Persistence versus Reversion of 3TC Resistance in HIV-1 Determine the Rate of Emergence of NVP Resistance

Abstract
:
1. Introduction
1.1. In vitro ‘Combination Therapy’
1.2. Lamivudine (3TC) Resistance—The M184V Substitution
1.3. Maintaining 3TC Pressure When NNRTI Are Introduced
1.4. Adding Adefovir (ADV)
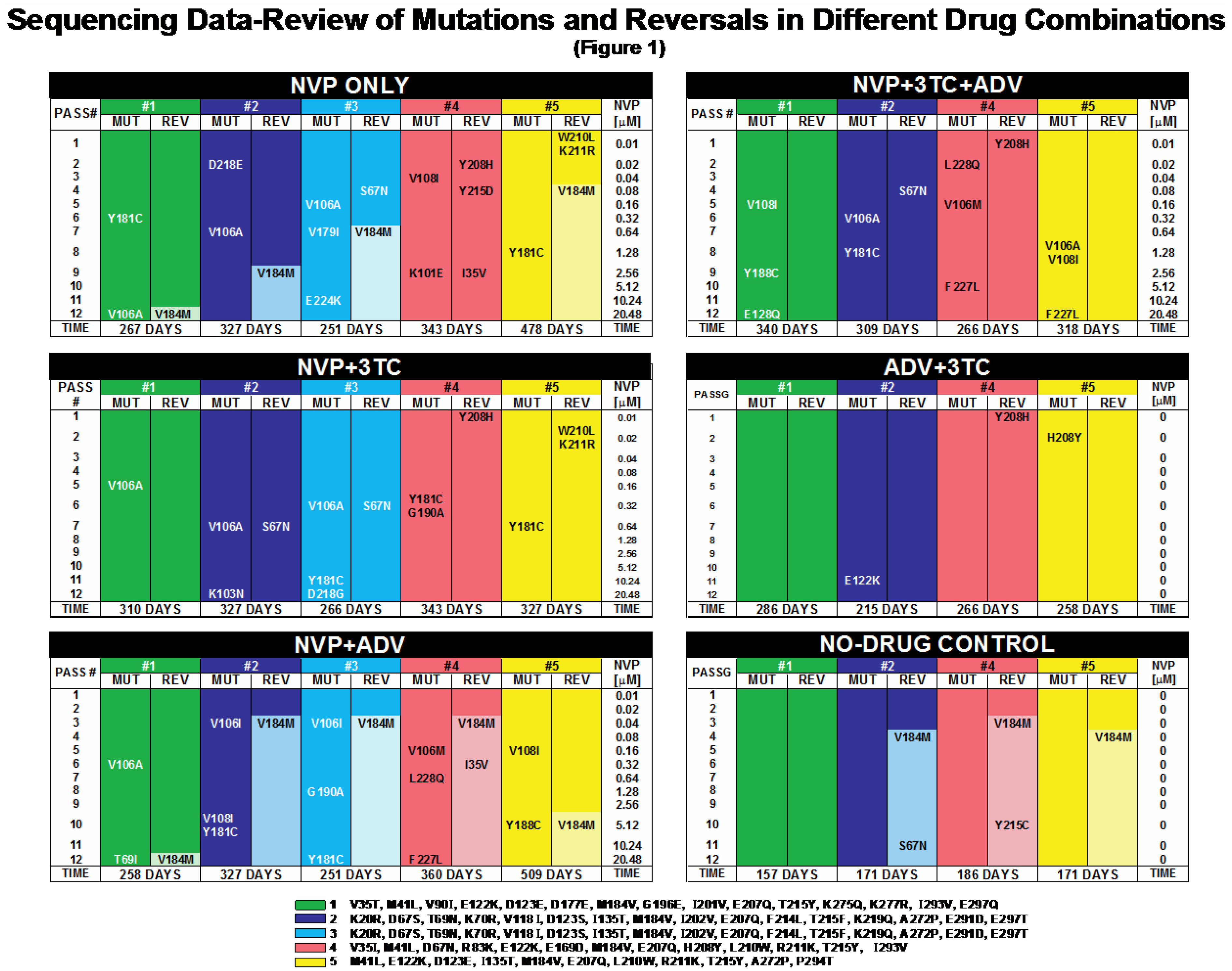
