Application of Live-Cell RNA Imaging Techniques to the Study of Retroviral RNA Trafficking

Abstract
:1. Introduction
2. Hybridization-Based RNA Labeling Techniques
2.1. Fluorescence in situ Hybridization (FISH)
2.2. Linear Oligonucleotide Probes

{kind=link}
| Method | Diagram | Recognition Sequence | Citations |
|---|---|---|---|
| Linear Oligonucleotide Probe |  | User defined | [6,7] |
| Linear FRET Probe |  | User defined | [8,9] |
| Autoligation FRET Probe | 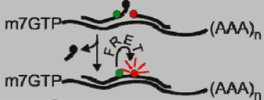 | User defined | [10,11] |
| Molecular Beacon | 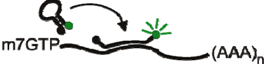 | User defined | [12] |
| MS2-GFP | 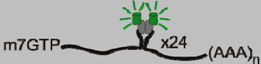 | 19 nucleotide stem-loop | [13,14] |
| Bgl-mCherry |  | 29 nucleotide stem-loop | [15] |
| λN-GFP |  | 15 nucleotide stem-loop | [16] |
| PUM-HD |  | User defined | [17,18] |
| Spinach |  | Varies based on desired fluorescence | [19] |
2.3 Molecular Beacons
3. RNA Binding Protein-Based RNA Labeling Techniques
3.1. MS2-GFP
3.2. Bgl-mCherry
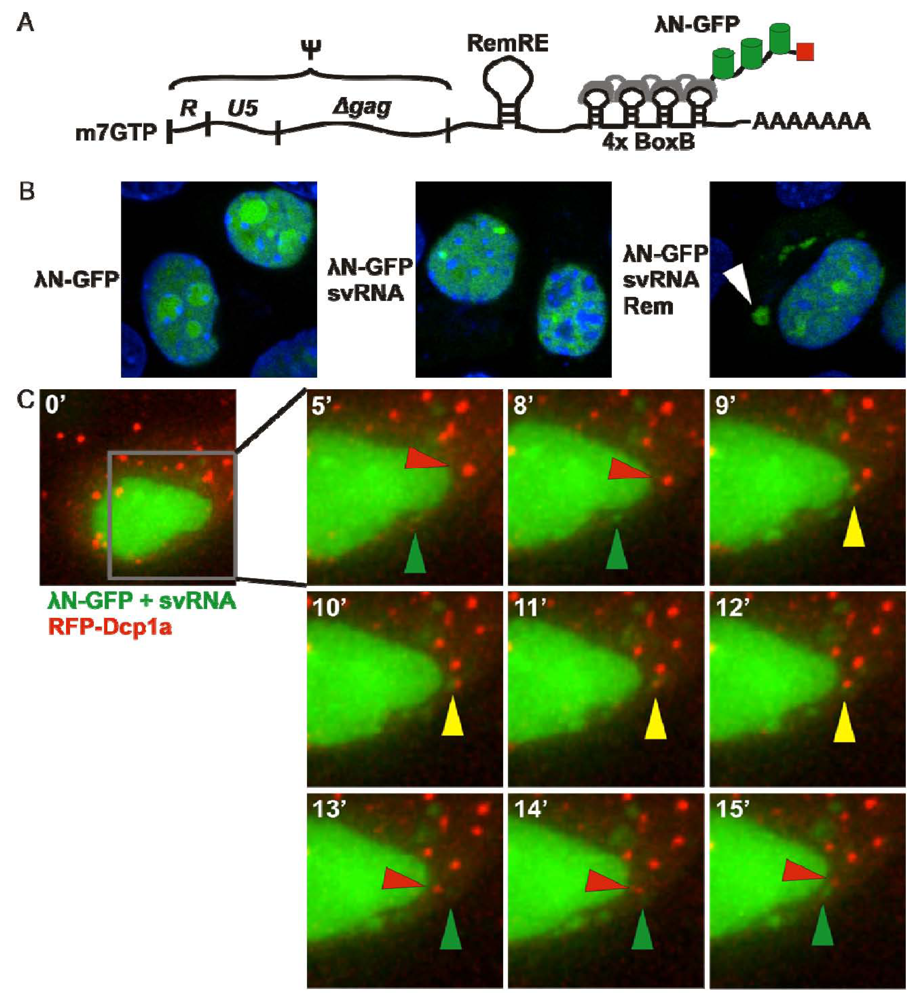
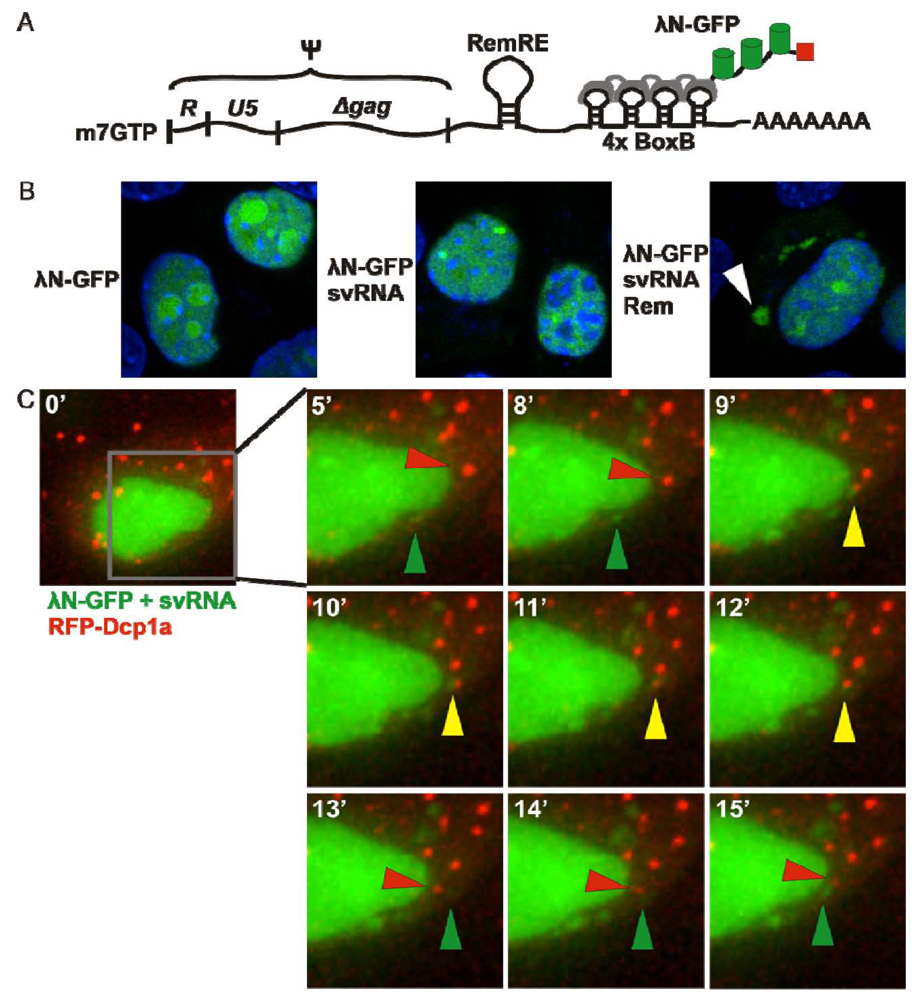
3.3. λN-GFP
3.4. PUM-HD
4. “Spinach” RNA Tracking System
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar]
- Inouye, S.; Tsuji, F.I. Aequorea green fluorescent protein. Expression of the gene and fluorescence characteristics of the recombinant protein. FEBS Lett. 1994, 341, 277–280. [Google Scholar] [CrossRef]
- Ainger, K.; Avossa, D.; Morgan, F.; Hill, S.J.; Barry, C.; Barbarese, E.; Carson, J.H. Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J. Cell Biol. 1993, 123, 431–441. [Google Scholar] [CrossRef]
- Butsch, M.; Boris-Lawrie, K. Destiny of unspliced retroviral RNA: ribosome and/or virion? J. Virol. 2002, 76, 3089–3094. [Google Scholar] [CrossRef]
- Gall, J.G.; Pardue, M.L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 63, 378–383. [Google Scholar] [CrossRef]
- Carmo-Fonseca, M.; Pepperkok, R.; Sproat, B.S.; Ansorge, W.; Swanson, M.S.; Lamond, A.I. In vivo detection of snRNP-rich organelles in the nuclei of mammalian cells. EMBO J. 1991, 10, 1863–1873. [Google Scholar]
- Molenaar, C.; Marras, S.A.; Slats, J.C.; Truffert, J.C.; Lemaitre, M.; Raap, A.K.; Dirks, R.W.; Tanke, H.J. Linear 2' O-Methyl RNA probes for the visualization of RNA in living cells. Nucleic Acids Res. 2001, 29, E89. [Google Scholar]
- Tsuji, A.; Koshimoto, H.; Sato, Y.; Hirano, M.; Sei-Iida, Y.; Kondo, S.; Ishibashi, K. Direct observation of specific messenger RNA in a single living cell under a fluorescence microscope. Biophys. J. 2000, 78, 3260–3274. [Google Scholar] [CrossRef]
