Recent Progress in Studies of Arterivirus- and Coronavirus-Host Interactions

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Sub-Family | Genera | Representative Animal Species | Host and Tissue Tropism |
|---|---|---|---|---|---|
| Nidovirales | Coronaviridae | Coronavirinae | Alphacoronavirus | Transmissible gastroenteritis virus | Pigs (GI) |
| Feline Coronavirus | Domestic cats (GI, Peritoneal) | ||||
| Bovine Coronavirus | Cattle (GI) | ||||
| Bat Coronavirus HKU2 and HKU8 | Bats (Carrier) | ||||
| Betacoronavirus | Mouse Hepatitis Virus | Mice (Res, GI, Hep, CNS) | |||
| Bat Coronavirus HKU9 | Bats (Carrier) | ||||
| Severe Acute Respiratory Syndrome Coronavrus# | Palm Civets (Carrier)Bats (Carrier) | ||||
| Gammacoronavirus | Avian Infectious Bronchitis Virus | Chickens (Res, Neph, Rep) | |||
| SW1 virus | Beluga Whale (Res, Hep) | ||||
| Torovirinae | Bafinivirus | White bream virus | Fish (GI, Hep) | ||
| Torovirus | Breda virus (bovine torovirus) | Cattle (GI) | |||
| Arteriviridae | - | Arterivirius | Porcine reproductive and respiratory syndrome virus | Pigs (Resp, Rep) | |
| Equine arteritis virus | Horses (Resp, Rep) | ||||
| Roniviridae | - | Okavirus | Yellow head virus | Crustaceans (prawns); cephalothorax |
1.1. Virus Infection and Host Responses
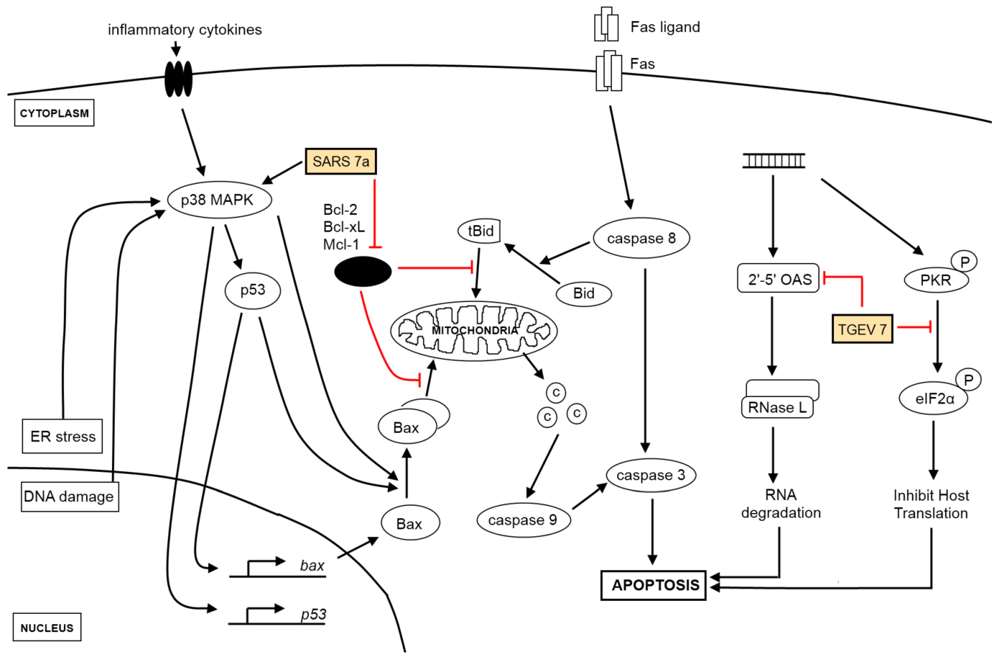
2. The Effect of Virus Infection on Apoptosis
2.1. Extrinsic and Intrinsic Apoptotic Pathways
2.2. Other Apoptotic Pathways
2.3. Viruses and Apoptosis

2.5. IBV-Induced Apoptosis and Its Regulation
2.6. Viral Genes Implicated in Apoptosis
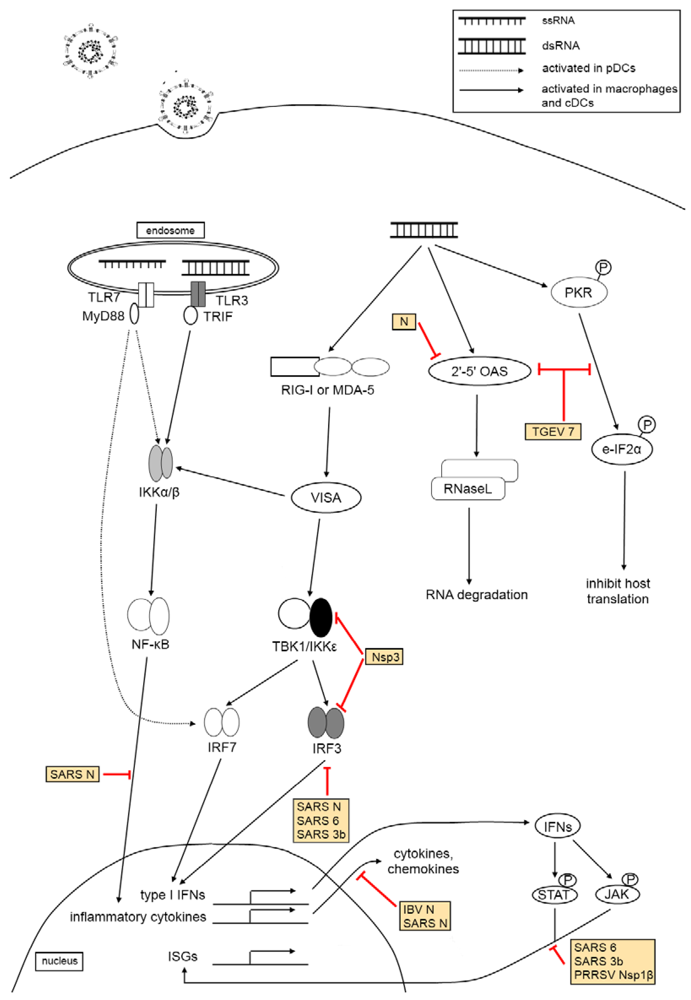
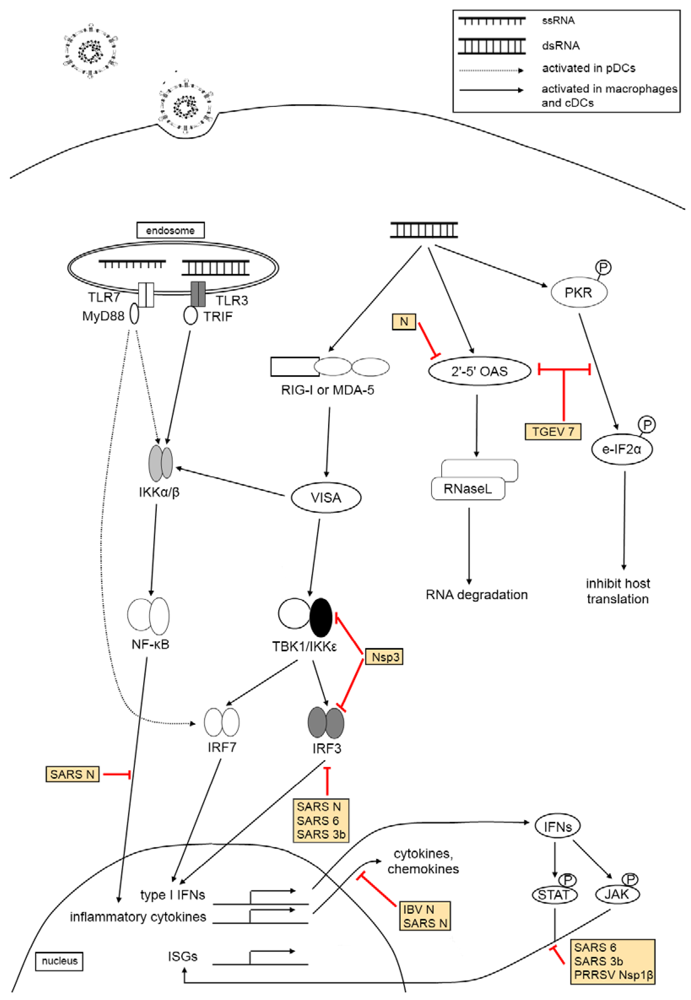
3. The Effect of Virus Infection on Host Innate Immunity
3.3. Pattern Recognition Receptor Families
3.4. Type I Interferon Response
3.5. The Effect of Coronaviruses and Arteriviruses on Host Innate Immune Responses
4. The Effect of Virus Infection on Other Host Innate Defenses
4.2. Virus Infection and the MAPK Signaling Pathway
4.3. Induction of Other Pro-Inflammatory Cytokines
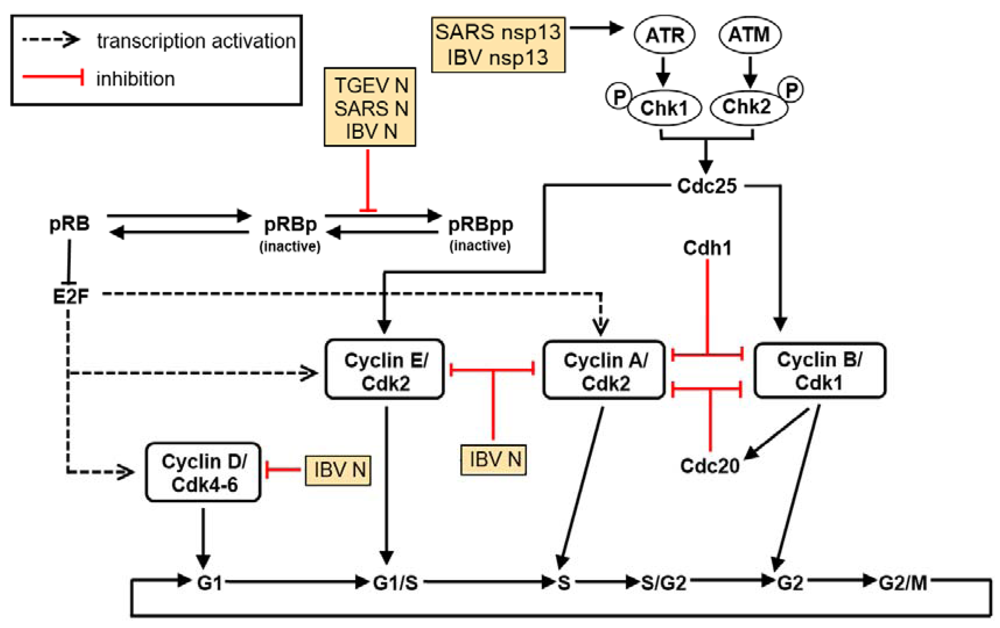
5. The Effect of Virus Infection on Cell Cycle Regulation
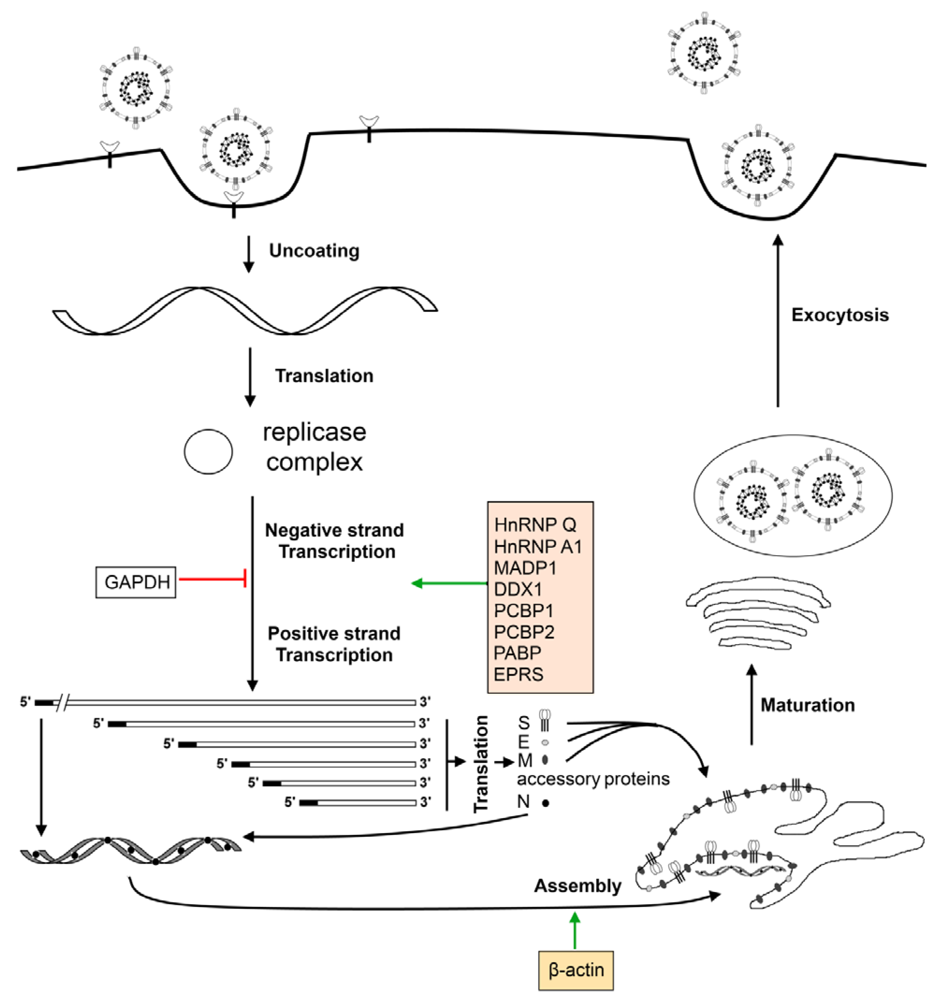
6. The Effect of Virus Infection on the Host Translation Machinery
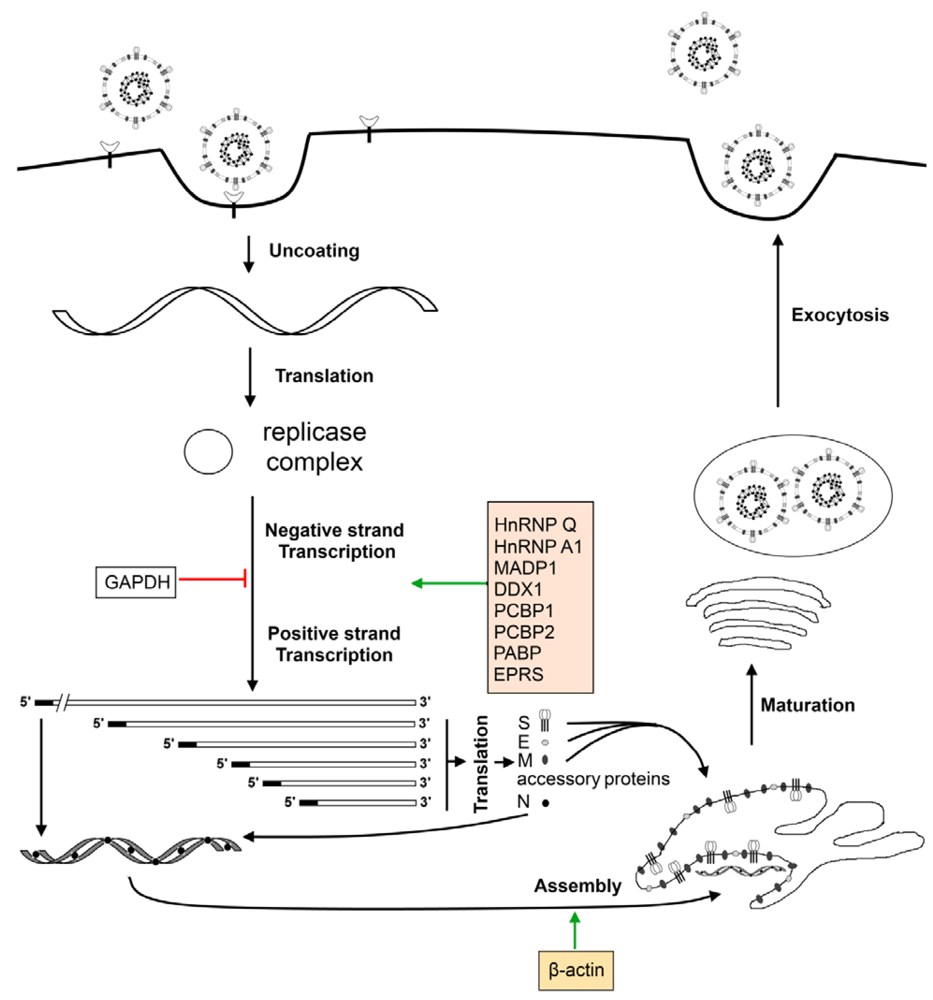
7. Cellular Factors Involved in the Virus Life Cycle
8. Conclusions
Conflict of Interest
References and Notes
- Ivanov, K.A.; Hertzig, T.; Rozanov, M.; Bayer, S.; Thiel, V.; Gorbalenya, A.E.; Ziebuhr, J. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12694–12699. [Google Scholar]
- Cavanagh, D. Coronaviruses in poultry and other birds. Avian Pathol. 2005, 34, 439–448. [Google Scholar] [CrossRef]
- Rowland, R.R. The interaction between PRRSV and the late gestation pig fetus. Virus Res. 2010, 154, 114–122. [Google Scholar] [CrossRef]
- Wevers, B.A.; van der Hoek, L. Recently discovered human coronaviruses. Clin. Lab. Med. 2009, 29, 715–724. [Google Scholar] [CrossRef]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef]
- Picault, J.P.; Drouin, P.; Guittet, M.; Bennejean, G.; Protais, J.; L'Hospitalier, R.; Gillet, J.P.; Lamande, J.; Bachelier, A.L. Isolation, characterisation and preliminary cross-protection studies with a new pathogenic avian infectious bronchitis virus (strain PL-84084). Avian Pathol. 1986, 15, 367–383. [Google Scholar] [CrossRef]
- King, D.J.; Cavanagh, D. Infectious bronchitis. In Disease of Poultry; Calnek, B.W., Barnes, H.J., Beard, C.W., Reid, W.M., Yoder, H.W.J., Eds.; Iowa State University Press: Ames, IA, USA, 1991; pp. 471–484. [Google Scholar]
