Genetics-Based Classification of Filoviruses Calls for Expanded Sampling of Genomic Sequences


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
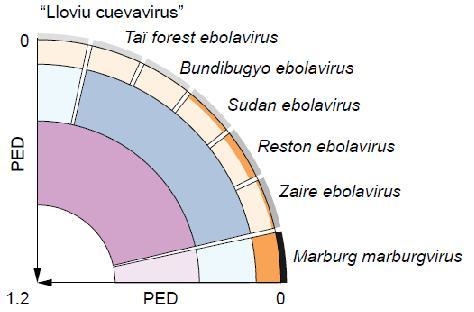
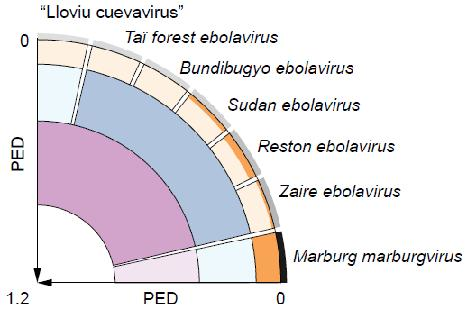
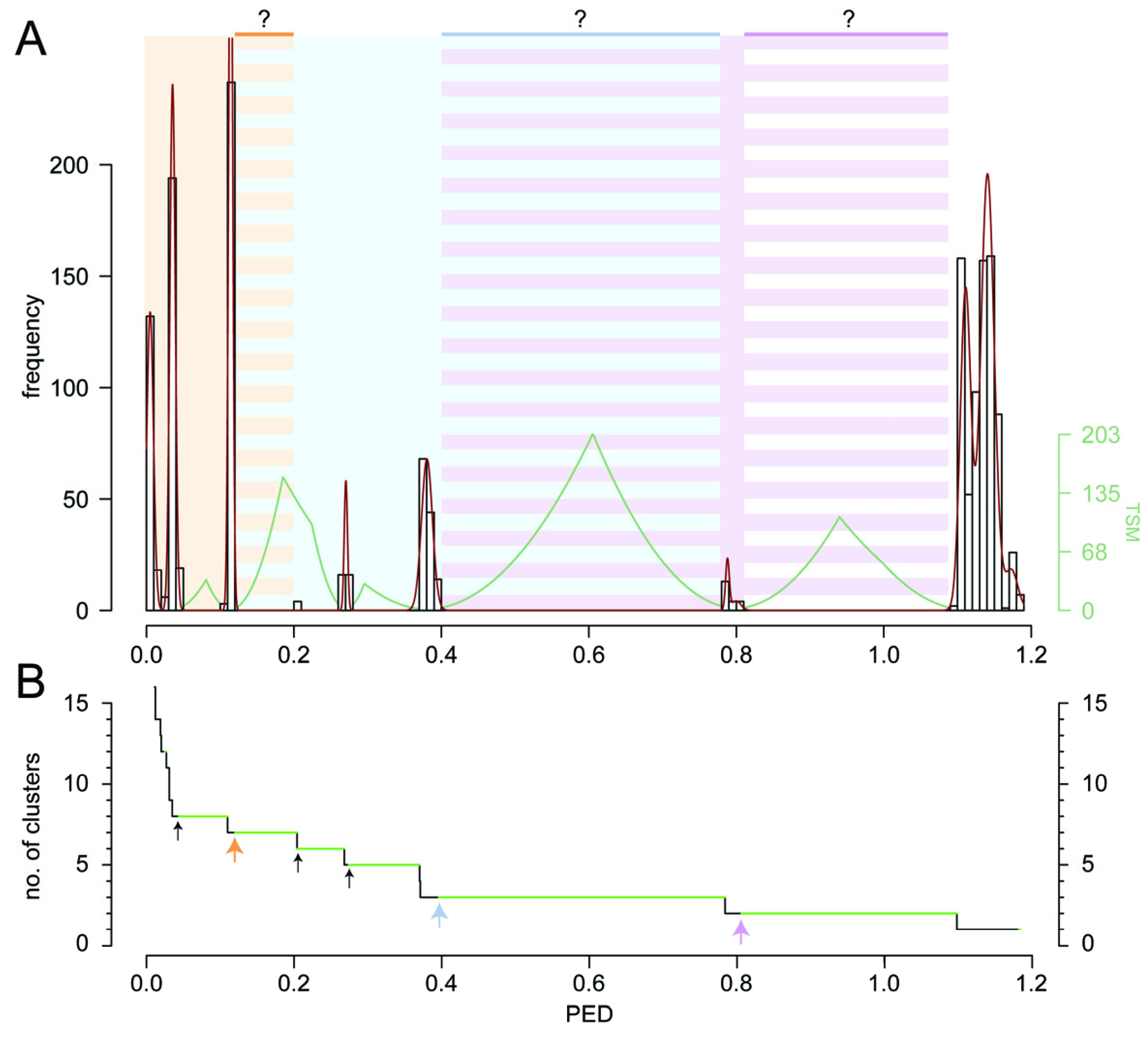
2. Results and Discussion
3. Experimental Section
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Lauber, C.; Gorbalenya, A.E. Partitioning the genetic diversity of a virus family: Approach and evaluation through a case study of picornaviruses. J. Virol. 2012, 86, 3890–3904. [Google Scholar] [CrossRef]
- Lauber, C.; Gorbalenya, A.E. Toward genetics-based virus taxonomy: Comparative analysis of a genetics-based classification and the taxonomy of picornaviruses. J. Virol. 2012, 86, 3905–3915. [Google Scholar]
- Knowles, N.J.; Hovi, T.; Hyypia, T.; King, A.M.Q.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Simmonds, P.; Skern, T.; Stanway, G.; et al. Family Picornaviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 855–880. [Google Scholar]
- Adams, M.J.; Antoniw, J.F.; Bar-Joseph, M.; Brunt, A.A.; Candresse, T.; Foster, G.D.; Martelli, G.P.; Milne, R.G.; Fauquet, C.M. The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch. Virol. 2004, 149, 1045–1060. [Google Scholar]
- Adams, M.J.; Antoniw, J.F.; Fauquet, C.M. Molecular criteria for genus and species discrimination within the family Potyviridae. Arch. Virol. 2005, 150, 459–479. [Google Scholar]
- Bao, Y.; Kapustin, Y.; Tatusova, T. Virus classification by Pairwise Sequence Comparison (PASC). In Encyclopedia of Virology; Mahy, B.W.J., van Regenmortel, M.H.V., Eds.; Elsevier: Oxford, UK, 2008; Volume 5, pp. 342–348. [Google Scholar]
- Bernard, H.U.; Burk, R.D.; Chen, Z.G.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef]
- Chan, Y.F.; Sam, I.C.; Abubakar, S. Phylogenetic designation of enterovirus 71 genotypes and subgenotypes using complete genome sequences. Inf. Genet. Evol. 2010, 10, 404–412. [Google Scholar] [CrossRef]
- Fauquet, C.M.; Bisaro, D.M.; Briddon, R.W.; Brown, J.K.; Harrison, B.D.; Rybicki, E.P.; Stenger, D.C.; Stanley, J. Revision of taxonomic criteria for species demarcation in the family Geminiviridae, and an updated list of begomovirus species. Arch. Virol. 2003, 148, 405–421. [Google Scholar] [CrossRef]
