Respiratory Syncytial Virus Persistence in Macrophages Alters the Profile of Cellular Gene Expression

{kind=link}
Abstract
:1. The Virus: Characteristics, Pathogenesis, and Epidemiology
2. RSV Persistence
3. Establishment and Characteristics of a Persistently RSV-Infected Macrophage-Like Culture
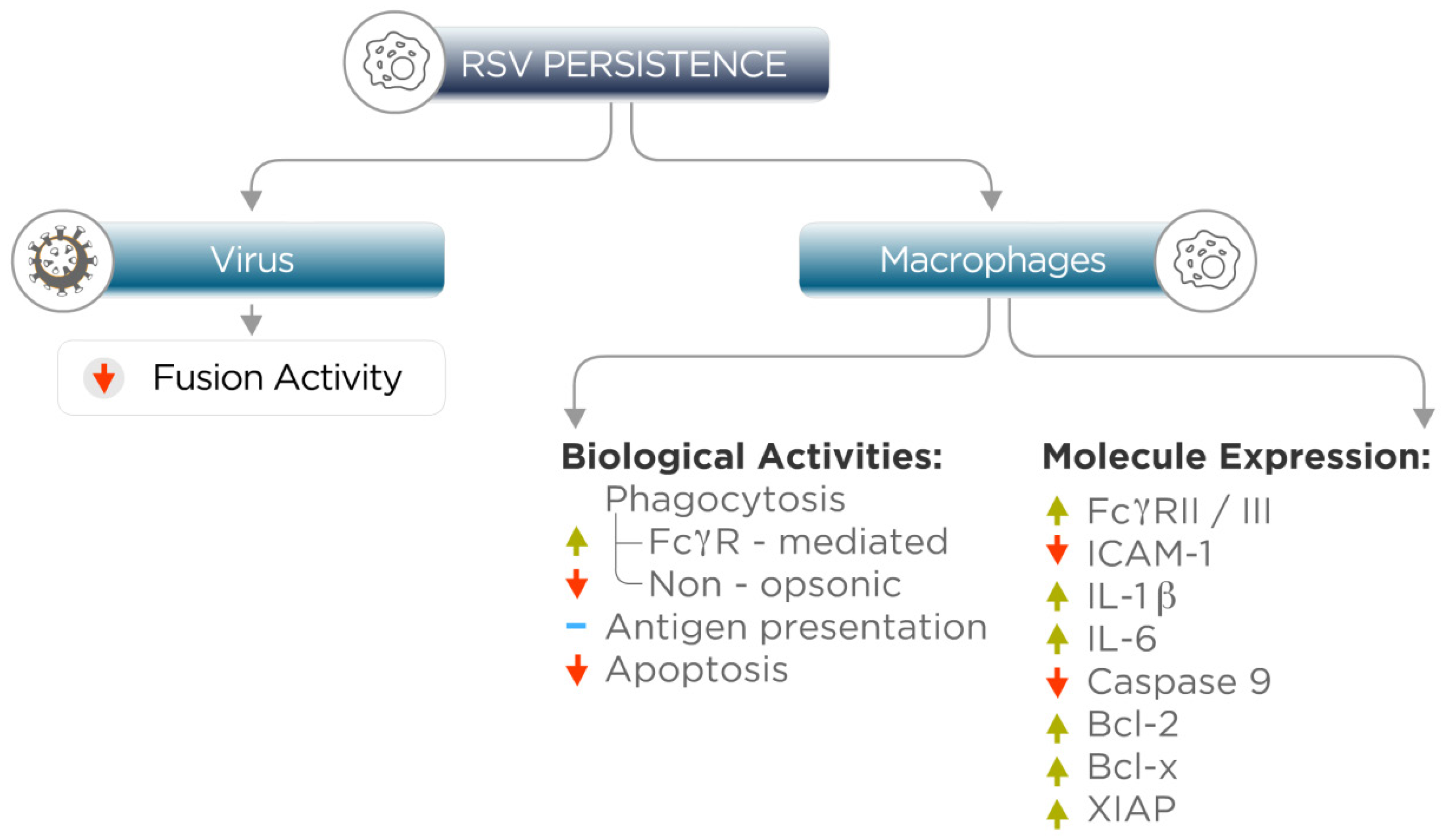
4. Persistent RSV Infection Alters Macrophage Gene Expression and Biological Activities
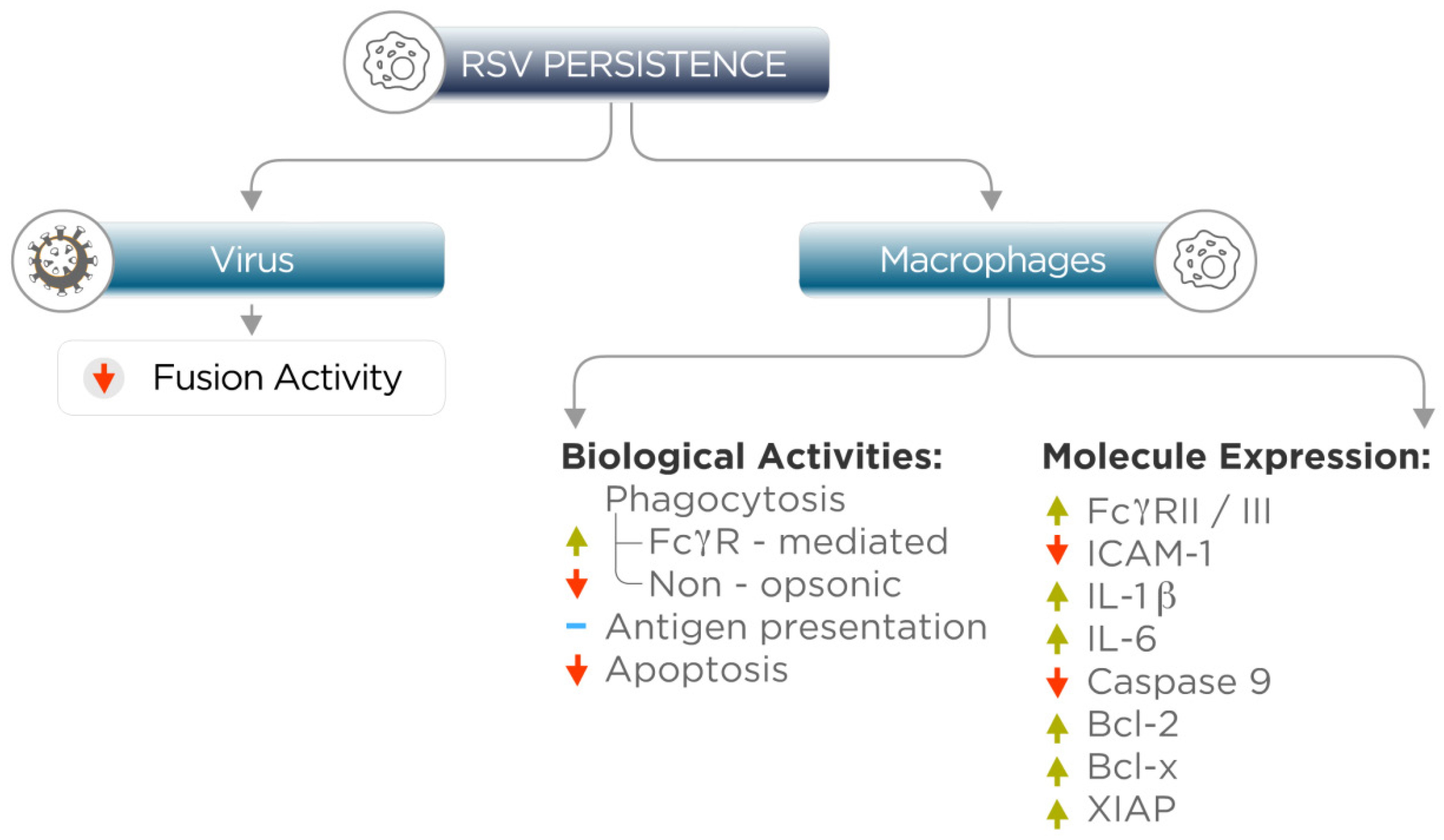
5. Relevance of RSV Persistence in Macrophages and Epithelial Cells
6. Conclusion
Acknowledgments
Conflict of Interest
References
- Cane, P.A. Molecular epidemiology of respiratory syncytial virus. Rev. Med. Virol. 2007, 11, 103–116. [Google Scholar] [CrossRef]
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef]
- Ogra, P.L. Respiratory syncytial virus: The virus, the disease and the immune response. Paediatr. Respir. Rev. 2004, 5, 119–126. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–98. [Google Scholar] [CrossRef]
- Law, B.J.; Carbonell-Estrany, X.; Simoes, E.A. An update on respiratory syncytial virus epidemiology: A developed country perspective. Respir. Med. 2002, 96, 1–7. [Google Scholar] [CrossRef]
- Hervás, D.; Reina, J.; Yañez, A.; del Valle, J.M.; Figuerola, J.; Hervás, J.A. Epidemiology of hospitalization for acute bronchiolitis in children: Differences between RSV and non-RSV bronchiolitis. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1975–1981. [Google Scholar] [CrossRef]
- Langley, G.F.; Anderson, L.J. Epidemiology and prevention of respiratory syncytial virus infections among infants and young children. Pediatr. Infect. Dis. J. 2011, 30, 510–517. [Google Scholar]
- Welliver, R.C. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J. Pediatr. 2003, 143, 112–117. [Google Scholar] [CrossRef]
- Mejías, A.; Chávez-Bueno, S.; Ramilo, O. Respiratory syncytial virus pneumonia: mechanisms of inflammation and prolonged airway hyperresponsiveness. Curr. Opin. Infect. Dis. 2005, 18, 199–204. [Google Scholar] [CrossRef]
- Sigurs, N.; Gustafsson, P.M.; Bjarnason, R.; Lundberg, F.; Schmidt, S.; Sigurbergsson, F.; Kjellman, B. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care. Med. 2005, 171, 137–141. [Google Scholar]
- Staat, M.A. Respiratory syncytial virus infections in children. Semin. Respir. Infect. 2002, 17, 15–20. [Google Scholar] [CrossRef]
