Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein

Abstract
:1. Introduction

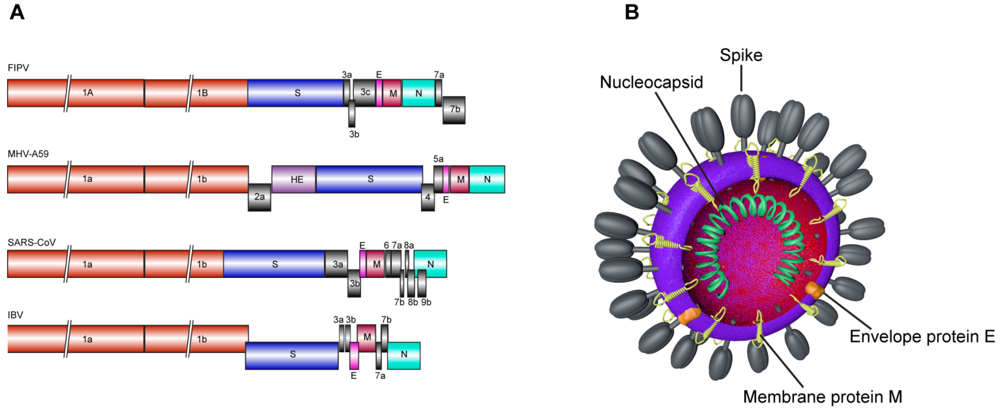
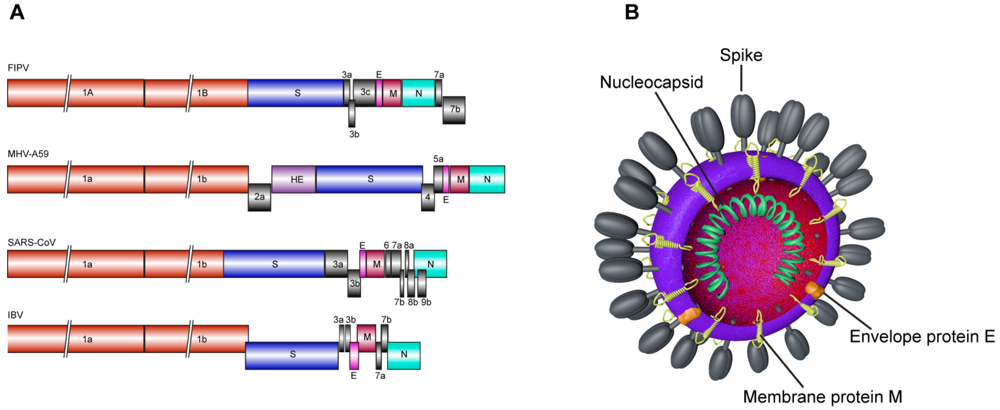{kind=link}
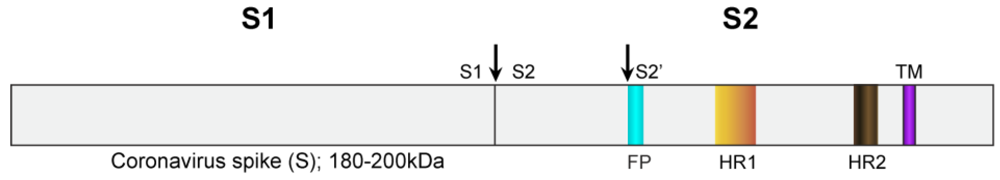
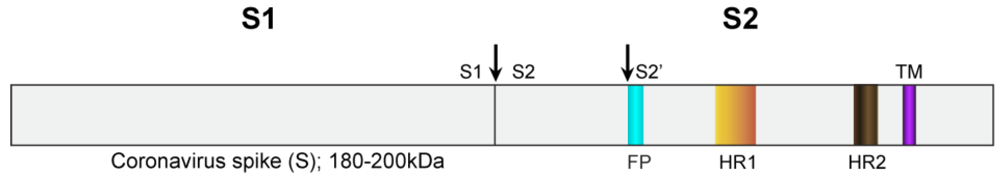{kind=link}
{kind=link}
| Genus | Species | Receptor | |
|---|---|---|---|
| Alphacoronavirus | • Alphacoronavirus 1 comprising: | ||
| Feline Coronavirus (FCoV) serotype 2 | Aminopeptidase N | ||
| Canine Coronavirus (CCoV) serotype 2 | Aminopeptidase N | ||
| Transmissible gastroenteritis virus (TGEV) | Aminopeptidase N | ||
| • Human coronavirus 229E | Aminopeptidase N | ||
| • Human coronavirus NL63 | ACE2 | ||
| • Porcine Epidemic Diarrhea Coronavirus (PEDV) | Aminopeptidase N | ||
| • Rhinolophus bat coronavirus HKU2 | |||
| • Scotophilus bat coronavirus 512/05 | |||
| • Miniopterus bat coronavirus 1 | |||
| • Miniopterus bat coronavirus HKU8 | |||
| Betacoronavirus | • Betacoronavirus 1 comprising: | ||
| Bovine coronavirus (BCoV) | Neu 5,9 Ac2 | ||
| Human coronavirus OC43 (HCoV-OC43) | Neu 5,9 Ac2 | ||
| Equine coronavirus (ECoV) | |||
| Human enteric coronavirus (HECoV) | |||
| Porcine haemagglutinating encephalomyelitis virus (PHEV) | |||
| Canine respiratory coronavirus (CrCoV) | |||
| • Murine coronavirus comprising: | |||
| Existing species of mouse hepatitis virus (MHV) | CEACAM1 | ||
| Rat coronavirus | |||
| Puffinosis virus | |||
| • Human coronavirus HKU9 | |||
| • Rousettus bat coronavirus HKU4 | |||
| • Tylonycteris bat coronvirus HKU5 | |||
| • SARSr-CoV (SARS related Coronavirus) comprising | |||
| Human SARS-CoV | ACE2 | ||
| Rhinolophus bat viruses | |||
| Gamma-coronavirus | • Avian coronavirus comprising: | ||
| IBV Various coronaviruses infecting turkey, pheasant, duck, goose and pigeon | |||
| • Beluga Whale coronavirus SW1 | |||
| Delta-coronavirus | • Bulbul coronavirus HKU11 | ||
| • Thrush coronavirus HKU12 | |||
| • Munia coronavirus HKU13 | |||
2. Spike Protein
3. Receptor Binding and Tropism
4. Entry and Fusion
| Protease | Cleavage site | Cell-cell fusion | Virus Entry |
|---|---|---|---|
| Cathepsin L [72,79] | S1/S2 HTVSLLRSTSQKSIVAYTMSL | - | + |
| Elastase [74,85] | S2’ LPDPLKPTKRSFIEDLLFNKV | + | + |
| HAT (TMPRSS11d) [84] | S1/S2 HTVSLLRSTSQKSIVAYTMSL | + | - |
| Plasmin [82] | S1/S2 HTVSLLRSTSQKSIVAYTMSL S2’ LPDPLKPTKRSFIEDLLFNKV | n.d. | + |
| TMPRSS11a [82] | S1/S2 HTVSLLRSTSQKSIVAYTMSL S2’ LPDPLKPTKRSFIEDLLFNKV | n.d. | + |
| TMPRSS2 [81,83,84] | Multiple sites | + | + |
| Trypsin [7373,75] | S1/S2 HTVSLLRSTSQKSIVAYTMSL S2’ LPDPLKPTKRSFIEDLLFNKV | + | + |
5. Fusion Peptide
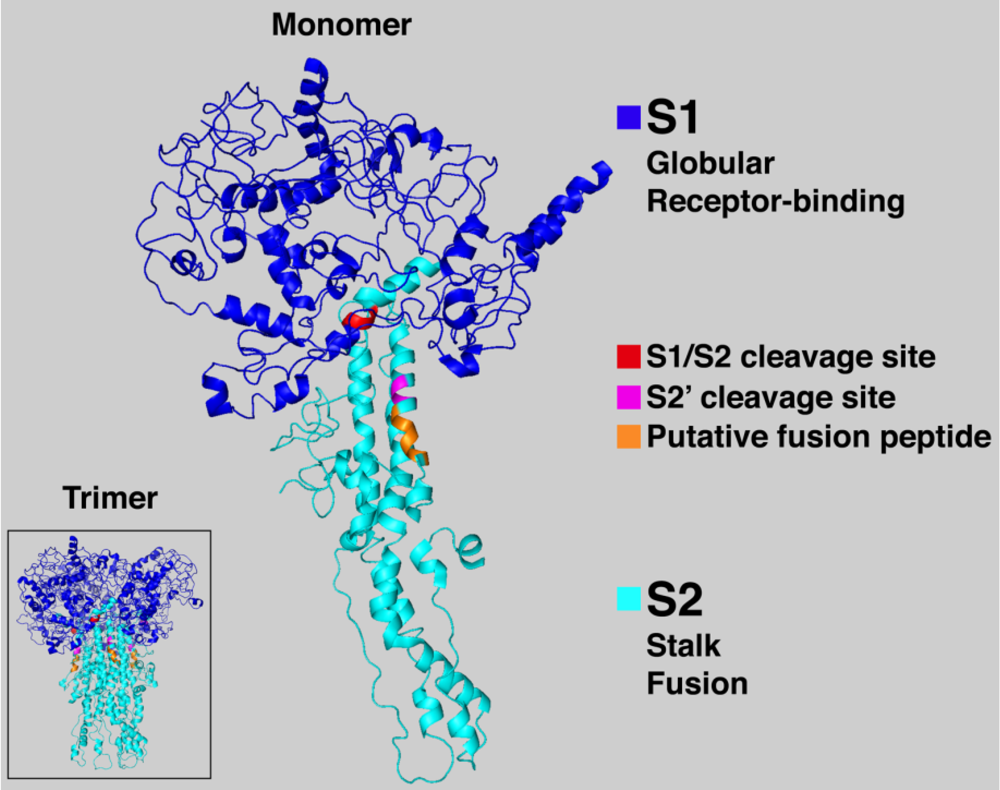
6. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Hudson, C.B.; Beaudette, F.R. Infection of the cloaca with the virus of infectious bronchitis. Science 1932, 76, 34. [Google Scholar]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Enjuanes, L.; Almazan, F.; Sola, I.; Zuniga, S. Biochemical aspects of coronavirus replication and virus-host interaction. Annu. Rev. Microbiol. 2006, 60, 211–230. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Xu, Y.; Cole, D.K.; Lou, Z.; Liu, Y.; Qin, L.; Li, X.; Bai, Z.; Yuan, F.; Rao, Z.; Gao, G.F. Construct design, biophysical, and biochemical characterization of the fusion core from mouse hepatitis virus (a coronavirus) spike protein. Protein Expr. Purif. 2004, 38, 116–122. [Google Scholar] [CrossRef]
- Supekar, V.M.; Bruckmann, C.; Ingallinella, P.; Bianchi, E.; Pessi, A.; Carfi, A. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17958–17963. [Google Scholar]
- Chan, W.E.; Chuang, C.K.; Yeh, S.H.; Chang, M.S.; Chen, S.S. Functional characterization of heptad repeat 1 and 2 mutants of the spike protein of severe acute respiratory syndrome coronavirus. J. Virol. 2006, 80, 3225–3237. [Google Scholar] [CrossRef]
- Luo, Z.; Matthews, A.M.; Weiss, S.R. Amino acid substitutions within the leucine zipper domain of the murine coronavirus spike protein cause defects in oligomerization and the ability to induce cell-to-cell fusion. J. Virol. 1999, 73, 8152–8159. [Google Scholar]
- Navas, S.; Seo, S.H.; Chua, M.M.; Das Sarma, J.; Lavi, E.; Hingley, S.T.; Weiss, S.R. Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J. Virol. 2001, 75, 2452–2457. [Google Scholar] [CrossRef]
- Navas, S.; Weiss, S.R. Murine coronavirus-induced hepatitis: JHM genetic background eliminates A59 spike-determined hepatotropism. J. Virol. 2003, 77, 4972–4978. [Google Scholar] [CrossRef]
- Hingley, S.T.; Gombold, J.L.; Lavi, E.; Weiss, S.R. MHV-A59 fusion mutants are attenuated and display altered hepatotropism. Virology 1994, 200, 1–10. [Google Scholar] [CrossRef]
- Leparc-Goffart, I.; Hingley, S.T.; Chua, M.M.; Phillips, J.; Lavi, E.; Weiss, S.R. Targeted recombination within the spike gene of murine coronavirus mouse hepatitis virus-A59: Q159 is a determinant of hepatotropism. J. Virol. 1998, 72, 9628–9636. [Google Scholar]
- Casais, R.; Dove, B.; Cavanagh, D.; Britton, P. Recombinant avian infectious bronchitis virus expressing a heterologous spike gene demonstrates that the spike protein is a determinant of cell tropism. J. Virol. 2003, 77, 9084–9089. [Google Scholar] [CrossRef]
- Hodgson, T.; Casais, R.; Dove, B.; Britton, P.; Cavanagh, D. Recombinant infectious bronchitis coronavirus Beaudette with the spike protein gene of the pathogenic M41 strain remains attenuated but induces protective immunity. J. Virol. 2004, 78, 13804–13811. [Google Scholar]
- Haijema, B.J.; Rottier, P.; de Groot, R.P. Feline coronaviruses: A tale of two-faced types. In Coronaviruses Molecular and Cellular Biology; Thiel, V., Ed.; Caister Academic Press: Norfolk, UK, 2007; pp. 183–204. [Google Scholar]
- Pedersen, N.C. Virologic and immunologic aspects of feline infectious peritonitis virus infection. Adv. Exp. Med. Biol. 1987, 218, 529–550. [Google Scholar] [CrossRef]
