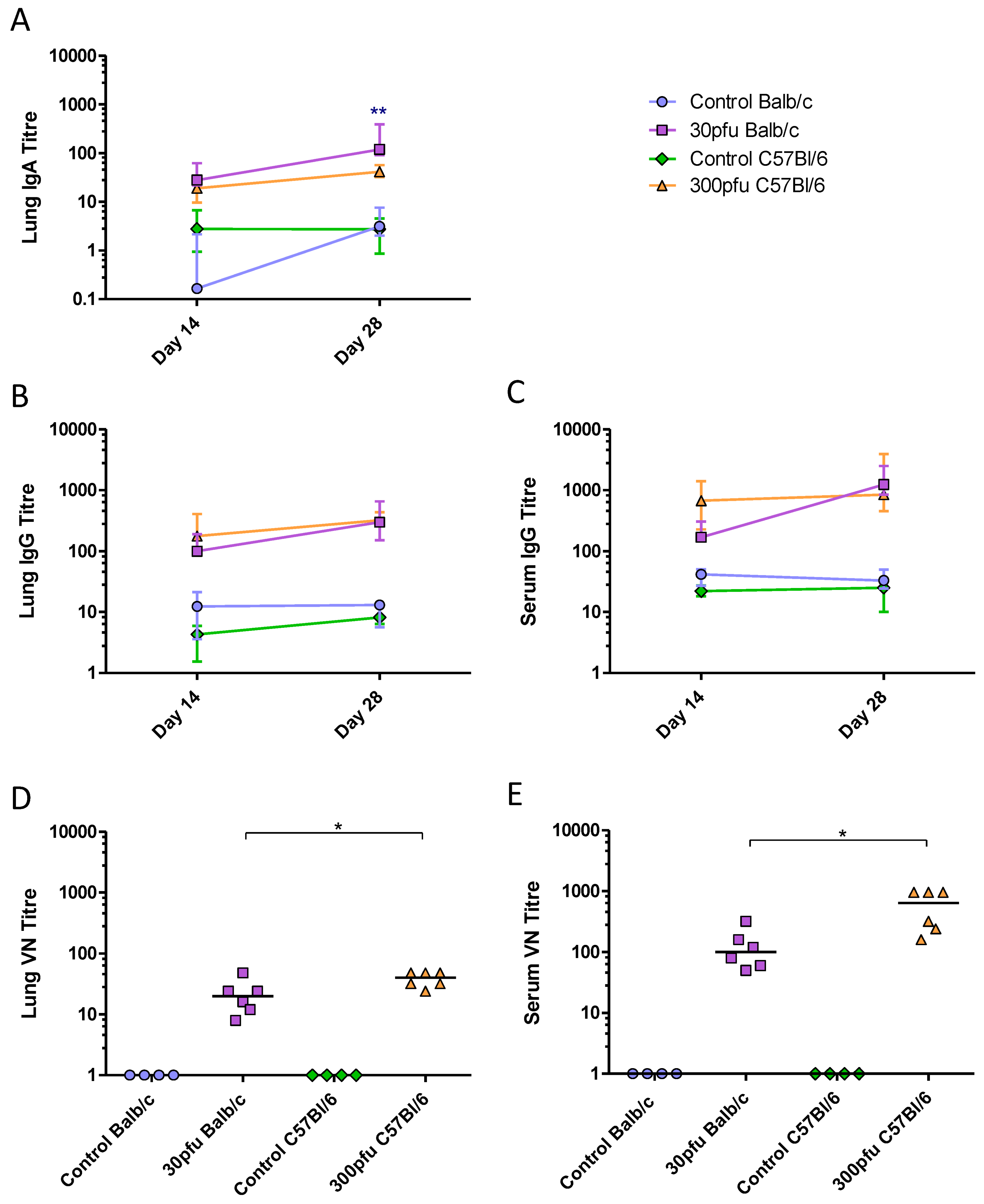
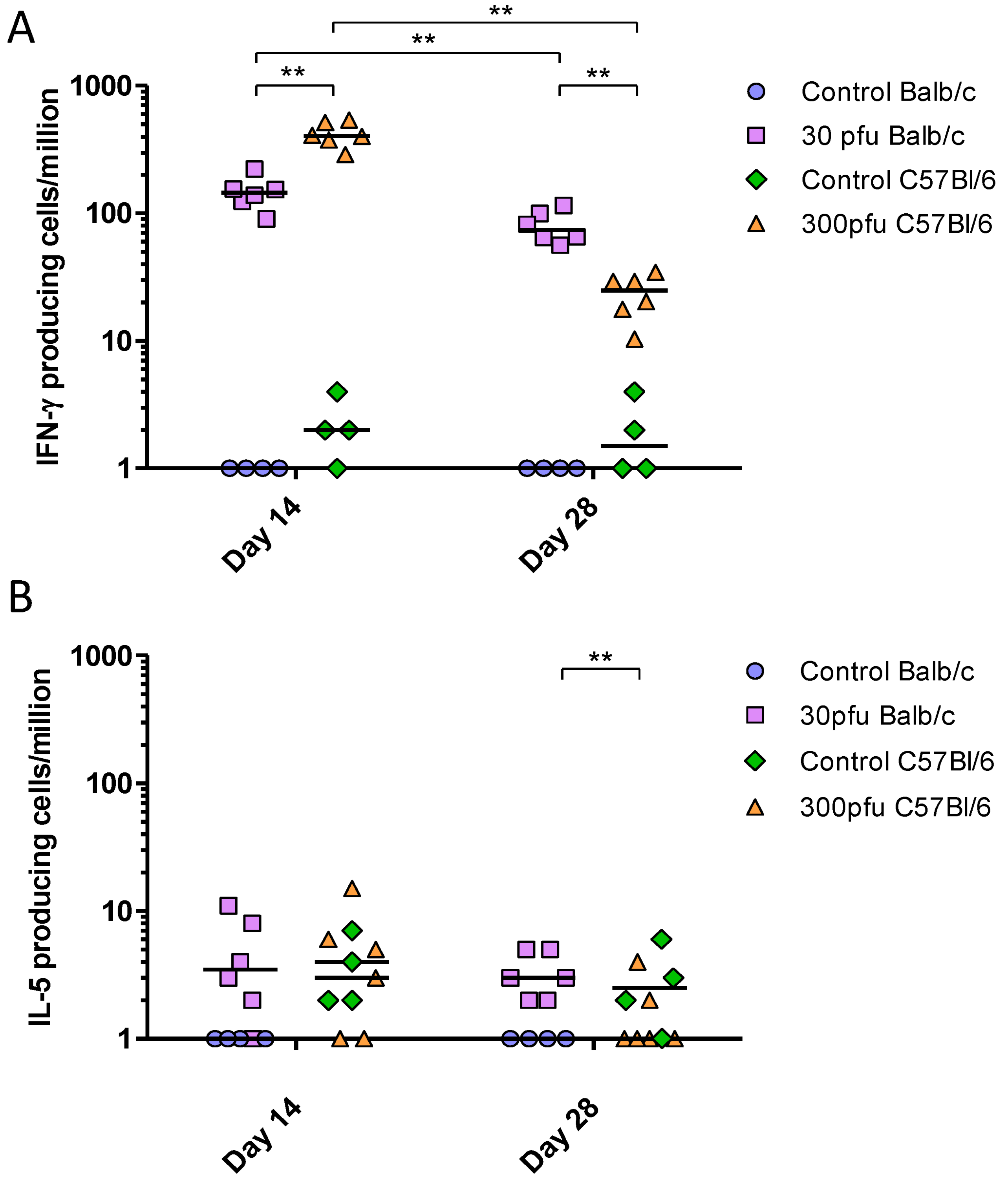
2.6. Balb/c and C57Bl/6 Mice Develop Similar, Th1-Biased Adaptive Immune Responses to a Sublethal Dose of PVM

3. Experimental Section
3.1. Cell Lines and Virus
3.2. Challenge of Mice with PVM
3.3. Lung Samples
3.4. Histology
3.5. Lung Fragment Cultures
3.6. Lung Cell Isolation and Flow Cytometry
3.7. PVM Quantification
3.8. Analysis of Chemokine and Cytokine mRNA Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target gene | Direction | Sequence | Source | Annealing Temp (°C) |
|---|---|---|---|---|
| β-actin | Forward | ACTGGGACGACATGGAG | [44] | 57.5 |
| Reverse | GTAGATGGGCACAGTGTGGG | |||
| GAPDH | Forward | AACTTTGGCATTGTGGAAGG | [45] | 57.5 |
| Reverse | ACACATTGGGGGTAGGAACA | |||
| CCL3/MIP-1α | Forward | CTTCTCTGTACCATGACACTC | [44] | 57.5 |
| Reverse | AGGTCTCTTTGGAGTCAGCG | |||
| CXCL8/MIP-2 | Forward | TGCGCCCAGACAGAAGTCATAGC | designed in house | 63.9 |
| Reverse | GCTCTAGAGTCAGTTAGCCTTGCCTTTG | |||
| CCL2/MCP-1 | Forward | CTTCTGGGCCTGCTGTTCA | [46] | 57.5 |
| Reverse | CCAGCCTACTCATTGGGATCA | |||
| IFN-γ | Forward | TCAAGTGGCATAGATGTGGAAGAA | [46] | 57.5 |
| Reverse | TGGCTCTGCAGGATTTTCATG | |||
| IL-4 | Forward | GGAGATGGATGTGCCAAACG | designed in house | 63.9 |
| Reverse | ACCTTGGAAGCCCTACAGAC | |||
| IFN-α | Forward | CCTGTGTGATGCAACAGGTC | [47] | 59.3 |
| Reverse | TCACTCCTCCTTGCTCAATC | |||
| IFN-β | Forward | ATCATGAACAACAGGTGGATCCTCC | [47] | 63.9 |
| Reverse | TTCAAGTGGAGAGCAGTTGAG | |||
| TNF-α | Forward | GAACTGGCAGAAGAGGCACT | [47] | 68.9 |
| Reverse | AGGGTCTGGGCCATAGAACT | |||
| CXCL10/IP-10 | Forward | GAGATCATTGCCACGATGAA | designed in house | 63.9 |
| Reverse | CACTGGGTAAAGGGGAGTGA | |||
| CCL5/RANTES | Forward | CTCACTGCAGCCGCCCTCTG | designed in house | 57.5 |
| Reverse | CCTTGACGTGGGCACGAGGC | |||
| CCL11/Eotaxin | Forward | AGAGGCTGAGATCCAAGCAG | designed in house | 63.9 |
| Reverse | CAGATCTCTTTGCCCAACCT |
3.9. PVM-Specific ELISA
3.10. Virus Neutralization Assay
3.11. IFN-γ and IL-5 ELISPOT Assays
3.12. Statistical Analysis
4. Conclusions
Acknowledgements
Supplementary Files
References
- Bonville, C.A.; Bennett, N.J.; Koehnlein, M.; Haines, D.M.; Ellis, J.A.; DelVecchio, A.M; Rosenberg, H.F.; Domachowske, J.B. Respiratory dysfunction and proinflammatory chemokines in the pneumonia virus of mice (PVM) model of viral bronchiolitis. Virology 2006, 349, 87–95. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Bonville, C.A.; Easton, A.J.; Domachowske, J.B. The pneumonia virus of mice infection model for severe respiratory syncytial virus infection: Identifying novel targets for therapeutic intervention. Pharmacol. Ther. 2005, 105, 1–6. [Google Scholar] [CrossRef]