2. Results and Discussion
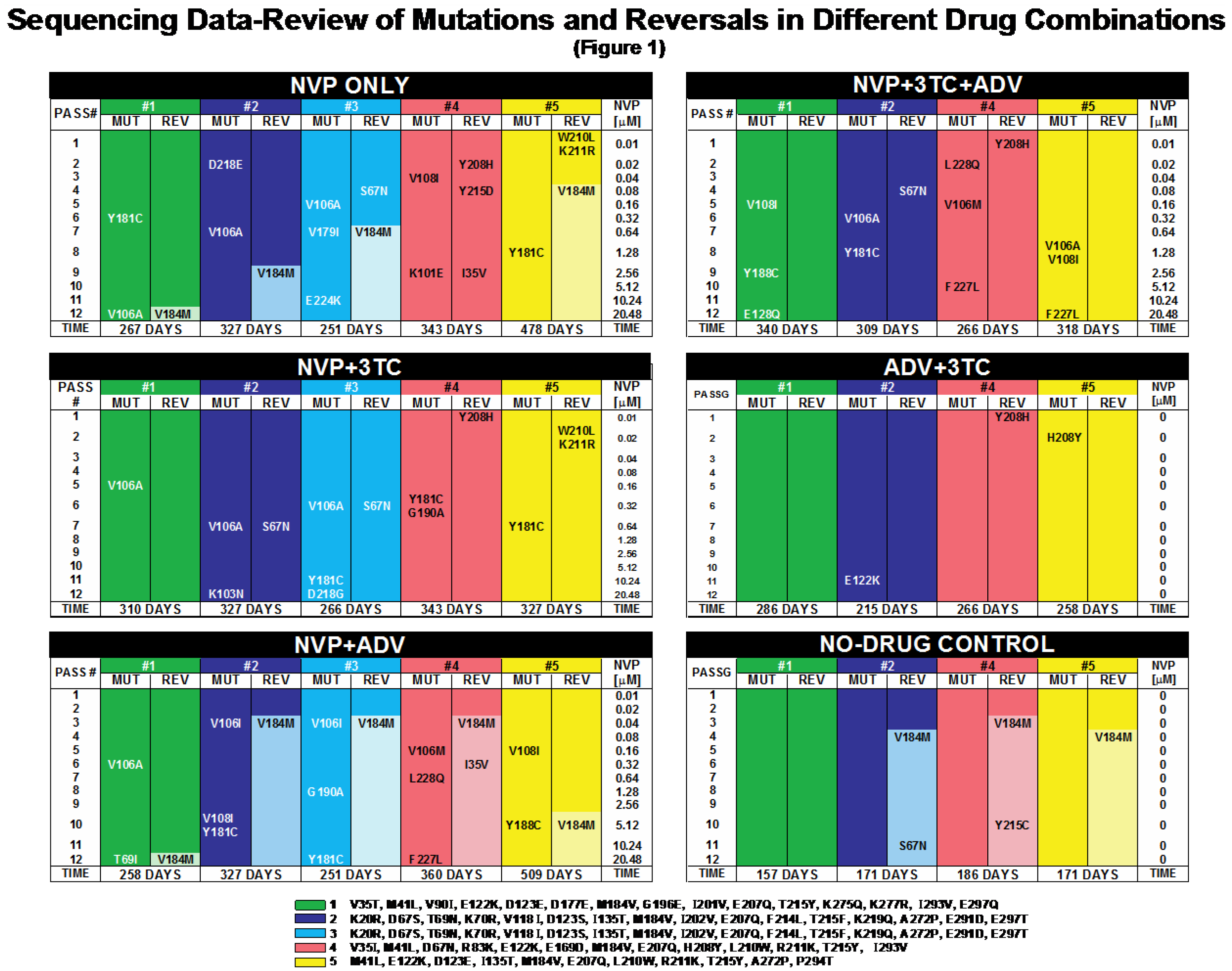
2.1. Mutations and Reversals in Different Drug Combinations
2.1.2. Significance of NNRTI Binding Pocket Mutations
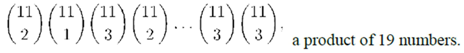
2.1.3. Maintaining versus Withdrawing 3TC Pressure
2.1.4. The Impact of Chance Effects
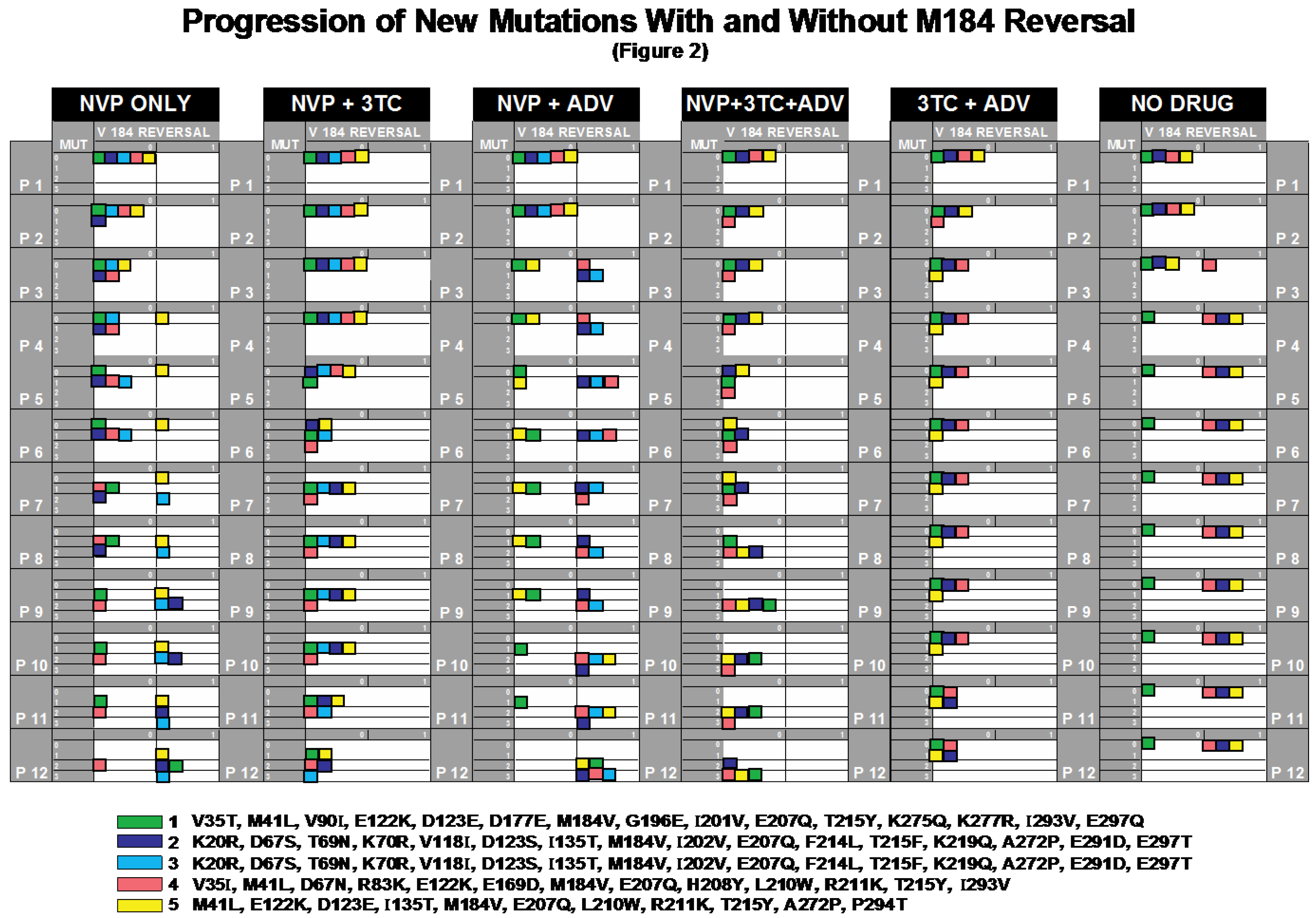
