Pathogenesis of Hepatitis C During Pregnancy and Childhood

{kind=link}
Abstract
:1. Introduction
2. Hepatitis C during Pregnancy
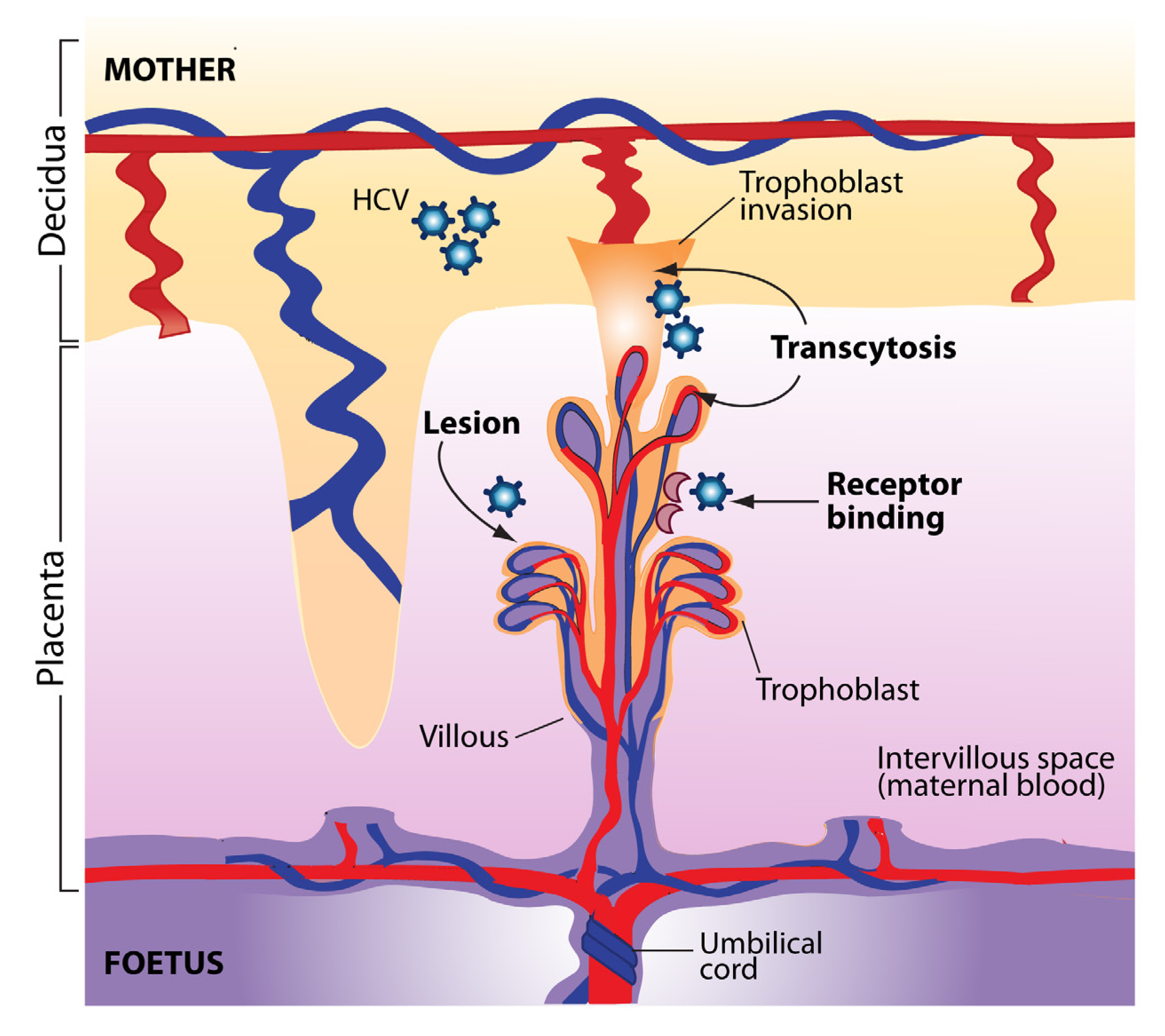
3. Transmission of HCV from Mother to Child
4. Pathogenesis of Hepatitis C in Childhood
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29 Suppl 1, 74–81. [Google Scholar] [CrossRef]
- Alter, M.J. Hepatitis C virus infection in the United States. J. Hepatol. 1999, 31 Suppl 1, 88–91. [Google Scholar] [CrossRef]
- Arshad, M.; El-Kamary, S.S.; Jhaveri, R. Hepatitis C virus infection during pregnancy and the newborn period--are they opportunities for treatment? J. Viral. Hepat. 2011, 18, 229–236. [Google Scholar] [CrossRef]
- Zahran, K.M.; Badary, M.S.; Agban, M.N.; Abdel Aziz, N.H. Pattern of hepatitis virus infection among pregnant women and their newborns at the Women's Health Center of Assiut University, Upper Egypt. Int. J. Gynaecol. Obstet. 2010, 111, 171–174. [Google Scholar] [CrossRef]
- Alter, M.J. Epidemiology of hepatitis C. Hepatology 1997, 26, 62S–65S. [Google Scholar] [CrossRef]
- Thomas, D.L.; Zenilman, J.M.; Shih, J.W.; Galai, N.; Carella, A.V.; Quinn, T.C. Sexual transmission of hepatitis C virus among patients attending sexually transmitted diseases clinics in Baltimore-an analysis of 309 sex partnerships. J. Infect. Dis. 1995, 171, 768–775. [Google Scholar] [CrossRef]
- Klevens, R.M.; Hu, D.J.; Jiles, R.; Holmberg, S.D. Evolving epidemiology of hepatitis C virus in the United States. Clin. Infect. Dis. 2012, 55 Suppl 1, S3–S9. [Google Scholar] [CrossRef]
- Conte, D.; Fraquelli, M.; Prati, D.; Colucci, A.; Minola, E. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology 2000, 31, 751–755. [Google Scholar] [CrossRef]
- Wesjtal, R.; Widell, A.; Mansson, A.S.; Hermodsson, S.; Nokrans, G. Mother-to-infant transmission of hepatitis C virus. Ann. Intern. Med. 1992, 117, 887–890. [Google Scholar]
- Ohto, H.; Terazawa, S.; Sasaki, N.; Hino, K.; Ishiwata, C.; Kako, M.; Ujiie, N.; Endo, C.; Matsui, A.; Okamoto, H.; Mishiro, S. Vertical Transmission of Hepatitis C Virus Collaborative Study Group. Transmission of hepatitis C virus from mothers to infants. N. Engl. J. Med. 1994, 330, 744–750. [Google Scholar] [CrossRef]
- Novati, R.; Thiers, V.; Monforte, A.D.; Maisonneuve, P.; Principi, N.; Conti, M.; Lazzarin, A.; Brechot, C. Mother-to-child transmission of hepatitis C virus detected by nested polymerase chain reaction. J. Infect. Dis. 1992, 165, 720–723. [Google Scholar] [CrossRef]
- Zanetti, A.R.; Tanzi, E.; Paccagnini, S.; Principi, N.; Pizzocolo, G.; Caccamo, M.L.; D’Amico, E.; Cambie, G.; Vecchi, L. Lombardy Study Group on Vertical HCV Transmission. Mother-to-infant transmission of hepatitis C. Lombardy study group on vertical HCV transmission. Lancet 1995, 345, 289–291. [Google Scholar]
- Agha, S.; Sherif, L.S.; Allam, M.A.; Fawzy, M. Transplacental transmission of hepatitis C virus in HIV-negative mothers. Res. Virol. 1998, 149, 229–234. [Google Scholar]
- Matsubara, T.; Sumazaki, R.; Takita, H. Mother-to-infant transmission of hepatitis C virus: a prospective study. Eur. J. Pediatr. 1995, 154, 973–978. [Google Scholar] [CrossRef]
- Lin, H.H.; Kao, J.H.; Hsu, H.Y.; Ni, Y.H.; Yeh, S.H.; Hwang, L.H.; Chang, M.H.; Hwang, S.C.; Chen, P.J.; Chen, D.S. Possible role of high-titer maternal viremia in perinatal transmission of hepatitis C virus. J. Infect. Dis. 1993, 169, 638–641. [Google Scholar]
- Thomas, S.L.; Newell, M.L.; Peckham, C.S.; Ades, A.E.; Hall, A.J. A review of hepatitis C virus (HCV) vertical transmission: risks of transmission to infants born to mothers with and without HCV viraemia or human immunodeficiency virus infection. Int. J. Epidemiol. 1998, 27, 108–117. [Google Scholar] [CrossRef]
- Akhtar, S.; Moatter, T. Intra-household clustering of hepatitis C virus infection in Karachi, Pakistan. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 535–539. [Google Scholar]
