Herpesvirus Exploitation of Host Immune Inhibitory Pathways

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mammalian and Viral Interleukin-10
2.1. EBV BCRF1
2.2. HCMV UL111A
2.3. Mammalian IL-10 and Herpesvirus Infections
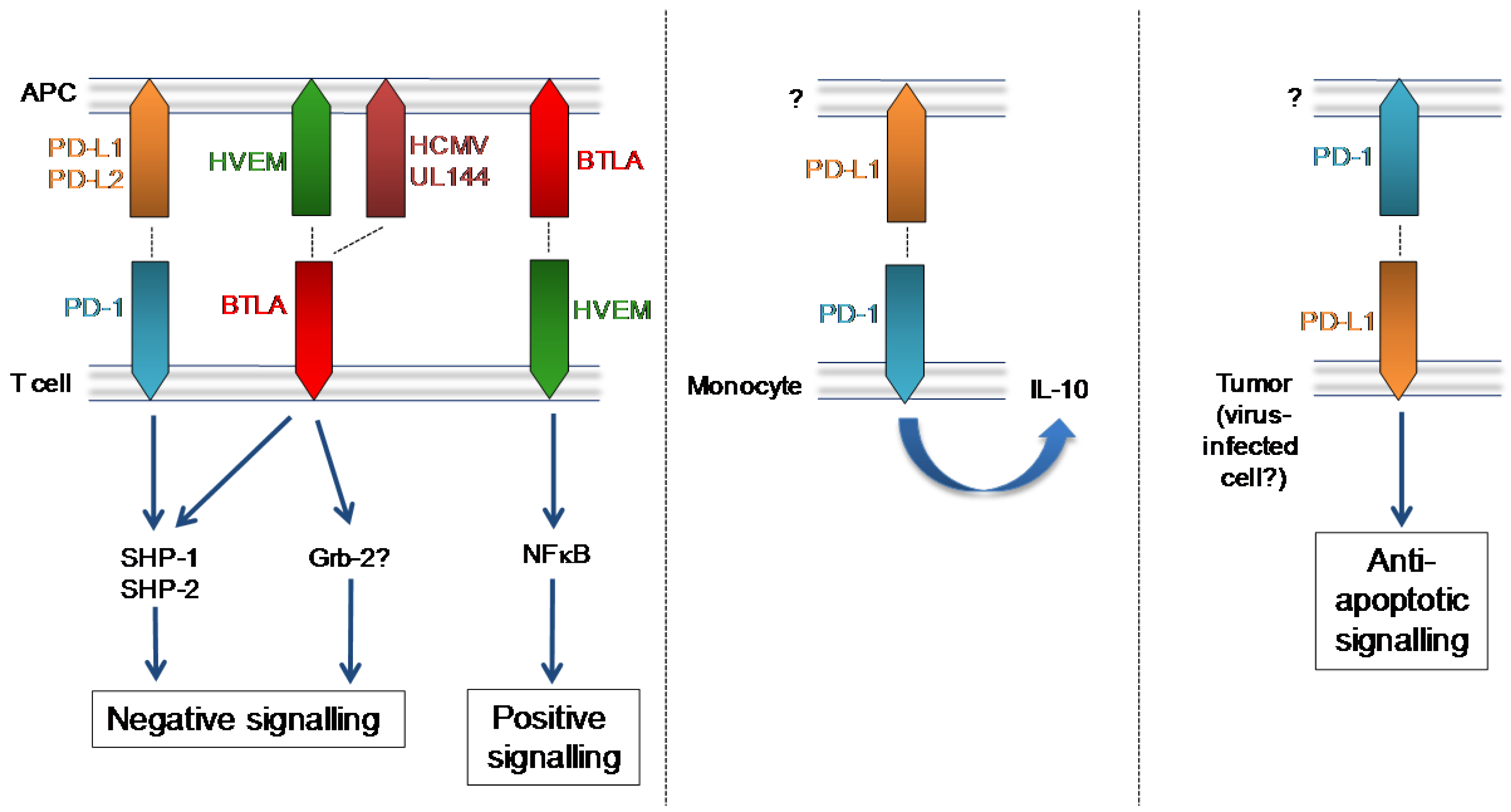
3. Programmed Death Receptor (PD-1)
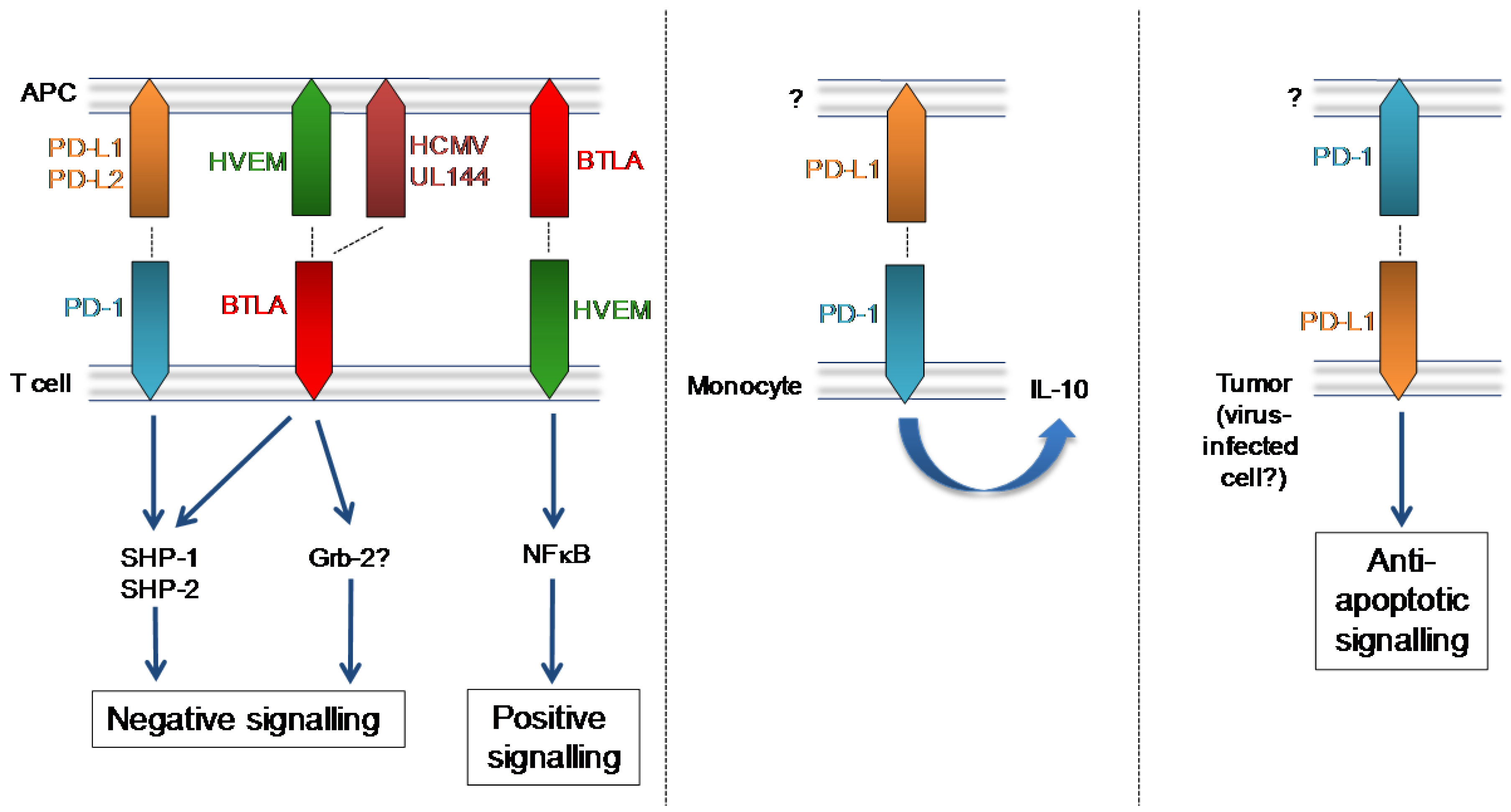