2.2. Progression of Mutations with and without M184V Reversal
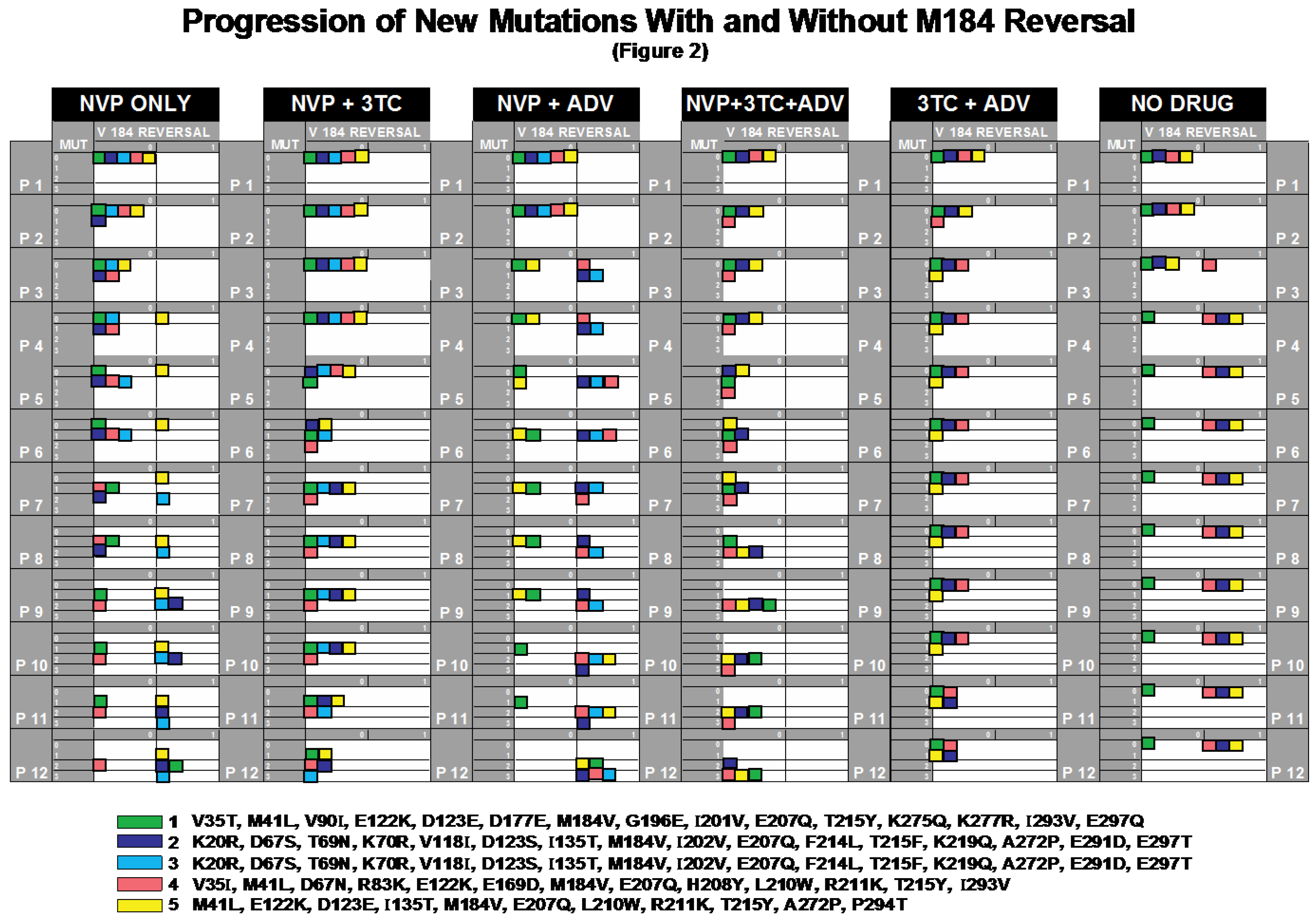
2.2.1. NVP_only
2.2.2. NVP+3TC
2.2.3. NVP+ADV
2.2.4. NVP+3TC+ADV
2.2.5. 3TC+ADV
2.2.6. No_Drug
2.2.7. Testing the 3TC-Effect
- (A) NVP_only has significantly more mutations than 3TC+ADV at passages 8 through 10 and 12.