- Landman, W.J.; Feberwee, A. Aerosol-induced Mycoplasma synoviae arthritis: The synergistic effect of infectious bronchitis virus infection. Avian Pathol. 2004, 33, 591–598. [Google Scholar] [CrossRef]
- Matthijs, M.G.; van Eck, J.H.; Landman, W.J.; Stegeman, J.A. Ability of Massachusetts-type infectious bronchitis virus to increase colibacillosis susceptibility in commercial broilers: A comparison between vaccine and virulent field virus. Avian Pathol. 2003, 32, 473–481. [Google Scholar] [CrossRef]
- Chang, H.W.; de Groot, R.J.; Egberink, H.F.; Rottier, P.J. Feline infectious peritonitis: Insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3c gene. J. Gen. Virol. 2010, 91, 415–420. [Google Scholar] [CrossRef]
- Pratelli, A. The evolutionary processes of canine coronaviruses. Adv. Virol. 2011, 2011, 562831. [Google Scholar]
- Dea, S.; Michaud, L.; Milane, G. Comparison of bovine coronavirus isolates associated with neonatal calf diarrhoea and winter dysentery in adult dairy cattle in Quebec. J. Gen. Virol. 1995, 76, 1263–1270. [Google Scholar] [CrossRef]
- Wang, L.F.; Eaton, B.T. Bats, civets and the emergence of SARS. Curr. Top. Microbiol. Immunol. 2007, 315, 325–344. [Google Scholar]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Muller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Laude, H.; Rasschaert, D.; Delmas, B.; Godet, M.; Gelfi, J.; Charley, B. Molecular biology of transmissible gastroenteritis virus. Vet. Microbiol. 1990, 23, 147–154. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Derbyshire, J.B. Binding of porcine transmissible gastroenteritis virus by enterocytes from newborn and weaned piglets. Vet. Microbiol. 1993, 35, 23–32. [Google Scholar] [CrossRef]
- Britton, P.; Armesto, M.; Cavanagh, D.; Keep, S. Modification of the avian coronavirus infectious bronchitis virus for vaccine development. Bioeng Bugs 2012, 3, 114–119. [Google Scholar]
- Miller, L.C.; Fox, J.M. Apoptosis and porcine reproductive and respiratory syndrome virus. Vet. Immunol. Immunopathol. 2004, 102, 131–142. [Google Scholar] [CrossRef]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef]
- de Haan, C.A.; Reggiori, F. Are nidoviruses hijacking the autophagy machinery? Autophagy 2008, 4, 276–279. [Google Scholar]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell E 2001, 3, E255–E263. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Mikhailov, V.; Mikhailova, M.; Degenhardt, K.; Venkatachalam, M.A.; White, E.; Saikumar, P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 2003, 278, 5367–5376. [Google Scholar]
- Adams, J.M.; Cory, S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 2001, 26, 61–66. [Google Scholar] [CrossRef]
- Galonek, H.L.; Hardwick, J.M. Upgrading the BCL-2 network. Nat. Cell Biol 2006, 8, 1317–1319. [Google Scholar] [CrossRef]
- Assuncao Guimaraes, C.; Linden, R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur. J. Biochem. 2004, 271, 1638–1650. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: Multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Danthi, P. Enter the kill zone: Initiation of death signaling during virus entry. Virology 2011, 411, 316–324. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. To kill or be killed: How viruses interact with the cell death machinery. J. Intern. Med. 2010, 267, 473–482. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef]
