Quantitative Live-Cell Imaging of Human Immunodeficiency Virus (HIV-1) Assembly

Abstract
:
1. Introduction
2. Methods
2.1. Fluorescence Labeling Strategies for HIV-1
2.2. Fluorescence Microscopy and Spectroscopy Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Potential Applications |
|---|---|
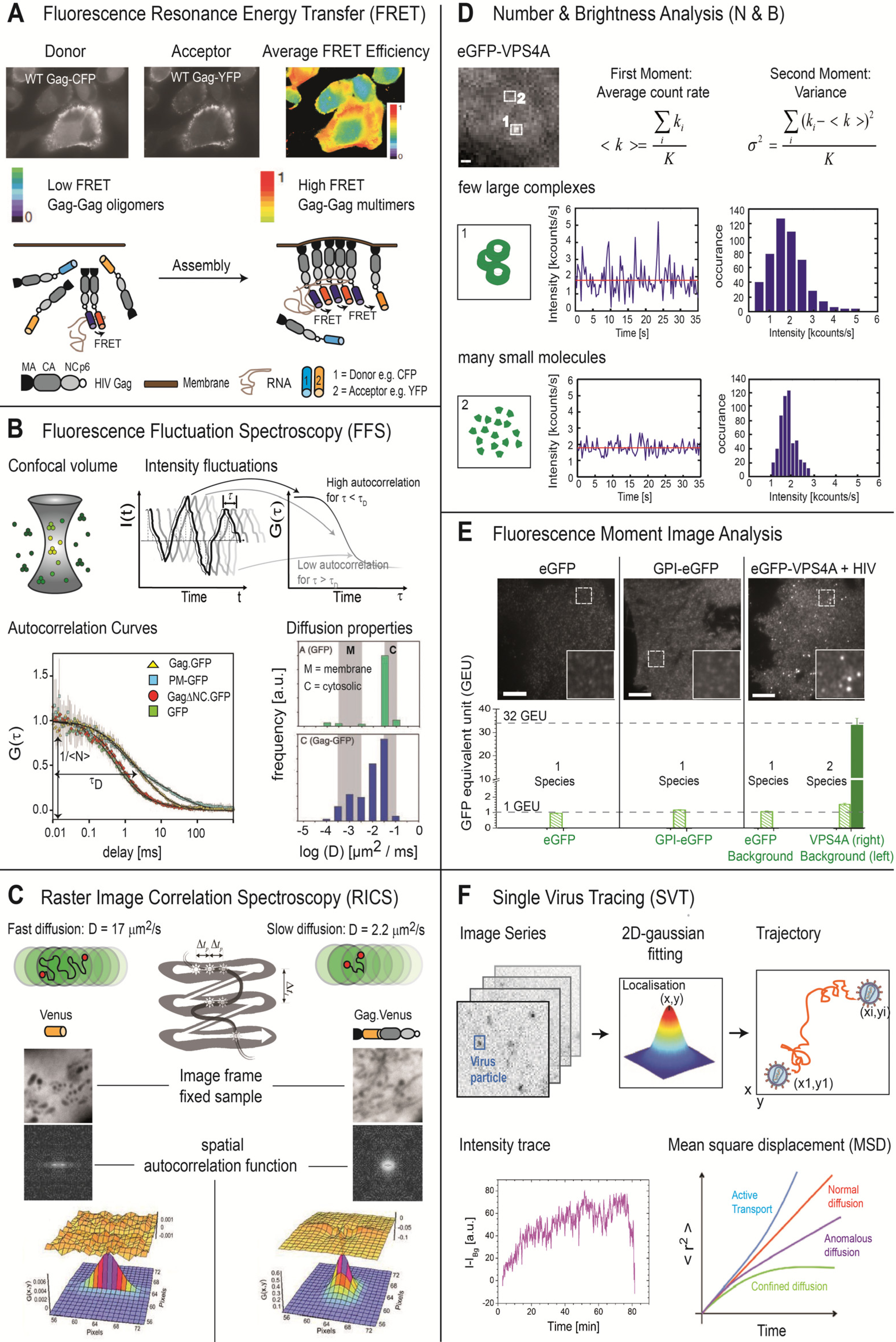
| Fluorescence Resonance Energy Transfer (Figure 1A) [22] | Spatial and temporal investigations of molecular interactions (Gag-Gag interactions, membrane microdomain clustering [18,19,23] |
| Conformational dynamics | |
| Fluorescence Fluctuation Spectroscopy (Figure 1B) [24,25,26] | Binding kinetics and affinities |
| Determination of the mobility of molecular complexes (Gag-Gag interactions in the cytosol [19]) | |
| Detection of molecular interactions | |
| Mapping of the local environment of the cell through its influence on the diffusion properties | |
| Determination of fluorophores per complex (Gag stoichiometry in VLPs [27]) | |
| Raster Image Correlation Spectroscopy (Figure 1C) [28,29,30] | Binding kinetics and affinities |
| Determination of the mobility of molecular complexes | |
| Detection of molecular interactions | |
| Number and Brightness Analysis (Figure 1D) [31] | Determination of molecular or complex concentration, brightness and stoichiometry at each pixel of an image from an image series [27] |
| Fluorescence Moment Image Analysis, Image Correlation Spectroscopy (Figure 1E) [32,33] | Determination of molecular or complex concentration, brightness and stoichiometry and presence of different subpopulations from the intensity distribution over a single image (cytoplasmic Gag-Gag interactions [34]) |
| Live cell imaging combined with Single Virus Tracing (Figure 1F) [35] | Analysis of entry and release pathways [36] |
| Kinetics of virus entry and assembly [20,37,38] | |
| Dynamics of intracellular trafficking of virus or viral proteins | |
| Interaction of viral proteins with host factors [20,39]. |
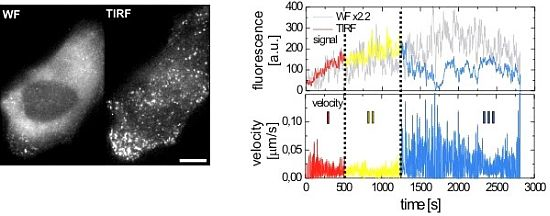
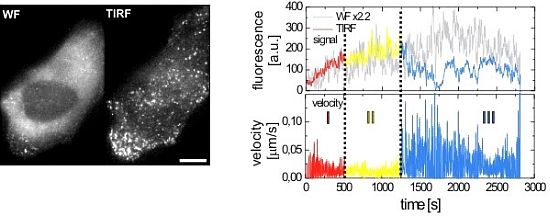
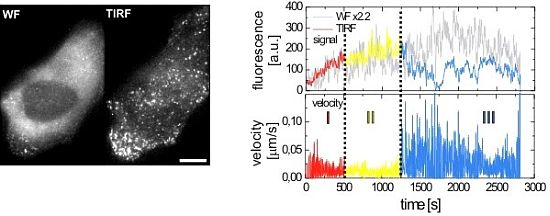
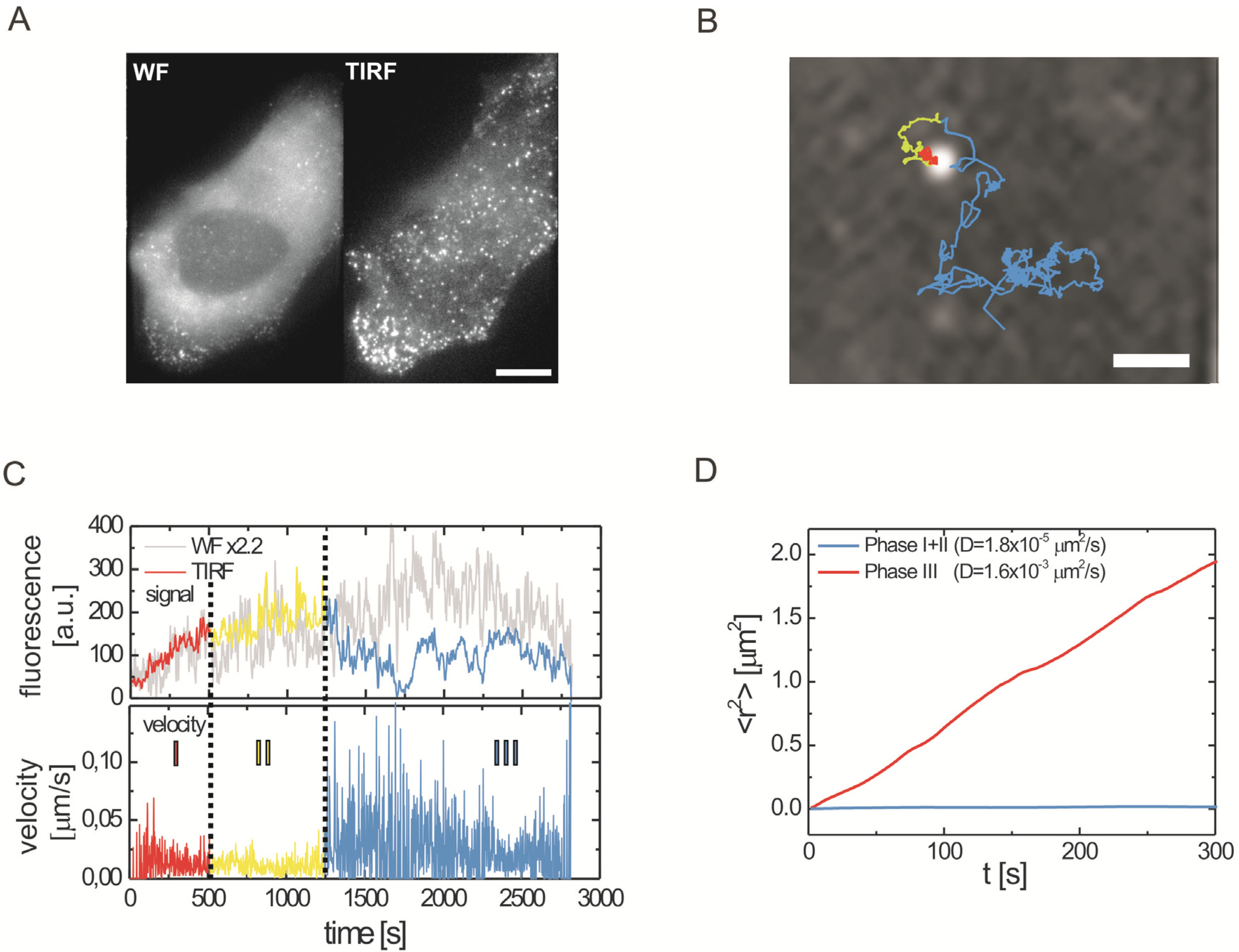
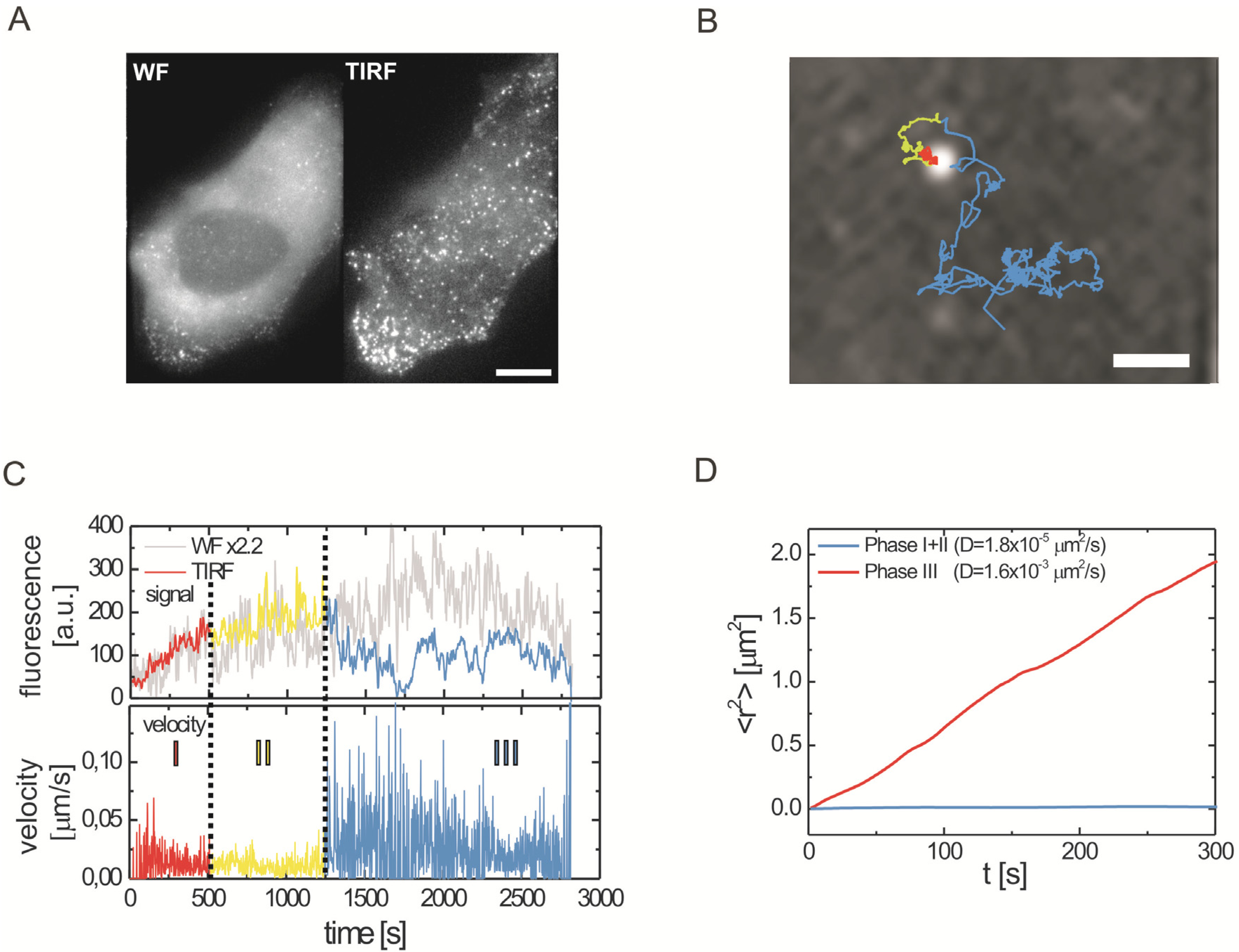
2.2.1. Wide-Field and TIRF Microscopy
2.2.2. Spinning Disc Confocal Microscopy
2.2.3. High Resolution Fluorescence Microscopy
2.2.4. Förster Resonance Energy Transfer
2.2.5. Fluorescence Fluctuation Spectroscopy
2.3. Single Virus Tracing