- (B) NVP+3TC has significantly fewer mutations than NVP+3TC+ADV at passages 9 and 10.
- (C) NVP+3TC has significantly more mutations than 3TC+ADV at passages 7 through 10.
- (D) NVP+ADV has significantly more mutations than 3TC+ADV at passages 6 through 12.
- (E) NVP+3TC+ADV has significantly more mutations than 3TC+ADV at passages 9 through 12.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Passage Number | ||||||
|---|---|---|---|---|---|---|---|
| 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A) NVP_only versus 3TC+ADV | 0.5238 | 0.1429 | 0.0317 | 0.0476 | 0.0476 | 0.0794 | 0.0317 |
| B) NVP+3TC versus NVP+3TC+ADV | 0.9762 | 1.000 | 0.2063 | 0.0476 | 0.0397 | 0.1190 | 0.1429 |
| C) NVP+3TC versus 3TC+ADV | 0.4444 | 0.0397 | 0.0397 | 0.0397 | 0.0397 | 0.1587 | 0.0794 |
| D) NVP+ADV versus 3TC+ADV | 0.0476 | 0.0397 | 0.0317 | 0.0317 | 0.0238 | 0.0317 | 0.0079 |
| E) NVP+3TC+ADV versus 3TC+ADV | 0.3714 | 0.3714 | 0.0571 | 0.0286 | 0.0286 | 0.0286 | 0.0286 |
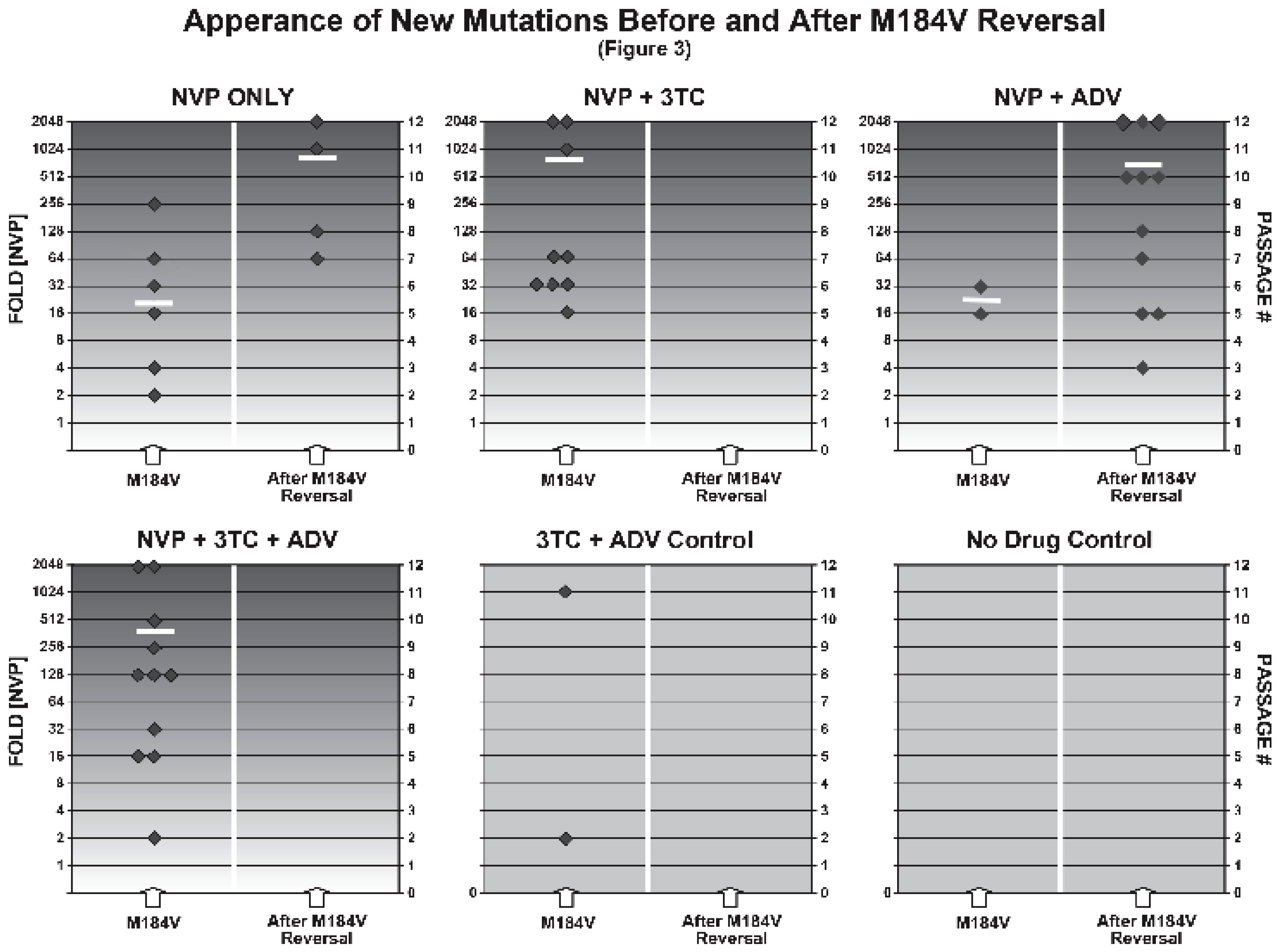
2.3. Appearance of New Mutations before and after 184 Reversal
2.4. Summary of Results
- 1. The presence of low-dose (1μM) 3TC prevented reversal to wild-type from an M184V mutant background.