- Okabe, K.; Harada, Y.; Zhang, J.; Tadakuma, H.; Tani, T.; Funatsu, T. Real time monitoring of endogenous cytoplasmic mRNA using linear antisense 2'-O-methyl RNA probes in living cells. Nucleic Acids Res. 2011, 39, e20. [Google Scholar]
- Xu, Y.; Karalkar, N.B.; Kool, E.T. Nonenzymaticautoligation in direct three-color detection of RNA and DNA point mutations. Nat. Biotechnol. 2001, 19, 148–152. [Google Scholar] [CrossRef]
- Abe, H.; Kool, E.T. Flow cytometric detection of specific RNAs in native human cells with quenched autoligating FRET probes. Proc. Natl. Acad. Sci. USA 2006, 103, 263–268. [Google Scholar] [CrossRef]
- Tyagi, S.; Kramer, F.R. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar] [CrossRef]
- Fusco, D.; Accornero, N.; Lavoie, B.; Shenoy, S.M.; Blanchard, J.M.; Singer, R.H.; Bertrand, E. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr. Biol. 2003, 13, 161–167. [Google Scholar] [CrossRef]
- Chen, J.; Nikolaitchik, O.; Singh, J.; Wright, A.; Bencsics, C.E.; Coffin, J.M.; Ni, N.; Lockett, S.; Pathak, V.K.; Hu, W.S. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 13535–13540. [Google Scholar]
- Daigle, N.; Ellenberg, J. [lambda]N-GFP: An RNA reporter system for live-cell imaging. Nat. Meth. 2007, 4, 633–636. [Google Scholar] [CrossRef]
- Ozawa, T.; Natori, Y.; Sato, M.; Umezawa, Y. Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat. Methods 2007, 4, 413–419. [Google Scholar]
- Yamada, T.; Yoshimura, H.; Inaguma, A.; Ozawa, T. Visualization of nonengineered single mRNAs in living cells using genetically encoded fluorescent probes. Anal. Chem. 2011, 83, 5708–5714. [Google Scholar] [CrossRef]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef]
- Bao, G.; Rhee, W.J.; Tsourkas, A. Fluorescent probes for live-cell RNA detection. Annu. Rev. Biomed. Eng. 2009, 11, 25–47. [Google Scholar] [CrossRef]
- Paillasson, S.; Robert-Nicoud, M.; Ronot, X. Specific detection of RNA molecules by fluorescent in situ hybridization in living cells. Cell Biol. Toxicol. 1996, 12, 359–361. [Google Scholar] [CrossRef]
- Okamura, Y.; Kondo, S.; Sase, I.; Suga, T.; Mise, K.; Furusawa, I.; Kawakami, S.; Watanabe, Y. Double-labeled donor probe can enhance the signal of fluorescence resonance energy transfer (FRET) in detection of nucleic acid hybridization. Nucleic Acids Res. 2000, 28, E107. [Google Scholar]
- Santangelo, P.J.; Nix, B.; Tsourkas, A.; Bao, G. Dual FRET molecular beacons for mRNA detection in living cells. Nucleic Acids Res. 2004, 32, e57. [Google Scholar] [CrossRef]