3. Applications of Quantitative Fluorescence Microscopy in Elucidating HIV-1 Assembly
3.1. Initiation of the Assembly Process
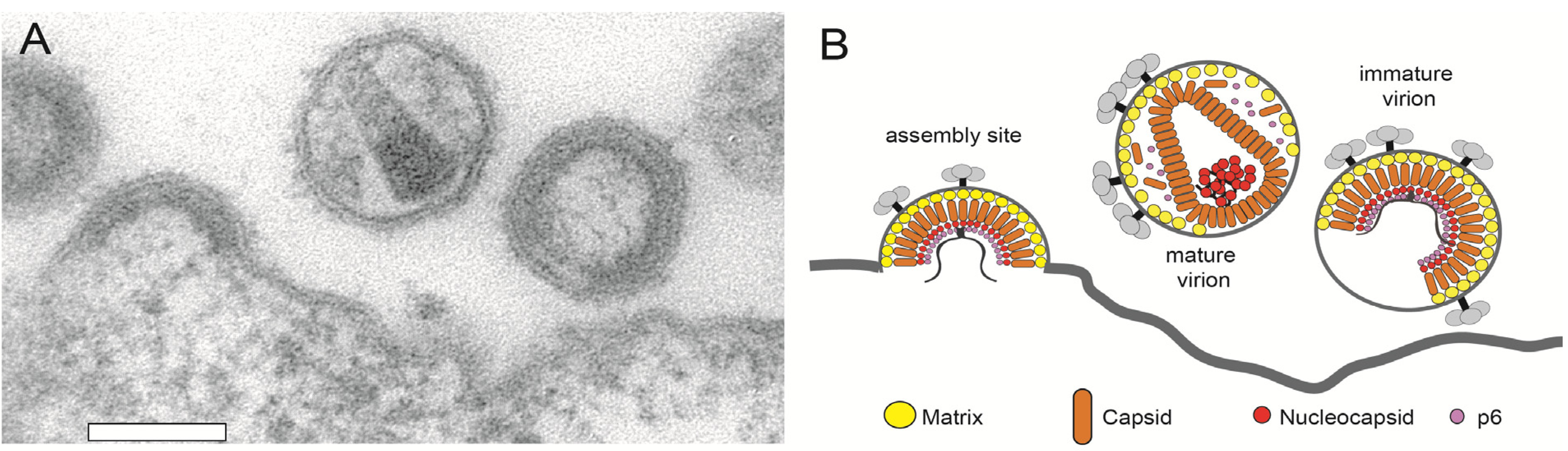
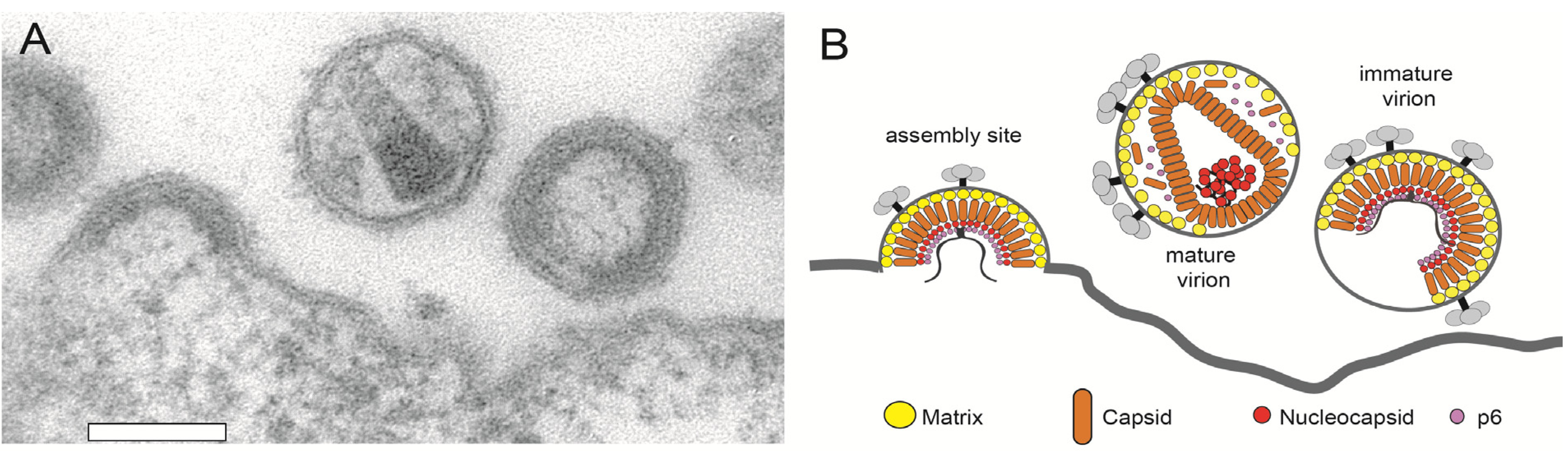
3.2. HIV-1 Assembly at the Plasma Membrane
3.3. ESCRT Recruitment and HIV-1 Release

4. Future Directions
Conflict of Interest
Acknowledgements
References
- Briggs, J.A.; Kräusslich, H.G. The molecular architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar]
- Weiss, E.R.; Göttlinger, H. The role of cellular factors in promoting HIV budding. J. Mol. Biol. 2011, 410, 525–533. [Google Scholar]
- Andresen, M.; Stiel, A.; Fölling, J.; Wenzel, D.; Schönle, A.; Egner, A.; Eggeling, C.; Hell, S.; Jakobs, S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat. Biotechnol. 2008, 26, 1035–1040. [Google Scholar]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar]
- Wiedenmann, J.; Oswald, F.; Nienhaus, G.U. Fluorescent proteins for live cell imaging: Opportunities, limitations, and challenge. IUBMB Life 2009, 61, 1029–1042. [Google Scholar]
- Carlson, L.A.; Briggs, J.A.; Glass, B.; Riches, J.D.; Simon, M.N.; Johnson, M.C.; Müller, B.; Grünewald, K.; Kräusslich, H.G. Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host Microbe 2008, 4, 592–599. [Google Scholar]
- Wright, E.R.; Schooler, J.B.; Ding, H.J.; Kieffer, C.; Fillmore, C.; Sundquist, W.I.; Jensen, G.J. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007, 26, 2218–2226. [Google Scholar]
- Hermida-Matsumoto, L.; Resh, M.D. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 2000, 74, 8670–8679. [Google Scholar] [CrossRef]
- Hübner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.; Chen, B. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar]
- Müller, B.; Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Zentgraf, H.; Kräusslich, H.G. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 2004, 78, 10803–10813. [Google Scholar]
- Griffin, B.A.; Adams, S.R.; Tsien, R.Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281, 269–272. [Google Scholar]
- Keppler, A.; Pick, H.; Arrivoli, C.; Vogel, H.; Johnsson, K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9955–9959. [Google Scholar]
- Eckhardt, M.; Anders, M.; Muranyi, W.; Heilemann, M.; Krijnse-Locker, J.; Müller, B. A SNAP-Tagged Derivative of HIV-1-A Versatile Tool to Study Virus-Cell Interactions. PLoS One 2011, 6, e22007. [Google Scholar]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar]