- Miller, F.D.; Abu-Raddad, L.J. Evidence of intense ongoing endemic transmission of hepatitis C virus in Egypt. Proc. Natl Acad. Sci. USA. 2010, 107, 14757–14762. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Balistreri, W.F. Transmission, natural history, and treatment of hepatitis C virus infection in the pediatric population. Semin. Liver Dis. 2000, 20, 37–46. [Google Scholar] [CrossRef]
- Jafri, W.; Jafri, N.; Yakoob, J.; Islam, M.; Tirmizi, S.F.; Jafar, T.; Akhtar, S.; Hamid, S.; Shah, H.A.; Nizami, S.Q. Hepatitis B and C: prevalence and risk factors associated with seropositivity among children in Karachi, Pakistan. BMC Infect. Dis. 2006, 6, 101. [Google Scholar] [CrossRef]
- Mor, G.; Cardenas, I. The immune system in pregnancy: a unique complexity. Am. J. Reprod. Immunol. 2010, 63, 425–433. [Google Scholar]
- Pergam, S.A.; Wang, C.C.; Gardella, C.M.; Sandison, T.G.; Phipps, W.T.; Hawes, S.E. Pregnancy complications associated with hepatitis C: Data from a 2003-2005 Washington state birth cohort. Am. J. Obstet. Gynecol. 2008, 199, 38.e1–38.e9. [Google Scholar] [CrossRef]
- Buresi, M.C.; Lee, J.; Gill, S.; Kong, J.M.; Money, D.M.; Yoshida, E.M. Hepatitis C vertical Transmission Study Group. The prevalence of gestational diabetes mellitus and glucose abnormalities in pregnant women with hepatitis C virus infection in British Columbia. J. Obstet. Gynaecol. Can. 2010, 32, 935–941. [Google Scholar]
- Reddick, K.L.; Jhaveri, R.; Gandhi, M.; James, A.H.; Swamy, G.K. Pregnancy outcomes associated with viral hepatitis. J. Viral. Hepat. 2011, 18, e394–e398. [Google Scholar] [CrossRef]
- Locatelli, A.; Roncaglia, N.; Arreghini, A.; Bellini, P.; Vergani, P.; Ghidini, A. Hepatitis C virus infection is associated with a higher incidence of cholestasis of pregnancy. BJOG. 1999, 106, 498–500. [Google Scholar] [CrossRef]
- Paternoster, D.M.; Fabris, F.; Palu, G.; Santarossa, C.; Bracciante, R.; Snijders, D.; Floreani, A. Intra-hepatic cholestasis of pregnancy in hepatitis C virus infection. Acta Obstet. Gynecol. Scand. 2002, 81, 99–103. [Google Scholar]
- Safir, A.; Levy, A.; Sikuler, E.; Sheiner, E. Maternal hepatitis B virus or hepatitis C virus carrier status as an independent risk factor for adverse perinatal outcome. Liver Int. 2010, 30, 765–770. [Google Scholar] [CrossRef]
- Floreani, A.; Paternoster, D.; Zappala, F.; Cusinato, R.; Bombi, G.; Grella, P.; Chiaramonte, M. Hepatitis C virus infection in pregnancy. BJOG. 1996, 103, 325–329. [Google Scholar] [CrossRef]
- Jabeen, T.; Cannon, B.; Hogan, J.; Crowley, M.; Devereux, C.; Fanning, L.; Kenny-Walsh, E.; Shanahan, F.; Whelton, M.J. Pregnancy and pregnancy outcome in hepatitis C type 1b. QJM. 2000, 93, 597–601. [Google Scholar] [CrossRef]
- Gervais, A.; Bacq, Y.; Bernuau, J.; Martinot, M.; Auperin, A.; Boyer, N.; Kilani, A.; Erlinger, S.; Valla, D.; Marcellin, P. Decrease in serum ALT and increase in serum HCV RNA during pregnancy in women with chronic hepatitis C. J. Hepatol. 2000, 32, 293–299. [Google Scholar]
- Paternoster, D.M.; Santarossa, C.; Grella, P.; Palu, G.; Baldo, V.; Boccagni, P.; Floreani, A. Viral load in HCV RNA-positive pregnant women. Am. J. Gastroenterol. 2001, 96, 2751–2754. [Google Scholar]
- Fontaine, H.; Nalpas, B.; Carnot, F.; Brechot, C.; Pol, S. Effect of pregnancy on chronic hepatitis C: a case-control study. Lancet 2000, 356, 1328–1329. [Google Scholar]
- Vento, S.; Longhi, M.S.; Cainelli, F.; Concia, E. Effect of pregnancy on chronic hepatitis C. Lancet 2001, 357, 389–390. [Google Scholar]
- Oketani, M.; Shibatou, T.; Yamashita, K.; Arima, T.; Arima, T. Postpartum acute exacerbation of chronic hepatitis C with complete response to interferon-α. J. Gastroenterol. 2002, 37, 658–662. [Google Scholar] [CrossRef]
- Latt, N.C.; Spencer, J.D.; Beeby, P.J.; McCaughan, G.W.; Saunders, J.B.; Collins, E.; Cossart, Y.E. Hepatitis C in injecting drug-using women during and after pregnancy. J. Gastroenterol. Hepatol. 2000, 15, 175–181. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Suárez-García, E.; Casanovas, J.; Nogales, M.C.; Vargas, J.; Otero, M.A.; Castro Fernández, M. Influencia del embarazo en la infección crónica por el virus C de la hepatitis [Influence of pregnancy in chronic hepatitis C virus infection]. Med. Clin. (Barc). 1998, 111, 641–644. [Google Scholar]