- Santangelo, P.; Nitin, N.; LaConte, L.; Woolums, A.; Bao, G. Live-cell characterization and analysis of a clinical isolate of bovine respiratory syncytial virus, using molecular beacons. J. Virol. 2006, 80, 682–688. [Google Scholar]
- Santangelo, P.J.; Bao, G. Dynamics of filamentous viral RNPs prior to egress. Nucleic Acids Res. 2007, 35, 3602–3611. [Google Scholar] [CrossRef]
- Yeh, H.Y.; Yates, M.V.; Mulchandani, A.; Chen, W. Molecular beacon-quantum dot-Au nanoparticle hybrid nanoprobes for visualizing virus replication in living cells. Chem. Commun. (Camb) 2010, 46, 3914–3916. [Google Scholar] [CrossRef]
- Cui, Z.Q.; Zhang, Z.P.; Zhang, X.E.; Wen, J.K.; Zhou, Y.F.; Xie, W.H. Visualizing the dynamic behavior of poliovirus plus-strand RNA in living host cells. Nucleic Acids Res. 2005, 33, 3245–3252. [Google Scholar]
- Wang, W.; Cui, Z.Q.; Han, H.; Zhang, Z.P.; Wei, H.P.; Zhou, Y.F.; Chen, Z.; Zhang, X.E. Imaging and characterizing influenza A virus mRNA transport in living cells. Nucleic Acids Res. 2008, 36, 4913–4928. [Google Scholar]
- Kozak, C.; Peters, G.; Pauley, R.; Morris, V.; Michalides, R.; Dudley, J.; Green, M.; Davisson, M.; Prakash, O.; Vaidya, A.; et al. A standardized nomenclature for endogenous mouse mammary tumor viruses. J. Virol. 1987, 61, 1651–1654. [Google Scholar]
- Mhlanga, M.M.; Vargas, D.Y.; Fung, C.W.; Kramer, F.R.; Tyagi, S. tRNA-linked molecular beacons for imaging mRNAs in the cytoplasm of living cells. Nucleic Acids Res. 2005, 33, 1902–1912. [Google Scholar]
- Chen, A.K.; Behlke, M.A.; Tsourkas, A. Avoiding false-positive signals with nuclease-vulnerable molecular beacons in single living cells. Nucleic Acids Res. 2007, 35, e105. [Google Scholar] [CrossRef]
- Ni, C.Z.; Syed, R.; Kodandapani, R.; Wickersham, J.; Peabody, D.S.; Ely, K.R. Crystal structure of the MS2 coat protein dimer: implications for RNA binding and virus assembly. Structure 1995, 3, 255–263. [Google Scholar] [CrossRef]
- Peabody, D.S.; Ely, K.R. Control of translational repression by protein-protein interactions. Nucleic Acids Res. 1992, 20, 1649–1655. [Google Scholar] [CrossRef]
- Lim, F.; Peabody, D.S. Mutations that increase the affinity of a translational repressor for RNA. Nucleic Acids Res. 1994, 22, 3748–3752. [Google Scholar] [CrossRef]
- Shav-Tal, Y.; Darzacq, X.; Shenoy, S.M.; Fusco, D.; Janicki, S.M.; Spector, D.L.; Singer, R.H. Dynamics of single mRNPs in nuclei of living cells. Science (New York) 2004, 304, 1797–1800. [Google Scholar]
- Boireau, S.; Maiuri, P.; Basyuk, E.; de la Mata, M.; Knezevich, A.; Pradet-Balade, B.; Backer, V.; Kornblihtt, A.; Marcello, A.; Bertrand, E. The transcriptional cycle of HIV-1 in real-time and live cells. J. Cell Biol. 2007, 179, 291–304. [Google Scholar] [CrossRef]