- Dowell, S.F.; Anderson, L.J.; Gary, H.E., Jr.; Erdman, D.D.; Plouffe, J.F.; File, T.M., Jr.; Marston, B.J.; Breiman, R.F. Respiratory syncytial virus is an important cause of community-acquired lower respiratory infection among hospitalized adults. J. Infect. Dis. 1996, 174, 456–462. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef]
- World Health Organization. Initiative for Vaccine Research: Respiratory Syncytial Virus. Available online: http://www.who.int/vaccine_research/diseases/ari/en/index3.html (accessed on 22 August 2012).
- Bhatt, J.M.; Everard, M.L. Do environmental pollutants influence the onset of respiratory syncytial virus epidemics or disease severity? Paediatr. Respir. Rev. 2004, 5, 333–338. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Seemungal, T.; Harper-Owen, R.; Bhowmik, A.; Moric, I.; Sanderson, G.; Message, S.; Maccallum, P.; Meade, T.W.; Jeffries, D.J.; Johnston, S.L.; Wedzicha, J.A. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2001, 164, 1618–1623. [Google Scholar]
- Sikkel, M.B.; Quint, J.K.; Mallia, P.; Wedzicha, J.A.; Johnston, S.L. Respiratory syncytial virus persistence in chronic obstructive pulmonary disease. Pediatr. Infect. Dis. J. 2008, 27, 63–70. [Google Scholar] [CrossRef]
- Kim, E.Y.; Battaile, J.T.; Patel, A.C.; You, Y.; Agapov, E.; Grayson, M.H.; Benoit, L.A.; Byers, D.E.; Alevy, Y.; Tucker, J.; et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 2008, 14, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Di Rosa, F.; Barnaba, V. Persisting viruses and chronic inflammation: Understanding their relation to autoimmunity. Immunol. Rev. 1998, 164, 17–27. [Google Scholar] [CrossRef]
- Wald, O.; Weiss, I.D.; Galun, E.; Peled, A. Chemokines in hepatitis C virus infection: Pathogenesis, prognosis and therapeutics. Cytokine. 2007, 39, 50–62. [Google Scholar] [CrossRef]
- Culley, F.J.; Pennycook, A.M.; Tregoning, J.S.; Hussell, T.; Openshaw, P.J. Differential chemokine expression following respiratory virus infection reflects Th1- or Th2-biased immunopathology. J. Virol. 2006, 80, 4521–4527. [Google Scholar]
- Krishnan, S.; Halonen, M.; Welliver, R.C. Innate immune responses in respiratory syncytial virus infections. Viral Immunol. 2004, 17, 220–233. [Google Scholar] [CrossRef]
- Mills, B.G.; Singer, F.R.; Weiner, L.P.; Holst, P.A. Immunohistological demonstration of respiratory syncytial virus antigens in Paget disease of bone. Proc. Natl. Acad. Sci. USA 1981, 78, 1209–1213. [Google Scholar] [CrossRef]
