Herpesviruses Placating the Unwilling Host: Manipulation of the MHC Class II Antigen Presentation Pathway

Abstract
:1. Introduction

{kind=link}
| Subfamily | Virus name | Nomenclature | Associated diseases |
|---|---|---|---|
| Alpha-herpesviruses | Herpes Simplex-1 | HSV-1 or HHV-1 | Gingivostomatosis, Cold sores, Encephalitis |
| Herpes Simplex-2 | HSV-2 or HHV-3 | Genital herpes, Cutaneous herpes, Encephalitis, Meningoencephalitis | |
| Varicella zoster virus | VZV or HHV-3 | Chickenpox, Shingles | |
| Beta-herpesviruses | Cytomegalovirus | HCMV or HHV-5 | Mononucleosis, Hepatititis, Pneumonitis |
| Human herpesvirus-6 | HHV-6 | Exanthum subitum, Mild febrile illness | |
| Human herpesvirus-7 | HHV-7 | Exanthum subitum, Mild febrile illness | |
| Gamma-herpesviruses | Epstein-Barr virus | EBV or HHV-4 | Mononucleosis, Burkitt’s lymphoma, post-transplant lymphoproliferative syndrome (PTLD), nasopharyngeal carcinoma, |
| Kaposi’s sarcoma-associated herpesvirus | KSHV or HHV-8 | Kaposi’s sarcoma, primary effusion lymphoma, some types of multicentric Castleman’s disease |
2. Why MHC-II Evasion Is Important
2.1. Herpesvirus Infections Generate Broad Range Anti-Viral CD4 T Cell Responses
2.1.1. HSV
2.1.2. HCMV
2.1.3. EBV
2.1.4. KSHV
2.1.5. VZV
2.1.6. HHV-6
2.2. Target Cells Expressing MHC-II and Presenting Peptide
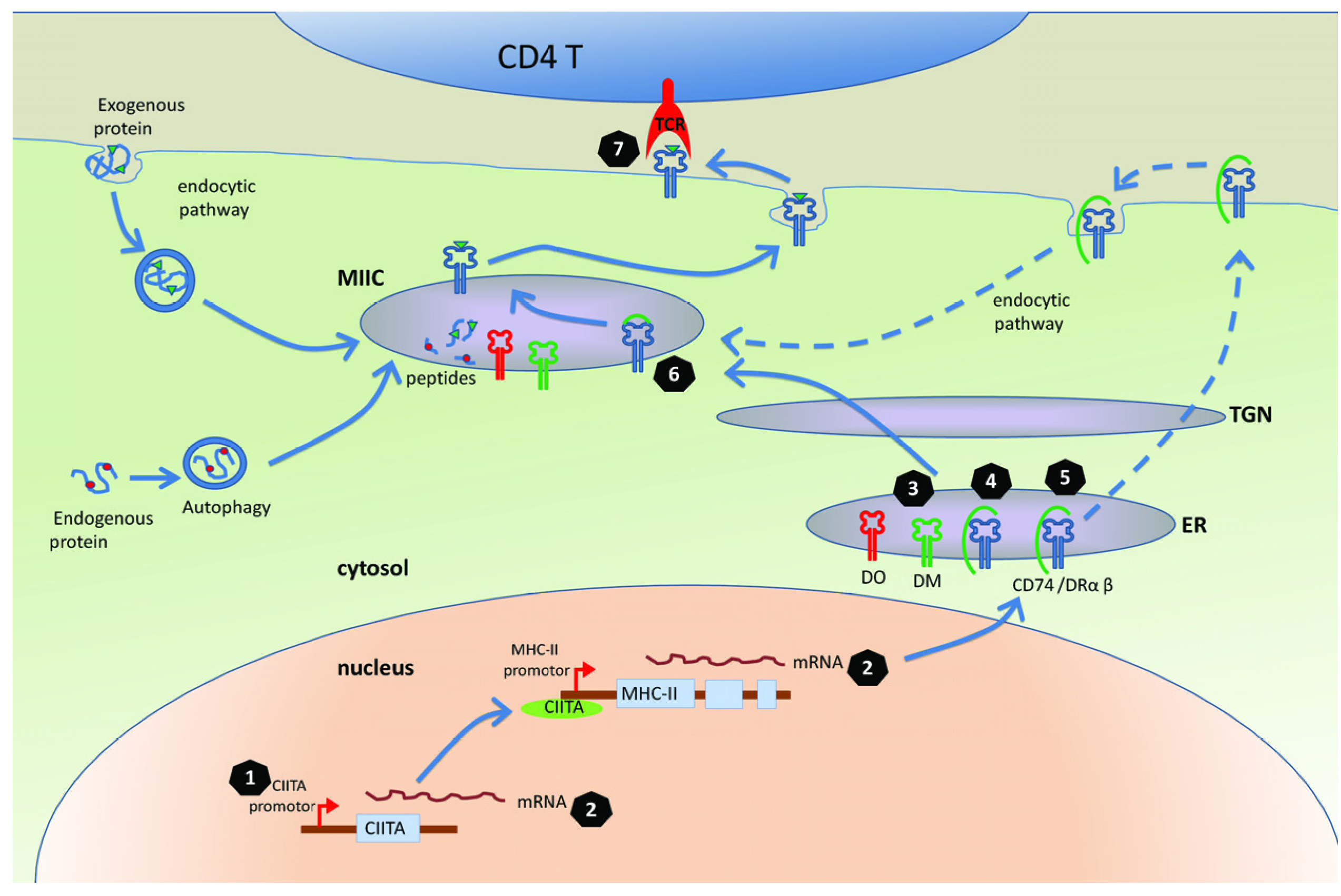
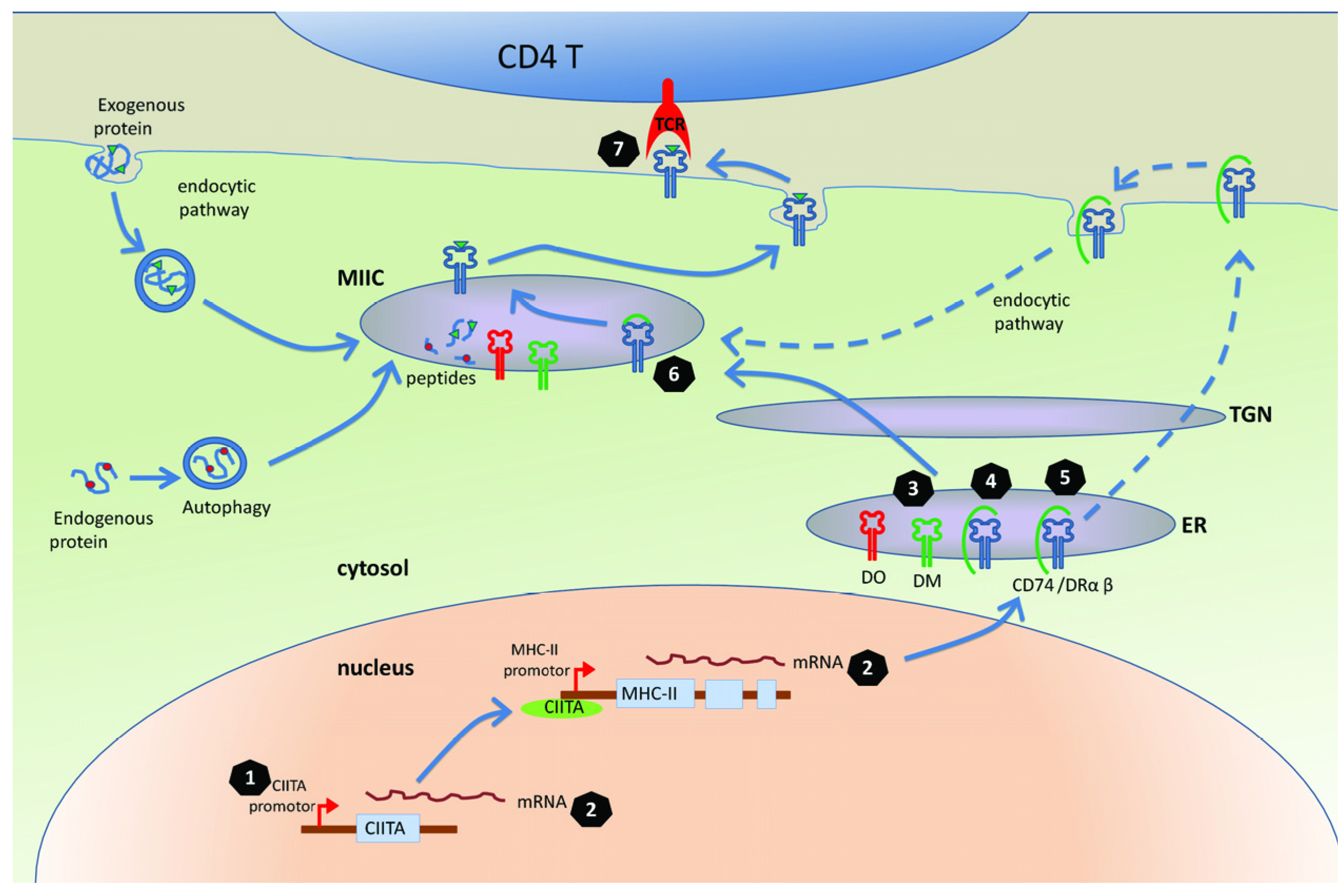
3. Mechanisms for Interfering with the MHC-II Antigen Presentation Pathway
3.1. The MHC-II Antigen Presentation Is Inhibited in the Cells Infected with Herpesviruses
3.2. Molecular Mechanisms of Herpesviruses’ Modulation of MHC-II Antigen Presentation
3.2.1. Targeting CIITA, the Master Regulator of MHC Class II Gene Expression
3.2.2. Targeting Transport of DR
3.2.3 Targeting CD74
3.2.4. Manipulation of T Cell Receptor (TCR) Recognition
3.2.5. Other Mechanisms of Manipulation of MHC-II Pathway
4. Conclusion
Acknowledgments
Conflict of Interest
References
- Arvin, A.; Fiume, G.C.; Mocarski, E.; Moore, P.S.; Roizman, B.; Whitley, R.; Yamanishi, K. Human Herpesviruses; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef]
- Strauch, B.; Siegel, N.; Andrews, L.-L.; Miller, G. Oropharyngeal excretion of Epstein-Barr virus by renal transplant recipients and other patients treated with immunosuppressive drugs. Lancet 1974, 303, 234–237. [Google Scholar]
- Emanuel, D.; Cunningham, I.; Jules-Elysee, K.; Brochstein, J.A.; Kernan, N.A.; Laver, J.; Stover, D.; White, D.A.; Fels, A.; Polsky, B.; et al. Cytomegalovirus pneumonia after bone marrow transplantation successfully treated with the combination of ganciclovir and high-dose intravenous immune globulin. Ann. Intern. Med. 1988, 109, 777–782. [Google Scholar]
- Emery, V.C. Investigation of CMV disease in immunocompromised patients. J. Clin. Pathol. 2001, 54, 84–88. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Hill, A.; Jugovic, P.; York, L.; Russ, G.; Bennink, J.; Yewdell, J.; Ploegh, H.; Johnson, D. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995, 375, 411–415. [Google Scholar]