- Querido, E.; Chartrand, P. Using fluorescent proteins to study mRNA trafficking in living cells. Meth. Cell Biol. 2008, 85, 273–292. [Google Scholar]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 19114–19119. [Google Scholar]
- Hogue, I.B.; Hoppe, A.; Ono, A. Quantitative fluorescence resonance energy transfer microscopy analysis of the human immunodeficiency virus type 1 Gag-Gag interaction: Relative contributions of the CA and NC domains and membrane binding. J. Virol. 2009, 83, 7322–7336. [Google Scholar]
- Larson, D.R.; Ma, Y.M.; Vogt, V.M.; Webb, W.W. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 2003, 162, 1233–1244. [Google Scholar]
- Baumgärtel, V.; Ivanchenko, S.; Dupont, A.; Sergeev, M.; Wiseman, P.W.; Kräusslich, H.G.; Bräuchle, C.; Müller, B.; Lamb, D.C. Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 2011, 469–474. [Google Scholar]
- Ruthardt, N.; Lamb, D.C.; Bräuchle, C. Single-particle tracking as a quantitative microscopy-based approach to unravel cell entry mechanisms of viruses and pharmaceutical nanoparticles. Mol. Ther. 2011, 19, 1199–1211. [Google Scholar]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol. 2003, 21, 1387–1395. [Google Scholar]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar]
- Digman, M.A.; Gratton, E. Lessons in fluctuation correlation spectroscopy. Annu. Rev. Phys. Chem. 2011, 62, 645–668. [Google Scholar]
- Ivanchenko, S.; Lamb, D.C. Fluorescence Correlation Spectroscopy: Principles and develeopments. In Supramolecular Structure and Function ; Brnjas-Kraljevic, J., Pifat-Mrzljak, G., Eds.; Springer: New York, NY, USA, 2011; Volume 10, pp. 1–30. [Google Scholar]
- Shi, X.; Wohland, T. Fluorescence Correlation Spectroscopy. In Nanoscopy and Multidimensional Optical Fluorescence Microscopy; Diaspro, A., Ed.; CRC Press, Taylor & Francis Group: New York, NY, USA, 2010. [Google Scholar]
- Chen, Y.; Wu, B.; Musier-Forsyth, K.; Mansky, L.M.; Mueller, J.D. Fluorescence fluctuation spectroscopy on viral-like particles reveals variable gag stoichiometry. Biophys. J. 2009, 96, 1961–1969. [Google Scholar]
- Digman, M.A.; Gratton, E. Analysis of diffusion and binding in cells using the RICS approach. Microsc. Res. Tech. 2009, 72, 323–332. [Google Scholar]