- den Boon, J.A.; Snijder, E.J.; Chirnside, E.D.; de Vries, A.A.; Horzinek, M.C.; Spaan, W.J. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J. Virol. 1991, 65, 2910–2920. [Google Scholar]
- St-Louis, M.C.; Archambault, D. The equine arteritis virus induces apoptosis via caspase-8 and mitochondria-dependent caspase-9 activation. Virology 2007, 367, 147–155. [Google Scholar] [CrossRef]
- van Reeth, K.; Nauwynck, H. Proinflammatory cytokines and viral respiratory disease in pigs. Vet. Res. 2000, 31, 187–213. [Google Scholar] [CrossRef]
- Costers, S.; Lefebvre, D.J.; Delputte, P.L.; Nauwynck, H.J. Porcine reproductive and respiratory syndrome virus modulates apoptosis during replication in alveolar macrophages. Arch. Virol. 2008, 153, 1453–1465. [Google Scholar] [CrossRef]
- Lee, S.M.; Kleiboeker, S.B. Porcine reproductive and respiratory syndrome virus induces apoptosis through a mitochondria-mediated pathway. Virology 2007, 365, 419–434. [Google Scholar] [CrossRef]
- Ding, L.; Xu, X.; Huang, Y.; Li, Z.; Zhang, K.; Chen, G.; Yu, G.; Wang, Z.; Li, W.; Tong, D. Transmissible gastroenteritis virus infection induces apoptosis through FasL- and mitochondria-mediated pathways. Vet. Microbiol. 2012, 158, 12–22. [Google Scholar] [CrossRef]
- Marfe, G.; Tafani, M.; Fiorito, F.; Pagnini, U.; Iovane, G.; De Martino, L. Involvement of FOXO transcription factors, TRAIL-FasL/Fas, and sirtuin proteins family in canine coronavirus type II-induced apoptosis. PLoS One 2011, 6, e27313. [Google Scholar]
- Shen, S.; Wen, Z.L.; Liu, D.X. Emergence of a coronavirus infectious bronchitis virus mutant with a truncated 3b gene: Functional characterization of the 3b protein in pathogenesis and replication. Virology 2003, 311, 16–27. [Google Scholar] [CrossRef]
- Fang, S.G.; Shen, S.; Tay, F.P.; Liu, D.X. Selection of and recombination between minor variants lead to the adaptation of an avian coronavirus to primate cells. Biochem. Biophys. Res. Commun. 2005, 336, 417–423. [Google Scholar] [CrossRef]
- Zhong, Y.; Liao, Y.; Fang, S.; Tam, J.P.; Liu, D.X. Up-regulation of mcl-1 and bak by coronavirus infection of human, avian and animal cells modulates apoptosis and viral replication. PLoS One 2012, 7, e30191. [Google Scholar]
- Liu, C.; Xu, H.Y.; Liu, D.X. Induction of caspase-dependent apoptosis in cultured cells by the avian coronavirus infectious bronchitis virus. J. Virol. 2001, 75, 6402–6409. [Google Scholar] [CrossRef]
- Li, F.Q.; Tam, J.P.; Liu, D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 2007, 365, 435–445. [Google Scholar] [CrossRef]
- Tan, Y.J.; Fielding, B.C.; Goh, P.Y.; Shen, S.; Tan, T.H.; Lim, S.G.; Hong, W. Overexpression of 7a, a protein specifically encoded by the severe acute respiratory syndrome coronavirus, induces apoptosis via a caspase-dependent pathway. J. Virol. 2004, 78, 14043–14047. [Google Scholar]
- de Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae; Elsevier: Oxford, UK, 2011. [Google Scholar]
- Cruz, J.L.; Sola, I.; Becares, M.; Alberca, B.; Plana, J.; Enjuanes, L.; Zuniga, S. Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathog. 2011, 7, e1002090. [Google Scholar] [CrossRef]
- Opferman, J.T.; Korsmeyer, S.J. Apoptosis in the development and maintenance of the immune system. Nat. Immunol. 2003, 4, 410–415. [Google Scholar] [CrossRef]