- El-Hajje, M.J.; Lambe, C.; Moulin, F.; Suremain, N.; Pons-Catalano, C.; Chalumeau, M.; Raymond, J.; Lebon, P.; Gendrel, D. The burden of respiratory viral disease in hospitalized children in Paris. Eu. Eur. J. Pediatr. 2008, 167, 435–436. [Google Scholar] [CrossRef]
- Deshpande, S.A.; Northern, V. The clinical and health economic burden of respiratory syncytial virus disease among children under 2 years of age in a defined geographical area. Arch. Dis. Child 2003, 88, 1065–1069. [Google Scholar] [CrossRef]
- Iwane, M.K.; Edwards, K.M.; Szilagyi, P.G.; Walker, F.J.; Griffin, M.R.; Weinberg, G.A.; Coulen, C.; Poehling, K.A.; Shone, L.P.; Balter, S.; et al. Population-based surveillance for hospitalizations associated with respiratory syncytial virus, influenza virus, and parainfluenza viruses among young children. Pediatrics 2004, 113, 1758–1764. [Google Scholar] [CrossRef]
- Glezen, W.P.; Taber, L.H.; Frank, A.L.; Kasel, J.A. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child 1986, 140, 543–546. [Google Scholar]
- Hall, C.B. Respiratory syncytial virus: its transmission in the hospital environment. Yale J. Biol. Med. 1982, 55, 219–223. [Google Scholar]
- Welliver, R.C. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J. Pediatrics 2003, 143, 112–117. [Google Scholar] [CrossRef]
- Openshaw, P.J.; Tregoning, J.S. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin. Microbiol. Rev. 2005, 18, 541–555. [Google Scholar]
- Byrd, L.G.; Prince, G.A. Animal models of respiratory syncytial virus infection. Clin. Infect. Dis. 1997, 25, 1363–1368. [Google Scholar]
- Easton, A.J.; Domachowske, J.B.; Rosenberg, H.F. Animal pneumoviruses: Molecular genetics and pathogenesis. Clin. Microbiol. Rev 2004, 17, 390–412. [Google Scholar] [CrossRef]
- Anh, D.B.; Faisca, P.; Desmecht, D.J. Differential resistance/susceptibility patterns to pneumovirus infection among inbred mouse strains. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L426–435. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Domachowske, J.B. Pneumonia virus of mice: severe respiratory infection in a natural host. Immunol. Lett. 2008, 118, 6–12. [Google Scholar] [CrossRef]