- Billingham, R.E.; Brent, L.; Medawar, P.B. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–606. [Google Scholar] [CrossRef]
- Munoz-Suano, A.; Hamilton, A.B.; Betz, A.G. Gimme shelter: the immune system during pregnancy. Immunol. Rev. 2011, 241, 20–38. [Google Scholar] [CrossRef]
- Lichtenstein, M.R. Tuberculin reaction in tuberculosis during pregnancy. Am. Rev. Tuberc. Pulm. Dis. 1942, 48, 89–93. [Google Scholar]
- Raghupathy, R.; Raghupathy, R.; Makhseed, M.; Azizieh, F.; Omu, A.; Gupta, M.; Farhat, R. Cytokine production by maternal lymphocytes during normal human pregnancy and in unexplained recurrent spontaneous abortion. Hum. Reprod. 2000, 15, 713–718. [Google Scholar] [CrossRef]
- Narita, M.; Yamada, S.; Kikuta, H.; Togashi, T. Reconstitution of humoral immunity during pregnancy. Am. J. Reprod. Immunol. 2000, 44, 148–152. [Google Scholar]
- Watanabe, M.; Iwatani, Y.; Kaneda, T.; Hidaka, Y.; Mitsuda, N.; Morimoto, Y.; Amino, N. Changes in T, B, and NK lymphocyte subsets during and after normal pregnancy. Am. J. Reprod. Immunol. 1997, 37, 368–377. [Google Scholar] [CrossRef]
- Okuyama, R.; Abo, T.; Seki, S.; Ohteki, T.; Sugiura, K.; Kusumi, A.; Kumagai, K. Estrogen administration activates extrathymic T cell differentiation in the liver. J. Exp. Med. 1992, 175, 661–669. [Google Scholar]
- Kimura, M.; Hanawa, H.; Watanabe, H.; Ogawa, M.; Abo, T. Synchronous expansion of intermediate TCR cells in the liver and uterus during pregnancy. Cell. Immunol. 1995, 162, 16–25. [Google Scholar] [CrossRef]
- Aluvihare, V.R.; Kallikourdis, M.; Betz, A.G. Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 2004, 5, 266–271. [Google Scholar] [CrossRef]
- Heikkinen, J.; Mottonen, M.; Alanen, A.; Lassila, O. Phenotypic characterization of regulatory T cells in the human decidua. Clin. Exp. Immunol. 2004, 136, 373–378. [Google Scholar] [CrossRef]
- Somerset, D.A.; Zheng, Y.; Kilby, M.D.; Sansom, D.M.; Drayson, M.T. Normal human pregnancy is associated with an elevation in the immune suppressive CD25+ CD4+ regulatory T-cell subset. Immunology 2004, 112, 38–43. [Google Scholar] [CrossRef]
- Rogerson, S.J.; Hviid, L.; Duffy, P.E.; Leke, R.F.; Taylor, D.W. Malaria in pregnancy: pathogenesis and immunity. Lancet Infect. Dis. 2007, 7, 105–117. [Google Scholar] [CrossRef]
- Da Silva, J.A.P.; Spector, T.D. The role of pregnancy in the course and etiology of rheumatoid arthritis. Clin. Rheumatol. 1992, 11, 189–194. [Google Scholar]
- Soldan, S.S.; Alvarez Retuerto, A.I.; Sicotte, N.L.; Voskuhi, R.R. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estradiol. J. Immunol. 2003, 171, 6267–6274. [Google Scholar]
- Nahmias, A.J.; Schollin, J.; Abramowsky, C. Evolutionary-developmental perspectives on immune system interactions among the pregnant woman, placenta, and fetus, and responses to sexually transmitted infectious agents. Ann. N. Y. Acad. Sci. 1230, 25–47. [Google Scholar]
- Constantin, C.M.; Masopust, D.; Gourley, T.; Grayson, J.; Strickland, O.L.; Ahmed, R.; Bonney, E.A. Normal establishment of virus-specific memory CD8 T cell pool following primary infection during pregnancy. J. Immunol. 2007, 179, 4383–4389. [Google Scholar]
- Norton, M.T.; Fortner, K.A.; Oppenheimer, K.H.; Bonney, E.A. Evidence that CD8 T-cell homeostasis and function remain intact during murine pregnancy. Immunology 2010, 131, 426–437. [Google Scholar] [CrossRef]
- Troesch, M.; Jalbert, E.; Canobio, S.; Boulassel, M.R.; Routy, J.P.; Bernard, N.F.; Bruneau, J.; Lapointe, N.; Boucher, M.; Soudeyns, H. Characterization of humoral and cell-mediated immune responses directed against hepatitis C virus F protein in subjects co-infected with hepatitis C virus and HIV-1. AIDS 2005, 19, 775–784. [Google Scholar] [CrossRef]
- Honegger, J.; Prasad, M.; Walker, C. Post-partum surge in T-cell immunity to the hepatitis C virus. In 47th Annual Meeting of the Infectious Diseases Society of America, October 29th-November 1st, 2009. Abstract no810..