- Lucas, M.; Stuart, L.M.; Savill, J.; Lacy-Hulbert, A. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. J. Immunol. 2003, 171, 2610–2615. [Google Scholar]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 2000, 21, 447–455. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. Interferons and viral infections. BioFactors 2009, 35, 14–20. [Google Scholar] [CrossRef]
- de Veer, M.J.; Holko, M.; Frevel, M.; Walker, E.; Der, S.; Paranjape, J.M.; Silverman, R.H.; Williams, B.R. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 2001, 69, 912–920. [Google Scholar]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 2009, 420, 1–16. [Google Scholar] [CrossRef]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Shaw, M.H.; Reimer, T.; Kim, Y.G.; Nunez, G. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Curr. Opin. Immunol. 2008, 20, 377–382. [Google Scholar]
- Creagh, E.M.; O'Neill, L.A. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef]
- Hong, M.; Yoon, S.I.; Wilson, I.A. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 2012, 36, 337–347. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar]
- Takeuchi, O.; Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008, 20, 17–22. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Virgin, H.W.; Colonna, M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Bowie, A.G.; Fitzgerald, K.A. RIG-I: Tri-ing to discriminate between self and non-self RNA. Trends Immunol. 2007, 28, 147–150. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antivirial responses to single-stranded RNA bearing 5'-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.; Fodor, E.; et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar]
- Davis, W.G.; Bowzard, J.B.; Sharma, S.D.; Wiens, M.E.; Ranjan, P.; Gangappa, S.; Stuchlik, O.; Pohl, J.; Donis, R.O.; Katz, J.M.; et al. The 3' untranslated regions of influenza genomic sequences are 5'PPP-independent ligands for RIG-I. PLoS One 2012, 7, e32661. [Google Scholar]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon Antivir. Res.ponse through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Peters, R.T.; Liao, S.M.; Maniatis, T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 2000, 5, 513–522. [Google Scholar] [CrossRef]
- Bonnard, M.; Mirtsos, C.; Suzuki, S.; Graham, K.; Huang, J.; Ng, M.; Itie, A.; Wakeham, A.; Shahinian, A.; Henzel, W.J.; et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000, 19, 4976–4985. [Google Scholar]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Genin, P.; Pitha, P.M.; Hiscott, J. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell Biol. 1999, 19, 959–966. [Google Scholar]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don't know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef]
- Frieman, M.B.; Chen, J.; Morrison, T.E.; Whitmore, A.; Funkhouser, W.; Ward, J.M.; Lamirande, E.W.; Roberts, A.; Heise, M.; Subbarao, K.; et al. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog. 2010, 6, e1000849. [Google Scholar]
- Overend, C.; Mitchell, R.; He, D.; Rompato, G.; Grubman, M.J.; Garmendia, A.E. Recombinant swine beta interferon protects swine alveolar macrophages and MARC-145 cells from infection with Porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 2007, 88, 925–931. [Google Scholar]
- Brockmeier, S.L.; Loving, C.L.; Nelson, E.A.; Miller, L.C.; Nicholson, T.L.; Register, K.B.; Grubman, M.J.; Brough, D.E.; Kehrli, M.E., Jr. Presence of interferon-alpha at the time of infection alters the innate and adaptive immune response to porcine reproductive and respiratory syndrome virus. Clin. Vaccine Immunol. 2012, 19, 508–514. [Google Scholar] [CrossRef]