- Digman, M.A.; Wiseman, P.W.; Horwitz, A.R.; Gratton, E. Detecting protein complexes in living cells from laser scanning confocal image sequences by the cross correlation raster image spectroscopy method. Biophys. J. 2009, 96, 707–716. [Google Scholar]
- Rossow, M.J.; Sasaki, J.M.; Digman, M.A.; Gratton, E. Raster image correlation spectroscopy in live cells. Nat. Protoc. 2010, 5, 1761–1774. [Google Scholar]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar]
- Kolin, D.L.; Wiseman, P.W. Advances in image correlation spectroscopy: Measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cell. Cell Biochem. Biophys. 2007, 49, 141–164. [Google Scholar]
- Sergeev, M.; Costantino, S.; Wiseman, P.W. Measurement of monomer-oligomer distributions via fluorescence moment image analysis. Biophys. J. 2006, 91, 3884–3896. [Google Scholar]
- Fogarty, K.H.; Chen, Y.; Grigsby, I.F.; Macdonald, P.J.; Smith, E.M.; Johnson, J.L.; Rawson, J.M.; Mansky, L.M.; Mueller, J.D. Characterization of cytoplasmic gag-gag interactions by dual-color z-scan fluorescence fluctuation spectroscopy. Biophys. J. 2011, 100, 1587–1595. [Google Scholar]
- Ruthardt, N.; Lamb, D.; Bräuchle, C. Single-particle tracking as a quantitative microscopy-based approach to unravel cell entry mechanisms of viruses and pharmaceutical nanoparticles. Mol. Ther. 2011, 19, 1199–1211. [Google Scholar]
- Endress, T.; Lampe, M.; Briggs, J.A.; Kräusslich, H.G.; Bräuchle, C.; Müller, B.; Lamb, D.C. HIV-1-cellular interactions analyzed by single virus tracing. Eur. Biophys. J. 2008, 37, 1291–1301. [Google Scholar]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar]
- Ivanchenko, S.; Godinez, W.J.; Lampe, M.; Kräusslich, H.-G.; Eils, R.; Rohr, K.; Bräuchle, C.; Müller, B.; Lamb, D.C. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009, 5, e1000652. [Google Scholar]
- Jouvenet, N.; Zhadina, M.; Bieniasz, P.D.; Simon, S.M. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 2011, 13, 394–401. [Google Scholar]
- Seisenberger, G.; Ried, M.U.; Endreß, T.; Büning, H.; Hallek, M.; Bräuchle, C. Real-time single molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929–1932. [Google Scholar]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 9280–9285. [Google Scholar]
- Axelrod, D.; Burghardt, T.P.; Thompson, N.L. Total internal reflection fluorescence. Annu. Rev. Biophys. Bioeng. 1984, 13, 247–268. [Google Scholar]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003, 361, 1–33. [Google Scholar]
