Cellular Prion Protein: From Physiology to Pathology

Abstract
:1. Introduction
2. Normal cellular prion protein, PrPC
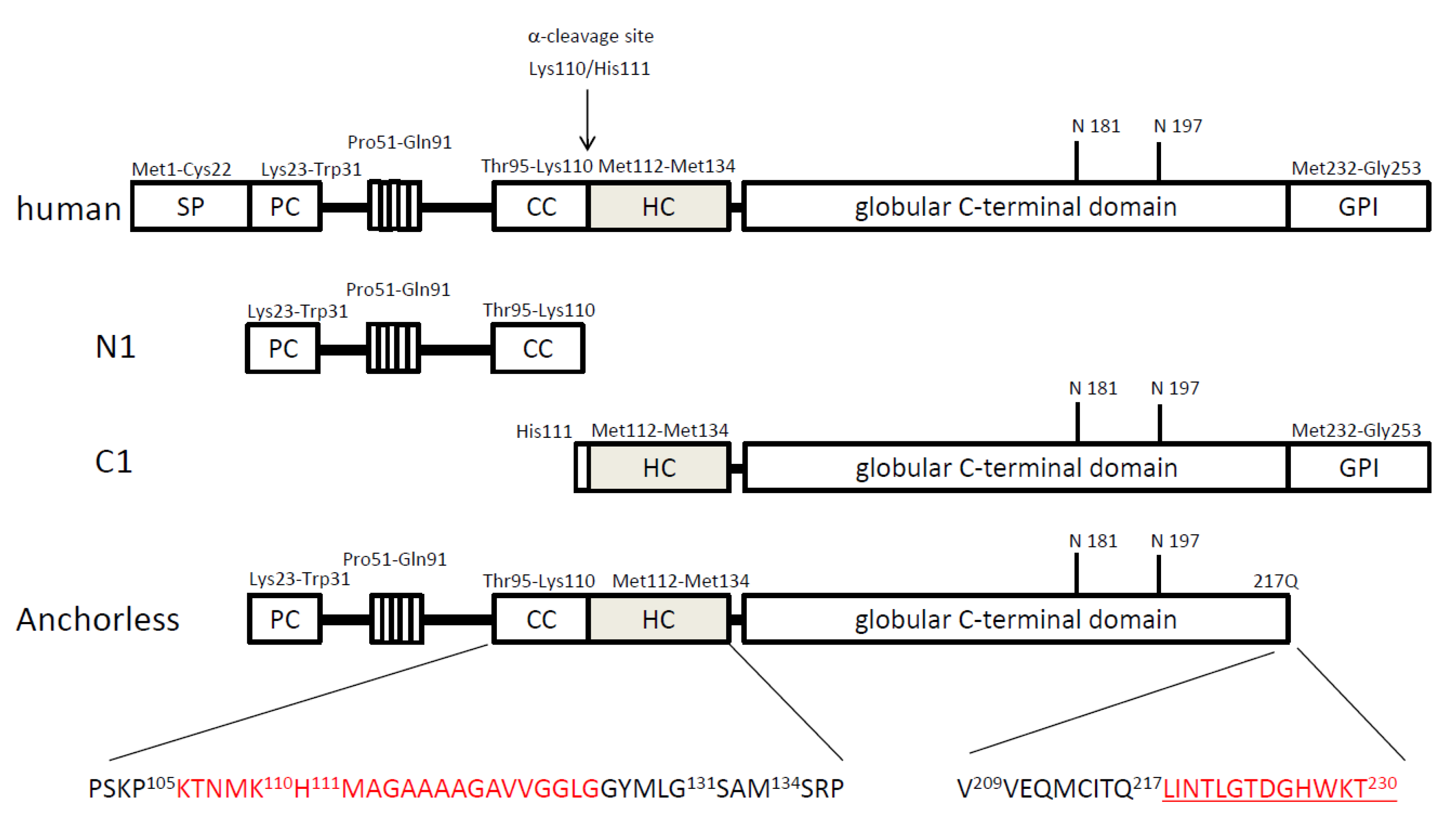
3. Transgenic mouse models
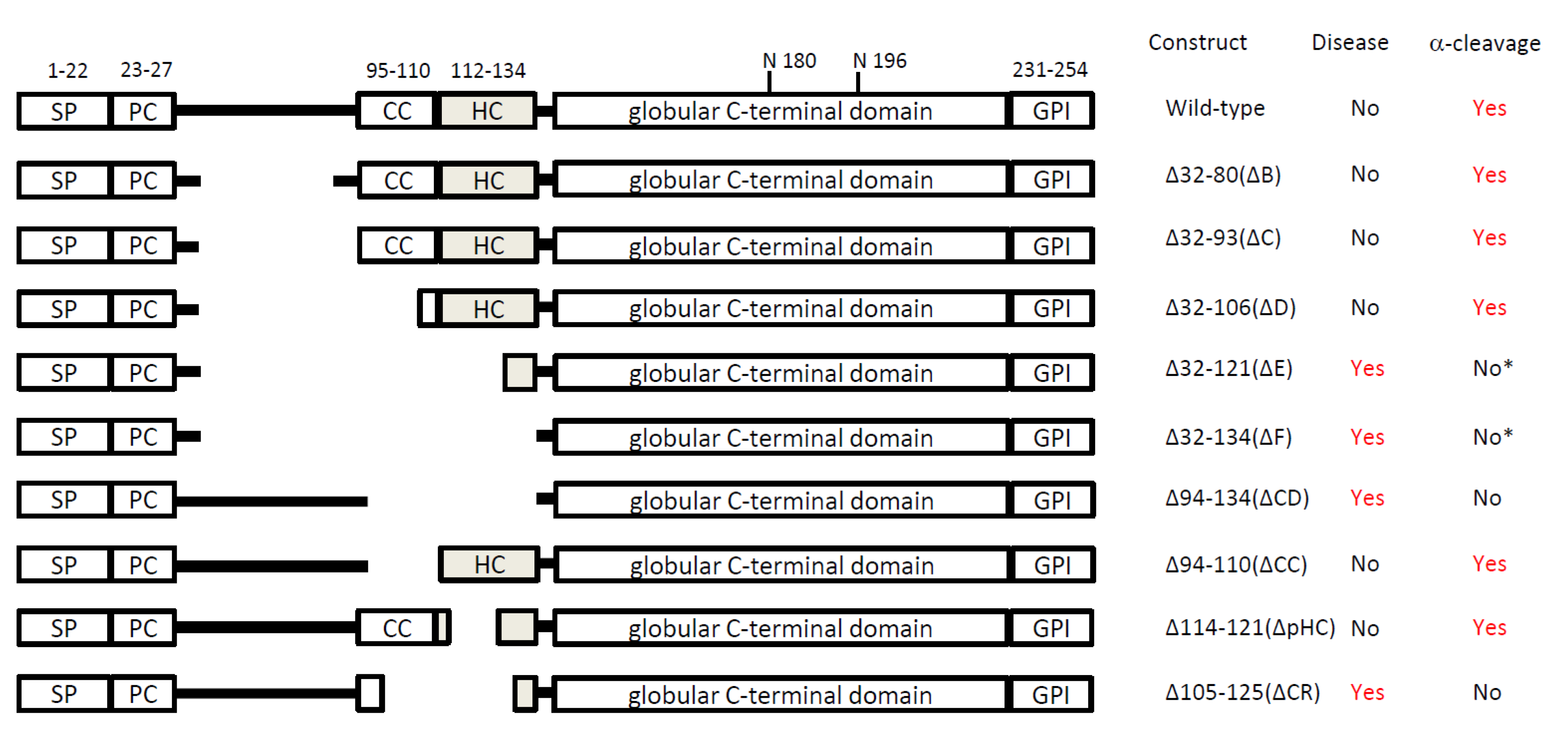

{kind=link}
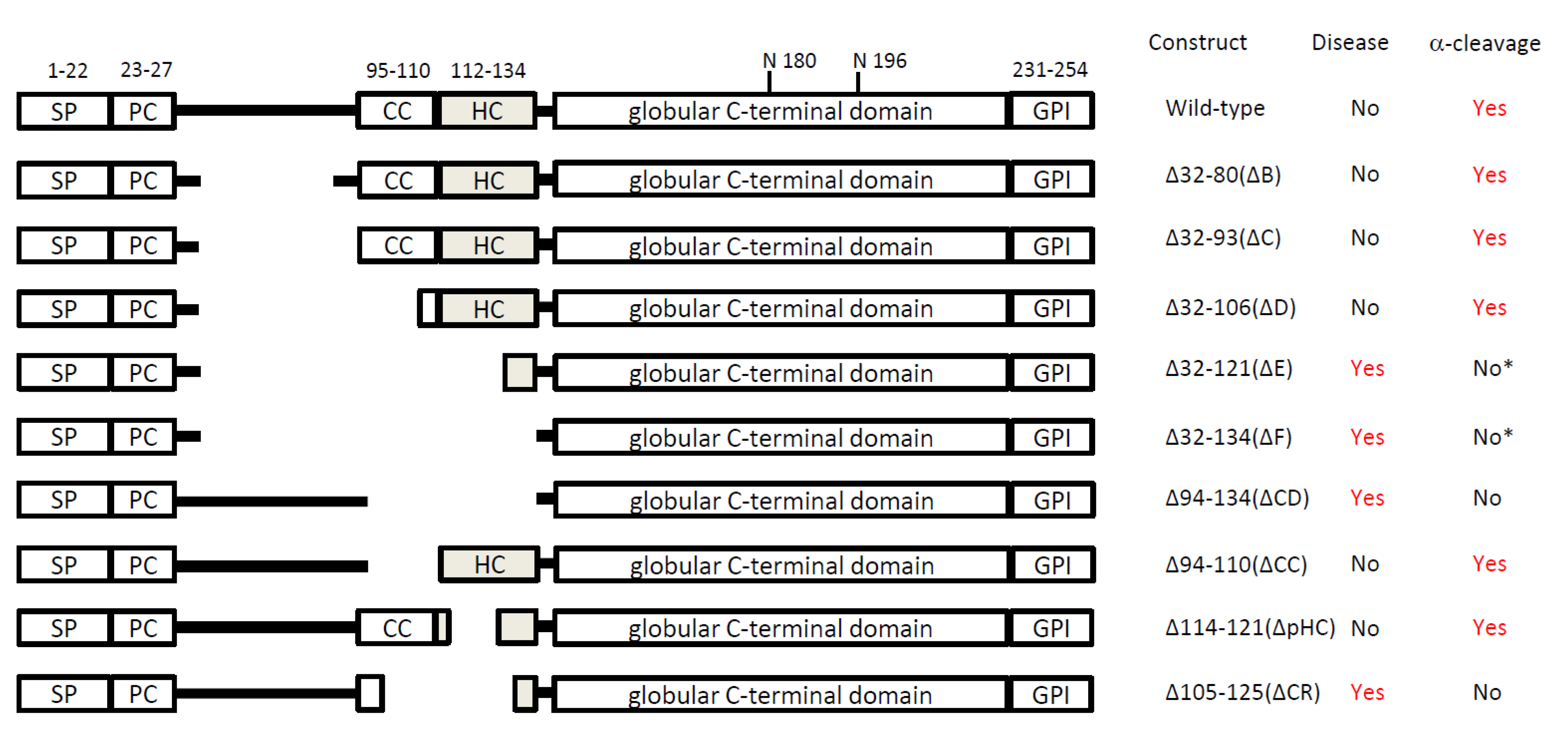{kind=link}
{kind=link}
{kind=link}
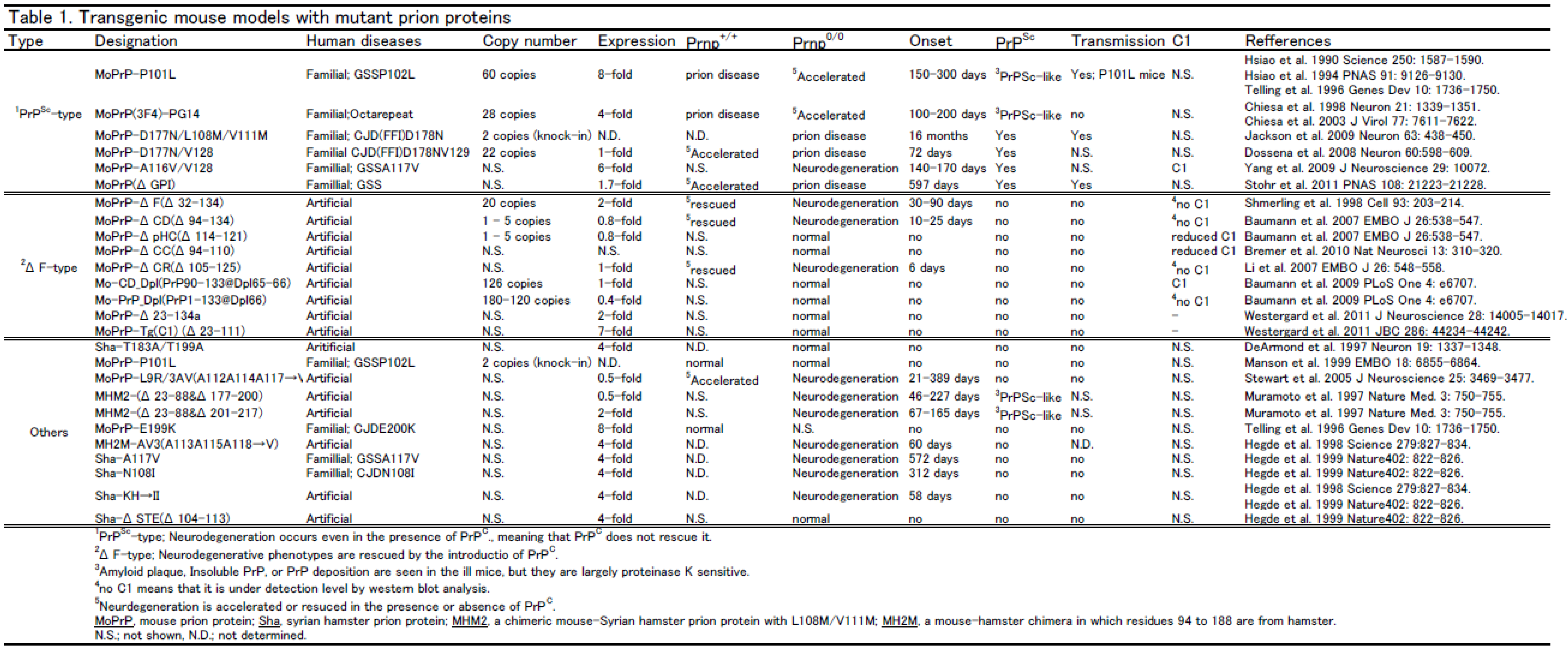 |
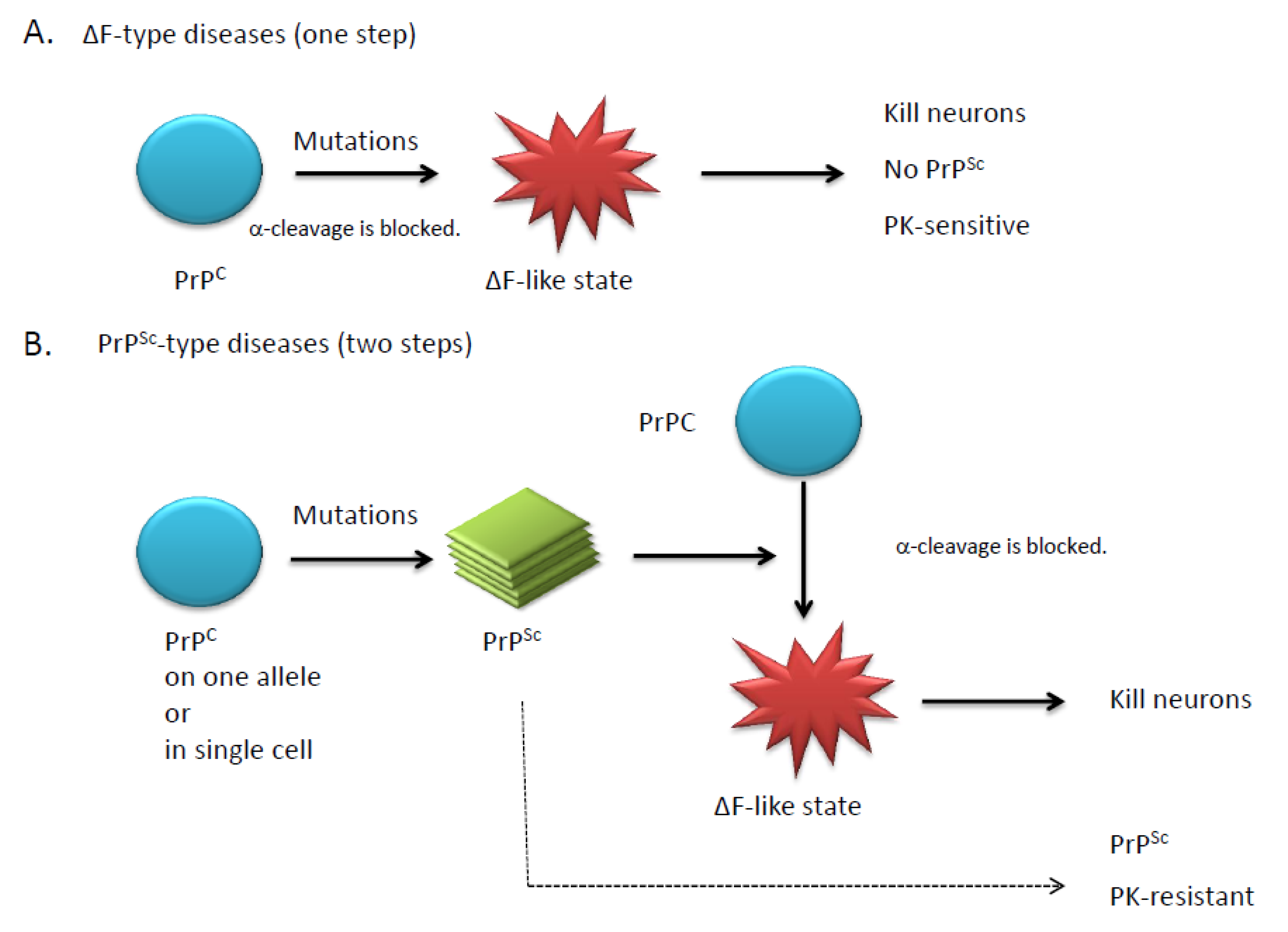
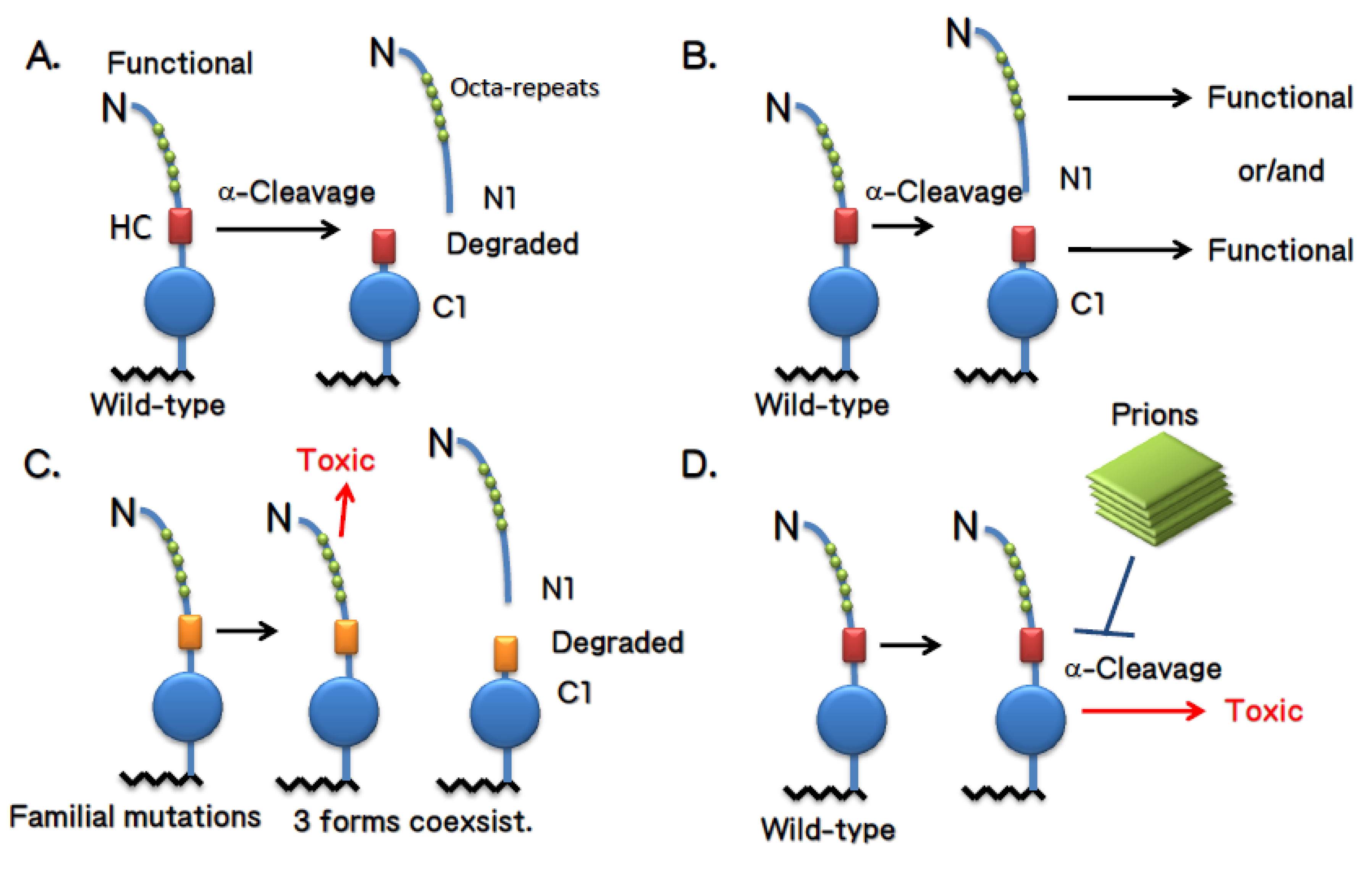
4. Doppel and Shadoo
4.1. Doppel
4.2. Shadoo
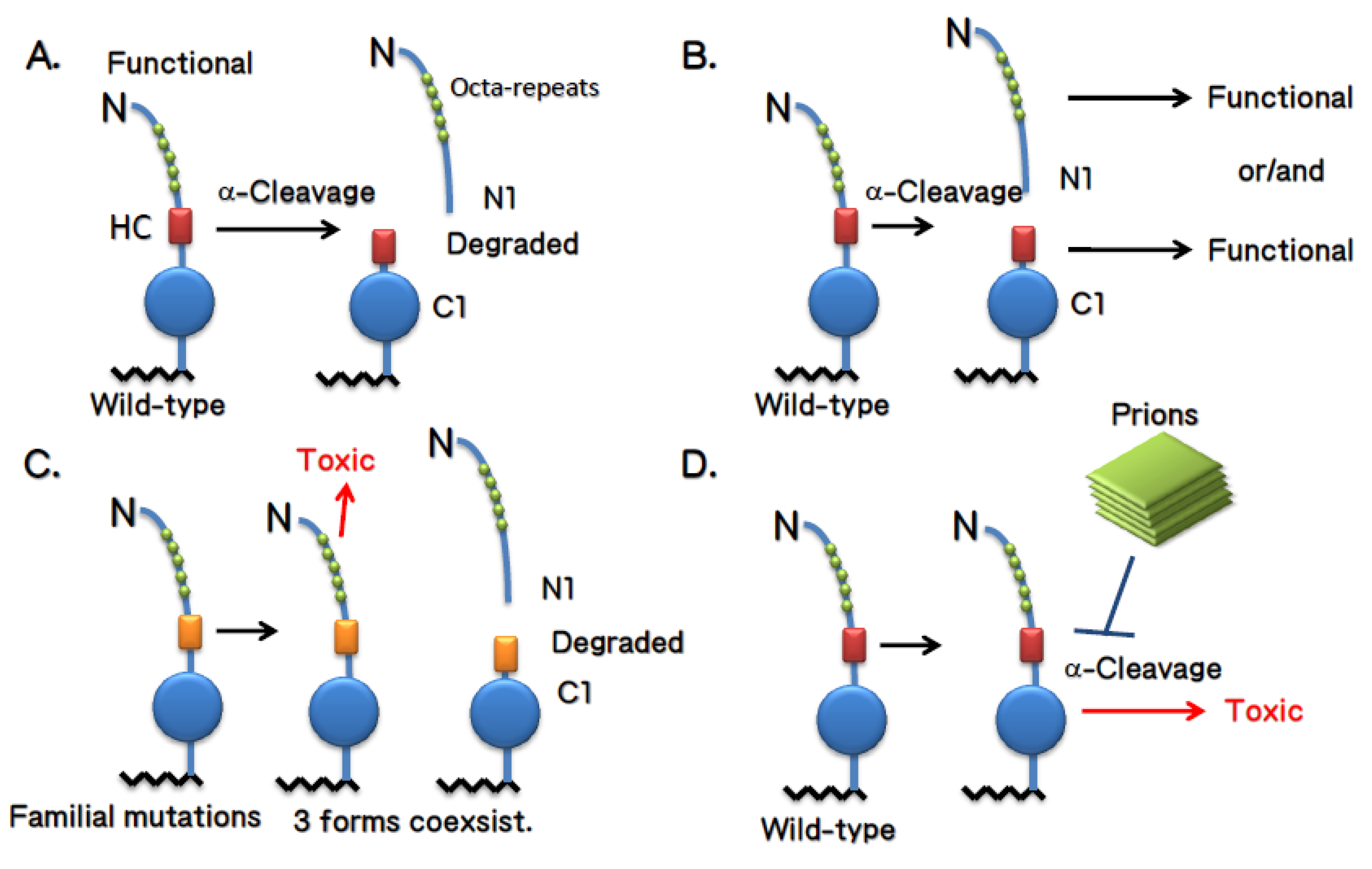
5. The cellular compartment in which α-cleavage occurs
6. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Oesch, B.; Westaway, D.; Walchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27-30 protein. cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar]
- Collinge, J.; Whittington, M.A.; Sidle, K.C.; Smith, C.J.; Palmer, M.S.; Clarke, A.R.; Jefferys, J.G. Prion protein is necessary for normal synaptic function. Nature 1994, 370, 295–297. [Google Scholar] [CrossRef]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef]
- Ovadia, H.; Rosenmann, H.; Shezen, E.; Halimi, M.; Ofran, I.; Gabizon, R. Effect of scrapie infection on the activity of neuronal nitric-oxide synthase in brain and neuroblastoma cells. J.Biol.Chem. 1996, 271, 16856–16861. [Google Scholar]
- Keshet, G.I.; Ovadia, H.; Taraboulos, A.; Gabizon, R. Scrapie-infected mice and PrP knockout mice share abnormal localization and activity of neuronal nitric oxide synthase. J.Neurochem. 1999, 72, 1224–1231. [Google Scholar]
- Brown, P.B. lymphocytes and neuroinvasion. Nature 1997, 390, 662–663. [Google Scholar] [CrossRef]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Ratte, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.; Collinge, J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002, 21, 202–210. [Google Scholar]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; Clarke, A.R.; Collinge, J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011, 470, 540–542. [Google Scholar]
- Basler, K.; Oesch, B.; Scott, M.; Westaway, D.; Walchli, M.; Groth, D.F.; McKinley, M.P.; Prusiner, S.B.; Weissmann, C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986, 46, 417–428. [Google Scholar] [CrossRef]