- Troesch, M.; Meunier, I.; Lapierre, P.; Lapointe, N.; Alvarez, F.; Boucher, M.; Soudeyns, H. Study of a novel hypervariable region in hepatitis C virus (HCV) E2 envelope glycoprotein. Virology 2006, 352, 357–367. [Google Scholar] [CrossRef]
- Cheynier, R.; Langlade-Demoyen, P.; Marescot, M.R.; Blanche, S.; Blondin, G.; Wain-Hobson, S.; Griscelli, C.; Vilmer, E.; Plata, F. Cytotoxic T lymphocyte responses in the peripheral blood of children born to human immunodeficiency virus-1-infected mothers. Eur. J. Immunol. 1992, 22, 2211–2217. [Google Scholar] [CrossRef]
- Rowland-Jones, S.; Nixon, D.F.; Aldhous, M.C.; Gotch, F.; Ariyoshi, K.; Hallam, N.; Kroll, J.S.; Froebel, K.; McMichael, A. HIV-specific cytotoxic T-cell activity in an HIV-exposed but uninfected infant. Lancet 1993, 341, 860–861. [Google Scholar]
- Dauby, N.; Goetghebuer, T.; Kollmann, T.R.; Levy, J.; Marchant, A. Uninfected but not unaffected: chronic maternal infections during pregnancy, fetal immunity, and susceptibility to postnatal infections. Lancet Infect. Dis. 2012, 12, 330–340. [Google Scholar] [CrossRef]
- Haynes, B.F.; Heinly, C.S. Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J. Exp. Med. 1995, 181, 1445–1458. [Google Scholar] [CrossRef]
- Marchant, A.; Appay, V.; Van Der Sande, M.; Dulphy, N.; Liesnard, C.; Kidd, M.; Kaye, S.; Ojuola, O.; Gillespie, G.M.; Vargas Cuero, A.L.; Cerundolo, V.; Callan, M.; McAdam, K.P.; Rowland-Jones, S.L.; Donner, C.; McMichael, A.J.; Whittle, H. Mature CD8(+) T lymphocyte response to viral infection during fetal life. J. Clin. Invest. 2003, 111, 1747–1755. [Google Scholar]
- Babik, J.M.; Cohan, D.; Monto, A.; Hartigan-O'Connor, D.J.; McCune, J.M. The human fetal immune response to hepatitis C virus exposure in utero. J. Infect. Dis. 2011, 203, 196–206. [Google Scholar] [CrossRef]
- Burns, D.N.; Nourjah, P.; Wright, D.J.; Minkoff, H.; Landesman, S.; Rubinstein, A.; Goedert, J.J.; Nugent, R.P. Changes in immune activation markers during pregnancy and postpartum. J. Reprod. Immunol. 1999, 42, 147–165. [Google Scholar]
- Hattori, Y.; Orito, E.; Ohno, T.; Sugauchi, F.; Suzuki, S.; Sugiura, M.; Suzumori, K.; Hattori, K.; Ueda, R.; Mizokami, M. Loss of hepatitis C virus RNA after parturition in female patients with chronic HCV infection. J. Med. Virol. 2003, 71, 205–211. [Google Scholar] [CrossRef]
- Resti, M.; Azzari, C.; Mannelli, F.; Moriondo, M.; Novembre, E.; de Martino, M.; Vierucci, A. Mother to child transmission of hepatitis C virus: prospective study of risk factors and timing of infection in children born to women seronegative for HIV-1. Tuscany Study Group on Hepatitis C Virus Infection. BMJ 1998, 317, 437–441. [Google Scholar]
- Indolfi, G.; Resti, M. Perinatal transmission of hepatitis C virus infection. J. Med. Virol. 2009, 81, 836–843. [Google Scholar]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Goncales, F.L.; Haussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; Craxi, A.; Lin, A.; Hoffman, J.; Yu, J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Manns, M.P.; Muir, A.J.; Terrault, N.A.; Jacobson, I.M.; Afdhal, N.H.; Heathcote, E.J.; Zeuzem, S.; Reesink, H.W.; Garg, J.; Bsharat, M.; George, S.; Kauffman, R.S.; Adda, N.; Di Bisceglie, A.M. PROVE3 Study Team. Telaprevir for previously treated chronic HCV infection. N. Engl. J. Med. 2010, 362, 1292–1303. [Google Scholar]
- Poordad, F.; McCone, J.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; DiNubile, M.J.; Sniukiene, V.; Brass, C.A.; Albrecht, J.K.; Bronowicki, J.P. SPRINT-2 Investigators. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar] [CrossRef]
- Connor, E.M.; Sperling, R.S.; Gelber, R.; Kiselev, P.; Scott, G.; O’Sullivan, M.J.; VanDyke, R.; Bey, M.; Shearer, W.; Jacobson, R.L.; Jimenez, E.; O’Neill, E.; Bazin, B.; Delfraissy, J.F.; Culnane, M.; Coombs, R.; Elkins, M.; Moye, J.; Stratton, P.; Balsley, J. Pediatric AIDS Clinical Trials Group Protocol 076 Study Group. Reduction of maternal-infant transmission of human immunodeficiency virus type 1 with zidovudine treatment. N. Engl. J. Med. 1994, 331, 1173–1180. [Google Scholar]