- Albina, E.; Carrat, C.; Charley, B. Interferon-alpha response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. J. Interferon. Cytokine Res. 1998, 18, 485–490. [Google Scholar] [CrossRef]
- Buddaert, W.; Van Reeth, K.; Pensaert, M. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV). Adv. Exp. Med. Biol. 1998, 440, 461–467. [Google Scholar] [CrossRef]
- Luo, R.; Xiao, S.; Jiang, Y.; Jin, H.; Wang, D.; Liu, M.; Chen, H.; Fang, L. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol. Immunol. 2008, 45, 2839–2846. [Google Scholar]
- Patel, D.; Nan, Y.; Shen, M.; Ritthipichai, K.; Zhu, X.; Zhang, Y.J. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2010, 84, 11045–11055. [Google Scholar]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795–1803. [Google Scholar]
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O'Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One 2009, 4, e5466. [Google Scholar]
- Zust, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2'-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2'-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar]
- Roth-Cross, J.K.; Bender, S.J.; Weiss, S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008, 82, 9829–9838. [Google Scholar]
- Li, J.; Liu, Y.; Zhang, X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J. Virol. 2010, 84, 6472–6482. [Google Scholar] [CrossRef]
- Zhou, H.; Perlman, S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 2007, 81, 568–574. [Google Scholar] [CrossRef]
- Rose, K.M.; Elliott, R.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weiss, S.R. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J. Virol. 2010, 84, 5656–5669. [Google Scholar]
- Cervantes-Barragan, L.; Kalinke, U.; Zust, R.; Konig, M.; Reizis, B.; Lopez-Macias, C.; Thiel, V.; Ludewig, B. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J. Immunol. 2009, 182, 1099–1106. [Google Scholar]
- Versteeg, G.A.; Bredenbeek, P.J.; van den Worm, S.H.; Spaan, W.J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology 2007, 361, 18–26. [Google Scholar] [CrossRef]
- Ye, Y.; Hauns, K.; Langland, J.O.; Jacobs, B.L.; Hogue, B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007, 81, 2554–2563. [Google Scholar]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Menard, R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar]
- Chen, Z.; Wang, Y.; Ratia, K.; Mesecar, A.D.; Wilkinson, K.D.; Baker, S.C. Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 2007, 81, 6007–6018. [Google Scholar] [CrossRef]
- Wojdyla, J.A.; Manolaridis, I.; van Kasteren, P.B.; Kikkert, M.; Snijder, E.J.; Gorbalenya, A.E.; Tucker, P.A. Papain-like protease 1 from transmissible gastroenteritis virus: Crystal structure and enzymatic activity toward viral and cellular substrates. J. Virol. 2010, 84, 10063–10073. [Google Scholar]
- Wang, G.; Chen, G.; Zheng, D.; Cheng, G.; Tang, H. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS One 2011, 6, e17192. [Google Scholar]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; Li, K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar]
- Ariaans, M.P.; van de Haar, P.M.; Hensen, E.J.; Vervelde, L. Infectious Bronchitis Virus induces acute interferon-gamma production through polyclonal stimulation of chicken leukocytes. Virology 2009, 385, 68–73. [Google Scholar] [CrossRef]