- Biasini, E.; Turnbaugh, J.A.; Unterberger, U.; Harris, D.A. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012, 35, 92–103. [Google Scholar] [CrossRef]
- Nieznanski, K. Interactions of prion protein with intracellular proteins: so many partners and no consequences? Cell Mol. Neurobiol. 2010, 30, 653–666. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol Rev. 2008, 88, 673–728. [Google Scholar]
- van, R.T.; Smolenaars, M.M.; Madsen, O.; de Jong, W.W. Molecular evolution of the mammalian prion protein. Mol.Biol.Evol. 2003, 20, 111–121. [Google Scholar] [CrossRef]
- Pan, K.M.; Stahl, N.; Prusiner, S.B. Purification and properties of the cellular prion protein from Syrian hamster brain. Protein Sci. 1992, 1, 1343–1352. [Google Scholar] [CrossRef]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J.Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef]
- Chen, S.G.; Teplow, D.B.; Parchi, P.; Teller, J.K.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J.Biol.Chem. 1995, 270, 19173–19180. [Google Scholar]
- Harris, D.A.; Huber, M.T.; van, D.P.; Shyng, S.L.; Chait, B.T.; Wang, R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 1993, 32, 1009–1016. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kakeya, T.; Nakajima, O.; Sakai, A.; Ikeda, K.; Yamaguchi, N.; Yamazaki, T.; Tanamoto, K.; Matsuda, H.; Sawada, J.; et al. Hypoxia induces expression of a GPI-anchorless splice variant of the prion protein. FEBS J. 2008, 275, 2965–2976. [Google Scholar]
- Syed, M.; Nourizadeh-Lillabadi, R.; Press, C.M.; Alestrom, P. Prion protein function and the disturbance of early embryonic development in zebrafish. Prion. 2011, 5, 88–92. [Google Scholar]
- Campana, V.; Caputo, A.; Sarnataro, D.; Paladino, S.; Tivodar, S.; Zurzolo, C. Characterization of the properties and trafficking of an anchorless form of the prion protein. J.Biol.Chem. 2007, 282, 22747–22756. [Google Scholar] [CrossRef]