- 2. Adding low-dose 3TC in the presence of NVP delayed the selection of NVP-associated mutations.
- 3. The presence of ADV, in addition to NVP, led to more rapid reversal to wild-type at position 184 than NVP alone.
- 4. ADV plus NVP selected for greater numbers of mutations than NVP alone.
2.5. Discussion
3. Experimental Section
3.1. Test Compounds
3.2. HIV Strains
3.3. Cells and Cell Culture
3.4. Passage Experiments
3.5. ABI Sequencing
3.6. Statistical Methods
3.6.1. Viral Particle Model
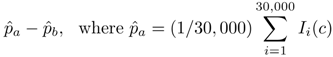
 is defined by analogy.
is defined by analogy.  ,
, 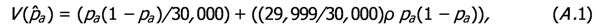

 by replacing pa on the right hand side of (A.1) by
by replacing pa on the right hand side of (A.1) by  .
. is estimated analogously. Because and
is estimated analogously. Because and  are clearly independent, our t-like statistic is now seen to be
are clearly independent, our t-like statistic is now seen to be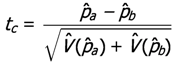
 = 0.8,
= 0.8,  = 0.2, and tc = 3.7482; and solve for ρ. The resulting ρ is 0.08.
= 0.2, and tc = 3.7482; and solve for ρ. The resulting ρ is 0.08.3.6.2. Population Models
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Camacho, R.; Teofilo, E. Antiretroviral therapy in treatment-naive patients with HIV infection. Current opinion in HIV and AIDS 2011, 6 Suppl 1, S3–S11. [Google Scholar] [CrossRef]
- Akanbi, M.O.; Scarci, K.; Taiwo, B.; Murphy, R.L. Combination nucleoside/nucleotide reverse transcriptase inhibitors for treatment of HIV infection. Expert Opin. Pharmacother. 2012, 13, 65–79. [Google Scholar] [CrossRef]
- Rouleau, D.; Fortin, C.; Trottier, B.; Lalonde, R.; Lapointe, N.; Cote, P.; Routy, J.P.; Matte, M.F.; Tsarevsky, I.; Baril, J.G. Antiretroviral therapy for adults infected with HIV: Guidelines for health care professionals from the Quebec HIV care committee. Can. J. Infect. Dis. Med. Microbiol. 2011, 22, 52–60. [Google Scholar]
- Sierra-Aragon, S.; Walter, H. Targets for inhibition of HIV replication: Entry, enzyme action, release and maturation. Intervirology 2012, 55, 84–97. [Google Scholar] [CrossRef]
- Van Rompay, K.K.; Matthews, T.B.; Higgins, J.; Canfield, D.R.; Tarara, R.P.; Wainberg, M.A.; Schinazi, R.F.; Pedersen, N.C.; North, T.W. Virulence and reduced fitness of simian immunodeficiency virus with the M184V mutation in reverse transcriptase. J. Virol. 2002, 76, 6083–6092. [Google Scholar]
- McColl, D.J.; Margot, N.; Chen, S.S.; Harris, J.; Borroto-Esoda, K.; Miller, M.D. Reduced emergence of the M184V/I resistance mutation when antiretroviral-naive subjects use emtricitabine versus lamivudine in regimens composed of two NRTIs plus the NNRTI efavirenz. HIV Clin. Trials 2011, 12, 61–70. [Google Scholar] [CrossRef]
- Svedhem, V.; Bergroth, T.; Lidman, K.; Sonnerborg, A. Presence of M184I/V in minor HIV-1 populations of patients with lamivudine and/or didanosine treatment failure. HIV Med. 2007, 8, 504–510. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Das, K.; Clark, A.D., Jr.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl. Acad. Sci. USA 1999, 96, 10027–10032. [Google Scholar]
- Wei, X.; Liang, C.; Gotte, M.; Wainberg, M.A. Negative effect of the M184V mutation in HIV-1 reverse transcriptase on initiation of viral DNA synthesis. Virology 2003, 311, 202–212. [Google Scholar] [CrossRef]
- Wei, X.; Liang, C.; Gotte, M.; Wainberg, M.A. The M184V mutation in HIV-1 reverse transcriptase reduces the restoration of wild-type replication by attenuated viruses. AIDS (London, England) 2002, 16, 2391–2398. [Google Scholar] [CrossRef]
- Diallo, K.; Gotte, M.; Wainberg, M.A. Molecular impact of the M184V mutation in human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 2003, 47, 3377–3383. [Google Scholar] [CrossRef]
- Diallo, K.; Brenner, B.; Oliveira, M.; Moisi, D.; Detorio, M.; Gotte, M.; Wainberg, M.A. The M184V substitution in human immunodeficiency virus type 1 reverse transcriptase delays the development of resistance to amprenavir and efavirenz in subtype B and C clinical isolates. Antimicrob. Agents Chemother. 2003, 47, 2376–2379. [Google Scholar]
- Gotte, M.; Arion, D.; Parniak, M.A.; Wainberg, M.A. The M184V mutation in the reverse transcriptase of human immunodeficiency virus type 1 impairs rescue of chain-terminated DNA synthesis. J. Virol. 2000, 74, 3579–3585. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Dapp, M.J.; Holtz, C.M.; Mansky, L.M. Concomitant lethal mutagenesis of human immunodeficiency virus type 1. J. Mol. Biol. 2012, 419, 158–170. [Google Scholar] [CrossRef]