- Isaia, G.; Teodosiu, O.; Popescu, G.; Athanasiu, P.; Sternberg, I.; Dumitriu, Z. Persistence of viruses in the nasopharynx of apparently healthy children aged 0-5 years. Results of investigations performed in 1982-83. Virologie 1985, 36, 175–179. [Google Scholar]
- Cubie, H.A.; Duncan, L.A.; Marshall, L.A.; Smith, N.M. Detection of respiratory syncytial virus nucleic acid in archival postmortem tissue from infants. Pediatr. Pathol. Lab. Med. 1997, 17, 927–938. [Google Scholar] [CrossRef]
- Rezaee, F.; Gibson, L.F.; Piktel, D.; Othumpangat, S.; Piedimonte, G. Respiratory syncytial virus infection in human bone marrow stromal cells. Am. J. Respir. Cell. Mol. Biol. 2011, 45, 277–286. [Google Scholar] [CrossRef]
- Hegele, R.G.; Hayashi, S.; Bramley, A.M.; Hogg, J.C. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest. 1994, 105, 1848–1854. [Google Scholar] [CrossRef]
- Schwarze, J.; O'Donnell, D.R.; Rohwedder, A.; Openshaw, P.J. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am. J. Respir. Crit. Care Med. 2004, 169, 801–805. [Google Scholar] [CrossRef]
- Sutton, T.C.; Tayyari, F.; Khan, M.A.; Manson, H.E.; Hegele, R.G. T helper 1 background protects against airway hyperresponsiveness and inflammation in guinea pigs with persistent respiratory syncytial virus infection. Pediatr. Res. 2007, 61, 525–529. [Google Scholar]
- Martínez, I.; Lombardía, L.; Herranz, C.; García-Barreno, B.; Domínguez, O.; Melero, J.A. Cultures of HEp-2 cells persistently infected by human respiratory syncytial virus differ in chemokine expression and resistance to apoptosis as compared to lytic infections of the same cell type. Virology 2009, 388, 31–41. [Google Scholar] [CrossRef]
- Sarmiento, R.E.; Tirado, R.; Gómez, B. Characteristics of a respiratory syncytial virus persistently infected macrophage-like culture. Virus Res. 2002, 84, 45–58. [Google Scholar] [CrossRef]
- Tirado, R.; Ortega, A.; Sarmiento, R.E.; Gómez, B. Interleukin-8 mRNA synthesis and protein secretion are continuously up-regulated by respiratory syncytial virus persistently infected cells. Cell Immunol. 2005, 233, 61–71. [Google Scholar] [CrossRef]
- Valdovinos, M.R.; Gómez, B. Establishment of respiratory syncytial virus persistence in cell lines: Association with defective interfering particles. Intervirology 2003, 46, 190–198. [Google Scholar] [CrossRef]