- Fruh, K.; Ahn, K.; Djaballah, H.; Sempe, P.; van Endert, P.M.; Tampe, R.; Peterson, P.A.; Yang, Y. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995, 375, 415–418. [Google Scholar]
- York, I.A.; Roop, C.; Andrews, D.W.; Riddell, S.R.; Graham, F.L.; Johnson, D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994, 77, 525–535. [Google Scholar]
- Tortorella, D.; Gewurz, B.E.; Furman, M.H.; Schust, D.J.; Ploegh, H.L. Viral subversion of the immune system. Annu. Rev. Immunol. 2000, 18, 861–926. [Google Scholar] [CrossRef]
- Vossen, M.; Westerhout, E.; Söderberg-Nauclér, C.; Wiertz, E. Viral immune evasion: A masterpiece of evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar]
- Yewdell, J.W.; Hill, A.B. Viral interference with antigen presentation. Nat. Immunol. 2002, 3, 1019–1025. [Google Scholar] [CrossRef]
- Rowe, M.; Zuo, J. Immune responses to Epstein-Barr virus: Molecular interactions in the virus evasion of CD8+ T cell immunity. Microbes Infect. 2010, 12, 173–181. [Google Scholar] [CrossRef]
- Ressing, M.E.; Horst, D.L.; Griffin, B.D.; Tellam, J.; Zuo, J.; Khanna, R.; Rowe, M.; Wiertz, E.J.H.J. Epstein-Barr virus evasion of CD8+ and CD4+ T cell immunity via concerted actions of multiple gene products. Semin. Cancer Biol. 2008, 18, 397–408. [Google Scholar] [CrossRef]
- Helen, E.; Heslop, M.D.; Malcolm, K.; Brenner, M.B.; Cliona, M.R.; Cliona, M.R. Donor T cells to treat EBV-associated lymphoma. N. Engl. J. Med. 1994, 331, 679–680. [Google Scholar] [CrossRef]
- Rooney, C.M.; Ng, C.Y.C.; Loftin, S.; Smith, C.A.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar]
- Haque, T.; Wilkie, G.M.; Jones, M.M.; Higgins, C.D.; Urquhart, G.; Wingate, P.; Burns, D.; McAulay, K.; Turner, M.; Bellamy, C.; et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: Results of a phase 2 multicenter clinical trial. Blood 2007, 110, 1123–1131. [Google Scholar]
- Stevenson, P.G.; Cardin, R.D.; Christensen, J.P.; Doherty, P.C. Immunological control of a murine gammaherpesvirus independent of CD8+ T cells. J. Gen. Virol. 1999, 80, 477–483. [Google Scholar]
- Christensen, J.P.; Cardin, R.D.; Branum, K.C.; Doherty, P.C. CD4+ T cell-mediated control of a γ-herpesvirus in B cell-deficient mice is mediated by IFN-γ. Proc. Natl. Acad. Sci. USA 1999, 96, 5135–5140. [Google Scholar] [CrossRef]
- Sparks-Thissen, R.L.; Braaten, D.C.; Kreher, S.; Speck, S.H.; Virgin, H.W. An optimized CD4 T-cell response can control productive and Latent gammaherpesvirus infection. J. Virol. 2004, 78, 6827–6835. [Google Scholar] [CrossRef]
- Stuller, K.A.; Cush, S.S.; Flano, E. Persistent γ-herpesvirus infection induces a CD4 T cell response containing functionally distinct effector populations. J. Immunol. 2010, 184, 3850–3856. [Google Scholar] [CrossRef]