- Gonzaalez, J.M.; Gomez-Puertas, P.; Cavanagh, D.; Gorbalenya, A.E.; Enjuanes, L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003, 148, 2207–2235. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, present and future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef]
- Maes, P.; Klempa, B.; Clement, J.; Matthijnssens, J.; Gajdusek, D.C.; Kruger, D.H.; van Ranst, M. A proposal for new criteria for the classification of hantaviruses, based on S and M segment protein sequences. Inf. Genet. Evol. 2009, 9, 813–820. [Google Scholar]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gomara, M.; Maes, P.; Patton, J.T.; et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar]
- Schuffenecker, I.; Ando, T.; Thouvenot, D.; Lina, B.; Aymard, M. Genetic classification of “Sapporo-like viruses”. Arch. Virol. 2001, 146, 2115–2132. [Google Scholar] [CrossRef]
- Shukla, D.D.; Ward, C.W. Amino-acid sequence homology of coat proteins as a basis for identification and classification of the potyvirus group. J. Gen. Virol. 1988, 69, 2703–2710. [Google Scholar] [CrossRef]
- Zheng, D.P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 806–828. [Google Scholar]
- Nga, P.T.; Parquet, M.D.; Lauber, C.; Parida, M.; Nabeshima, T.; Yu, F.X.; Thuy, N.T.; Inoue, S.; Ito, T.; Okamoto, K.; et al. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. 2011, 7, e1002215. [Google Scholar] [CrossRef]
- Zirkel, F.; Kurth, A.; Quan, P.L.; Briese, T.; Ellerbrok, H.; Pauli, G.; Leendertz, F.H.; Lipkin, W.I.; Ziebuhr, J.; Drosten, C.; et al. An insect nidovirus emerging from a primary tropical rainforest. mBio 2011, 2, e00077-11. [Google Scholar]
- Lauber, C.; Ziebuhr, J.; Junglen, S.; Drosten, C.; Zirkel, F.; Nga, P.T.; Morita, K.; Snijder, E.J.; Gorbalenya, A.E. Mesoniviridae: A proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch. Virol. 2012. [Google Scholar] [CrossRef]
- Easton, A.J.; Pringle, C.R. Order Mononegavirales. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 653–657. [Google Scholar]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T.; et al. Proposal for a revised taxonomy of the family Filoviridae: Classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar]
- Kuhn, J.K.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Jahrling, P.B.; Kawaoka, Y.; Netesov, S.V.; Nichol, S.T.; Peters, C.J.; Volchkov, V.E.; et al. Familiy Filoviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 665–671. [Google Scholar]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Stroher, U.; et al. Marburgvirus Genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar]