4. B and T Lymphocyte Attenuator (BTLA)
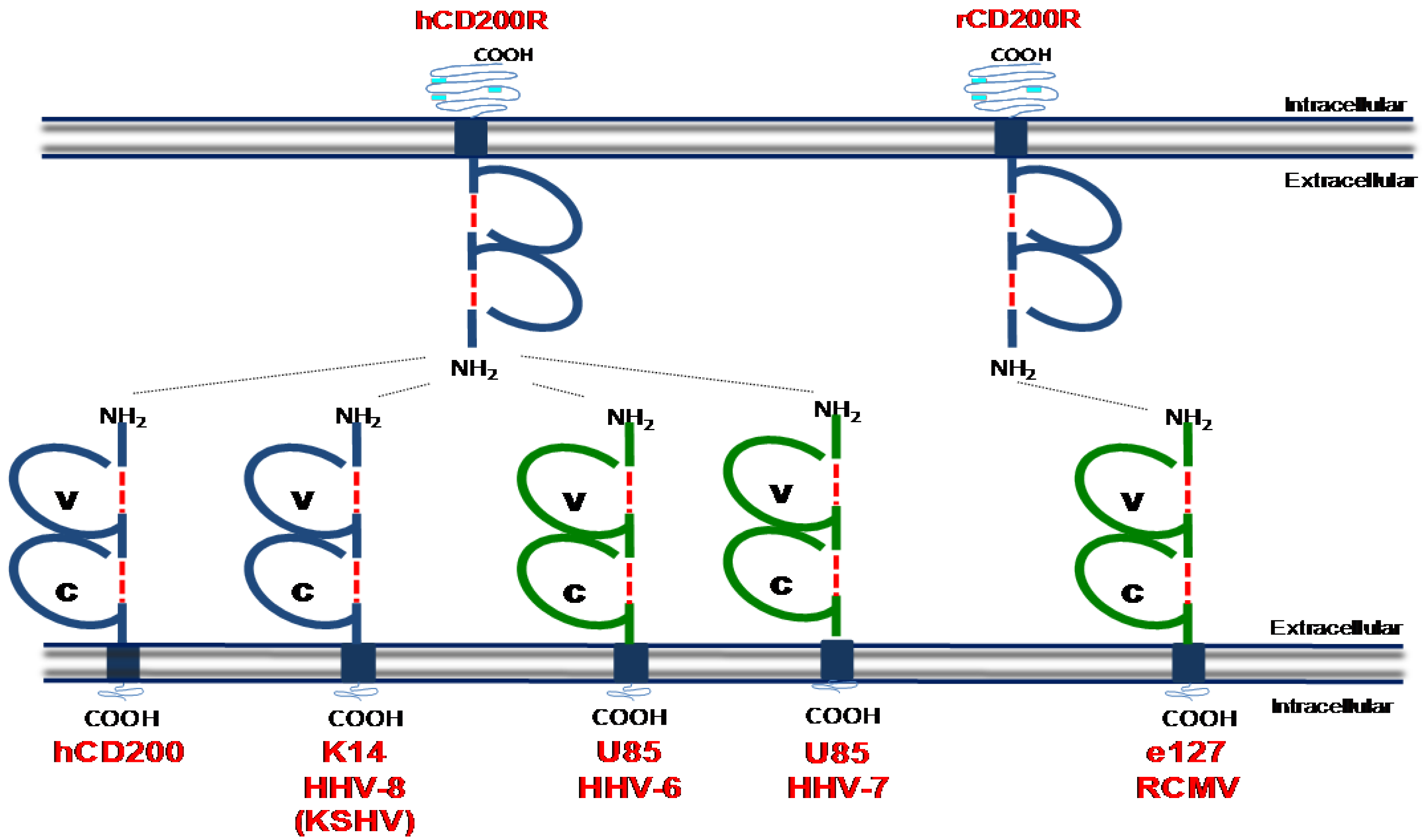
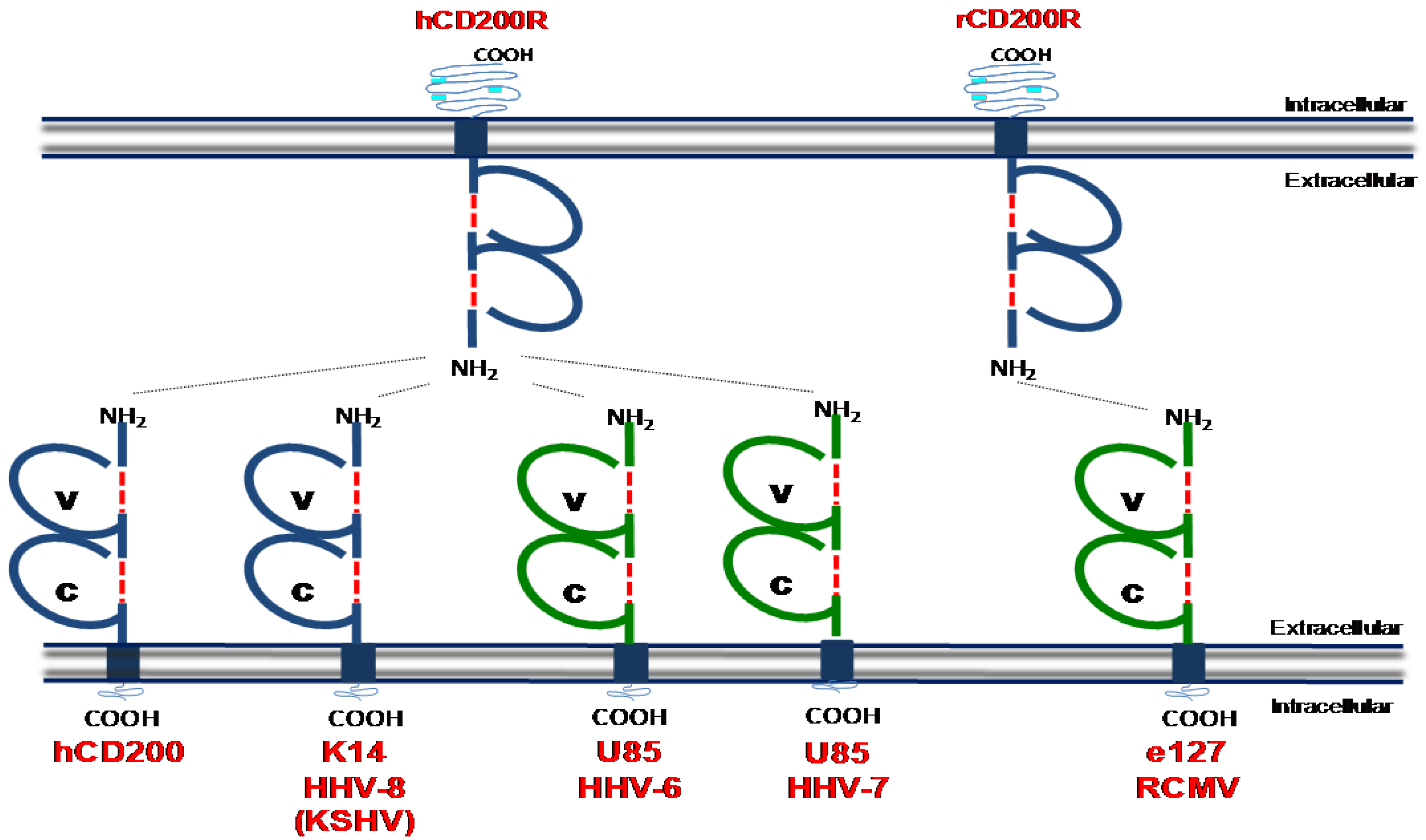
5. CD200:CD200 Receptor Pathway
Viral CD200 Homologues (vCD200s)