- Townsend, C.L.; Cortina-Borja, M.; Peckham, C.S.; de Ruiter, A.; Lyall, H.; Tookey, P.A. Low rates of mother-to-child transmission of HIV following effective pregnancy interventions in the United Kingdom and Ireland, 2000-2006. AIDS 2008, 22, 973–981. [Google Scholar]
- Ferm, V.H.; Willhite, C.; Kilham, L. Teratogenic effects of ribavirin on hamster and rat embryos. Teratology 1978, 17, 93–101. [Google Scholar] [CrossRef]
- Kilham, L.; Ferm, V.H. Congenital anomalies induced in hamster embryos with ribavirin. Science 1977, 195, 413–414. [Google Scholar]
- Roberts, S,S.; Miller, R.K.; Jones, J.K.; Lindsay, K.L.; Greene, M.F.; Maddrey, W.C.; Williams, I.T.; Liu, J.; Spiegel, R.J. The Ribavirin Pregnancy Registry: Findings after 5 years of enrollment, 2003-2009. Birth Defects Res. A. Clin. Mol. Teratol. 2010, 88, 551–559. [Google Scholar] [CrossRef]
- Trotter, J.F.; Zygmunt, A.J. Conception and pregnancy during interferon-alpha therapy for chronic hepatitis C. J. Clin. Gastroenterol. 2001, 32, 76–78. [Google Scholar] [CrossRef]
- Labarga, P.; Pinilla, J.; Cachorro, I.; Ruiz, Y. Infant of 22 months of age with no anomalies born from a HCV- and HIV-infected mother under treatment with pegylated interferon, ribavirin and antiretroviral therapy during the first 16 weeks of pregnancy. Reprod. Toxicol. 2007, 24, 414–416. [Google Scholar] [CrossRef]
- Dubois, J.; Hershon, L.; Carmant, L.; Belanger, S.; Leclerc, J.M.; David, M. Toxicity profile of interferon alfa-2b in children: A prospective evaluation. J. Pediatr. 1999, 135, 782–785. [Google Scholar] [CrossRef]
- Marine-Barjoan, E.; Berrebi, A.; Giordanengo, V.; Favre, S.F.; Haas, H.; Moreigne, M.; Izopet, J.; Tricoire, J.; Tran, A.; Pradier, C.; Bongain, A. HCV/HIV co-infection, HCV viral load and mode of delivery: risk factors for mother-to-child transmission of hepatitis C virus? AIDS 2007, 21, 1811–1815. [Google Scholar] [CrossRef]
- Gibb, D.M.; Goodall, R.L.; Dunn, D.T.; Healy, M.; Neave, P.; Cafferkey, M.; Butler, K. Mother-to-child transmission of hepatitis C virus: evidence for preventable peripartum transmission. Lancet 2000, 356, 904–907. [Google Scholar] [CrossRef]
- European Paediatric Hepatitis C Virus Network. A significant sex--but not elective cesarean section--effect on mother-to-child transmission of hepatitis C virus infection. J. Infect. Dis. 2005, 192, 1872–1879. [CrossRef]
- Ghamar Chehreh, M.E.; Tabatabaei, S.V.; Khazanehdari, S.; Alavian, S.M. Effect of cesarean section on the risk of perinatal transmission of hepatitis C virus from HCV-RNA+/HIV- mothers: a meta-analysis. Arch. Gynecol Obstet. 2011, 283, 255–260. [Google Scholar] [CrossRef]
- Arshad, M.; El-Kamary, S.S.; Jhaveri, R. Hepatitis C virus infection during pregnancy and the newborn period--are they opportunities for treatment? J. Viral. Hepat. 2011, 18, 229–236. [Google Scholar] [CrossRef]
- Mok, J.; Pembrey, L.; Tovo, P.A.; Newell, M.L. European Paediatric Hepatitis C Virus Network. When does mother to child transmission of hepatitis C virus occur? Arch. Dis. Child. 2005, 90, F156–F160. [Google Scholar]
- Pollack, H.; Hou, Z.; Hughes, A.L.; Borkowsky, W. Perinatal transmission and viral evolution of hepatitis C virus quasispecies in infants coinfected with HIV. J. Acquir. Immune Defic. Syndr. 2004, 36, 890–899. [Google Scholar] [CrossRef]
- Hayashida, A.; Inaba, N.; Oshima, K.; Nishikawa, M.; Shoda, A.; Hayashida, S.; Negishi, M.; Inaba, F.; Inaba, M.; Fukasawa, I.; Watanabe, H.; Takamizawa, H. Re-evaluation of the true rate of hepatitis C virus mother-to-child transmission and its novel risk factors based on our two prospective studies. J. Obstet. Gynaecol. Res. 2007, 33, 417–422. [Google Scholar] [CrossRef]
- Indolfi, G.; Azzari, C.; Moriondo, M.; Lippi, F.; de Martino, M.; Resti, M. Alanine transaminase levels in the year before pregnancy predict the risk of hepatitis C virus vertical transmission. J. Med. Virol. 2006, 78, 911–914. [Google Scholar]