- Shmerling, D.; Hegyi, I.; Fischer, M.; Blattler, T.; Brandner, S.; Gotz, J.; Rulicke, T.; Flechsig, E.; Cozzio, A.; von, M.C.; et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. cell 1998, 93, 203–214. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Scott, M.; Foster, D.; Groth, D.F.; DeArmond, S.J.; Prusiner, S.B. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990, 250, 1587–1590. [Google Scholar]
- Hsiao, K.K.; Groth, D.; Scott, M.; Yang, S.L.; Serban, H.; Rapp, D.; Foster, D.; Torchia, M.; Dearmond, S.J.; Prusiner, S.B. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc.Natl.Acad.Sci.U.S.A 1994, 91, 9126–9130. [Google Scholar]
- Telling, G.C.; Haga, T.; Torchia, M.; Tremblay, P.; DeArmond, S.J.; Prusiner, S.B. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev. 1996, 10, 1736–1750. [Google Scholar] [CrossRef]
- Oliveira-Martins, J.B.; Yusa, S.; Calella, A.M.; Bridel, C.; Baumann, F.; Dametto, P.; Aguzzi, A. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS.One. 2010, 5, e9107. [Google Scholar]
- Borchelt, D.R.; Davis, J.; Fischer, M.; Lee, M.K.; Slunt, H.H.; Ratovitsky, T.; Regard, J.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet.Anal. 1996, 13, 159–163. [Google Scholar] [CrossRef]
- Fischer, M.; Rulicke, T.; Raeber, A.; Sailer, A.; Moser, M.; Oesch, B.; Brandner, S.; Aguzzi, A.; Weissmann, C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996, 15, 1255–1264. [Google Scholar]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H.; Heikenwalder, M.; Rulicke, T.; Burkle, A.; Aguzzi, A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007, 26, 538–547. [Google Scholar] [CrossRef]
- Muramoto, T.; Scott, M.; Cohen, F.E.; Prusiner, S.B. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc.Natl.Acad.Sci.U.S.A 1996, 93, 15457–15462. [Google Scholar]
- Holscher, C.; Delius, H.; Burkle, A. Overexpression of nonconvertible PrPc delta114-121 in scrapie-infected mouse neuroblastoma cells leads to trans-dominant inhibition of wild-type PrP(Sc) accumulation. J.Virol. 1998, 72, 1153–1159. [Google Scholar]
- Li, A.; Christensen, H.M.; Stewart, L.R.; Roth, K.A.; Chiesa, R.; Harris, D.A. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J. 2007, 26, 548–558. [Google Scholar] [CrossRef]