- Hobson, L.; Everard, M.L. Persistent of respiratory syncytial virus in human dendritic cells and influence of nitric oxide. Clin. Exp. Immunol. 2008, 151, 359–366. [Google Scholar] [CrossRef]
- Midulla, F.; Villani, A.; Panuska, J.R.; Dab, I.; Kolls, J.K.; Merolla, R.; Ronchetti, R. Respiratory syncytial virus lung infection in infants: Immunoregulatory role of infected alveolar macrophages. J. Infect. Dis. 1993, 168, 1515–1519. [Google Scholar] [CrossRef]
- Castleman, W.L.; Lay, J.C.; Dubovi, E.J.; Slauson, D.O. Experimental bovine respiratory syncytial virus infection in conventional calves: light microscopic lesions, microbiology, and studies on lavaged lung cells. Am. J. Vet. Res. 1985, 46, 547–553. [Google Scholar]
- Panuska, J.R.; Cirino, N.M.; Midulla, F.; Despot, J.E.; McFadden, E.R., Jr.; Huang, Y.T. Productive infection of isolated human alveolar macrophages by respiratory syncytial virus. J. Clin. Invest. 1990, 86, 113–119. [Google Scholar] [CrossRef]
- Oldstone, M.B. Viral persistence: parameters, mechanisms and future predictions. Virology 2006, 344, 111–118. [Google Scholar] [CrossRef]
- Koren, H.S.; Handwerger, B.S.; Wunderlich, J.R. Identification of macrophage-like characteristics in a cultured murine tumor line. J. Immunol. 1975, 114, 894–897. [Google Scholar]
- Arrevillaga, G.; Gaona, J.; Sánchez, C.; Rosales, V.; Gómez, B. Respiratory syncytial virus persistence in macrophages downregulates intercellular adhesion molecule-1 expression and reduces adhesion of non-typeable haemophilus influenzae. Intervirology 2012, 55, 442–450. [Google Scholar] [CrossRef]
- Sarmiento, R.E.; Arias, C.F.; Méndez, E.; Gómez, B. Characterization of a persistent respiratory syncytial virus showing a low-fusogenic activity associated to an impaired F protein. Virus Res. 2009, 139, 39–47. [Google Scholar] [CrossRef]
- Matsuyama, S.; Delos, S.E.; White, J.M. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J. Virol. 2004, 78, 8201–8209. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Thoennes, S.; Li, Z.N.; Lee, B.J.; Langley, W.A.; Skehel, J.J.; Russell, R.J.; Steinhauer, D.A. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 2008, 370, 403–414. [Google Scholar] [CrossRef]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef]
- Swanson, J.A.; Hoppe, A.D. The coordination of signaling during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 2004, 76, 1093–1103. [Google Scholar] [CrossRef]