- Azzari, C.; Resti, M.; Moriondo, M.; Ferrari, R.; Lionetti, P.; Vierucci, A. Vertical transmission of HCV is related to maternal peripheral blood mononuclear cell infection. Blood 2000, 96, 2045–2048. [Google Scholar]
- Azzari, C.; Moriondo, M.; Indolfi, G.; Betti, L.; Gambineri, E.; de Martino, M.; Resti, M. Higher risk of hepatitis C virus perinatal transmission from drug user mothers is mediated by peripheral blood mononuclear cell infection. J. Med. Virol. 2008, 80, 65–71. [Google Scholar] [CrossRef]
- Tovo, P.A.; Palomba, E.; Ferraris, G.; Principi, N.; Ruga, E.; Dallacasa, P.; Maccabruni, A. Increased risk of maternal-infant hepatitis C virus transmission for women coinfected with human immunodeficiency virus type 1. Italian Study Group for HCV Infection in Children. Clin. Infect. Dis. 1997, 25, 1121–1124. [Google Scholar]
- Mast, E.E.; Hwang, L.Y.; Seto, D.S.; Nolte, F.S.; Nainan, O.V.; Wurtzel, H.; Alter, M.J. Risk factors for perinatal transmission of hepatitis C virus (HCV) and the natural history of HCV infection acquired in infancy. J. Infect. Dis. 2005, 192, 1880–1889. [Google Scholar]
- Polis, C.B.; Shah, S.N.; Johnson, K.E.; Gupta, A. Impact of maternal HIV coinfection on the vertical transmission of hepatitis C virus: a meta-analysis. Clin. Infect. Dis. 2007, 44, 1123–1131. [Google Scholar] [CrossRef]
- Papaevangelou, V.; Pollack, H.; Rochford, G.; Kokka, R.; Hou, Z.; Chernoff, D.; Hanna, B.; Krasinski, K.; Borkowsky, W. Increased transmission of vertical hepatitis C virus (HCV) infection to human immunodeficiency virus (HIV)-infected infants of HIV- and HCV-coinfected women. J. Infect. Dis. 1998, 178, 1047–1052. [Google Scholar] [CrossRef]
- Blackard, J.T.; Smeaton, L.; Hiasa, Y.; Horiike, N.; Onji, M.; Jamieson, D.J.; Rodriguez, I.; Mayer, K.H.; Chung, R.T. Detection of hepatitis C virus (HCV) in serum and peripheral-blood mononuclear cells from HCV-monoinfected and HIV/HCV-coinfected persons. J. Infect. Dis. 2005, 192, 258–265. [Google Scholar] [CrossRef]
- Vidricaire, G.; Gauthier, S.; Tremblay, M.J. HIV-1 infection of trophoblasts is independent of gp120/CD4 Interactions but relies on heparan sulfate proteoglycans. J. Infect. Dis. 2007, 195, 1461–1471. [Google Scholar] [CrossRef]
- Kwiek, J.J.; Mwapasa, V.; Milner, D.A., Jr; Alker, A.P.; Miller, W.C.; Tadesse, E.; Molyneux, M.E.; Rogerson, S.J.; Meshnick, S.R. Maternal-fetal microtransfusions and HIV-1 mother-to-child transmission in Malawi. PLoS Med. 2006, 3, e10. [Google Scholar] [CrossRef]
- Gammill, H.S.; Nelson, J.L. Naturally acquired microchimerism. Int. J. Dev. Biol. 2010, 54, 531–543. [Google Scholar] [CrossRef]
- Mold, J.E.; Michaelsson, J.; Burt, T.D.; Muench, M.O.; Beckerman, K.P.; Busch, M.P.; Lee, T.H.; Nixon, D.F.; McCune, J.M. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 2008, 322, 1562–1565. [Google Scholar]
- Delamare, C.; Carbonne, B.; Heim, N.; Berkane, N.; Petit, J.C.; Uzan, S.; Grange, J.D. Detection of hepatitis C virus RNA (HCV RNA) in amniotic fluid: A prospective study. J. Hepatol. 1999, 31, 416–420. [Google Scholar]
- Pereira, L.; Maidji, E.; McDonagh, S.; Tabata, T. Insights into viral transmission at the uterine-placental interface. Trends Microbiol. 2005, 13, 164–174. [Google Scholar] [CrossRef]
- Bhat, P.; Anderson, D.A. Hepatitis B virus translocates across a trophoblastic barrier. J. Virol. 2007, 81, 7200–7207. [Google Scholar] [CrossRef] [Green Version]
- Lagaye, S.; Derrien, M.; Menu, E.; Coïto, C.; Tresoldi, E.; Mauclère, P.; Scarlatti, G.; Chaouat, G.; Barré-Sinoussi, F.; Bomsel, M. European Network for the Study of In Utero Transmission of HIV-1. Cell-to-cell contact results in a selective translocation of maternal human immunodeficiency virus type 1 quasispecies across a trophoblastic barrier by both transcytosis and infection. J. Virol. 2001, 75, 4780–4791. [Google Scholar]
- Hygino, J.; Lima, P.G.; Filho, R.G.; Silva, A.A.; Saramago, C.S.; Andrade, R.M.; Andrade, D.M.; Andrade, A.F.; Brindeiro, R.; Tanuri, A.; Bento, C.A. Altered immunological reactivity in HIV-1-exposed uninfected neonates. Clin. Immunol. 2008, 127, 340–347. [Google Scholar] [CrossRef]