- Vennema, H.; Poland, A.; Foley, J.; Pedersen, N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology 1998, 243, 150–157. [Google Scholar] [CrossRef]
- Rottier, P.J.M.; Nakamura, K.; Schellen, P.; Volders, H.; Haijema, B.J. Acquisition of macrophage tropism during the pathogenesis of feline infectious peritonitis is determined by mutations in the feline coronavirus spike protein. J. Virol. 2005, 79, 14122–14130. [Google Scholar]
- Haijema, B.J.; Volders, H.; Rottier, P.J. Switching species tropism: An effective way to manipulate the feline coronavirus genome. J. Virol. 2003, 77, 4528–4538. [Google Scholar]
- Dveksler, G.S.; Pensiero, M.N.; Cardellichio, C.B.; Williams, R.K.; Jiang, G.S.; Holmes, K.V.; Dieffenbach, C.W. Cloning of the mouse hepatitis virus (MHV) receptor: Expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 1991, 65, 6881–6891. [Google Scholar]
- Taguchi, F.; Hirai-Yuki, A. Mouse hepatitis virus receptor as a determinant of the mouse susceptibility to MHV infection. Front. Microbiol. 2012, 3, 68. [Google Scholar]
- Dveksler, G.S.; Pensiero, M.N.; Dieffenbach, C.W.; Cardellichio, C.B.; Basile, A.A.; Elia, P.E.; Holmes, K.V. Mouse hepatitis virus strain A59 and blocking antireceptor monoclonal antibody bind to the N-terminal domain of cellular receptor. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 1716–1720. [Google Scholar]
- Miura, T.A.; Travanty, E.A.; Oko, L.; Bielefeldt-Ohmann, H.; Weiss, S.R.; Beauchemin, N.; Holmes, K.V. The spike glycoprotein of murine coronavirus MHV-JHM mediates receptor-independent infection and spread in the central nervous systems of Ceacam1a-/- Mice. J. Virol. 2008, 82, 755–763. [Google Scholar]
- Nakagaki, K.; Taguchi, F. Receptor-independent spread of a highly neurotropic murine coronavirus JHMV strain from initially infected microglial cells in mixed neural cultures. J. Virol. 2005, 79, 6102–6110. [Google Scholar] [CrossRef]
- Hemmila, E.; Turbide, C.; Olson, M.; Jothy, S.; Holmes, K.V.; Beauchemin, N. Ceacam1a-/- mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J. Virol. 2004, 78, 10156–10165. [Google Scholar]
- Schickli, J.H.; Zelus, B.D.; Wentworth, D.E.; Sawicki, S.G.; Holmes, K.V. The murine coronavirus mouse hepatitis virus strain A59 from persistently infected murine cells exhibits an extended host range. J. Virol. 1997, 71, 9499–9507. [Google Scholar]
- de Haan, C.A.; Te Lintelo, E.; Li, Z.; Raaben, M.; Wurdinger, T.; Bosch, B.J.; Rottier, P.J. Cooperative involvement of the S1 and S2 subunits of the murine coronavirus spike protein in receptor binding and extended host range. J. Virol. 2006, 80, 10909–10918. [Google Scholar]
- de Haan, C.A.; Li, Z.; te Lintelo, E.; Bosch, B.J.; Haijema, B.J.; Rottier, P.J. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J. Virol. 2005, 79, 14451–14456. [Google Scholar]
- Phillips, J.J.; Chua, M.; Seo, S.H.; Weiss, S.R. Multiple regions of the murine coronavirus spike glycoprotein influence neurovirulence. J. Neurovirol. 2001, 7, 421–431. [Google Scholar] [CrossRef]