- Johnson, A.J.; Chu, C.-F.; Milligan, G.N. Effector CD4+ T-Cell involvement in clearance of infectious herpes simplex virus type 1 from sensory ganglia and spinal cords. J. Virol. 2008, 82, 9678–9688. [Google Scholar] [CrossRef]
- Ishikawa, T.; Yamada, H.; Oyamada, A.; Goshima, F.; Nishiyama, Y.; Yoshikai, Y. Protective role of fas-fasl signaling in lethal infection with herpes simplex virus type 2 in mice. J. Virol. 2009, 83, 11777–11783. [Google Scholar] [CrossRef]
- Haberthur, K.; Engelmann, F.; Park, B.; Barron, A.; Legasse, A.; Dewane, J.; Fischer, M.; Kerns, A.; Brown, M.; Messaoudi, I. CD4 T cell immunity is critical for the control of simian varicella virus infection in a nonhuman primate model of VZV infection. PLoS Pathog. 2011, 7, e1002367. [Google Scholar] [CrossRef]
- Brown, D.M.; Dilzer, A.M.; Meents, D.L.; Swain, S.L. CD4 T cell-mediated protection from lethal influenza: Perforin and antibody-mediated mechanisms give a one-two punch. J. Immunol. 2006, 177, 2888–2898. [Google Scholar]
- Mahon, B.P.; Katrak, K.; Nomoto, A.; Macadam, A.J.; Minor, P.D.; Mills, K.H. Poliovirus-specific CD4+ Th1 clones with both cytotoxic and helper activity mediate protective humoral immunity against a lethal poliovirus infection in transgenic mice expressing the human poliovirus receptor. J. Exp. Med. 1995, 181, 1285–1292. [Google Scholar] [CrossRef]
- Brien, J.D.; Uhrlaub, J.L.; Nikolich-Zugich, J. West nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J. Immunol. 2008, 181, 8568–8575. [Google Scholar]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef]
- Nikiforow, S.; Bottomly, K.; Miller, G. CD4+ T-cell effectors inhibit Epstein-Barr virus-induced B-cell proliferation. J. Virol. 2001, 75, 3740–3752. [Google Scholar] [CrossRef]
- Bickham, K.; Goodman, K.; Paludan, C.; Nikiforow, S.; Tsang, M.L.; Steinman, R.M.; Münz, C. Dendritic cells initiate immune control of Epstein-Barr virus transformation of B Lymphocytes in vitro. J. Exp. Med. 2003, 198, 1653–1663. [Google Scholar] [CrossRef]
- Nikiforow, S.; Bottomly, K.; Miller, G.; Münz, C. Cytolytic CD4+-T-cell clones reactive to EBNA1 inhibit Epstein-Barr virus-induced B-cell proliferation. J. Virol. 2003, 77, 12088–12104. [Google Scholar]
- Omiya, R.; Buteau, C.; Kobayashi, H.; Paya, C.V.; Celis, E. Inhibition of EBV-induced lymphoproliferation by CD4+ T cells specific for an MHC class II promiscuous epitope. J. Immunol. 2002, 169, 2172–2179. [Google Scholar]
- Haigh, T.A.; Lin, X.; Jia, H.; Hui, E.P.; Chan, A.T.C.; Rickinson, A.B.; Taylor, G.S. EBV Latent Membrane Proteins (LMPs) 1 and 2 as immunotherapeutic targets: LMP-specific CD4+ cytotoxic T cell recognition of EBV-transformed B cell lines. J. Immunol. 2008, 180, 1643–1654. [Google Scholar]
- Robertson, K.A.; Usherwood, E.J.; Nash, A.A. Regression of a murine gammaherpesvirus 68-positive B-Cell lymphoma mediated by CD4 T lymphocytes. J. Virol. 2001, 75, 3480–3482. [Google Scholar] [CrossRef]