- Saffarian, S.; Kirchhausen, T. Differential evanescence nanometry: Live-cell fluorescence measurements with 10-nm axial resolution on the plasma membrane. Biophys. J. 2008, 94, 2333–2342. [Google Scholar]
- Arhel, N.; Genovesio, A.; Kim, K.A.; Miko, S.; Perret, E.; Olivo-Marin, J.C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar]
- Hübner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 2009, 323, 1743–1747. [Google Scholar]
- Jin, J.; Sherer, N.; Heidecker, G.; Derse, D.; Mothes, W. Assembly of the murine leukemia virus is directed towards sites of cell-cell contact. PLoS Biol. 2009, 7, e1000163. [Google Scholar]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Optic. Lett. 1994, 19, 780–782. [Google Scholar]
- Hell, S.W.; Dyba, M.; Jakobs, S. Concepts for nanoscale resolution in fluorescence microscopy. Curr. Opin. Neurobiol. 2004, 14, 599–609. [Google Scholar]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar]
- Hess, S.T.; Girirajan, T.P.; Mason, M. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar]
- Willig, K.I.; Kellner, R.R.; Medda, R.; Hein, B.; Jakobs, S.; Hell, S.W. Nanoscale resolution in GFP-based microscopy. Nat. Methods 2006, 3, 721–723. [Google Scholar]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar]
- Lehmann, M.; Rocha, S.; Mangeat, B.; Blanchet, F.; Uji, I.H.; Hofkens, J.; Piguet, V. Quantitative multicolor super-resolution microscopy reveals tetherin HIV-1 interaction. PLoS Pathog. 2011, 7, e1002456. [Google Scholar]
- Grotjohann, T.; Testa, I.; Leutenegger, M.; Bock, H.; Urban, N.T.; Lavoie-Cardinal, F.; Willig, K.I.; Eggeling, C.; Jakobs, S.; Hell, S.W. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature 2011, 478, 204–208. [Google Scholar]
- Förster, T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 6, 166–175. [Google Scholar]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. 1948. [Google Scholar]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. U. S. A. 1967, 58, 719–726. [Google Scholar]
- Derdowski, A.; Ding, L.; Spearman, P. A novel fluorescence resonance energy transfer assay demonstrates that the human immunodeficiency virus type 1 Pr55Gag I domain mediates Gag-Gag interactions. J. Virol. 2004, 78, 1230–1242. [Google Scholar]
- Dale, B.M.; McNerney, G.P.; Thompson, D.L.; Hubner, W.; de Los Reyes, K.; Chuang, F.Y.; Huser, T.; Chen, B.K. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 2011, 10, 551–562. [Google Scholar]