- Krueger, D.K.; Kelly, S.M.; Lewicki, D.N.; Ruffolo, R.; Gallagher, T.M. Variations in disparate regions of the murine coronavirus spike protein impact the initiation of membrane fusion. J. Virol. 2001, 75, 2792–2802. [Google Scholar] [CrossRef]
- Tusell, S.M.; Schittone, S.A.; Holmes, K.V. Mutational analysis of aminopeptidase N, a receptor for several group 1 coronaviruses, identifies key determinants of viral host range. J. Virol. 2007, 81, 1261–1273. [Google Scholar]
- Rasschaert, D.; Duarte, M.; Laude, H. Porcine respiratory coronavirus differs from transmissible gastroenteritis virus by a few genomic deletions. J. Gen. Virol. 1990, 71, 2599–2607. [Google Scholar] [CrossRef]
- Schultze, B.; Krempl, C.; Ballesteros, M.L.; Shaw, L.; Schauer, R.; Enjuanes, L.; Herrler, G. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (N-glycolylneuraminic acid) binding activity. J. Virol. 1996, 70, 5634–5637. [Google Scholar]
- Krempl, C.; Schultze, B.; Laude, H.; Herrler, G. Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J. Virol. 1997, 71, 3285–3287. [Google Scholar]
- Schwegmann-Wessels, C.; Zimmer, G.; Schroder, B.; Breves, G.; Herrler, G. Binding of transmissible gastroenteritis coronavirus to brush border membrane sialoglycoproteins. J. Virol. 2003, 77, 11846–11848. [Google Scholar] [CrossRef]
- Krempl, C.; Ballesteros, M.L.; Zimmer, G.; Enjuanes, L.; Klenk, H.D.; Herrler, G. Characterization of the sialic acid binding activity of transmissible gastroenteritis coronavirus by analysis of haemagglutination-deficient mutants. J. Gen. Virol. 2000, 81, 489–496. [Google Scholar]
- Schwegmann-Wessels, C.; Herrler, G. Sialic acids as receptor determinants for coronaviruses. Glycoconj. J. 2006, 23, 51–58. [Google Scholar] [CrossRef]
- Peng, G.; Sun, D.; Rajashankar, K.R.; Qian, Z.; Holmes, K.V.; Li, F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10696–10701. [Google Scholar]
- Madu, I.G.; Chu, V.C.; Lee, H.; Regan, A.D.; Bauman, B.E.; Whittaker, G.R. Heparan sulfate is a selective attachment factor for the avian coronavirus infectious bronchitis virus Beaudette. Avian. Dis. 2007, 51, 45–51. [Google Scholar] [CrossRef]
- de Haan, C.A.M.; Haijema, B.J.; Schellen, P.; Schreur, P.W.; te Lintelo, E.; Vennema, H.; Rottier, P.J.M. Cleavage of group 1 coronavirus spike proteins: How furin cleavage is traded off against heparan sulfate binding upon cell culture adaptation. J. Virol. 2008, 82, 6078–6083. [Google Scholar]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus . Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar]
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by SARS coronavirus. J. Biol. Chem. 2012, 287, 8904–8911. [Google Scholar]
- Ren, W.; Qu, X.; Li, W.; Han, Z.; Yu, M.; Zhou, P.; Zhang, S.Y.; Wang, L.F.; Deng, H.; Shi, Z. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 2008, 82, 1899–1907. [Google Scholar]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15748–15753. [Google Scholar]