- Koelle, D.M.; Schomogyi, M.; McClurkan, C.; Reymond, S.N.; Chen, H.B. CD4 T-cell responses to herpes simplex virus type 2 major capsid protein VP5: Comparison with responses to tegument and envelope glycoproteins. J. Virol. 2000, 74, 11422–11425. [Google Scholar] [CrossRef]
- Koelle, D.M.; Reymond, S.N.; Chen, H.; Kwok, W.W.; McClurkan, C.; Gyaltsong, T.; Petersdorf, E.W.; Rotkis, W.; Talley, A.R.; Harrison, D.A. Tegument-specific, virus-reactive CD4 T cells localize to the cornea in herpes simplex virus interstitial keratitis in humans. J. Virol. 2000, 74, 10930–10938. [Google Scholar] [CrossRef]
- Jing, L.; Haas, J.R.; Chong, T.M.; Bruckner, J.J.; Dann, G.C.; Dong, L.; Marshak, J.O.; McClurkan, C.L.; Yamamoto, T.N.; Bailer, S.M.; et al. Cross-presentation and genome-wide screening reveal candidate T cells antigens for a herpes simplex virus type 1 vaccine. J. Clin. Invest. 2012, 122, 654–673. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Landais, E.; Saulquin, X.; Scotet, E.; Trautmann, L.; Peyrat, M.-A.; Yates, J.L.; Kwok, W.W.; Bonneville, M.; Houssaint, E. Direct killing of Epstein-Barr virus (EBV)-infected B cells by CD4 T cells directed against the EBV lytic protein BHRF1. Blood 2004, 103, 1408–1416. [Google Scholar]
- Su, Z.; Peluso, M.V.; Raffegerst, S.H.; Schendel, D.J.; Roskrow, M.A. The generation of LMP2a-specific cytotoxic T lymphocytes for the treatment of patients with Epstein-Barr virus-positive Hodgkin disease. Eur. J. Immunol. 2001, 31, 947–958. [Google Scholar] [CrossRef]
- Paludan, C.; Bickham, K.; Nikiforow, S.; Tsang, M.L.; Goodman, K.; Hanekom, W.A.; Fonteneau, J.-F.; Stevanovic, S.; Munz, C. Epstein-barr nuclear antigen 1-specific CD4+ Th1 cells kill Burkitt's lymphoma cells. J. Immunol. 2002, 169, 1593–1603. [Google Scholar]
- Long, H.M.; Haigh, T.A.; Gudgeon, N.H.; Leen, A.M.; Tsang, C.-W.; Brooks, J.; Landais, E.; Houssaint, E.; Lee, S.P.; Rickinson, A.B.; et al. CD4+ T-cell responses to Epstein-Barr virus (EBV) latent-cycle antigens and the recognition of EBV-transformed lymphoblastoid cell lines. J. Virol. 2005, 79, 4896–4907. [Google Scholar]
- Wallace, L.E.; Wright, J.; Ulaeto, D.O.; Morgan, A.J.; Rickinson, A.B. Identification of two T-cell epitopes on the candidate Epstein-Barr virus vaccine glycoprotein gp340 recognized by CD4+ T-cell clones. J. Virol. 1991, 65, 3821–3828. [Google Scholar]
- Adhikary, D.; Behrends, U.; Moosmann, A.; Witter, K.; Bornkamm, G.W.; Mautner, J. Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J. Exp. Med. 2006, 203, 995–1006. [Google Scholar] [CrossRef]
- Long, H.M.; Leese, A.M.; Chagoury, O.L.; Connerty, S.R.; Quarcoopome, J.; Quinn, L.L.; Shannon-Lowe, C.; Rickinson, A.B. Cytotoxic CD4+ T cell responses to EBV contrast with CD8 responses in breadth of lytic cycle antigen choice and in lytic cycle recognition. J. Immunol. 2011, 187, 92–101. [Google Scholar] [CrossRef]