- Bechill, J.; Chen, Z.; Brewer, J.W.; Baker, S.C. Coronavirus infection modulates the unfolded protein response and mediates sustained translational repression. J. Virol. 2008, 82, 4492–4501. [Google Scholar] [CrossRef]
- Liang, S.H.; Zhang, W.; McGrath, B.C.; Zhang, P.; Cavener, D.R. PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem. J. 2006, 393, 201–209. [Google Scholar] [CrossRef]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 2004, 319, 1228–1234. [Google Scholar] [CrossRef]
- Banerjee, S.; Narayanan, K.; Mizutani, T.; Makino, S. Murine coronavirus replication-induced p38 mitogen-activated protein kinase activation promotes interleukin-6 production and virus replication in cultured cells. J. Virol. 2002, 76, 5937–5948. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Alvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar]
- Liao, Y.; Wang, X.; Huang, M.; Tam, J.P.; Liu, D.X. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 2011, 420, 106–116. [Google Scholar] [CrossRef]
- Mizutani, T. Signal transduction in SARS-CoV-infected cells. Ann. N. Y. Acad. Sci. 2007, 1102, 86–95. [Google Scholar] [CrossRef]
- Xiao, H.; Xu, L.H.; Yamada, Y.; Liu, D.X. Coronavirus spike protein inhibits host cell translation by interaction with eIF3f. PLoS One 2008, 3, e1494. [Google Scholar]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, C. Porcine reproductive and respiratory syndrome virus replication is suppressed by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. Virus Res. 2010, 152, 50–58. [Google Scholar] [CrossRef]
- Yoo, D.; Song, C.; Sun, Y.; Du, Y.; Kim, O.; Liu, H.C. Modulation of host cell responses and evasion strategies for porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 48–60. [Google Scholar] [CrossRef]
- Moore, B.D.; Balasuriya, U.B.; Watson, J.L.; Bosio, C.M.; MacKay, R.J.; MacLachlan, N.J. Virulent and avirulent strains of equine arteritis virus induce different quantities of TNF-alpha and other proinflammatory cytokines in alveolar and blood-derived equine macrophages. Virology 2003, 314, 662–670. [Google Scholar] [CrossRef]
- Charley, B.; Riffault, S.; Van Reeth, K. Porcine innate and adaptative immune responses to influenza and coronavirus infections. Ann. N. Y. Acad. Sci. 2006, 1081, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Crawford, A.; Angelosanto, J.M.; Nadwodny, K.L.; Blackburn, S.D.; Wherry, E.J. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011, 7, e1002098. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, R.; Fang, L.; Wang, D.; Bi, J.; Chen, H.; Xiao, S. Porcine reproductive and respiratory syndrome virus (PRRSV) infection activates chemokine RANTES in MARC-145 cells. Mol. Immunol. 2011, 48, 586–591. [Google Scholar] [CrossRef]
- Jung, K.; Renukaradhya, G.J.; Alekseev, K.P.; Fang, Y.; Tang, Y.; Saif, L.J. Porcine reproductive and respiratory syndrome virus modifies innate immunity and alters disease outcome in pigs subsequently infected with porcine respiratory coronavirus: Implications for respiratory viral co-infections. J. Gen. Virol. 2009, 90, 2713–2723. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Bortoluci, K.R.; Medzhitov, R. Control of infection by pyroptosis and autophagy: Role of TLR and NLR. Cell Mol. Life Sci. 2010, 67, 1643–1651. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Davy, C.; Doorbar, J. G2/M cell cycle arrest in the life cycle of viruses. Virology 2007, 368, 219–226. [Google Scholar] [CrossRef]