6. Mammalian and Viral Immune Inhibitory Molecules: Therapeutic Targets or Necessary Evils?
Supplementary Files
Acknowledgements
Conflicts of Interest
References
- Zamora, M.R. DNA viruses (CMV, EBV, and the herpesviruses). Semin. Respir. Crit. Care Med. 2011, 32, 454–470. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Gatherer, D. Integrating reptilian herpesviruses into the family herpesviridae. J. Virol. 2005, 79, 725–731. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef]
- O'Garra, A.; Barrat, F.J.; Castro, A.G.; Vicari, A.; Hawrylowicz, C. Strategies for use of IL-10 or its antagonists in human disease. Immunol. Rev. 2008, 223, 114–131. [Google Scholar] [CrossRef]
- Moore, K.W.; O'Garra, A.; de Waal Malefyt, R.; Vieira, P.; Mosmann, T.R. Interleukin-10. Annu. Rev. Immunol. 1993, 11, 165–190. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar]
- Sun, J.; Cardani, A.; Sharma, A.K.; Laubach, V.E.; Jack, R.S.; Muller, W.; Braciale, T.J. Autocrine regulation of pulmonary inflammation by effector T-cell derived IL-10 during infection with respiratory syncytial virus. PLoS Pathog. 2011, 7, e1002173. [Google Scholar] [CrossRef]
- Loebbermann, J.; Schnoeller, C.; Thornton, H.; Durant, L.; Sweeney, N.P.; Schuijs, M.; O'Garra, A.; Johansson, C.; Openshaw, P.J. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS One 2012, 7, e32371. [Google Scholar]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar]
- McKinstry, K.K.; Strutt, T.M.; Buck, A.; Curtis, J.D.; Dibble, J.P.; Huston, G.; Tighe, M.; Hamada, H.; Sell, S.; Dutton, R.W.; et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J. Immunol. 2009, 182, 7353–7363. [Google Scholar]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef]
- Lockridge, K.M.; Zhou, S.S.; Kravitz, R.H.; Johnson, J.L.; Sawai, E.T.; Blewett, E.L.; Barry, P.A. Primate cytomegaloviruses encode and express an IL-10-like protein. Virology 2000, 268, 272–280. [Google Scholar] [CrossRef]
- Lee, H.J.; Essani, K.; Smith, G.L. The genome sequence of Yaba-like disease virus, a yatapoxvirus. Virology 2001, 281, 170–192. [Google Scholar] [CrossRef]
- Rode, H.J.; Janssen, W.; Rosen-Wolff, A.; Bugert, J.J.; Thein, P.; Becker, Y.; Darai, G. The genome of equine herpesvirus type 2 harbors an interleukin 10 (IL10)-like gene. Virus Genes 1993, 7, 111–116. [Google Scholar] [CrossRef]
- Taus, N.S.; Herndon, D.R.; Traul, D.L.; Stewart, J.P.; Ackermann, M.; Li, H.; Knowles, D.P.; Lewis, G.S.; Brayton, K.A. Comparison of ovine herpesvirus 2 genomes isolated from domestic sheep (Ovis aries) and a clinically affected cow (Bos bovis). J. Gen. Virol. 2007, 88, 40–45. [Google Scholar] [CrossRef]
- Telford, E.A.; Watson, M.S.; Aird, H.C.; Perry, J.; Davison, A.J. The DNA sequence of equine herpesvirus 2. J. Mol. Biol. 1995, 249, 520–528. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Saccani, S.; Izotova, L.S.; Mirochnitchenko, O.V.; Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 1695–1700. [Google Scholar]
- Hsu, D.H.; de Waal Malefyt, R.; Fiorentino, D.F.; Dang, M.N.; Vieira, P.; de Vries, J.; Spits, H.; Mosmann, T.R.; Moore, K.W. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science 1990, 250, 830–832. [Google Scholar]
- Stewart, J.P.; Behm, F.G.; Arrand, J.R.; Rooney, C.M. Differential expression of viral and human interleukin-10 (IL-10) by primary B cell tumors and B cell lines. Virology 1994, 200, 724–732. [Google Scholar] [CrossRef]
- Touitou, R.; Cochet, C.; Joab, I. Transcriptional analysis of the Epstein-Barr virus interleukin-10 homologue during the lytic cycle. J. Gen. Virol. 1996, 77, 1163–1168. [Google Scholar] [CrossRef]
- de Waal Malefyt, R.; Haanen, J.; Spits, H.; Roncarolo, M.G.; te Velde, A.; Figdor, C.; Johnson, K.; Kastelein, R.; Yssel, H.; de Vries, J.E. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 1991, 174, 915–924. [Google Scholar] [CrossRef]
- Salek-Ardakani, S.; Arrand, J.R.; Mackett, M. Epstein-Barr virus encoded interleukin-10 inhibits HLA-class I, ICAM-1, and B7 expression on human monocytes: Implications for immune evasion by EBV. Virology 2002, 304, 342–351. [Google Scholar] [CrossRef]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampe, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar]
- Vieira, P.; de Waal-Malefyt, R.; Dang, M.N.; Johnson, K.E.; Kastelein, R.; Fiorentino, D.F.; deVries, J.E.; Roncarolo, M.G.; Mosmann, T.R.; Moore, K.W. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: Homology to Epstein-Barr virus open reading frame BCRFI. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 1172–1176. [Google Scholar]
- Defrance, T.; Vanbervliet, B.; Briere, F.; Durand, I.; Rousset, F.; Banchereau, J. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J. Exp. Med. 1992, 175, 671–682. [Google Scholar] [CrossRef]
- Go, N.F.; Castle, B.E.; Barrett, R.; Kastelein, R.; Dang, W.; Mosmann, T.R.; Moore, K.W.; Howard, M. Interleukin 10, a novel B cell stimulatory factor: Unresponsiveness of X chromosome-linked immunodeficiency B cells. J. Exp. Med. 1990, 172, 1625–1631. [Google Scholar] [CrossRef]