- Maiuri, P.; Knezevich, A.; Bertrand, E.; Marcello, A. Real-time imaging of the HIV-1 transcription cycle in single living cells. Methods 2011, 53, 62–67. [Google Scholar] [CrossRef]
- Moore, M.D.; Nikolaitchik, O.A.; Chen, J.; Hammarskjold, M.L.; Rekosh, D.; Hu, W.S. Probing the HIV-1 genomic RNA trafficking pathway and dimerization by genetic recombination and single virion analyses. PLoS Pathog. 2009, 5, e1000627. [Google Scholar] [CrossRef]
- Mor, A.; Suliman, S.; Ben-Yishay, R.; Yunger, S.; Brody, Y.; Shav-Tal, Y. Dynamics of single mRNPnucleocytoplasmic transport and export through the nuclear pore in living cells. Nat. Cell Biol. 2010, 12, 543–552. [Google Scholar] [CrossRef]
- Kemler, I.; Meehan, A.; Poeschla, E.M. Live-cell coimaging of the genomic RNAs and Gag proteins of two lentiviruses. J. Virol. 2010, 84, 6352–6366. [Google Scholar] [CrossRef]
- Basyuk, E.; Galli, T.; Mougel, M.; Blanchard, J.M.; Sitbon, M.; Bertrand, E. Retroviral genomic RNAs are transported to the plasma membrane by endosomal vesicles. Dev. Cell 2003, 5, 161–174. [Google Scholar] [CrossRef]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar]
- Rackham, O.; Brown, C.M. Visualization of RNA-protein interactions in living cells: FMRP and IMP1 interact on mRNAs. EMBO J. 2004, 23, 3346–3355. [Google Scholar] [CrossRef]
- Milev, M.P.; Brown, C.M.; Mouland, A.J. Live cell visualization of the interactions between HIV-1 Gag and the cellular RNA-binding protein Staufen1. Retrovirology 2010, 7, 41. [Google Scholar] [CrossRef]
- Manival, X.; Yang, Y.; Strub, M.P.; Kochoyan, M.; Steinmetz, M.; Aymerich, S. From genetic to structural characterization of a new class of RNA-binding domain within the SacY/BglG family of antiterminator proteins. EMBO J. 1997, 16, 5019–5029. [Google Scholar] [CrossRef]
- Dilley, K.A.; Ni, N.; Nikolaitchik, O.A.; Chen, J.; Galli, A.; Hu, W.S. Determining the frequency and mechanisms of HIV-1 and HIV-2 RNA copackaging by single-virion analysis. J. Virol. 2011, 85, 10499–10508. [Google Scholar]
- Ni, N.; Nikolaitchik, O.A.; Dilley, K.A.; Chen, J.; Galli, A.; Fu, W.; Prasad, V.V.; Ptak, R.G.; Pathak, V.K.; Hu, W.S. Mechanisms of human immunodeficiency virus type 2 RNA packaging: Efficient trans packaging and selection of RNA copackaging partners. J. Virol. 2011, 85, 7603–7612. [Google Scholar]
- Hu, W.S.; Temin, H.M. Retroviral recombination and reverse transcription. Science 1990, 250, 1227–1233. [Google Scholar]
- Al Dhaheri, N.S.; Phillip, P.S.; Ghazawi, A.; Ali, J.; Beebi, E.; Jaballah, S.A.; Rizvi, T.A. Cross-packaging of genetically distinct mouse and primate retroviral RNAs. Retrovirology 2009, 6, 66. [Google Scholar] [CrossRef]