- Elson, E.L.; Magde, D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 1974, 13, 1–27. [Google Scholar] [CrossRef]
- Magde, D.; Elson, E.L.; Webb, W.W. Thermodynamic fluctuations in a reacting system—Measurement by Fluorescence Correlation Spectroscopy. Phys. Rev. Lett. 1972, 29, 705–708. [Google Scholar] [CrossRef]
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 1974, 13, 29–61. [Google Scholar] [CrossRef]
- Digman, M.A.; Brown, C.M.; Sengupta, P.; Wiseman, P.W.; Horwitz, A.R.; Gratton, E. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 2005, 89, 1317–1327. [Google Scholar]
- Petersen, N.O.; Hoddelius, P.L.; Wiseman, P.W.; Seger, O.; Magnusson, K.E. Quantitation of membrane receptor distributions by image correlation spectroscopy: Concept and application. Biophys. J. 1993, 65, 1135–1146. [Google Scholar]
- Wiseman, P.W.; Petersen, N.O. Image correlation spectroscopy. II. Optimization for ultrasensitive detection of preexisting platelet-derived growth factor-beta receptor oligomers on intact cells. Biophys. J. 1999, 76, 963–977. [Google Scholar] [CrossRef]
- Dupont, A.; Lamb, D. Nanoscale three-dimensional single particle tracking. Nanoscale 2011, 3, 4532–4541. [Google Scholar]
- Godinez, W.J.; Lampe, M.; Wörz, S.; Müller, B.; Eils, R.; Rohr, K. Deterministic and probabilistic approaches for tracking virus particles in time-lapse fluorescence microscopy image sequences. Med. Image Anal. 2009, 13, 325–342. [Google Scholar]
- Meijering, E.; Dzyubachyk, O.; Smal, I.; van Cappellen, W.A. Tracking in cell and developmental biology. Semin. Cell Dev. Biol. 2009, 20, 894–902. [Google Scholar]
- Wu, P.H.; Agarwal, A.; Hess, H.; Khargonekar, P.P.; Tseng, Y. Analysis of video-based microscopic particle trajectories using Kalman filtering. Biophys. J. 2010, 98, 2822–2830. [Google Scholar]
- Yoon, J.W.; Bruckbauer, A.; Fitzgerald, W.J.; Klenerman, D. Bayesian inference for improved single molecule fluorescence tracking. Biophys. J. 2008, 94, 4932–4947. [Google Scholar]
- Saxton, M.J. Lateral diffusion in an archipelago. Single-particle diffusion. Biophys. J. 1993, 64, 1766–1780. [Google Scholar] [CrossRef]
- Saxton, M.J. Single-particle tracking: Models of directed transport. Biophys. J. 1994, 67, 2110–2119. [Google Scholar]
- Saxton, M.J. Single-particle tracking: Effects of corrals. Biophys. J. 1995, 69, 389–398. [Google Scholar]