- Rousset, F.; Garcia, E.; Defrance, T.; Peronne, C.; Vezzio, N.; Hsu, D.H.; Kastelein, R.; Moore, K.W.; Banchereau, J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 1890–1893. [Google Scholar]
- Ding, Y.; Qin, L.; Kotenko, S.V.; Pestka, S.; Bromberg, J.S. A single amino acid determines the immunostimulatory activity of interleukin 10. J. Exp. Med. 2000, 191, 213–224. [Google Scholar] [CrossRef]
- Liu, Y.; de Waal Malefyt, R.; Briere, F.; Parham, C.; Bridon, J.M.; Banchereau, J.; Moore, K.W.; Xu, J. The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J. Immunol. 1997, 158, 604–613. [Google Scholar]
- Miyazaki, I.; Cheung, R.K.; Dosch, H.M. Viral interleukin 10 is critical for the induction of B cell growth transformation by Epstein-Barr virus. J. Exp. Med. 1993, 178, 439–447. [Google Scholar] [CrossRef]
- Jones, B.C.; Logsdon, N.J.; Josephson, K.; Cook, J.; Barry, P.A.; Walter, M.R. Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9404–9409. [Google Scholar]
- Slobedman, B.; Barry, P.A.; Spencer, J.V.; Avdic, S.; Abendroth, A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J. Virol. 2009, 83, 9618–9629. [Google Scholar]
- Spencer, J.V.; Lockridge, K.M.; Barry, P.A.; Lin, G.; Tsang, M.; Penfold, M.E.; Schall, T.J. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J. Virol. 2002, 76, 1285–1292. [Google Scholar]
- Raftery, M.J.; Wieland, D.; Gronewald, S.; Kraus, A.A.; Giese, T.; Schonrich, G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J. Immunol. 2004, 173, 3383–3391. [Google Scholar]
- Chang, W.L.; Baumgarth, N.; Yu, D.; Barry, P.A. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J. Virol. 2004, 78, 8720–8731. [Google Scholar] [CrossRef]
- Jenkins, C.; Garcia, W.; Godwin, M.J.; Spencer, J.V.; Stern, J.L.; Abendroth, A.; Slobedman, B. Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J. Virol. 2008, 82, 3736–3750. [Google Scholar]
- Jaworowski, A.; Cheng, W.J.; Westhorpe, C.L.; Abendroth, A.; Crowe, S.M.; Slobedman, B. Enhanced monocyte Fc phagocytosis by a homologue of interleukin-10 encoded by human cytomegalovirus. Virology 2009, 391, 20–24. [Google Scholar] [CrossRef]
- Cheung, A.K.; Gottlieb, D.J.; Plachter, B.; Pepperl-Klindworth, S.; Avdic, S.; Cunningham, A.L.; Abendroth, A.; Slobedman, B. The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: Implications for virus elimination during latency. Blood 2009, 114, 4128–4137. [Google Scholar]
- Lee, S.H.; Kim, K.S.; Fodil-Cornu, N.; Vidal, S.M.; Biron, C.A. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J. Exp. Med. 2009, 206, 2235–2251. [Google Scholar] [CrossRef]
- Stacey, M.A.; Marsden, M.; Wang, E.C.; Wilkinson, G.W.; Humphreys, I.R. IL-10 restricts activation-induced death of NK cells during acute murine cytomegalovirus infection. J. Immunol. 2011, 187, 2944–2952. [Google Scholar]
- Oakley, O.R.; Garvy, B.A.; Humphreys, S.; Qureshi, M.H.; Pomeroy, C. Increased weight loss with reduced viral replication in interleukin-10 knock-out mice infected with murine cytomegalovirus. Clin. Exp. Immunol. 2008, 151, 155–164. [Google Scholar]
- Mandaric, S.; Oxenius, A. Personal communication, 2012. Eidgenössische Technische Hochschule Zürich, Zurich, Switzerland..
- Cheeran, M.C.; Mutnal, M.B.; Hu, S.; Armien, A.; Lokensgard, J.R. Reduced lymphocyte infiltration during cytomegalovirus brain infection of interleukin-10-deficient mice. J. Neurovirol. 2009, 15, 334–342. [Google Scholar] [CrossRef]
- Sarangi, P.P.; Sehrawat, S.; Suvas, S.; Rouse, B.T. IL-10 and natural regulatory T cells: Two independent anti-inflammatory mechanisms in herpes simplex virus-induced ocular immunopathology. J. Immunol. 2008, 180, 6297–6306. [Google Scholar]
- Humphreys, I.R.; de Trez, C.; Kinkade, A.; Benedict, C.A.; Croft, M.; Ware, C.F. Cytomegalovirus exploits IL-10-mediated immune regulation in the salivary glands. J. Exp. Med. 2007, 204, 1217–1225. [Google Scholar] [CrossRef]
- Tessmer, M.S.; Reilly, E.C.; Brossay, L. Salivary gland NK cells are phenotypically and functionally unique. PLoS Pathog. 2011, 7, e1001254. [Google Scholar] [CrossRef]
- Jones, M.; Ladell, K.; Wynn, K.K.; Stacey, M.A.; Quigley, M.F.; Gostick, E.; Price, D.A.; Humphreys, I.R. IL-10 restricts memory T cell inflation during cytomegalovirus infection. J. Immunol. 2010, 185, 3583–3592. [Google Scholar]
- Molloy, M.J.; Zhang, W.; Usherwood, E.J. Suppressive CD8+ T cells arise in the absence of CD4 help and compromise control of persistent virus. J. Immunol. 2011, 186, 6218–6226. [Google Scholar] [CrossRef]
- Chang, W.L.; Barry, P.A. Attenuation of innate immunity by cytomegalovirus IL-10 establishes a long-term deficit of adaptive antiviral immunity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22647–22652. [Google Scholar]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Messal, N.; Serriari, N.E.; Pastor, S.; Nunes, J.A.; Olive, D. PD-L2 is expressed on activated human T cells and regulates their function. Mol. Immunol. 2011, 48, 2214–2219. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Brown, K.E.; Freeman, G.J.; Wherry, E.J.; Sharpe, A.H. Role of PD-1 in regulating acute infections. Curr. Opin. Immunol. 2010, 22, 397–401. [Google Scholar]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef]