- Towner, J.S.; Sealy, T.K.; Khristova, M.L.; Albarino, C.G.; Conlan, S.; Reeder, S.A.; Quan, P.L.; Lipkin, W.I.; Downing, R.; Tappero, J.W.; et al. Newly Discovered Ebola Virus Associated with Hemorrhagic Fever Outbreak in Uganda. PLoS Pathog. 2008, 4, e1000212. [Google Scholar]
- Ferron, F.; Longhi, S.; Henrissat, B.; Canard, B. Viral RNA-polymerases—A predicted 2 '-O-ribose methyltransferase domain shared by all Mononegavirales. Trends Biochem. Sci. 2002, 27, 222–224. [Google Scholar]
- Feldmann, H.; Muhlberger, E.; Randolf, A.; Will, C.; Kiley, M.P.; Sanchez, A.; Klenk, H.D. Marburg Virus, A Filovirus—Messenger-RNAs, gene order, and regulatory elements of the replication cycle. Virus Res. 1992, 24, 1–19. [Google Scholar] [CrossRef]
- Negredo, A.; Palacios, G.; Vazquez-Moron, S.; Gonzalez, F.; Dopazo, H.; Molero, F.; Juste, J.; Quetglas, J.; Savji, N.; Martinez, M.D.; et al. Discovery of an ebolavirus-like filovirus in Europe. PLoS Pathog. 2011, 7, e1002304. [Google Scholar]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.J.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 3602–3607. [Google Scholar]
- Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H.D. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 1995, 214, 421–430. [Google Scholar] [CrossRef]
- Volchkova, V.A.; Klenk, H.D.; Volchkov, V.E. Delta-peptide is the carboxy-terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 1999, 265, 164–171. [Google Scholar] [CrossRef]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Miranda, M.E.; Ksiazek, T.G.; Retuya, T.J.; Khan, A.S.; Sanchez, A.; Fulhorst, C.F.; Rollin, P.E.; Calaor, A.B.; Manalo, D.L.; Roces, M.C.; et al. Epidemiology of Ebola (subtype Reston) virus in the Philippines, 1996. J. Infect. Dis. 1999, 179, S115–S119. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Lieutaud, P.; Harris, M.R.; Coutard, B.; Canard, B.; Kleywegt, G.J.; Kravchenko, A.A.; Samborskiy, D.V.; Sidorov, I.A.; Leontovich, A.M.; et al. Practical application of bioinformatics by the multidisciplinary VIZIER consortium. Antivir. Res. 2010, 87, 95–110. [Google Scholar]
- Lauber, C.; Gorbalenya, A.E. Genetics-based classification of coronaviruses. Molecular Virology Laboratory, Department of Medical Microbiology, Leiden University Medical Center: 2333 ZA Leiden, The Netherlands, Unpublished work, to be submitted for publication, 2012.