- Kuhn, L.; Coutsoudis, A.; Moodley, D.; Mngqundaniso, N.; Trabattoni, D.; Shearer, G.M.; Clerici, M.; Coovadia, H.M. Interferon-gamma and interleukin-10 production among HIV-1-infected and uninfected infants of HIV-1-infected mothers. Ped. Res. 2001, 50, 412–416. [Google Scholar] [CrossRef]
- McDonagh, S.; Maidji, E.; Ma, W.; Chang, H.T.; Fisher, S.; Pereira, L. Viral and bacterial pathogens at the maternal-fetal interface. J. Infect. Dis. 2004, 190, 826–834. [Google Scholar]
- Dye, J.F.; Jablenska, R.; Donnelly, J.L.; Lawrence, L.; Leach, L.; Clark, P.; Firth, J.A. Phenotype of the endothelium in the human term placenta. Placenta 2001, 22, 32–43. [Google Scholar]
- Ethier-Chiasson, M.; Duchesne, A.; Forest, J.C.; Giguere, Y.; Masse, A.; Mounier, C.; Lafond, J. Influence of maternal lipid profile on placental protein expression of LDLr and SR-BI. Biochem. Biophys. Res. Comm. 2007, 359, 8–14. [Google Scholar]
- Soilleux, E.J.; Barten, R.; Trowsdale, J. DC-SIGN; a related gene, DC-SIGNR; and CD23 form a cluster on 19p13. J. Immunol. 2000, 165, 2937–2942. [Google Scholar]
- Nie, Q.H.; Gao, L.H.; Cheng, Y.Q.; Huang, X.F.; Zhang, Y.F.; Luo, X.D.; Wang, J.Q.; Wang, Y.Y. Hepatitis C virus infection of human cytotrophoblasts cultured in vitro. J. Med. Virol. 2012, 84, 1586–1592. [Google Scholar] [CrossRef]
- Hurtado, C.W.; Golden-Mason, L.; Brocato, M.; Krull, M.; Narkewicz, M.R.; Rosen, H.R. Innate immune function in placenta and cord blood of hepatitis C-seropositive mother-infant dyads. PloS One 2010, 5, e12232. [Google Scholar]
- Pelletier, S.; Drouin, C.; Bédard, N.; Khakoo, S.I.; Bruneau, J.; Shoukry, N.H. Increased degranulation of natural killer cells during acute HCV correlates with the magnitude of virus-specific T cell responses. J. Hepatol. 2010, 53, 805–816. [Google Scholar] [Green Version]
- Alter, G.; Jost, S.; Rihn, S.; Reyor, L.L.; Nolan, B.E.; Ghebremichael, M.; Bosch, R.; Altfeld, M.; Lauer, G.M. Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+ NK cells in acute HCV infection may predict viral clearance. J Hepatol. 2011, 55, 278–288. [Google Scholar] [CrossRef]
- Durante-Mangoni, E.; Wang, R.; Shaulov, A.; He, Q.; Nasser, I.; Afdhal, N.; Koziel, M.J.; Exley, M.A. Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J. Immunol. 2004, 173, 2159–2166. [Google Scholar]
- Farci, P.; Quinti, I.; Farci, S.; Alter, H.J.; Strazzera, R.; Palomba, E.; Coiana, A.; Cao, D.; Casadei, A.M.; Ledda, R.; Iorio, R.; Vegnente, A.; Diaz, G.; Tovo, P.A. Evolution of hepatitis C viral quasispecies and hepatic injury in perinatally infected children followed prospectively. Proc. Natl Acad. Sci. USA. 2006, 103, 8475–8480. [Google Scholar]
- Gerotto, M.; Resti, M.; Dal Pero, F.; Migliorato, I.; Alberti, A.; Bortolotti, F. Evolution of hepatitis C virus quasispecies in children with chronic hepatitis C. Infection 2006, 34, 62–65. [Google Scholar]
- Manzin, A.; Solforosi, L.; Debiaggi, M.; Zara, F.; Tanzi, E.; Romano, L.; Zanetti, A.R.; Clementi, M. Dominant role of host selective pressure in driving hepatitis C virus evolution in perinatal infection. J. Virol. 2000, 74, 4327–4334. [Google Scholar] [CrossRef]
- Dowd, K.A.; Hershow, R.C.; Yawetz, S.; Larussa, P.; Diaz, C.; Landesman, S.H.; Paul, M.E.; Read, J.S.; Lu, M.; Thomas, D.L.; Netski, D.M.; Ray, S.C. Maternal neutralizing antibody and transmission of hepatitis C virus to infants. J. Infect. Dis. 2008, 198, 1651–1655. [Google Scholar] [CrossRef]
- Meunier, J.C.; Bukh, J.; Diaz, G.; Tovo, P.A.; Casadei, A.M.; Quinti, I.; Iorio, R.; Emerson, S.; Purcell, R.H.; Farci, P. Neutralizing antibodies to hepatitis C virus in perinatally infected children followed up prospectively. J. Infect. Dis. 2011, 204, 1741–1745. [Google Scholar]
- Yeung, L.T.; King, S.M.; Roberts, E.A. Mother-to-infant transmission of hepatitis C virus. Hepatology 2001, 34, 223–229. [Google Scholar]
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR 1998, 47. No. RR-19..
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR 2012, 61. No. RR-4..