- Jeffers, S.A.; Hemmila, E.M.; Holmes, K.V. Human coronavirus 229E can use CD209L (L-SIGN) to enter cells. Adv. Exp. Med. Biol. 2006, 581, 265–269. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.-P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus Is Mediated by the Spike Glycoprotein and Enhanced by Dendritic Cell Transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar]
- Han, D.P.; Lohani, M.; Cho, M.W. Specific asparagine-linked glycosylation sites are critical for DC-SIGN- and L-SIGN-mediated severe acute respiratory syndrome coronavirus entry. J. Virol. 2007, 81, 12029–12039. [Google Scholar]
- Regan, A.D.; Whittaker, G.R. Utilization of DC-SIGN for entry of feline coronaviruses into host cells. J. Virol. 2008, 82, 11992–11996. [Google Scholar] [CrossRef]
- Regan, A.D.; Ousterout, D.G.; Whittaker, G.R. Feline lectin activity is critical for the cellular entry of feline infectious peritonitis virus. J. Virol. 2010, 84, 7917–7921. [Google Scholar] [CrossRef]
- Zhang, Y.; Buckles, E.; Whittaker, G.R. Expression of the C-type lectins DC-SIGN or L-SIGN alters host cell susceptibility for the avian coronavirus, infectious bronchitis virus. Vet. Microbiol. 2012, 157, 285–293. [Google Scholar] [CrossRef]
- Nash, T.C.; Buchmeier, M.J. Entry of mouse hepatitis virus into cells by endosomal and nonendosomal pathways. Virology 1997, 233, 1–8. [Google Scholar] [CrossRef]
- Matsuyama, S.; Taguchi, F. Receptor-induced conformational changes of murine coronavirus spike protein. J. Virol. 2002, 76, 11819–11826. [Google Scholar] [CrossRef]
- Sturman, L.S.; Ricard, C.S.; Holmes, K.V. Conformational change of the coronavirus peplomer glycoprotein at pH 8.0 and 37 degrees C correlates with virus aggregation and virus-induced cell fusion. 1990. [Google Scholar]
- Zelus, B.D.; Schickli, J.H.; Blau, D.M.; Weiss, S.R.; Holmes, K.V. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees C either by soluble murine CEACAM1 receptors or by pH 8. J. Virol. 2003, 77, 830–840. [Google Scholar]
- Taguchi, F.; Matsuyama, S. Soluble receptor potentiates receptor-independent infection by murine coronavirus. J. Virol. 2002, 76, 950–958. [Google Scholar] [CrossRef]
- Gallagher, T.M.; Escarmis, C.; Buchmeier, M.J. Alteration of the pH dependence of coronavirus-induced cell fusion: Effect of mutations in the spike glycoprotein. J. Virol. 1991, 65, 1916–1928. [Google Scholar]
- Qiu, Z.; Hingley, S.T.; Simmons, G.; Yu, C.; Das Sarma, J.; Bates, P.; Weiss, S.R. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006, 80, 5768–5776. [Google Scholar]
- Eifart, P.; Ludwig, K.; Bottcher, C.; de Haan, C.A.; Rottier, P.J.; Korte, T.; Herrmann, A. Role of endocytosis and low pH in murine hepatitis virus strain A59 cell entry. J. Virol. 2007, 81, 10758–10768. [Google Scholar]