- Talay, O.; Shen, C.H.; Chen, L.; Chen, J. B7-H1 (PD-L1) on T cells is required for T-cell-mediated conditioning of dendritic cell maturation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2741–2746. [Google Scholar]
- Rowe, J.H.; Johanns, T.M.; Ertelt, J.M.; Way, S.S. PDL-1 blockade impedes T cell expansion and protective immunity primed by attenuated Listeria monocytogenes. J. Immunol. 2008, 180, 7553–7557. [Google Scholar]
- Lazar-Molnar, E.; Chen, B.; Sweeney, K.A.; Wang, E.J.; Liu, W.; Lin, J.; Porcelli, S.A.; Almo, S.C.; Nathenson, S.G.; Jacobs, W.R., Jr. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13402–13407. [Google Scholar]
- Lafon, M.; Megret, F.; Meuth, S.G.; Simon, O.; Velandia Romero, M.L.; Lafage, M.; Chen, L.; Alexopoulou, L.; Flavell, R.A.; Prehaud, C.; et al. Detrimental contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J. Immunol. 2008, 180, 7506–7515. [Google Scholar]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar]
- Greenough, T.C.; Campellone, S.C.; Brody, R.; Jain, S.; Sanchez-Merino, V.; Somasundaran, M.; Luzuriaga, K. Programmed Death-1 expression on Epstein Barr virus specific CD8+ T cells varies by stage of infection, epitope specificity, and T-cell receptor usage. PLoS One 2010, 5, e12926. [Google Scholar]
- Larsen, M.; Sauce, D.; Deback, C.; Arnaud, L.; Mathian, A.; Miyara, M.; Boutolleau, D.; Parizot, C.; Dorgham, K.; Papagno, L.; et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog. 2011, 7, e1002328. [Google Scholar]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar]
- Zhang, J.Y.; Zhang, Z.; Jin, B.; Zhang, S.Y.; Zhou, C.B.; Fu, J.L.; Wang, F.S. Cutting edge: Programmed death-1 up-regulation is involved in the attrition of cytomegalovirus-specific CD8+ T cells in acute self-limited hepatitis B virus infection. J. Immunol. 2008, 181, 3741–3744. [Google Scholar]
- Campbell, A.E.; Cavanaugh, V.J.; Slater, J.S. The salivary glands as a privileged site of cytomegalovirus immune evasion and persistence. Med. Microbiol. Immunol. 2008, 197, 205–213. [Google Scholar] [CrossRef]
- Walton, S.M.; Mandaric, S.; Torti, N.; Zimmermann, A.; Hengel, H.; Oxenius, A. Absence of cross-presenting cells in the salivary gland and viral immune evasion confine cytomegalovirus immune control to effector CD4 T cells. PLoS Pathog. 2011, 7, e1002214. [Google Scholar]
- Dias, P.; Giannoni, F.; Lee, L.N.; Han, D.; Yoon, S.; Yagita, H.; Azuma, M.; Sarawar, S.R. CD4 T-cell help programs a change in CD8 T-cell function enabling effective long-term control of murine gammaherpesvirus 68: Role of PD-1-PD-L1 interactions. J. Virol. 2010, 84, 8241–8249. [Google Scholar] [CrossRef]
- Mintern, J.D.; Klemm, E.J.; Wagner, M.; Paquet, M.E.; Napier, M.D.; Kim, Y.M.; Koszinowski, U.H.; Ploegh, H.L. Viral interference with B7–1 costimulation: A new role for murine cytomegalovirus fc receptor-1. J. Immunol. 2006, 177, 8422–8431. [Google Scholar]
- Benedict, C.A.; Loewendorf, A.; Garcia, Z.; Blazar, B.R.; Janssen, E.M. Dendritic cell programming by cytomegalovirus stunts naive T cell responses via the PD-L1/PD-1 pathway. J. Immunol. 2008, 180, 4836–4847. [Google Scholar]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O'Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar]
- Gavrieli, M.; Watanabe, N.; Loftin, S.K.; Murphy, T.L.; Murphy, K.M. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem. Biophys. Res. Commun. 2003, 312, 1236–1243. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Annu. Rev. Immunol. 2010, 28, 389–411. [Google Scholar] [CrossRef]
- Shui, J.W.; Steinberg, M.W.; Kronenberg, M. Regulation of inflammation, autoimmunity, and infection immunity by HVEM-BTLA signaling. J. Leukoc. Biol. 2011, 89, 517–523. [Google Scholar] [CrossRef]
- Sun, Y.; Brown, N.K.; Ruddy, M.J.; Miller, M.L.; Lee, Y.; Wang, Y.; Murphy, K.M.; Pfeffer, K.; Chen, L.; Kaye, J.; et al. B and T lymphocyte attenuator tempers early infection immunity. J. Immunol. 2009, 183, 1946–1951. [Google Scholar]
- Adler, G.; Steeg, C.; Pfeffer, K.; Murphy, T.L.; Murphy, K.M.; Langhorne, J.; Jacobs, T. B and T lymphocyte attenuator restricts the protective immune response against experimental malaria. J. Immunol. 2011, 187, 5310–5319. [Google Scholar]
- Cheung, T.C.; Steinberg, M.W.; Oborne, L.M.; Macauley, M.G.; Fukuyama, S.; Sanjo, H.; D'Souza, C.; Norris, P.S.; Pfeffer, K.; Murphy, K.M.; et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6244–6249. [Google Scholar]
- Benedict, C.A.; Butrovich, K.D.; Lurain, N.S.; Corbeil, J.; Rooney, I.; Schneider, P.; Tschopp, J.; Ware, C.F. Cutting edge: A novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J. Immunol. 1999, 162, 6967–6970. [Google Scholar]