- Krempl, C.D.; Lamirande, E.W.; Collins, P.L. Complete sequence of the RNA genome of pneumonia virus of mice (PVM). Virus Gene. 2005, 30, 237–249. [Google Scholar] [CrossRef]
- Domachowske, J.B.; Bonville, C.A.; Easton, A.J.; Rosenberg, H.F. Differential expression of proinflammatory cytokine genes in vivo in response to pathogenic and nonpathogenic pneumovirus infections. J. Infect. Dis. 2002, 186, 8–14. [Google Scholar] [CrossRef]
- Krempl, C.D.; Collins, P.L. Reevaluation of the virulence of prototypic strain 15 of pneumonia virus of mice. J. Virol. 2004, 78, 13362–13365. [Google Scholar] [CrossRef]
- Horsfall, F.L.; Hahn, R.G. A latent virus in normal mice capable of producing pneumonia in its natural host. J. Exp. Med. 1940, 71, 391–408. [Google Scholar] [CrossRef]
- Kuroda, E.; Kito, T.; Yamashita, U. Reduced expression of STAT4 and IFN-gamma in macrophages from BALB/c mice. J. Immunol. 2002, 168, 5477–5482. [Google Scholar]
- Launois, P.; Swihart, K.G.; Milon, G.; Louis, J.A. Early production of IL-4 in susceptible mice infected with Leishmania major rapidly induces IL-12 unresponsiveness. J. Immunol. 1997, 158, 3317–3324. [Google Scholar]
- Liu, T.; Matsuguchi, T.; Tsuboi, N.; Yajima, T.; Yoshikai, Y. Differences in expression of toll-like receptors and their reactivities in dendritic cells in BALB/c and C57BL/6 mice. Infect. Immun. 2002, 70, 6638–6645. [Google Scholar] [CrossRef]
- Scharton-Kersten, T.; Afonso, L.C.; Wysocka, M.; Trinchieri, G.; Scott, P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J. Immunol. 1995, 154, 5320–5330. [Google Scholar]
- Srikiatkhachorn, A.; Chang, W.; Braciale, T.J. Induction of Th-1 and Th-2 responses by respiratory syncytial virus attachment glycoprotein is epitope and major histocompatibility complex independent. J. Virol. 1999, 73, 6590–6597. [Google Scholar]
- Srikiatkhachorn, A.; Braciale, T.J. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 1997, 186, 421–432. [Google Scholar] [CrossRef]
- Garvey, T.L.; Dyer, K.D.; Ellis, J.A.; Bonville, C.A.; Foster, B.; Prussin, C.; Easton, A.J.; Domachowske, J.B.; Rosenberg, H.F. Inflammatory responses to pneumovirus infection in IFN-alpha beta R gene-deleted mice. J. Immunol. 2005, 175, 4735–4744. [Google Scholar]
- Bonville, C.A.; Lau, V.K.; DeLeon, J.M.; Gao, J.L.; Easton, A.J.; Rosenberg, H.F.; Domachowske, J.B. Functional antagonism of chemokine receptor CCR1 reduces mortality in acute pneumovirus infection in vivo. J. Virol. 2004, 78, 7984–7089. [Google Scholar] [CrossRef]