- Pu, Y.; Zhang, X. Mouse hepatitis virus type 2 enters cells through a clathrin-mediated endocytic pathway independent of Eps15. J. Virol. 2008, 82, 8112–8123. [Google Scholar] [CrossRef]
- Chu, V.C.; McElroy, L.J.; Chu, V.; Bauman, B.E.; Whittaker, G.R. The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J. Virol. 2006, 80, 3180–3188. [Google Scholar]
- Yamada, Y.; Liu, D.X. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J. Virol. 2009, 83, 8744–8758. [Google Scholar] [CrossRef]
- Steinhauer, D.A. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 1999, 258, 1–20. [Google Scholar] [CrossRef]
- Leparc-Goffart, I.; Hingley, S.T.; Chua, M.M.; Jiang, X.; Lavi, E.; Weiss, S.R. Altered pathogenesis of a mutant of the murine coronavirus MHV-A59 is associated with a Q159L amino acid substitution in the spike protein. Virology 1997, 239, 1–10. [Google Scholar] [CrossRef]
- Gombold, J.L.; Hingley, S.T.; Weiss, S.R. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 1993, 67, 4504–4512. [Google Scholar]
- de Haan, C.A.; Stadler, K.; Godeke, G.J.; Bosch, B.J.; Rottier, P.J. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J. Virol. 2004, 78, 6048–6054. [Google Scholar]
- Hingley, S.T.; Leparc-Goffart, I.; Weiss, S.R. The spike protein of murine coronavirus mouse hepatitis virus strain A59 is not cleaved in primary glial cells and primary hepatocytes. J. Virol. 1998, 72, 1606–1609. [Google Scholar]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Seidah, N.G. The proprotein convertases, 20 years later. Methods Mol. Biol. 2011, 768, 23–57. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11876–11881. [Google Scholar]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4240–4245. [Google Scholar]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12543–12547. [Google Scholar]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5871–5876. [Google Scholar]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the SARS coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar]
- Callan, R.J.; Hartmann, F.A.; West, S.E.; Hinshaw, V.S. Cleavage of influenza A virus H1 hemagglutinin by swine respiratory bacterial proteases. J. Virol. 1997, 71, 7579–7585. [Google Scholar]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Kam, Y.W.; Okumura, Y.; Kido, H.; Ng, L.F.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One 2009, 4, e7870. [Google Scholar]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar]
- Bertram, S.; Glowacka, I.; Muller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; et al. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef]
- Regan, A.D.; Shraybman, R.; Cohen, R.D.; Whittaker, G.R. Differential role for low pH and cathepsin-mediated cleavage of the viral spike protein during entry of serotype II feline coronaviruses. Vet. Microbiol. 2008, 132, 235–248. [Google Scholar] [CrossRef]
- Earp, L.J.; Delos, S.E.; Park, H.E.; White, J.M. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 2005, 285, 25–66. [Google Scholar]