- Van Lookeren Campagne, M.; Wiesmann, C.; Brown, E.J. Macrophage complement receptors and pathogen clearance. Cell. Microbiol. 2007, 9, 2095–2102. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Ortega, E.; Gomez, B. Persistence of respiratory syncytial virus in macrophages alters phagocytosis and pro-inflammatory cytokine production. Viral Immunol. 2001, 14, 19–30. [Google Scholar] [CrossRef]
- Liu, K.; Nussenzweig, M.C. Origin and development of dendritic cells. Immunol. Rev. 2010, 234, 45–54. [Google Scholar] [CrossRef]
- Guth, A.M.; Janssen, W.J.; Bosio, C.M.; Crouch, E.C.; Henson, P.M.; Dow, S.W. Lung environment determines unique phenotype of alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, 936–946. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Ortega, E.; Ortíz-Navarrete, V.; Gómez, B. Antigen presentation by a macrophage-like cell line persistently infected with respiratory syncytial virus. Virus Res. 2004, 99, 95–100. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Coffer, P.J.; Schellens, I.M.; de Graaff, P.M.; Kimpen, J.L.; Koenderman, L. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J. Immunol. 2006, 176, 5529–5537. [Google Scholar]
- Monick, M.M.; Cameron, K.; Staber, J.; Powers, L.S.; Yarovinsky, T.O.; Koland, J.G.; Hunninghake, G.W. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J. Biol. Chem. 2005, 280, 2147–2158. [Google Scholar]
- Nakamura-López, Y.; Villegas-Sepúlveda, N.; Sarmiento-Silva, R.E.; Gómez, B. Intrinsic apoptotic pathway is subverted in mouse macrophages persistently infected by RSV. Virus Res. 2011, 158, 98–107. [Google Scholar] [CrossRef]
- Nakamura-López, Y.; Sarmiento-Silva, R.E.; Moran-Andrade, J.; Gómez-García, B. Staurosporine-induced apoptosis in P388D1 macrophages involves both extrinsic and intrinsic pathways. Cell Biol. Int. 2009, 33, 1026–1031. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rivera-Toledo, E.; Gómez, B. Respiratory Syncytial Virus Persistence in Macrophages Alters the Profile of Cellular Gene Expression. Viruses 2012, 4, 3270-3280. https://doi.org/10.3390/v4123270
Rivera-Toledo E, Gómez B. Respiratory Syncytial Virus Persistence in Macrophages Alters the Profile of Cellular Gene Expression. Viruses. 2012; 4(12):3270-3280. https://doi.org/10.3390/v4123270
Chicago/Turabian StyleRivera-Toledo, Evelyn, and Beatríz Gómez. 2012. "Respiratory Syncytial Virus Persistence in Macrophages Alters the Profile of Cellular Gene Expression" Viruses 4, no. 12: 3270-3280. https://doi.org/10.3390/v4123270