- Resti, M.; Jara, P.; Hierro, L.; Azzari, C.; Giacchino, R.; Zuin, G.; Zancan, L.; Pedditzi, S.; Bortolotti, F. Clinical features and progression of perinatally acquired hepatitis C virus infection. J. Med. Virol. 2003, 70, 373–377. [Google Scholar]
- Tovo, P.A.; Pembrey, L.J.; Newell, M.L. Persistence rate and progression of vertically acquired hepatitis C infection. European Paediatric Hepatitis C Virus Infection. J. Infect. Dis. 2000, 181, 419–424. [Google Scholar] [CrossRef]
- Yeung, L.T.; To, T.; King, S.M.; Roberts, E.A. Spontaneous clearance of childhood hepatitis C virus infection. J. Viral Hepat. 2007, 14, 797–805. [Google Scholar] [CrossRef]
- Alter, H.J.; Seeff, L.B. Recovery, persistence, and sequelae in hepatitis C virus infection: A perspective on long-term outcome. Semin. Liv. Dis. 2000, 20, 17–35. [Google Scholar] [CrossRef]
- Vogt, M.; Lang, T.; Frosner, G.; Klingler, C.; Sendl, A.F.; Zeller, A.; Wiebecke, B.; Langer, B.; Meisner, H.; Hess, J. Prevalence and clinical outcome of hepatitis C infection in children who underwent cardiac surgery before the implementation of blood-donor screening. New Engl. J. Med. 1999, 341, 866–870. [Google Scholar] [CrossRef]
- El-Kamary, S.S.; Hashem, M.; Saleh, D.A.; Abdelwahab, S.F.; Sobhy, M.; Shebl, F.M.; Shardell, M.D.; Strickland, G.T.; Shata, M.T. Hepatitis C virus-specific cell-mediated immune responses in children born to mothers infected with hepatitis C virus. J. Pediatr. 2012, in press.. [Google Scholar]
- European Paediatric Hepatitis C Virus Network. Three broad modalities in the natural history of vertically acquired hepatitis C virus infection. Clin. Infect. Dis. 2005, 41, 45–51. [CrossRef]
- Jonas, M.M. Children with hepatitis C. Hepatology 2002, 36, S173–S178. [Google Scholar] [CrossRef]
- Palomba, E.; Manzini, P.; Fiammengo, P.; Maderni, P.; Saracco, G.; Tovo, P.A. Natural history of perinatal hepatitis C virus infection. Clin. Infect. Dis. 1996, 23, 47–50. [Google Scholar] [CrossRef]
- Gismondi, M.I.; Becker, P.D.; Diaz Carrasco, J.M.; Guzman, C.A.; Campos, R.H.; Preciado, M.V. Evolution of hepatitis C virus hypervariable region 1 in immunocompetent children born to HCV-infected mothers. J. Viral Hepat. 2009, 16, 332–339. [Google Scholar] [CrossRef]
- Guilbert, C.; Troesch, M.; Samson, J.; Lemay, M.; Pelletier, V.A.; Bernard-Bonnin, A.C.; Kozielski, R.; Lapointe, N.; Martin, S.R.; Soudeyns, H. Differing patterns of liver disease progression and hepatitis C virus (HCV) quasispecies evolution in children vertically coinfected with HCV and human immunodeficiency virus type 1. J. Clin. Microbiol. 2004, 42, 4365–4369. [Google Scholar] [CrossRef]
- Quesnel-Vallières, M.; Lemay, M.; Lapointe, N.; Martin, S.R.; Soudeyns, H. HCV quasispecies evolution during treatment with interferon alfa-2b and ribavirin in two children coinfected with HCV and HIV-1. J. Clin. Virol. 2008, 43, 236–240. [Google Scholar] [CrossRef]
- Gaud, U.; Langer, B.; Petropoulou, T.; Thomas, H.C.; Karayiannis, P. Changes in hypervariable region 1 of the envelope 2 glycoprotein of hepatitis C virus in children and adults with humoral immune defects. J. Med. Virol. 2003, 69, 350–356. [Google Scholar]
- Larouche, A.; Gaëtan, G.; El-Bilali, N.; Quesnel-Vallières, M.; Martin, S.R.; Alvarez, F.; Shoukry, N.H.; Soudeyns, H. A case of seronegative HCV infection in a child infected via mother-to-child transmission. J. Clin. Microbiol. 2012, 50, 2515–2519. [Google Scholar] [CrossRef]
- Castello, G.; Scala, S.; Palmieri, G.; Curley, S.A.; Izzo, F. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin. Immunol. 2010, 134, 237–250. [Google Scholar] [CrossRef]
- Wirth, S.; Ribes-Koninckx, C.; Calzado, M.A.; Bortolotti, F.; Zancan, L.; Jara, P.; Shelton, M.; Kerkar, N.; Galoppo, M.; Pedreira, A.; Rodriguez-Baez, N.; Ciocca, M.; Lachaux, A.; Lacaille, F.; Lang, T.; Kullmer, U.; Huber, W.D.; Gonzalez, T.; Pollack, H.; Alonso, E.; Broue, P.; Ramakrishna, J.; Neigut, D.; Valle-Segarra, A.D.; Hunter, B.; Goodman, Z.; Xu, C.R.; Zheng, H.; Noviello, S.; Sniukiene, V.; Brass, C.; Albrecht, J.K. High sustained virologic response rates in children with chronic hepatitis C receiving peginterferon alfa-2b plus ribavirin. J. Hepatol. 2010, 52, 501–507. [Google Scholar]
- Schwarz, K.B.; Gonzalez-Peralta, R.P.; Murray, K.F.; Molleston, J.P.; Haber, B.A.; Jonas, M.M.; Rosenthal, P.; Mohan, P.; Balistreri, W.F.; Narkewicz, M.R.; Smith, L.; Lobritto, S.J.; Rossi, S.; Valsamakis, A.; Goodman, Z.; Robuck, P.R.; Barton, B.A. Peds-C Clinical Research Network. The combination of ribavirin and peginterferon is superior to peginterferon and placebo for children and adolescents with chronic hepatitis C. Gastroenterology 2011, 140, 450–458. [Google Scholar]
- Mack, C.L.; Gonzalez-Peralta, R.P.; Gupta, N.; Leung, D.; Narkewicz, M.R.; Roberts, E.A.; Rosenthal, P.; Schwarz, K.B. North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition. NASPGHAN practice guidelines: Diagnosis and management of hepatitis C infection in infants, children, and adolescents. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 838–855. [Google Scholar] [CrossRef]
- Pharmacokinetics of Boceprevir in Pediatric Subjects With Chronic Hepatitis C Genotype 1 (SCH503034-P07614 AM4). Available online: http://clinicaltrials.gov/ct2/show/NCT01425190 (accessed on 29 September 2012).
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Le Campion, A.; Larouche, A.; Fauteux-Daniel, S.; Soudeyns, H. Pathogenesis of Hepatitis C During Pregnancy and Childhood. Viruses 2012, 4, 3531-3550. https://doi.org/10.3390/v4123531
Le Campion A, Larouche A, Fauteux-Daniel S, Soudeyns H. Pathogenesis of Hepatitis C During Pregnancy and Childhood. Viruses. 2012; 4(12):3531-3550. https://doi.org/10.3390/v4123531
Chicago/Turabian StyleLe Campion, Armelle, Ariane Larouche, Sébastien Fauteux-Daniel, and Hugo Soudeyns. 2012. "Pathogenesis of Hepatitis C During Pregnancy and Childhood" Viruses 4, no. 12: 3531-3550. https://doi.org/10.3390/v4123531