- Gopinathan, L.; Ratnacaram, C.K.; Kaldis, P. Established and novel Cdk/cyclin complexes regulating the cell cycle and development. Results Probl. Cell Differ. 2011, 53, 365–389. [Google Scholar] [CrossRef]
- Nurse, P. Checkpoint pathways come of age. Cell 1997, 91, 865–867. [Google Scholar] [CrossRef]
- Pines, J. Four-dimensional control of the cell cycle. Nat. Cell Biol. 1999, 1, E73–E79. [Google Scholar] [CrossRef]
- Kastan, M.B. Cell cycle. Checking two steps. Nature 2001, 410, 766–767. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell Biol. 2002, 22, 8552–8561. [Google Scholar] [CrossRef]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef]
- Xu, L.H.; Huang, M.; Fang, S.G.; Liu, D.X. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase delta. J. Biol. Chem. 2011, 286, 39546–39559. [Google Scholar]
- Chen, H.; Wurm, T.; Britton, P.; Brooks, G.; Hiscox, J.A. Interaction of the coronavirus nucleoprotein with nucleolar antigens and the host cell. J. Virol. 2002, 76, 5233–5250. [Google Scholar] [CrossRef]
- Wurm, T.; Chen, H.; Hodgson, T.; Britton, P.; Brooks, G.; Hiscox, J.A. Localization to the nucleolus is a common feature of coronavirus nucleoproteins, and the protein may disrupt host cell division. J. Virol. 2001, 75, 9345–9356. [Google Scholar] [CrossRef]
- Li, F.Q.; Xiao, H.; Tam, J.P.; Liu, D.X. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Lett. 2005, 579, 2387–2396. [Google Scholar] [CrossRef]
- Dove, B.K.; You, J.H.; Reed, M.L.; Emmett, S.R.; Brooks, G.; Hiscox, J.A. Changes in nucleolar morphology and proteins during infection with the coronavirus infectious bronchitis virus. Cell Microbiol. 2006, 8, 1147–1157. [Google Scholar] [CrossRef]
- Harrison, S.M.; Dove, B.K.; Rothwell, L.; Kaiser, P.; Tarpey, I.; Brooks, G.; Hiscox, J.A. Characterisation of cyclin D1 down-regulation in coronavirus infected cells. FEBS Lett. 2007, 581, 1275–1286. [Google Scholar]
- Williams, B.R. PKR; A sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef]
- Kuntziger, T.; Landsverk, H.B.; Collas, P.; Syljuasen, R.G. Protein phosphatase 1 regulators in DNA damage signaling. Cell Cycle 2011, 10, 1356–1362. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Wang, X.; Liao, Y.; Yap, P.L.; Png, K.J.; Tam, J.P.; Liu, D.X. Inhibition of protein kinase R activation and upregulation of GADD34 expression play a synergistic role in facilitating coronavirus replication by maintaining de novo protein synthesis in virus-infected cells. J. Virol. 2009, 83, 12462–12472. [Google Scholar]
- Okunola, H.L.; Krainer, A.R. Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol. Cell Biol. 2009, 29, 5620–5631. [Google Scholar] [CrossRef]
- He, Y.; Smith, R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol. Life Sci. 2009, 66, 1239–1256. [Google Scholar] [CrossRef]
- Li, H.P.; Zhang, X.; Duncan, R.; Comai, L.; Lai, M.M. Heterogeneous nuclear ribonucleoprotein A1 binds to the transcription-regulatory region of mouse hepatitis virus RNA. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 9544–9549. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.P.; Xue, W.; Lai, M.M. Cellular protein hnRNP-A1 interacts with the 3'-end and the intergenic sequence of mouse hepatitis virus negative-strand RNA to form a ribonucleoprotein complex. Adv. Exp. Med. Biol. 1998, 440, 227–234. [Google Scholar]
- Wang, Y.; Zhang, X. The nucleocapsid protein of coronavirus mouse hepatitis virus interacts with the cellular heterogeneous nuclear ribonucleoprotein A1 in vitro and in vivo. Virology 1999, 265, 96–109. [Google Scholar] [CrossRef]