- Sabbah, S.; Jagne, Y.J.; Zuo, J.; de Silva, T.; Ahasan, M.M.; Brander, C.; Rowland-Jones, S.; Flanagan, K.L.; Hislop, A.D. T-cell immunity to Kaposi's sarcoma-associated herpesvirus: Recognition of primary effusion lymphoma with LANA-specific CD4+ T cells. Blood 2012, 119, 2083–2092. [Google Scholar]
- Jones, L.; Black, A.P.; Malavige, G.N.; Ogg, G.S. Persistent high frequencies of varicella-zoster virus ORF4 protein-specific CD4+ T cells after primary infection. J. Virol. 2006, 80, 9772–9778. [Google Scholar] [CrossRef]
- Malavige, G.N.; Jones, L.; Black, A.P.; Ogg, G.S. Rapid effector function of varicella-zoster virus glycoprotein I-specific CD4+ T cells many decades after primary infection. J. Infect. Dis. 2007, 195, 660–664. [Google Scholar] [CrossRef]
- Arvin, A.; Sharp, M.; Smith, S.; Koropchak, C.; Diaz, P.; Kinchington, P.; Ruyechan, W.; Hay, J. Equivalent recognition of a varicella-zoster virus immediate early protein (IE62) and glycoprotein I by cytotoxic T lymphocytes of either CD4+ or CD8+ phenotype. J. Immunol. 1991, 146, 257–264. [Google Scholar]
- Asanuma, H.; Sharp, M.; Maecker, H.T.; Vernon, C.M.; Arvin, A.M. Frequencies of memory T cells specific for varicella-zoster virus, herpes simplex virus, and cytomegalovirus by intracellular detection of cytokine expression. J. Infect. Dis. 2000, 181, 859–866. [Google Scholar] [CrossRef]
- Nastke, M.-D.; Becerra, A.; Yin, L.; Dominguez-Amorocho, O.; Gibson, L.; Stern, L.J.; Calvo-Calle, J.M. Human CD4+ T cell response to human herpesvirus 6. J. Virol. 2012, 86, 4776–4792. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Haigh, T.A.; Larsen, M.; Brooks, J.; Rickinson, A.B. A role for intercellular antigen transfer in the recognition of EBV-transformed B cell lines by EBV nuclear antigen-specific CD4+ T cells. J. Immunol. 2006, 177, 3746–3756. [Google Scholar]
- Mackay, L.K.; Long, H.M.; Brooks, J.M.; Taylor, G.S.; Leung, C.S.; Chen, A.; Wang, F.; Rickinson, A.B. T cell detection of a B-cell tropic virus infection: Newly-synthesised versus mature viral proteins as antigen sources for CD4 and CD8 epitope display. PLoS Pathog. 2009, 5, e1000699. [Google Scholar] [CrossRef]
- Landais, E.; Saulquin, X.; Bonneville, M.; Houssaint, E. Long-term MHC class II presentation of the EBV lytic protein BHRF1 by EBV latently infected B cells following capture of BHRF1 antigen. J. Immunol. 2005, 175, 7939–7946. [Google Scholar]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC Class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar]
- Hegde, N.R.; Dunn, C.; Lewinsohn, D.M.; Jarvis, M.A.; Nelson, J.A.; Johnson, D.C. Endogenous human cytomegalovirus gB is presented efficiently by MHC class II molecules to CD4+ CTL. J. Exp. Med. 2005, 202, 1109–1119. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Devlin, R.; Collins, H.L.; Ressing, M.E. Herpesvirus interference with major histocompatibility complex class II-Restricted T-Cell activation. J. Virol. 2007, 81, 4389–4396. [Google Scholar] [CrossRef]