- Domachowske, J.B.; Bonville, C.A.; Gao, J.L.; Murphy, P.M.; Easton, A.J.; Rosenberg, H.F. The chemokine macrophage-inflammatory protein-1 alpha and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J. Immunol. 2000, 165, 2677–2682. [Google Scholar]
- Bonville, C.A.; Easton, A.J.; Rosenberg, H.F.; Domachowske, J.B. Altered pathogenesis of severe pneumovirus infection in response to combined antiviral and specific immunomodulatory agents. J. Virol. 2003, 77, 1237–1244. [Google Scholar]
- Bonville, C.A.; Bennett, N.J.; Percopo, C.M.; Branigan, P.J.; Del Vecchio, A.M.; Rosenberg, H.F.; Domachowske, J.B. Diminished inflammatory responses to natural pneumovirus infection among older mice. Virology 2007, 368, 182–190. [Google Scholar] [CrossRef]
- Domachowske, J.B.; Bonville, C.A.; Dyer, K.D.; Easton, A.J.; Rosenberg, H.F. Pulmonary Eosinophilia and Production of MIP-1[alpha] Are Prominent Responses to Infection with Pneumonia Virus of Mice. Cellular Immunol. 2000, 200, 98–104. [Google Scholar] [CrossRef]
- Rigaux, P.; Killoran, K.E.; Qiu, Z.; Rosenberg, H.F. Depletion of alveolar macrophages prolongs survival in response to acute pneumovirus infection. Virology 2012, 422, 338–345. [Google Scholar] [CrossRef]
- Noah, T.L.; Becker, S. Chemokines in nasal secretions of normal adults experimentally infected with respiratory syncytial virus. Clin. Immunol. 2000, 97, 43–49. [Google Scholar] [CrossRef]
- Sheeran, P.; Jafri, H.; Carubelli, C.; Saavedra, J.; Johnson, C.; Krisher, K.; Sánchez, P.J.; Ramilo, O. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr. Infect. Dis. J. 1999, 18, 115–122. [Google Scholar] [CrossRef]
- Abu-Harb, M.; Bell, F.; Finn, A.; Rao, W.H.; Nixon, L.; Shale, D.; Everard, M.L. IL-8 and neutrophil elastase levels in the respiratory tract of infants with RSV bronchiolitis. Eur. Resp. J. 1999, 14, 139–143. [Google Scholar] [CrossRef]
- Bont, L.; Heijnen, C.J.; Kavelaars, A.; van Aalderen, W.M.; Brus, F.; Draaisma, J.T.; Geelen, S.M.; van Vught, H.J. Peripheral blood cytokine responses and disease severity in respiratory syncytial virus bronchiolitis. Eur. Respir. J. 1999, 14, 144–149. [Google Scholar] [CrossRef]
- Bonville, C.A.; Percopo, C.M.; Dyer, K.D.; Gao, J.; Prussin, C.; Foster, B.; Rosenberg, H.F.; Domachowske, J.B. Interferon-gamma coordinates CCL3-mediated neutrophil recruitment in vivo. BMC Immunol. 2009, 10, 14. [Google Scholar] [CrossRef]