- Chiesa, R.; Piccardo, P.; Quaglio, E.; Drisaldi, B.; Si-Hoe, S.L.; Takao, M.; Ghetti, B.; Harris, D.A. Molecular distinction between pathogenic and infectious properties of the prion protein. J.Virol. 2003, 77, 7611–7622. [Google Scholar]
- Chiesa, R.; Piccardo, P.; Ghetti, B.; Harris, D.A. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998, 21, 1339–1351. [Google Scholar]
- Jackson, W.S.; Borkowski, A.W.; Faas, H.; Steele, A.D.; King, O.D.; Watson, N.; Jasanoff, A.; Lindquist, S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 2009, 63, 438–450. [Google Scholar] [CrossRef]
- Dossena, S.; Imeri, L.; Mangieri, M.; Garofoli, A.; Ferrari, L.; Senatore, A.; Restelli, E.; Balducci, C.; Fiordaliso, F.; Salio, M.; et al. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 2008, 60, 598–609. [Google Scholar] [CrossRef]
- Yang, W.; Cook, J.; Rassbach, B.; Lemus, A.; DeArmond, S.J.; Mastrianni, J.A. A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. J.Neurosci. 2009, 29, 10072–10080. [Google Scholar]
- Bian, J.; Nazor, K.E.; Angers, R.; Jernigan, M.; Seward, T.; Centers, A.; Green, M.; Telling, G.C. GFP-tagged PrP supports compromised prion replication in transgenic mice. Biochem. Biophys. Res. Commun. 2006, 340, 894–900. [Google Scholar]
- Yadavalli, R.; Guttmann, R.P.; Seward, T.; Centers, A.P.; Williamson, R.A.; Telling, G.C. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J.Biol.Chem. 2004, 279, 21948–21956. [Google Scholar]
- Nazor, K.E.; Kuhn, F.; Seward, T.; Green, M.; Zwald, D.; Purro, M.; Schmid, J.; Biffiger, K.; Power, A.M.; Oesch, B.; et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J. 2005, 24, 2472–2480. [Google Scholar]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef]
- Hegde, R.S.; Tremblay, P.; Groth, D.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature 1999, 402, 822–826. [Google Scholar]
- Westergard, L.; Turnbaugh, J.A.; Harris, D.A. A nine amino acid domain is essential for mutant prion protein toxicity. J.Neurosci. 2011, 31, 14005–14017. [Google Scholar] [CrossRef]
- Li, A.; Piccardo, P.; Barmada, S.J.; Ghetti, B.; Harris, D.A. Prion protein with an octapeptide insertion has impaired neuroprotective activity in transgenic mice. EMBO J. 2007, 26, 2777–2785. [Google Scholar] [CrossRef]