- Van Regenmortel, M.H.V. Virus species and virus identification: Past and current controversies. Inf. Genet. Evol. 2007, 7, 133–144. [Google Scholar] [CrossRef]
- Mayo, M.A.; Pringle, C.R. Virus taxonomy—1997. J. Gen. Virol. 1998, 79, 649–657. [Google Scholar]
- Dimmic, M.W.; Rest, J.S.; Mindell, D.P.; Goldstein, R.A. rtREV: An amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J. Mol. Evol. 2002, 55, 65–73. [Google Scholar]
- Hraber, P.; Kuiken, C.; Waugh, M.; Geer, S.; Bruno, W.J.; Leitner, T. Classification of hepatitis C virus and human immunodeficiency virus-1 sequences with the branching index. J. Gen. Virol. 2008, 89, 2098–2107. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Jiang, P.; Faase, J.A.J.; Toyoda, H.; Paul, A.; Wimmer, E.; Gorbalenya, A.E. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 9457–9462. [Google Scholar]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar]
- Holmes, E.C. The evolutionary genetics of emerging viruses. Annu. Rev. Ecol. Evol. Systemat. 2009, 40, 353–372. [Google Scholar] [CrossRef]
- Archer, A.M.; Rico-Hesse, R. High genetic divergence and recombination in arenaviruses from the Americas. Virology 2002, 304, 274–281. [Google Scholar] [CrossRef]
- Charrel, R.N.; Feldmann, H.; Fulhorst, C.F.; Khelifa, R.; de Chesse, R.; de Lamballerie, X. Phylogeny of New World arenaviruses based on the complete coding sequences of the small genomic segment identified an evolutionary lineage produced by intrasegmental recombination. Biochem. Biophys. Res. Comm. 2002, 296, 1118–1124. [Google Scholar] [CrossRef]
- Hao, W.L. Evidence of intra-segmental homologous recombination in influenza A virus. Gene 2011, 481, 57–64. [Google Scholar] [CrossRef]
- Wittmann, T.J.; Biek, R.; Hassanin, A.; Rouquet, P.; Reed, P.; Yaba, P.; Pourrut, X.; Real, L.A.; Gonzalez, J.P.; Leroy, E.M. Isolates of Zaire ebolavirus from wild apes reveal genetic lineage and recombinants. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17123–17127. [Google Scholar]
- Chare, E.R.; Holmes, E.C. A phylogenetic survey of recombination frequency in plant RNA viruses. Arch. Virol. 2006, 151, 933–946. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Antonov, I.V.; Leontovich, A.M.; Gorbalenya, A.E. BAGG (Blocks Accepting Gaps Generator). 2008. Available online: http://www.genebee.msu.su/~antonov/bagg/cgi/bagg.cgi (accessed on 16 February 2012).
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lauber, C.; Gorbalenya, A.E. Genetics-Based Classification of Filoviruses Calls for Expanded Sampling of Genomic Sequences. Viruses 2012, 4, 1425-1437. https://doi.org/10.3390/v4091425
Lauber C, Gorbalenya AE. Genetics-Based Classification of Filoviruses Calls for Expanded Sampling of Genomic Sequences. Viruses. 2012; 4(9):1425-1437. https://doi.org/10.3390/v4091425
Chicago/Turabian StyleLauber, Chris, and Alexander E. Gorbalenya. 2012. "Genetics-Based Classification of Filoviruses Calls for Expanded Sampling of Genomic Sequences" Viruses 4, no. 9: 1425-1437. https://doi.org/10.3390/v4091425