- Chen, R.; Quinones-Mateu, M.E.; Mansky, L.M. Drug resistance, virus fitness and HIV-1 mutagenesis. Curr. Pharm. Des. 2004, 10, 4065–4070. [Google Scholar] [CrossRef]
- Stumpp, S.N.; Heyn, B.; Brakmann, S. Activity-based selection of HIV-1 reverse transcriptase variants with decreased polymerization fidelity. Biol. Chem. 2010, 391, 665–674. [Google Scholar] [CrossRef]
- Wainberg, M.A. The impact of the M184V substitution on drug resistance and viral fitness. Expert Rev. Anti Infect. Ther. 2004, 2, 147–151. [Google Scholar] [CrossRef]
- Turner, D.; Brenner, B.G.; Routy, J.P.; Petrella, M.; Wainberg, M.A. Rationale for maintenance of the M184v resistance mutation in human immunodeficiency virus type 1 reverse transcriptase in treatment experienced patients. New Microbiol. 2004, 27, 31–39. [Google Scholar]
- Hauser, A.; Mugenyi, K.; Kabasinguzi, R.; Kuecherer, C.; Harms, G.; Kunz, A. Emergence and persistence of minor drug-resistant HIV-1 variants in Ugandan women after nevirapine single-dose prophylaxis. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Petrella, M.; Wainberg, M.A. Might the M184V substitution in HIV-1 RT confer clinical benefit? AIDS Rev. 2002, 4, 224–232. [Google Scholar]
- Mansky, L.M.; Cunningham, K.S. Virus mutators and antimutators: Roles in evolution, pathogenesis and emergence. Trends Genet. 2000, 16, 512–517. [Google Scholar] [CrossRef]
- Palmer, S.; Margot, N.; Gilbert, H.; Shaw, N.; Buckheit, R., Jr.; Miller, M. Tenofovir, adefovir, and zidovudine susceptibilities of primary human immunodeficiency virus type 1 isolates with non-B subtypes or nucleoside resistance. AIDS Res. Hum. Retrovir. 2001, 17, 1167–1173. [Google Scholar] [CrossRef]
- Miller, M.D.; Anton, K.E.; Mulato, A.S.; Lamy, P.D.; Cherrington, J.M. Human immunodeficiency virus type 1 expressing the lamivudine-associated M184V mutation in reverse transcriptase shows increased susceptibility to adefovir and decreased replication capability in vitro. J. Infect. Dis. 1999, 179, 92–100. [Google Scholar] [CrossRef]
- Lehmann, E.; D’Abrera, H. Nonparametrics: Statistical Methods based on Ranks; Mcgraw-Hill: New York, NY, USA, 1975; pp. 240–247. [Google Scholar]
- Efron, B. Large-Scale Inference: Empirical Bayes Methods for Estimation, Testing, and Prediction; Cambridge University Press: Cambridge, United Kingdom, 2010; Volume 1. [Google Scholar]
- Kekitiinwa, A.; Asiimwe, A.R.; Kasirye, P.; Korutaro, V.; Kitaka, S.; Maganda, A.; Madalon, M.; Graziano, F.M. Prospective long-term outcomes of a cohort of ugandan children with laboratory monitoring during antiretroviral therapy. Pediatr. Infect. Dis. J. 2012, 31, e117–e125. [Google Scholar] [CrossRef]
- Prosperi, M.C.; De Luca, A. Computational models for prediction of response to antiretroviral therapies. AIDS Rev. 2012, 14, 145–153. [Google Scholar]
- Baxter, J.D.; Mayers, D.L.; Wentworth, D.N.; Neaton, J.D.; Hoover, M.L.; Winters, M.A.; Mannheimer, S.B.; Thompson, M.A.; Abrams, D.I.; Brizz, B.J.; et al. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. AIDS (London, England) 2000, 14, F83–F93. [Google Scholar] [CrossRef]
- Hill, A.L.; Rosenbloom, D.I.; Nowak, M.A. Evolutionary dynamics of HIV at multiple spatial and temporal scales. J. Mol. Med. (Berl. Ger.) 2012, 90, 543–561. [Google Scholar] [CrossRef]
- Fox, Z.; Dragsted, U.B.; Gerstoft, J.; Phillips, A.N.; Kjaer, J.; Mathiesen, L.; Youle, M.; Katlama, C.; Hill, A.; Bruun, J.N.; et al. A randomized trial to evaluate continuation versus discontinuation of lamivudine in individuals failing a lamivudine-containing regimen: The COLATE trial. Antivir. Ther. 2006, 11, 761–770. [Google Scholar]
- Quan, Y.; Brenner, B.G.; Oliveira, M.; Wainberg, M.A. Lamivudine can exert a modest antiviral effect against human immunodeficiency virus type 1 containing the M184V mutation. Antimicrob. Agents Chemother. 2003, 47, 747–754. [Google Scholar]
- Rezende, L.F.; Prasad, V.R. Nucleoside-analog resistance mutations in HIV-1 reverse transcriptase and their influence on polymerase fidelity and viral mutation rates. Int. J. Biochem. Cell Biol. 2004, 36, 1716–1734. [Google Scholar]
- Keulen, W.; van Wijk, A.; Schuurman, R.; Berkhout, B.; Boucher, C.A. Increased polymerase fidelity of lamivudine-resistant HIV-1 variants does not limit their evolutionary potential. AIDS (London, England) 1999, 13, 1343–1349. [Google Scholar] [CrossRef]