- Jude, B.A.; Pobezinskaya, Y.; Bishop, J.; Parke, S.; Medzhitov, R.M.; Chervonsky, A.V.; Golovkina, T.V. Subversion of the innate immune system by a retrovirus. Nat. Immunol. 2003, 4, 573–578. [Google Scholar]
- Austin, R.J.; Xia, T.; Ren, J.; Takahashi, T.T.; Roberts, R.W. Designed arginine-rich RNA-binding peptides with picomolar affinity. J. Am. Chem. Soc. 2002, 124, 10966–10967. [Google Scholar]
- Rizvi, T.A.; Ali, J.; Phillip, P.S.; Ghazawi, A.; Jayanth, P.; Mustafa, F. Role of a heterologous retroviral transport element in the development of genetic complementation assay for mouse mammary tumor virus (MMTV) replication. Virology 2009, 385, 464–472. [Google Scholar] [CrossRef]
- Indik, S.; Gunzburg, W.H.; Salmons, B.; Rouault, F. A novel, mouse mammary tumor virus encoded protein with Rev-like properties. Virology 2005, 337, 1–6. [Google Scholar] [CrossRef]
- Mertz, J.A.; Simper, M.S.; Lozano, M.M.; Payne, S.M.; Dudley, J.P. Mouse mammary tumor virus encodes a self-regulatory RNA export protein and is a complex retrovirus. J. Virol. 2005, 79, 14737–14747. [Google Scholar] [CrossRef]
- Mertz, J.A.; Chadee, A.B.; Byun, H.; Russell, R.; Dudley, J.P. Mapping of the functional boundaries and secondary structure of the mouse mammary tumor virus Rem-responsive element. J. Biol. Chem. 2009, 284, 25642–25652. [Google Scholar]
- Franks, T.M.; Lykke-Andersen, J. The control of mRNA decapping and P-body formation. Mol. Cell 2008, 32, 605–615. [Google Scholar] [CrossRef]
- Zamore, P.D.; Williamson, J.R.; Lehmann, R. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 1997, 3, 1421–1433. [Google Scholar]
- Wang, X.; McLachlan, J.; Zamore, P.D.; Hall, T.M. Modular recognition of RNA by a human pumilio-homology domain. Cell 2002, 110, 501–512. [Google Scholar] [CrossRef]
- Lu, G.; Dolgner, S.J.; Hall, T.M. Understanding and engineering RNA sequence specificity of PUF proteins. Curr. Opin. Struct. Biol. 2009, 19, 110–115. [Google Scholar] [CrossRef]
- Cheong, C.G.; Hall, T.M. Engineering RNA sequence specificity of Pumilio repeats. Proc. Natl. Acad. Sci. USA 2006, 103, 13635–13639. [Google Scholar] [CrossRef]
- Tilsner, J.; Linnik, O.; Christensen, N.M.; Bell, K.; Roberts, I.M.; Lacomme, C.; Oparka, K.J. Live-cell imaging of viral RNA genomes using a Pumilio-based reporter. Plant J. 2009, 57, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Buxbaum, A.R.; Singer, R.H. Single mRNA tracking in live cells. Methods Enzymol. 2010, 472, 387–406. [Google Scholar]