- Claassen, E.A.W.; van der Kant, P.A.A.; Rychnavska, Z.S.; van Bleek, G.M.; Easton, A.J.; van der Most, R.G. Activation and Inactivation of Antiviral CD8 T Cell Responses during Murine Pneumovirus Infection. J. Immunol. 2005, 175, 6597–604. [Google Scholar]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annual Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef]
- Campbell, J.J.; Qin, S.; Unutmaz, D.; Soler, D.; Murphy, K.E.; Hodge, M.R.; Wu, L.; Butcher, EC. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J. Immunol. 2001, 166, 6477–6482. [Google Scholar]
- Moretta, A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat. Rev. Immunol. 2002, 2, 957–964. [Google Scholar] [CrossRef]
- Li, F.; Zhu, H.; Sun, R.; Wei, H.; Tian, Z. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J. Virol. 2012, 86, 2251–2258. [Google Scholar] [CrossRef]
- van Helden, M.J.; van Kooten, P.J.; Bekker, C.P.; Grone, A.; Topham, D.J.; Easton, A.J.; Boog, C.J.; Busch, D.H.; Zaiss, D.M.; Sijts, A.J. Pre-existing virus-specific CD8(+) T-cells provide protection against pneumovirus-induced disease in mice. Vaccine 2012, 30, 6382–6388. [Google Scholar] [CrossRef]
- Morton, D.B.; Griffiths, P.H. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet. Rec. 1985, 116, 431–436. [Google Scholar]
- Labiuk, S.L.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Major tegument protein VP8 of bovine herpesvirus 1 is phosphorylated by viral US3 and cellular CK2 protein kinases. J. Gen. Virol. 2009, 90, 2829–2839. [Google Scholar] [CrossRef]
- Park, J.K.; Cho, K.; Johnson, J.; Perez, R.V. Induction of MIP-1alpha in Kupffer cell by portal venous transfusion. Transplant. Immunol. 2004, 13, 33–38. [Google Scholar] [CrossRef]
- Vogel, C.F.; Nishimura, N.; Sciullo, E.; Wong, P.; Li, W.; Matsumura, F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch. Biochem. Biophys. 2007, 461, 169–175. [Google Scholar] [CrossRef]
- Overbergh, L.; Giulietti, A.; Valckx, D.; Decallonne, R.; Bouillon, R.; Mathieu, C. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J. Biomol. Tech. 2003, 14, 33–43. [Google Scholar]
- Pascual, M.; Fernandez-Lizarbe, S.; Guerri, C. Role of TLR4 in ethanol effects on innate and adaptive immune responses in peritoneal macrophages. Immunol. Cell Biol. 2011, 89, 716–727. [Google Scholar] [CrossRef]