- Abendroth, A.; Slobedman, B.; Lee, E.; Mellins, E.; Wallace, M.; Arvin, A.M. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J. Virol. 2000, 74, 1900–1907. [Google Scholar] [CrossRef]
- Cebulla, C.M.; Miller, D.M.; Zhang, Y.; Rahill, B.M.; Zimmerman, P.; Robinson, J.M.; Sedmak, D.D. Human cytomegalovirus disrupts constitutive MHC Class II expression. J. Immunol. 2002, 169, 167–176. [Google Scholar]
- Barcy, S.; Corey, L. Herpes simplex inhibits the capacity of lymphoblastoid B cell lines to stimulate CD4+ T cells. J. Immunol. 2001, 166, 6242–6249. [Google Scholar]
- Zuo, J.; Thomas, W.A.; Haigh, T.A.; Fitzsimmons, L.; Long, H.M.; Hislop, A.D.; Taylor, G.S.; Rowe, M. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog. 2011, 7, e1002455. [Google Scholar] [CrossRef]
- Cresswell, P. Assembly, transport, and function of MHC Class II molecules. Annu. Rev. Immunol. 1994, 12, 259–291. [Google Scholar] [CrossRef]
- Van den Hoorn, T.; Paul, P.; Jongsma, M.L.M.; Neefjes, J. Routes to manipulate MHC class II antigen presentation. Curr. Opin. Immunol. 2010, 23, 88–95. [Google Scholar]
- Ting, J.P.-Y.; Trowsdale, J. Genetic control of MHC Class II expression. Cell 2002, 109, S21–S33. [Google Scholar] [CrossRef]
- Li, D.; Qian, L.; Chen, C.; Shi, M.; Yu, M.; Hu, M.; Song, L.; Shen, B.; Guo, N. Down-regulation of MHC Class II expression through inhibition of CIITA transcription by lytic transactivator Zta during Epstein-Barr virus reactivation. J. Immunol. 2009, 182, 1799–1809. [Google Scholar] [CrossRef]
- Schmidt, K.; Wies, E.; Neipel, F. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits gamma interferon and major histocompatibility complex Class II expression. J. Virol. 2011, 85, 4530–4537. [Google Scholar] [CrossRef]
- Le Roy, E.; Muhlethaler-Mottet, A.; Davrinche, C.; Mach, B.; Davignon, J.-L. Escape of human cytomegalovirus from HLA-DR-restricted CD4+ T-cell response is mediated by repression of gamma interferon-induced Class II transactivator expression. J. Virol. 1999, 73, 6582–6589. [Google Scholar]
- Wiertz, E.J.H.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec6l-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef]
- Tomazin, R.; Boname, J.; Hegde, N.R.; Lewinsohn, D.M.; Altschuler, Y.; Jones, T.R.; Cresswell, P.; Nelson, J.A.; Riddell, S.R.; Johnson, D.C. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat. Med. 1999, 5, 1039–1043. [Google Scholar] [CrossRef]
- Chevalier, M.S.; Daniels, G.M.; Johnson, D.C. Binding of human cytomegalovirus US2 to major histocompatibility complex Class I and II proteins is not sufficient for their degradation. J. Virol. 2002, 76, 8265–8275. [Google Scholar] [CrossRef]
- Chevalier, M.S.; Johnson, D.C. Human cytomegalovirus US3 chimeras containing US2 cytosolic residues acquire major histocompatibility Class I and II protein degradation properties. J. Virol. 2003, 77, 4731–4738. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Wang, E.W.; Tortorella, D.; Schust, D.J.; Ploegh, H.L. Human cytomegalovirus US2 endoplasmic reticulum-lumenal domain dictates association with major histocompatibility complex class I in a locus-specific manner. J. Virol. 2001, 75, 5197–5204. [Google Scholar] [CrossRef]