- Jonckheere, H.; Witvrouw, M.; de Clercq, E.; Anne, J. Lamivudine resistance of HIV type 1 does not delay development of resistance to nonnucleoside HIV type 1-specific reverse transcriptase inhibitors as compared with wild-type HIV type 1. AIDS Res. Hum. Retrovir. 1998, 14, 249–253. [Google Scholar] [CrossRef]
- Balzarini, J.; Pelemans, H.; Karlsson, A.; de Clerc, Q.E.; Kleim, J.P. Concomitant combination therapy for HIV infection preferable over sequential therapy with 3TC and non-nucleoside reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. USA 1996, 93, 13152–13157. [Google Scholar] [CrossRef]
- Shafer, R.W.; Hertogs, K.; Zolopa, A.R.; Warford, A.; Bloor, S.; Betts, B.J.; Merigan, T.C.; Harrigan, R.; Larder, B.A. High degree of interlaboratory reproducibility of human immunodeficiency virus type 1 protease and reverse transcriptase sequencing of plasma samples from heavily treated patients. J. Clin. Microbiol. 2001, 39, 1522–1529. [Google Scholar] [CrossRef]
- Winters, M.A.; Holodniy, M.; Katzenstein, D.A.; Merigan, T.C. Quantitative RNA and DNA gene amplification can rapidly monitor HIV infection and antiviral activity in cell cultures. PCR Methods Appl. 1992, 1, 257–262. [Google Scholar]
- Holodniy, M.; Katzenstein, D.A.; Sengupta, S.; Wang, A.M.; Casipit, C.; Schwartz, D.H.; Konrad, M.; Groves, E.; Merigan, T.C. Detection and quantification of human immunodeficiency virus RNA in patient serum by use of the polymerase chain reaction. J. Infect. Dis. 1991, 163, 862–866. [Google Scholar]
- Tang, M.W.; Shafer, R.W. HIV-1 Antiretroviral resistance: Scientific principles and clinical applications. Drugs 2012, 72, e1–e25. [Google Scholar]
- Melikian, G.L.; Rhee, S.Y.; Taylor, J.; Fessel, W.J.; Kaufman, D.; Towner, W.; Troia-Cancio, P.V.; Zolopa, A.; Robbins, G.K.; Kagan, R.; Israelski, D.; Shafer, R.W. Standardized comparison of the relative impacts of HIV-1 reverse transcriptase (RT) mutations on nucleoside RT inhibitor susceptibility. Antimicrob. Agents Chemother. 2012, 56, 2305–2313. [Google Scholar] [CrossRef]
- Palmer, S.; Shafer, R.W.; Merigan, T.C. Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 1999, 13, 661–667. [Google Scholar]
- Paxton, W.B.; Coombs, R.W.; McElrath, M.J.; Keefer, M.C.; Hughes, J.; Sinangil, F.; Chernoff, D.; Demeter, L.; Williams, B.; Corey, L. Longitudinal analysis of quantitative virologic measures in human immunodeficiency virus-infected subjects with > or = 400 CD4 lymphocytes: Implications for applying measurements to individual patients. National Institute of Allergy and Infectious Diseases AIDS Vaccine Evaluation Group. J. Infect. Dis. 1997, 175, 247–254. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Tirado-Rives, J.; Jorgensen, W.L. Estimation of binding affinities for HEPT and nevirapine analogues with HIV-1 reverse transcriptase via Monte Carlo simulations. J. Med. Chem. 2001, 44, 145–154. [Google Scholar] [CrossRef]
- Delaugerre, C.; Gallien, S.; Flandre, P.; Mathez, D.; Amarsy, R.; Ferret, S.; Timsit, J.; Molina, J.M.; de Truchis, P. Impact of low-level-viremia on HIV-1 drug-resistance evolution among antiretroviral treated-patients. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Messiaen, P.; Verhofstede, C.; Vandenbroucke, I.; Dinakis, S.; Van Eygen, V.; Thys, K.; Winters, B.; Aerssens, J.; Vogelaers, D.; Stuyver, L.J.; Vandekerckhove, L. Ultra-deep sequencing of HIV-1 reverse transcriptase before start of an NNRTI-based regimen in treatment-naive patients. Virology 2012, 426, 7–11. [Google Scholar] [CrossRef]
- Murry, J.P.; Higgins, J.; Matthews, T.B.; Huang, V.Y.; van Rompay, K.K.; Pedersen, N.C.; North, T.W. Reversion of the M184V mutation in simian immunodeficiency virus reverse transcriptase is selected by tenofovir, even in the presence of lamivudine. J. Virol. 2003, 77, 1120–1130. [Google Scholar]
- Diallo, K.; Oliveira, M.; Moisi, D.; Brenner, B.; Wainberg, M.A.; Gotte, M. Pressure of zidovudine accelerates the reversion of lamivudine resistance-conferring M184V mutation in the reverse transcriptase of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2002, 46, 2254–2256. [Google Scholar] [CrossRef]
- Mansky, L.M.; Bernard, L.C. 3'-Azido-3'-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J. Virol. 2000, 74, 9532–9539. [Google Scholar] [CrossRef]
- Chen, R.; Yokoyama, M.; Sato, H.; Reilly, C.; Mansky, L.M. Human immunodeficiency virus mutagenesis during antiviral therapy: Impact of drug-resistant reverse transcriptase and nucleoside and nonnucleoside reverse transcriptase inhibitors on human immunodeficiency virus type 1 mutation frequencies. J. Virol. 2005, 79, 12045–12057. [Google Scholar] [CrossRef]