- Muramoto, T.; DeArmond, S.J.; Scott, M.; Telling, G.C.; Cohen, F.E.; Prusiner, S.B. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an alpha-helix. Nat.Med. 1997, 3, 750–755. [Google Scholar] [CrossRef]
- Atarashi, R.; Nishida, N.; Shigematsu, K.; Goto, S.; Kondo, T.; Sakaguchi, S.; Katamine, S. Deletion of N-terminal residues 23-88 from prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced Neurodegeneration. J. Biol.Chem. 2003, 278, 28944–28949. [Google Scholar]
- Race, B.; Meade-White, K.; Race, R.; Baumann, F.; Aguzzi, A.; Chesebro, B. Prion protein on astrocytes or in extracellular fluid impedes neurodegeneration induced by truncated prion protein. Exp. Neurol. 2009, 217, 347–352. [Google Scholar] [CrossRef]
- Stohr, J.; Watts, J.C.; Legname, G.; Oehler, A.; Lemus, A.; Nguyen, HO.; Sussman, J.; Wille, H.; DeArmond, S.J.; Prusiner, S.B.; et al. Sontaneous generation of anchorless prions in transgenic mice. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 21223–21228. [Google Scholar]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol.Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Katamine, S.; Nishida, N.; Moriuchi, R.; Shigematsu, K.; Sugimoto, T.; Nakatani, A.; Kataoka, Y.; Houtani, T.; Shirabe, S.; et al. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996, 380, 528–531. [Google Scholar] [CrossRef]
- Kuwahara, C.; Takeuchi, A.M.; Nishimura, T.; Haraguchi, K.; Kubosaki, A.; Matsumoto, Y.; Saeki, K.; Matsumoto, Y.; Yokoyama, T.; Itohara, S.; et al. Prions prevent neuronal cell-line death. Nature 1999, 400, 225–226. [Google Scholar]
- Rossi, D.; Cozzio, A.; Flechsig, E.; Klein, M.A.; Rulicke, T.; Aguzzi, A.; Weissmann, C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001, 20, 694–702. [Google Scholar]
- Behrens, A.; Genoud, N.; Naumann, H.; Rulicke, T.; Janett, F.; Heppner, F.L.; Ledermann, B.; Aguzzi, A. Absence of the prion protein homologue Doppel causes male sterility. EMBO J. 2002, 21, 3652–3658. [Google Scholar] [CrossRef]
- Paisley, D.; Banks, S.; Selfridge, J.; McLennan, N.F.; Ritchie, A.M.; McEwan, C.; Irvine, D.S.; Saunders, P.T.; Manson, J.C.; Melton, D.W. Male infertility and DNA damage in Doppel knockout and prion protein/Doppel double-knockout mice. Am.J.Pathol. 2004, 164, 2279–2288. [Google Scholar]