- Hegde, N.R.; Tomazin, R.A.; Wisner, T.W.; Dunn, C.; Boname, J.M.; Lewinsohn, D.M.; Johnson, D.C. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: A novel mechanism for evading major histocompatibility complex Class II antigen presentation. J. Virol. 2002, 76, 10929–10941. [Google Scholar] [CrossRef]
- Odeberg, J.; Soderberg-Naucler, C. Reduced expression of HLA Class II molecules and interleukin-10- and transforming growth factor β1-Independent suppression of T-Cell proliferation in human cytomegalovirus-infected macrophage cultures. J. Virol. 2001, 75, 5174–5181. [Google Scholar] [CrossRef]
- Odeberg, J.; Plachter, B.; Branden, L.; Soderberg-Naucler, C. Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR α-chain. Blood 2003, 101, 4870–4877. [Google Scholar] [CrossRef]
- Sievers, E.; Neumann, J.; Raftery, M.; SchÖnrich, G.; Eis-Hübinger, A.M.; Koch, N. Glycoprotein B from strain 17 of herpes simplex virus type I contains an invariant chain homologous sequence that binds to MHC class II molecules. Immunology 2002, 107, 129–135. [Google Scholar] [CrossRef]
- Neumann, J.R.; Eis-Hubinger, A.M.; Koch, N. Herpes simplex virus type 1 targets the MHC Class II processing pathway for immune evasion. J. Immunol. 2003, 171, 3075–3083. [Google Scholar]
- Temme, S.; Eis-Hubinger, A.M.; McLellan, A.D.; Koch, N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J. Immunol. 2010, 184, 236–243. [Google Scholar] [CrossRef]
- Lantner, F.; Starlets, D.; Gore, Y.; Flaishon, L.; Yamit-Hezi, A.; Dikstein, R.; Leng, L.; Bucala, R.; Machluf, Y.; Oren, M.; et al. CD74 induces TAp63 expression leading to B-cell survival. Blood 2007, 110, 4303–4311. [Google Scholar] [CrossRef]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef]
- Li, Q.; Turk, S.M.; Hutt-Fletcher, L.M. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 1995, 69, 3987–3994. [Google Scholar]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar]
- Spriggs, M.K.; Armitage, R.J.; Comeau, M.R.; Strockbine, L.; Farrah, T.; Macduff, B.; Ulrich, D.; Alderson, M.R.; Mullberg, J.; Cohen, J.I. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J. Virol. 1996, 70, 5557–5563. [Google Scholar]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.W.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr Virus gp42 Protein Bound to the MHC Class II Receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.W.; Keating, S.; Gomez, R.; Franken, K.L.M.C.; Ottenhoff, T.H.M.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar]
- Kwong, A.D.; Kruper, J.A.; Frenkel, N. Herpes simplex virus virion host shutoff function. J. Virol. 1988, 62, 912–921. [Google Scholar]