- Tamm, L.K.; Han, X. Viral fusion peptides: A tool set to disrupt and connect biological membranes. Biosci. Rep. 2000, 20, 501–518. [Google Scholar] [CrossRef]
- Lai, A.L.; Li, Y.; Tamm, L.K. Interplay of proteins and lipids in virus entry by membrane fusion. In Protein–Lipid Interactions; Tamm, L.K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 279–303. [Google Scholar]
- Martin, I.; Ruysschaert, J.M. Common properties of fusion peptides from diverse systems. Biosci. Rep. 2000, 20, 483–500. [Google Scholar] [CrossRef]
- Han, X.; Bushweller, J.H.; Cafiso, D.S.; Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001, 8, 715–720. [Google Scholar] [CrossRef]
- Gomara, M.J.; Mora, P.; Mingarro, I.; Nieva, J.L. Roles of a conserved proline in the internal fusion peptide of Ebola glycoprotein. FEBS Lett. 2004, 569, 261–266. [Google Scholar]
- Durell, S.R.; Martin, I.; Ruysschaert, J.M.; Shai, Y.; Blumenthal, R. What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion (review). Mol. Membr. Biol. 1997, 14, 97–112. [Google Scholar] [CrossRef]
- Bosch, B.J.; Rottier, P.J. Nidovirus entry into cells. In Nidoviruses; Perlman, S., Gallagher, T., Snijder, E.J., Eds.; ASM Press: Washington, DC, USA, 2008; pp. 157–178. [Google Scholar]
- Guillen, J.; Kinnunen, P.K.; Villalain, J. Membrane insertion of the three main membranotropic sequences from SARS-CoV S2 glycoprotein. Biochim. Biophys. Acta. 2008, 1778, 2765–2774. [Google Scholar] [CrossRef]
- Guillen, J.; Perez-Berna, A.J.; Moreno, M.R.; Villalain, J. Identification of the membrane-active regions of the severe acute respiratory syndrome coronavirus spike membrane glycoprotein using a 16/18-mer peptide scan: Implications for the viral fusion mechanism. J. Virol. 2005, 79, 1743–1752. [Google Scholar]
- Chambers, P.; Pringle, C.R.; Easton, A.J. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990, 71, 3075–3080. [Google Scholar] [CrossRef]
- Petit, C.M.; Melancon, J.M.; Chouljenko, V.N.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Genetic analysis of the SARS-coronavirus spike glycoprotein functional domains involved in cell-surface expression and cell-to-cell fusion. Virology 2005, 341, 215–230. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Rausch, J.M.; Gallaher, W.R.; Garry, R.F.; Wimley, W.C. Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J. Virol. 2005, 79, 7195–7206. [Google Scholar]
- Guillen, J.; Perez-Berna, A.J.; Moreno, M.R.; Villalain, J. A second SARS-CoV S2 glycoprotein internal membrane-active peptide Biophysical characterization and membrane interaction. 2008, 47, 8214–8224. [Google Scholar]
- Guillen, J.; de Almeida, R.F.; Prieto, M.; Villalain, J. Structural and dynamic characterization of the interaction of the putative fusion peptide of the S2 SARS-CoV virus protein with lipid membranes. J. Phys. Chem. B 2008, 112, 6997–7007. [Google Scholar]