- Mo, H.; Moore, R.C.; Cohen, F.E.; Westaway, D.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Two different neurodegenerative diseases caused by proteins with similar structures. Proc.Natl.Acad.Sci.U.S.A 2001, 98, 2352–2357. [Google Scholar]
- Silverman, G.L.; Qin, K.; Moore, R.C.; Yang, Y.; Mastrangelo, P.; Tremblay, P.; Prusiner, S.B.; Cohen, F.E.; Westaway, D. Doppel is an N-glycosylated, glycosylphosphatidylinositol-anchored protein. Expression in testis and ectopic production in the brains of Prnp(0/0) mice predisposed to Purkinje cell loss. J.Biol.Chem. 2000, 275, 26834–26841. [Google Scholar]
- Peoc'h, K.; Serres, C.; Frobert, Y.; Martin, C.; Lehmann, S.; Chasseigneaux, S.; Sazdovitch, V.; Grassi, J.; Jouannet, P.; Launay, J.M.; et al. The human "prion-like" protein Doppel is expressed in both Sertoli cells and spermatozoa. J.Biol.Chem. 2002, 277, 43071–43078. [Google Scholar]
- Rognoni, P.; Chiarelli, L.R.; Comincini, S.; Azzalin, A.; Miracco, C.; Valentini, G. Biochemical signatures of doppel protein in human astrocytomas to support prediction in tumor malignancy. J.Biomed.Biotechnol. 2010, 2010, 301067. [Google Scholar]
- Baumann, F.; Pahnke, J.; Radovanovic, I.; Rulicke, T.; Bremer, J.; Tolnay, M.; Aguzzi, A. Functionally relevant domains of the prion protein identified in vivo. PLoS.One. 2009, 4, e6707. [Google Scholar]
- Premzl, M.; Sangiorgio, L.; Strumbo, B.; Marshall Graves, J.A.; Simonic, T.; Gready, J.E. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003, 314, 89–102. [Google Scholar] [CrossRef]
- Uboldi, C.; Paulis, M.; Guidi, E.; Bertoni, A.; Meo, G.P.; Perucatti, A.; Iannuzzi, L.; Raimondi, E.; Brunner, R.M.; Eggen, A.; et al. Cloning of the bovine prion-like Shadoo (SPRN) gene by comparative analysis of the predicted genomic locus. Mamm.Genome 2006, 17, 1130–1139. [Google Scholar] [CrossRef]
- Watts, J.C.; Drisaldi, B.; Ng, V.; Yang, J.; Strome, B.; Horne, P.; Sy, M.S.; Yoong, L.; Young, R.; Mastrangelo, P.; et al. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J. 2007, 26, 4038–4050. [Google Scholar] [CrossRef]