- Lurain, N.S.; Kapell, K.S.; Huang, D.D.; Short, J.A.; Paintsil, J.; Winkfield, E.; Benedict, C.A.; Ware, C.F.; Bremer, J.W. Human cytomegalovirus UL144 open reading frame: Sequence hypervariability in low-passage clinical isolates. J. Virol. 1999, 73, 10040–10050. [Google Scholar]
- Serriari, N.E.; Gondois-Rey, F.; Guillaume, Y.; Remmerswaal, E.B.; Pastor, S.; Messal, N.; Truneh, A.; Hirsch, I.; van Lier, R.A.; Olive, D. B and T lymphocyte attenuator is highly expressed on CMV-specific T cells during infection and regulates their function. J. Immunol. 2010, 185, 3140–3148. [Google Scholar] [CrossRef]
- Barclay, A.N.; Wright, G.J.; Brooke, G.; Brown, M.H. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002, 23, 285–290. [Google Scholar] [CrossRef]
- Wright, G.J.; Jones, M.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology 2001, 102, 173–179. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar]
- Barclay, A.N. Different reticular elements in rat lymphoid tissue identified by localization of Ia, Thy-1 and MRC OX 2 antigens. Immunology 1981, 44, 727–736. [Google Scholar]
- Barclay, A.N.; Clark, M.J.; McCaughan, G.W. Neuronal/lymphoid membrane glycoprotein MRC OX-2 is a member of the immunoglobulin superfamily with a light-chain-like structure. Biochem. Soc. Symp. 1986, 51, 149–157. [Google Scholar]
- Preston, S.; Wright, G.J.; Starr, K.; Barclay, A.N.; Brown, M.H. The leukocyte/neuron cell surface antigen OX2 binds to a ligand on macrophages. Eur. J. Immunol. 1997, 27, 1911–1918. [Google Scholar] [CrossRef]
- Rijkers, E.S.; de Ruiter, T.; Baridi, A.; Veninga, H.; Hoek, R.M.; Meyaard, L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol. Immunol. 2008, 45, 1126–1135. [Google Scholar] [CrossRef]
- Wright, G.J.; Cherwinski, H.; Foster-Cuevas, M.; Brooke, G.; Puklavec, M.J.; Bigler, M.; Song, Y.; Jenmalm, M.; Gorman, D.; McClanahan, T.; et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J. Immunol. 2003, 171, 3034–3046. [Google Scholar]
- Wright, G.J.; Puklavec, M.J.; Willis, A.C.; Hoek, R.M.; Sedgwick, J.D.; Brown, M.H.; Barclay, A.N. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity 2000, 13, 233–242. [Google Scholar] [CrossRef]
- Mihrshahi, R.; Barclay, A.N.; Brown, M.H. Essential roles for Dok2 and RasGAP in CD200 receptor-mediated regulation of human myeloid cells. J. Immunol. 2009, 183, 4879–4886. [Google Scholar] [CrossRef]
- Gorczynski, L.; Chen, Z.; Hu, J.; Kai, Y.; Lei, J.; Ramakrishna, V.; Gorczynski, R.M. Evidence that an OX-2-positive cell can inhibit the stimulation of type 1 cytokine production by bone marrow-derived B7–1 (and B7–2)-positive dendritic cells. J. Immunol. 1999, 162, 774–781. [Google Scholar]
- Gorczynski, R.M.; Lee, L.; Boudakov, I. Augmented induction of CD4+CD25+ Treg using monoclonal antibodies to CD200R. Transplantation 2005, 79, 488–491. [Google Scholar]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef]
- Bain, C.C.; Mowat, A.M. CD200 receptor and macrophage function in the intestine. Immunobiology 2012, 217, 643–651. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Pluddemann, A.; Hoe, J.C.; Williams, K.J.; Varin, A.; Makepeace, K.; Aknin, M.L.; Bowdish, D.M.; Smale, S.T.; Barclay, A.N.; et al. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host Microbe 2010, 8, 236–247. [Google Scholar] [CrossRef]
- Sarangi, P.P.; Woo, S.R.; Rouse, B.T. Control of viral immunoinflammatory lesions by manipulating CD200:CD200 receptor interaction. Clin. Immunol. 2009, 131, 31–40. [Google Scholar] [CrossRef]
- Foster-Cuevas, M.; Wright, G.J.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 2004, 78, 7667–7676. [Google Scholar] [CrossRef]
- Rezaee, S.A.; Gracie, J.A.; McInnes, I.B.; Blackbourn, D.J. Inhibition of neutrophil function by the Kaposi's sarcoma-associated herpesvirus vOX2 protein. AIDS 2005, 19, 1907–1910. [Google Scholar] [CrossRef]
- Shiratori, I.; Yamaguchi, M.; Suzukawa, M.; Yamamoto, K.; Lanier, L.L.; Saito, T.; Arase, H. Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 2005, 175, 4441–4449. [Google Scholar]
- Misstear, K.; Chanas, S.A.; Rezaee, S.A.; Colman, R.; Quinn, L.L.; Long, H.M.; Goodyear, O.; Lord, J.M.; Hislop, A.D.; Blackbourn, D.J. Suppression of antigen-specific T cell responses by the Kaposi's sarcoma-associated herpesvirus viral OX2 protein and its cellular orthologue, CD200. J. Virol. 2011, 86, 6246–6257. [Google Scholar]
- Salata, C.; Curtarello, M.; Calistri, A.; Sartori, E.; Sette, P.; de Bernard, M.; Parolin, C.; Palu, G. vOX2 glycoprotein of human herpesvirus 8 modulates human primary macrophages activity. J. Cell Physiol. 2009, 219, 698–706. [Google Scholar] [CrossRef]
- Chung, Y.H.; Means, R.E.; Choi, J.K.; Lee, B.S.; Jung, J.U. Kaposi's sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 2002, 76, 4688–4698. [Google Scholar] [CrossRef]
- Uniprot. Available online: http://www.uniprot.org/ (accessed on 1 April 2012).