- Opravil, M.; Klimkait, T.; Louvel, S.; Wolf, E.; Battegay, M.; Fux, C.A.; Bernasconi, E.; Vogel, M.; Speck, R.; Weber, R. Prior therapy influences the efficacy of lamivudine monotherapy in patients with lamivudine-resistant HIV-1 infection. J. Acquir. Immune Defic. Syndr. (1999) 2010, 54, 51–58. [Google Scholar]
- Deeks, S.G.; Wrin, T.; Liegler, T.; Hoh, R.; Hayden, M.; Barbour, J.D.; Hellmann, N.S.; Petropoulos, C.J.; McCune, J.M.; Hellerstein, M.K.; Grant, R.M. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 2001, 344, 472–480. [Google Scholar]
- Metzner, K.J.; Leemann, C.; di Giallonardo, F.; Grube, C.; Scherrer, A.U.; Braun, D.; Kuster, H.; Weber, R.; Guenthard, H.F. Reappearance of minority K103N HIV-1 variants after interruption of ART initiated during primary HIV-1 infection. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Paquet, A.C.; Baxter, J.; Weidler, J.; Lie, Y.; Lawrence, J.; Kim, R.; Bates, M.; Coakley, E.; Chappey, C. Differences in reversion of resistance mutations to wild-type under structured treatment interruption and related increase in replication capacity. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Campbell, T.B.; Shulman, N.S.; Johnson, S.C.; Zolopa, A.R.; Young, R.K.; Bushman, L.; Fletcher, C.V.; Lanier, E.R.; Merigan, T.C.; Kuritzkes, D.R. Antiviral activity of lamivudine in salvage therapy for multidrug-resistant HIV-1 infection. Clin. Infect. Dis. 2005, 41, 236–242. [Google Scholar]
- Lecossier, D.; Shulman, N.S.; Morand-Joubert, L.; Shafer, R.W.; Joly, V.; Zolopa, A.R.; Clavel, F.; Hance, A.J. Detection of minority populations of HIV-1 expressing the K103N resistance mutation in patients failing nevirapine. J. Acquir. Immune Defic. Syndr. (1999) 2005, 38, 37–42. [Google Scholar] [CrossRef]
- Shulman, N.S.; Zolopa, A.R.; Passaro, D.J.; Murlidharan, U.; Israelski, D.M.; Brosgart, C.L.; Miller, M.D.; Van Doren, S.; Shafer, R.W.; Katzenstein, D.A. Efavirenz- and adefovir dipivoxil-based salvage therapy in highly treatment-experienced patients: Clinical and genotypic predictors of virologic response. J. Acquir. Immune Defic. Syndr. (1999) 2000, 23, 221–226. [Google Scholar]
- Winters, M.A.; Schapiro, J.M.; Lawrence, J.; Merigan, T.C. Human immunodeficiency virus type 1 protease genotypes and in vitro protease inhibitor susceptibilities of isolates from individuals who were switched to other protease inhibitors after long-term saquinavir treatment. J. Virol. 1998, 72, 5303–5306. [Google Scholar]
- Nijhuis, M.; Boucher, C.A.; Schuurman, R. Sensitive procedure for the amplification of HIV-1 RNA using a combined reverse-transcription and amplification reaction. Biotechniques 1995, 19, 178–180, 182. [Google Scholar]
- Schapiro, J.M.; Winters, M.A.; Stewart, F.; Efron, B.; Norris, J.; Kozal, M.J.; Merigan, T.C. The effect of high-dose saquinavir on viral load and CD4+ T-cell counts in HIV-infected patients. Ann. Internal Med. 1996, 124, 1039–1050. [Google Scholar]
- Winters, M.A.; Coolley, K.L.; Girard, Y.A.; Levee, D.J.; Hamdan, H.; Shafer, R.W.; Katzenstein, D.A.; Merigan, T.C. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J. Clin. Invest. 1998, 102, 1769–1775. [Google Scholar] [CrossRef]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; Chapman & Hall/CRC: New York, NY, USA, 1993; Volume 57. [Google Scholar]
- Rice, J.A. Mathematical Statistics and Data Analysis, 3rd ed; Thompson Higher Education: Belmont, CA, USA, 2006. [Google Scholar]
- Raikov, D. On the decomposition of Gauss and Poisson laws (in Russian). Izv. Ross. Akad. Nauk. Ser. Mat. 1938, 2, 91–124. [Google Scholar]
- Fisher, R. Statistical Methods for Research Workers, 19th ed; Oliver and Boyd: Edinburgh, Scotland, UK, 1963. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rath, B.A.; Olshen, R.A.; Halpern, J.; Merigan, T.C. Persistence versus Reversion of 3TC Resistance in HIV-1 Determine the Rate of Emergence of NVP Resistance. Viruses 2012, 4, 1212-1234. https://doi.org/10.3390/v4081212
Rath BA, Olshen RA, Halpern J, Merigan TC. Persistence versus Reversion of 3TC Resistance in HIV-1 Determine the Rate of Emergence of NVP Resistance. Viruses. 2012; 4(8):1212-1234. https://doi.org/10.3390/v4081212
Chicago/Turabian StyleRath, Barbara A., Richard A. Olshen, Jerry Halpern, and Thomas C. Merigan. 2012. "Persistence versus Reversion of 3TC Resistance in HIV-1 Determine the Rate of Emergence of NVP Resistance" Viruses 4, no. 8: 1212-1234. https://doi.org/10.3390/v4081212