- Muriaux, D.; Darlix, J.L. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010, 7, 744–753. [Google Scholar]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar]
- Rein, A.; Datta, S.A.; Jones, C.P.; Musier-Forsyth, K. Diverse interactions of retroviral Gag proteins with RNAs. Trends Biochem. Sci. 2011, 36, 373–380. [Google Scholar]
- Datta, S.A.; Heinrich, F.; Raghunandan, S.; Krueger, S.; Curtis, J.E.; Rein, A.; Nanda, H. HIV-1 Gag extension: Conformational changes require simultaneous interaction with membrane and nucleic acid. J. Mol. Biol. 2011, 406, 205–214. [Google Scholar]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar]
- Hurley, J.H. The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 463–487. [Google Scholar]
- Martin-Serrano, J.; Neil, S.J. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 2011, 9, 519–531. [Google Scholar]
- Fabrikant, G.; Lata, S.; Riches, J.D.; Briggs, J.A.; Weissenhorn, W.; Kozlov, M.M. Computational model of membrane fission catalyzed by ESCRT-III. PLoS Comput. Biol. 2009, 5, e1000575. [Google Scholar]
- Saksena, S.; Wahlman, J.; Teis, D.; Johnson, A.E.; Emr, S.D. Functional reconstitution of ESCRT-III assembly and disassembly. Cell 2009, 136, 97–109. [Google Scholar]
- Babst, M.; Wendland, B.; Estepa, E.J.; Emr, S.D. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 1998, 17, 2982–2993. [Google Scholar]
- Davies, B.A.; Azmi, I.F.; Payne, J.; Shestakova, A.; Horazdovsky, B.F.; Babst, M.; Katzmann, D.J. Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 2010, 21, 3396–3408. [Google Scholar]
- Langelier, C.; von Schwedler, U.K.; Fisher, R.D.; De Domenico, I.; White, P.L.; Hill, C.P.; Kaplan, J.; Ward, D.; Sundquist, W.I. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 2006, 80, 9465–9480. [Google Scholar]
- Morita, E.; Sandrin, V.; McCullough, J.; Katsuyama, A.; Baci Hamilton, I.; Sundquist, W.I. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 2011, 9, 235–242. [Google Scholar]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar]
- Wollert, T.; Wunder, C.; Lippincott-Schwartz, J.; Hurley, J.H. Membrane scission by the ESCRT-III complex. Nature 2009, 458, 172–177. [Google Scholar]
- Baumgärtel, V.; Ivanchenko, S.; Müller, B.; Lamb, D.C. Investigating the Life cycle of HIV with Fluorescent Proteins. In Fluorescent Proteins II: Application of Fluorescent Protein Technology; Jung, G., Ed.; Springer: Berlin, Germany, 2012; p. 280. [Google Scholar]