- Zelus, B.D.; Stewart, R.S.; Ross, J. The virion host shutoff protein of herpes simplex virus type 1: Messenger ribonucleolytic activity in vitro. J. Virol. 1996, 70, 2411–2419. [Google Scholar]
- Taddeo, B.; Zhang, W.; Roizman, B. The UL41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 2827–2832. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [CrossRef]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.H.J.; Ressing, M.E.; Rowe, M. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar]
- Buisson, M.; Geoui, T.; Flot, D.; Tarbouriech, N.; Ressing, M.E.; Wiertz, E.J.; Burmeister, W.P. A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J. Mol. Biol. 2009, 391, 717–728. [Google Scholar] [CrossRef]
- Bagneris, C.; Briggs, L.C.; Savva, R.; Ebrahimi, B.; Barrett, T.E. Crystal structure of a KSHV-SOX-DNA complex: Insights into the molecular mechanisms underlying DNase activity and host shutoff. Nucleic Acids Res. 2011, 39, 5744–5756. [Google Scholar] [CrossRef]
- Trgovcich, J.; Johnson, D.; Roizman, B. Cell surface major histocompatibility complex Class II proteins are regulated by the products of the γ134.5 and UL41 genes of herpes simplex virus 1. J. Virol. 2002, 76, 6974–6986. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Saccani, S.; Izotova, L.S.; Mirochnitchenko, O.V.; Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. USA 2000, 97, 1695–1700. [Google Scholar] [CrossRef]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampe, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar]
- Koppelman, B.; Neefjes, J.J.; de Vries, J.E.; de Waal Malefyt, R. Interleukin-10 down-regulates MHC Class II α β peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity 1997, 7, 861–871. [Google Scholar] [CrossRef]
- Spencer, J.V.; Lockridge, K.M.; Barry, P.A.; Lin, G.; Tsang, M.; Penfold, M.E.T.; Schall, T.J. Potent immunosuppressive activities of cytomegalovirus- encoded interleukin-10. J. Virol. 2002, 76, 1285–1292. [Google Scholar]
- Butler, L.M.; Jeffery, H.C.; Wheat, R.L.; Long, H.M.; Rae, P.C.; Nash, G.B.; Blackbourn, D.J. Kaposi's sarcoma-associated herpesvirus inhibits expression and function of endothelial cell major histocompatibility complex Class II via suppressor of cytokine signaling 3. J. Virol. 2012, 86, 7158–7166. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (Lhttp://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zuo, J.; Rowe, M. Herpesviruses Placating the Unwilling Host: Manipulation of the MHC Class II Antigen Presentation Pathway. Viruses 2012, 4, 1335-1353. https://doi.org/10.3390/v4081335
Zuo J, Rowe M. Herpesviruses Placating the Unwilling Host: Manipulation of the MHC Class II Antigen Presentation Pathway. Viruses. 2012; 4(8):1335-1353. https://doi.org/10.3390/v4081335
Chicago/Turabian StyleZuo, Jianmin, and Martin Rowe. 2012. "Herpesviruses Placating the Unwilling Host: Manipulation of the MHC Class II Antigen Presentation Pathway" Viruses 4, no. 8: 1335-1353. https://doi.org/10.3390/v4081335