- Luo, H.; Chen, Q.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. The nucleocapsid protein of SARS coronavirus has a high binding affinity to the human cellular heterogeneous nuclear ribonucleoprotein A1. FEBS Lett. 2005, 579, 2623–2628. [Google Scholar] [CrossRef]
- Zuniga, S.; Cruz, J.L.; Sola, I.; Mateos-Gomez, P.A.; Palacio, L.; Enjuanes, L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010, 84, 2169–2175. [Google Scholar] [CrossRef]
- Grossoehme, N.E.; Li, L.; Keane, S.C.; Liu, P.; Dann, C.E., 3rd; Leibowitz, J.L.; Giedroc, D.P. Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J. Mol. Biol. 2009, 394, 544–557. [Google Scholar] [CrossRef]
- Shi, S.T.; Huang, P.; Li, H.P.; Lai, M.M. Heterogeneous nuclear ribonucleoprotein A1 regulates RNA synthesis of a cytoplasmic virus. EMBO J. 2000, 19, 4701–4711. [Google Scholar] [CrossRef]
- Galan, C.; Sola, I.; Nogales, A.; Thomas, B.; Akoulitchev, A.; Enjuanes, L.; Almazan, F. Host cell proteins interacting with the 3' end of TGEV coronavirus genome influence virus replication. Virology 2009, 391, 304–314. [Google Scholar] [CrossRef]
- Archambault, D.; St-Louis, M.C.; Martin, S. Binding of cellular proteins to the leader RNA of equine arteritis virus. Virus Genes 2005, 30, 121–125. [Google Scholar] [CrossRef]
- Seay, D.; Hook, B.; Evans, K.; Wickens, M. A three-hybrid screen identifies mRNAs controlled by a regulatory protein. RNA 2006, 12, 1594–1600. [Google Scholar] [CrossRef]
- Tan, Y.W.; Hong, W.; Liu, D.X. Binding of the 5'-untranslated region of coronavirus RNA to zinc finger CCHC-type and RNA-binding motif 1 enhances viral replication and transcription. Nucleic Acids Res. 2012, 40, 5065–5077. [Google Scholar] [CrossRef]
- Xu, L.; Khadijah, S.; Fang, S.; Wang, L.; Tay, F.P.; Liu, D.X. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010, 84, 8571–8583. [Google Scholar] [CrossRef]
- Wang, J.; Fang, S.; Xiao, H.; Chen, B.; Tam, J.P.; Liu, D.X. Interaction of the coronavirus infectious bronchitis virus membrane protein with beta-actin and its implication in virion assembly and budding. PLoS One 2009, 4, e4908. [Google Scholar]
- Kong, Q.; Xue, C.; Ren, X.; Zhang, C.; Li, L.; Shu, D.; Bi, Y.; Cao, Y. Proteomic analysis of purified coronavirus infectious bronchitis virus particles. Proteome. Sci. 2010, 8, 29. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Liu, P.; Leibowitz, J.L.; Kao, C.C. The Coronavirus Endoribonuclease Nsp15 Interacts with Retinoblastoma Tumor Suppressor Protein. J. Virol. 2012, 86, 4294–4304. [Google Scholar] [CrossRef]
- Beura, L.K.; Dinh, P.X.; Osorio, F.A.; Pattnaik, A.K. Cellular poly(c) binding proteins 1 and 2 interact with porcine reproductive and respiratory syndrome virus nonstructural protein 1beta and support viral replication. J. Virol. 2011, 85, 12939–12949. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhong, Y.; Tan, Y.W.; Liu, D.X. Recent Progress in Studies of Arterivirus- and Coronavirus-Host Interactions. Viruses 2012, 4, 980-1010. https://doi.org/10.3390/v4060980
Zhong Y, Tan YW, Liu DX. Recent Progress in Studies of Arterivirus- and Coronavirus-Host Interactions. Viruses. 2012; 4(6):980-1010. https://doi.org/10.3390/v4060980
Chicago/Turabian StyleZhong, Yanxin, Yong Wah Tan, and Ding Xiang Liu. 2012. "Recent Progress in Studies of Arterivirus- and Coronavirus-Host Interactions" Viruses 4, no. 6: 980-1010. https://doi.org/10.3390/v4060980