- Santangelo, P.; Nitin, N.; Bao, G. Nanostructured probes for RNA detection in living cells. Ann. Biomed. Eng. 2006, 34, 39–50. [Google Scholar] [CrossRef]
- Tsourkas, A.; Behlke, M.A.; Rose, S.D.; Bao, G. Hybridization kinetics and thermodynamics of molecular beacons. Nucleic Acids Res. 2003, 31, 1319–1330. [Google Scholar] [CrossRef]
- Haim, L.; Zipor, G.; Aronov, S.; Gerst, J.E. A genomic integration method to visualize localization of endogenous mRNAs in living yeast. Nat. Methods 2007, 4, 409–412. [Google Scholar]
- Lionnet, T.; Czaplinski, K.; Darzacq, X.; Shav-Tal, Y.; Wells, A.L.; Chao, J.A.; Park, H.Y.; de Turris, V.; Lopez-Jones, M.; Singer, R.H. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat. Methods 2011, 8, 165–170. [Google Scholar]
- Beckham, C.J.; Parker, R. P bodies, stress granules, and viral life cycles. Cell Host Microbe 2008, 3, 206–212. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar]
- Yedavalli, V.S.; Neuveut, C.; Chi, Y.H.; Kleiman, L.; Jeang, K.T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 2004, 119, 381–392. [Google Scholar] [CrossRef]
- Furtak, V.; Mulky, A.; Rawlings, S.A.; Kozhaya, L.; Lee, K.; Kewalramani, V.N.; Unutmaz, D. Perturbation of the P-body component Mov10 inhibits HIV-1 infectivity. PLoSOne 2010, 5, e9081. [Google Scholar]
- Burdick, R.; Smith, J.L.; Chaipan, C.; Friew, Y.; Chen, J.; Venkatachari, N.J.; Delviks-Frankenberry, K.A.; Hu, W.S.; Pathak, V.K. P body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J. Virol. 2010, 84, 10241–10253. [Google Scholar]
- Wang, X.; Han, Y.; Dang, Y.; Fu, W.; Zhou, T.; Ptak, R.G.; Zheng, Y.H. Moloney leukemia virus 10 (MOV10) protein inhibits retrovirus replication. J. Biol. Chem. 2010, 285, 14346–14355. [Google Scholar]
- Bouttier, M.; Saumet, A.; Peter, M.; Courgnaud, V.; Schmidt, U.; Cazevieille, C.; Bertrand, E.; Lecellier, C.H. Retroviral GAG proteins recruit AGO2 on viral RNAs without affecting RNA accumulation and translation. Nucleic Acids Res. 2012, 40, 775–786. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bann, D.V.; Parent, L.J. Application of Live-Cell RNA Imaging Techniques to the Study of Retroviral RNA Trafficking. Viruses 2012, 4, 963-979. https://doi.org/10.3390/v4060963
Bann DV, Parent LJ. Application of Live-Cell RNA Imaging Techniques to the Study of Retroviral RNA Trafficking. Viruses. 2012; 4(6):963-979. https://doi.org/10.3390/v4060963
Chicago/Turabian StyleBann, Darrin V., and Leslie J. Parent. 2012. "Application of Live-Cell RNA Imaging Techniques to the Study of Retroviral RNA Trafficking" Viruses 4, no. 6: 963-979. https://doi.org/10.3390/v4060963