- Young, R.; Passet, B.; Vilotte, M.; Cribiu, E.P.; Beringue, V.; Le, P.F.; Laude, H.; Vilotte, J.L. The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS Lett. 2009, 583, 3296–3300. [Google Scholar] [CrossRef]
- Daude, N.; Wohlgemuth, S.; Brown, R.; Pitstick, R.; Gapeshina, H.; Yang, J.; Carlson, G.A.; Westaway, D. Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrPC-deficiency. Proc.Natl.Acad.Sci.U.S.A 2012, 109, 9035–9040. [Google Scholar]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar]
- Borchelt, D.R.; Scott, M.; Taraboulos, A.; Stahl, N.; Prusiner, S.B. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J.Cell Biol. 1990, 110, 743–752. [Google Scholar]
- Taraboulos, A.; Serban, D.; Prusiner, S.B. Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J.Cell Biol. 1990, 110, 2117–2132. [Google Scholar]
- Caughey, B.; Raymond, G.J. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J.Biol.Chem. 1991, 266, 18217–18223. [Google Scholar]
- Klohn, P.C.; Stoltze, L.; Flechsig, E.; Enari, M.; Weissmann, C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc.Natl.Acad.Sci.U.S.A 2003, 100, 11666–11671. [Google Scholar]
- Prado, M.A.; Alves-Silva, J.; Magalhaes, A.C.; Prado, V.F.; Linden, R.; Martins, V.R.; Brentani, R.R. PrPc on the road: trafficking of the cellular prion protein. J.Neurochem. 2004, 88, 769–781. [Google Scholar] [CrossRef]
- Shyng, S.L.; Huber, M.T.; Harris, D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J.Biol.Chem. 1993, 268, 15922–15928. [Google Scholar]
- Hachiya, N.S.; Watanabe, K.; Sakasegawa, Y.; Kaneko, K. Microtubules-associated intracellular localization of the NH2-terminal cellular prion protein fragment. Biochem.Biophys.Res.Commun. 2004, 313, 818–823. [Google Scholar] [CrossRef]
- Sunyach, C.; Jen, A.; Deng, J.; Fitzgerald, K.T.; Frobert, Y.; Grassi, J.; McCaffrey, M.W.; Morris, R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003, 22, 3591–3601. [Google Scholar] [CrossRef]
- Goold, R.; Rabbanian, S.; Sutton, L.; Andre, R.; Arora, P.; Moonga, J.; Clarke, A.R.; Schiavo, G.; Jat, P.; Collinge, J.; et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat.Commun. 2011, 2, 281. [Google Scholar] [CrossRef]