- Gompels, U.A.; Nicholas, J.; Lawrence, G.; Jones, M.; Thomson, B.J.; Martin, M.E.; Efstathiou, S.; Craxton, M.; Macaulay, H.A. The DNA sequence of human herpesvirus-6: Structure, coding content, and genome evolution. Virology 1995, 209, 29–51. [Google Scholar] [CrossRef]
- Nicholas, J. Determination and analysis of the complete nucleotide sequence of human herpesvirus. J. Virol. 1996, 70, 5975–5989. [Google Scholar]
- Voigt, S.; Sandford, G.R.; Hayward, G.S.; Burns, W.H. The English strain of rat cytomegalovirus (CMV) contains a novel captured CD200 (vOX2) gene and a spliced CC chemokine upstream from the major immediate-early region: Further evidence for a separate evolutionary lineage from that of rat CMV Maastricht. J. Gen. Virol. 2005, 86, 263–274. [Google Scholar] [CrossRef]
- Foster-Cuevas, M.; Westerholt, T.; Ahmed, M.; Brown, M.H.; Barclay, A.N.; Voigt, S. Cytomegalovirus e127 protein interacts with the inhibitory CD200 receptor. J. Virol. 2011, 85, 6055–6059. [Google Scholar]
- Stack, G.; Humphreys, I. 2012; Cardiff University, Cardiff, UK. Unpublished observations.
- Smith, L.M.; McWhorter, A.R.; Masters, L.L.; Shellam, G.R.; Redwood, A.J. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J. Virol. 2008, 82, 6689–6696. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar]
- Akkaya, M.; Barclay, A.N. Heterogeneity in the CD200R paired receptor family. Immunogenetics 2011, 62, 15–22. [Google Scholar] [CrossRef]
- Hatherley, D.; Cherwinski, H.M.; Moshref, M.; Barclay, A.N. Recombinant CD200 protein does not bind activating proteins closely related to CD200 receptor. J. Immunol. 2005, 175, 2469–2474. [Google Scholar]
- Cameron, C.M.; Barrett, J.W.; Liu, L.; Lucas, A.R.; McFadden, G. Myxoma virus M141R expresses a viral CD200 (vOX-2) that is responsible for down-regulation of macrophage and T-cell activation in vivo. J. Virol. 2005, 79, 6052–6067. [Google Scholar]
- Yue, Y.; Kaur, A.; Eberhardt, M.K.; Kassis, N.; Zhou, S.S.; Tarantal, A.F.; Barry, P.A. Immunogenicity and protective efficacy of DNA vaccines expressing rhesus cytomegalovirus glycoprotein B, phosphoprotein 65–2, and viral interleukin-10 in rhesus macaques. J. Virol. 2007, 81, 1095–1109. [Google Scholar]
- Eberhardt, M.K.; Chang, W.L.; Logsdon, N.J.; Yue, Y.; Walter, M.R.; Barry, P.A. Host immune responses to a viral immune modulating protein: Immunogenicity of viral interleukin-10 in rhesus cytomegalovirus-infected rhesus macaques. PLoS One 2012, 7, e37931. [Google Scholar]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 2001, 344, 1366–1371. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stack, G.; Stacey, M.A.; Humphreys, I.R. Herpesvirus Exploitation of Host Immune Inhibitory Pathways. Viruses 2012, 4, 1182-1201. https://doi.org/10.3390/v4081182
Stack G, Stacey MA, Humphreys IR. Herpesvirus Exploitation of Host Immune Inhibitory Pathways. Viruses. 2012; 4(8):1182-1201. https://doi.org/10.3390/v4081182
Chicago/Turabian StyleStack, Gabrielle, Maria A. Stacey, and Ian R. Humphreys. 2012. "Herpesvirus Exploitation of Host Immune Inhibitory Pathways" Viruses 4, no. 8: 1182-1201. https://doi.org/10.3390/v4081182
APA StyleStack, G., Stacey, M. A., & Humphreys, I. R. (2012). Herpesvirus Exploitation of Host Immune Inhibitory Pathways. Viruses, 4(8), 1182-1201. https://doi.org/10.3390/v4081182