- Mapletoft, J.W.; Latimer, L.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Intranasal immunization of mice with a bovine respiratory syncytial virus vaccine induces superior immunity and protection compared to those by subcutaneous delivery or combinations of intranasal and subcutaneous prime-boost strategies. Clin. Vaccine Immunol. 2010, 17, 23–35. [Google Scholar] [CrossRef]
- Dyer, K.D.; Percopo, C.M.; Fischer, E.R.; Gabryszewski, S.J.; Rosenberg, H.F. Pneumoviruses infect eosinophils and elicit MyD88-dependent release of chemoattractant cytokines and interleukin-6. Blood 2009, 114, 2649–2656. [Google Scholar]
- Heinze, B.; Frey, S.; Mordstein, M.; Schmitt-Graff, A.; Ehl, S.; Buchholz, U.J.; Collins, P.L.; Staeheli, P.; Krempl, C.D. Both nonstructural proteins NS1 and NS2 of pneumonia virus of mice are inhibitors of the interferon type I and type III responses in vivo. J. Virol. 2011, 85, 4071–4084. [Google Scholar]
- Atochina, E.N.; Beers, M.F.; Tomer, Y.; Scanlon, S.T.; Russo, S.J.; Panettieri, R.A., Jr.; Haczku, A. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir. Res. 2003, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Martinez, S.; Cancino-Diaz, M.E.; Jimenez-Zamudio, L.; Garcia-Latorre, E.; Cancino-Diaz, J.C. TLRs and NODs mRNA expression pattern in healthy mouse eye. British J. Ophthalmol. 2005, 89, 904–910. [Google Scholar] [CrossRef]
- Glineur, S.; Tran Anh, D.B.; Sarlet, M.; Michaux, C.; Desmecht, D. Characterization of the resistance of SJL/J mice to pneumonia virus of mice, a model for infantile bronchiolitis due to a respiratory syncytial virus. PloS One 2012, 7, e44581. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watkiss, E.R.T.; Shrivastava, P.; Arsic, N.; Gomis, S.; Van Drunen Littel-van den Hurk, S. Innate and Adaptive Immune Response to Pneumonia Virus of Mice in a Resistant and a Susceptible Mouse Strain. Viruses 2013, 5, 295-320. https://doi.org/10.3390/v5010295
Watkiss ERT, Shrivastava P, Arsic N, Gomis S, Van Drunen Littel-van den Hurk S. Innate and Adaptive Immune Response to Pneumonia Virus of Mice in a Resistant and a Susceptible Mouse Strain. Viruses. 2013; 5(1):295-320. https://doi.org/10.3390/v5010295
Chicago/Turabian StyleWatkiss, Ellen R. T., Pratima Shrivastava, Natasa Arsic, Susantha Gomis, and Sylvia Van Drunen Littel-van den Hurk. 2013. "Innate and Adaptive Immune Response to Pneumonia Virus of Mice in a Resistant and a Susceptible Mouse Strain" Viruses 5, no. 1: 295-320. https://doi.org/10.3390/v5010295