- Sainz, B., Jr.; Rausch, J.M.; Gallaher, W.R.; Garry, R.F.; Wimley, W.C. The aromatic domain of the coronavirus class I viral fusion protein induces membrane permeabilization: Putative role during viral entry. Biochemistry 2005, 44, 947–958. [Google Scholar]
- Howard, M.W.; Travanty, E.A.; Jeffers, S.A.; Smith, M.K.; Wennier, S.T.; Thackray, L.B.; Holmes, K.V. Aromatic amino acids in the juxtamembrane domain of severe acute respiratory syndrome coronavirus spike glycoprotein are important for receptor-dependent virus entry and cell-cell fusion. J. Virol. 2008, 82, 2883–2894. [Google Scholar]
- Lu, Y.; Neo, T.L.; Liu, D.X.; Tam, J.P. Importance of SARS-CoV spike protein Trp-rich region in viral infectivity. Biochem. Biophys. Res. Commun. 2008, 371, 356–360. [Google Scholar] [CrossRef]
- Guillen, J.; Moreno, M.R.; Perez-Berna, A.J.; Bernabeu, A.; Villalain, J. Interaction of a peptide from the pre-transmembrane domain of the severe acute respiratory syndrome coronavirus spike protein with phospholipid membranes. J. Phys. Chem. B 2007, 111, 13714–13725. [Google Scholar]
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J. Virol. 2009, 83, 7411–7421. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, G.; Li, J.; Nie, Y.; Shi, X.; Lian, G.; Wang, W.; Yin, X.; Zhao, Y.; Qu, X.; et al. Identification of an antigenic determinant on the S2 domain of the severe acute respiratory syndrome coronavirus spike glycoprotein capable of inducing neutralizing antibodies. J. Virol. 2004, 78, 6938–6945. [Google Scholar]
- Daniel, C.; Anderson, R.; Buchmeier, M.J.; Fleming, J.O.; Spaan, W.J.; Wege, H.; Talbot, P.J. Identification of an immunodominant linear neutralization domain on the S2 portion of the murine coronavirus spike glycoprotein and evidence that it forms part of complex tridimensional structure. J. Virol. 1993, 67, 1185–1194. [Google Scholar]
- Bernini, A.; Spiga, O.; Ciutti, A.; Chiellini, S.; Bracci, L.; Yan, X.; Zheng, B.; Huang, J.; He, M.L.; Song, H.D.; et al. Prediction of quaternary assembly of SARS coronavirus peplomer. Biochem. Biophys. Res. Commun. 2004, 325, 1210–1214. [Google Scholar] [CrossRef]
- Bale, S.; Liu, T.; Li, S.; Wang, Y.; Abelson, D.; Fusco, M.; Woods, V.L., Jr.; Saphire, E.O. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl. Trop. Dis. 2011, 5, e1395. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011-1033. https://doi.org/10.3390/v4061011
Belouzard S, Millet JK, Licitra BN, Whittaker GR. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses. 2012; 4(6):1011-1033. https://doi.org/10.3390/v4061011
Chicago/Turabian StyleBelouzard, Sandrine, Jean K. Millet, Beth N. Licitra, and Gary R. Whittaker. 2012. "Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein" Viruses 4, no. 6: 1011-1033. https://doi.org/10.3390/v4061011
APA StyleBelouzard, S., Millet, J. K., Licitra, B. N., & Whittaker, G. R. (2012). Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses, 4(6), 1011-1033. https://doi.org/10.3390/v4061011