- Waheed, A.A.; Freed, E.O. Lipids and membrane microdomains in HIV-1 replication. Virus Res. 2009, 143, 162–176. [Google Scholar]
- Ono, A. Relationships between plasma membrane microdomains and HIV-1 assembly. Biol. Cell 2010, 102, 335–350. [Google Scholar]
- Larson, D.R.; Johnson, M.C.; Webb, W.W.; Vogt, V.M. Visualization of retrovirus budding with correlated light and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15453–15458. [Google Scholar]
- Kukulski, W.; Schorb, M.; Welsch, S.; Picco, A.; Kaksonen, M.; Briggs, J.A. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J. Cell Biol. 2011, 192, 111–119. [Google Scholar]
- Jun, S.; Ke, D.; Debiec, K.; Zhao, G.; Meng, X.; Ambrose, Z.; Gibson, G.A.; Watkins, S.C.; Zhang, P. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure 2011, 19, 1573–1581. [Google Scholar]
- Verveer, P.J.; Swoger, J.; Pampaloni, F.; Greger, K.; Marcello, M.; Stelzer, E.H. High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. Nat. Methods 2007, 4, 311–313. [Google Scholar]
- Keller, P.; Schmidt, A.; Wittbrodt, J.; Stelzer, E. Reconstruction of Zebrafish Early Embryonic Development by Scanned Light Sheet Microscopy. Science 2008, 322, 1065–1069. [Google Scholar]
- Santi, P.A. Light sheet Fluorescence Microscopy: A review. J. Histochem. Cytochem. 2011, 59, 129–138. [Google Scholar]
- Blanchet, F.; Moris, A.; Mitchell, J.P.; Piguet, V. A look at HIV journey: From dendritic cells to infection spread in CD4 T cells. Curr. Opin. HIV AIDS 2011, 6, 391–397. [Google Scholar]
- Haller, C.; Fackler, O.T. HIV-1 at the immunological and T-lymphocytic virological synapse. Biol. Chem. 2008, 389, 1253–1260. [Google Scholar]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar]
- Piguet, V.; Sattentau, Q. Dangerous liaisons at the virological synapse. J. Clin. Invest. 2004, 114, 605–610. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baumgärtel, V.; Müller, B.; Lamb, D.C. Quantitative Live-Cell Imaging of Human Immunodeficiency Virus (HIV-1) Assembly. Viruses 2012, 4, 777-799. https://doi.org/10.3390/v4050777
Baumgärtel V, Müller B, Lamb DC. Quantitative Live-Cell Imaging of Human Immunodeficiency Virus (HIV-1) Assembly. Viruses. 2012; 4(5):777-799. https://doi.org/10.3390/v4050777
Chicago/Turabian StyleBaumgärtel, Viola, Barbara Müller, and Don C. Lamb. 2012. "Quantitative Live-Cell Imaging of Human Immunodeficiency Virus (HIV-1) Assembly" Viruses 4, no. 5: 777-799. https://doi.org/10.3390/v4050777