- Maas, E.; Geissen, M.; Groschup, M.H.; Rost, R.; Onodera, T.; Schatzl, H.; Vorberg, I.M. Scrapie infection of prion protein-deficient cell line upon ectopic expression of mutant prion proteins. J.Biol.Chem. 2007, 282, 18702–18710. [Google Scholar]
- Arellano-Anaya, Z.E.; Savistchenko, J.; Mathey, J.; Huor, A.; Lacroux, C.; Andreoletti, O.; Vilette, D. A simple, versatile and sensitive cell-based assay for prions from various species. PLoS.One. 2011, 6, e20563. [Google Scholar]
- Jimenez-Huete, A.; Lievens, P.M.; Vidal, R.; Piccardo, P.; Ghetti, B.; Tagliavini, F.; Frangione, B.; Prelli, F. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am.J.Pathol. 1998, 153, 1561–1572. [Google Scholar] [CrossRef]
- Vincent, B.; Paitel, E.; Saftig, P.; Frobert, Y.; Hartmann, D.; De, S.B.; Grassi, J.; Lopez-Perez, E.; Checler, F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J.Biol.Chem. 2001, 276, 37743–37746. [Google Scholar]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Kluth, M.A.; Bernreuther, C.; Thurm, D.; Jorissen, E.; Petrowitz, B.; Bartsch, U.; De, S.B.; et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol.Neurodegener. 2011, 6, 36. [Google Scholar] [CrossRef]
- Taylor, D.R.; Parkin, E.T.; Cocklin, S.L.; Ault, J.R.; Ashcroft, A.E.; Turner, A.J.; Hooper, N.M. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J.Biol.Chem. 2009, 284, 22590–22600. [Google Scholar]
- Liang, J.; Wang, W.; Sorensen, D.; Medina, S.; Ilchenko, S.; Kiselar, J.; Surewicz, W.K.; Booth, S.A.; Kong, Q. Cellular prion protein regulates its own alpha-cleavage through ADAM8 in skeletal muscle. J.Biol.Chem. 2012, 287, 16510–16520. [Google Scholar]
- Kelly, K.; Hutchinson, G.; Nebenius-Oosthuizen, D.; Smith, A.J.; Bartsch, J.W.; Horiuchi, K.; Rittger, A.; Manova, K.; Docherty, A.J.; Blobel, C.P. Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev.Dyn. 2005, 232, 221–231. [Google Scholar] [CrossRef]
- Hachiya, N.; Komata, Y.; Harguem, S.; Nishijima, K.; Kaneko, K. Possible involvement of calpain-like activity in normal processing of cellular prion protein. Neurosci.Lett. 2011, 490, 150–155. [Google Scholar] [CrossRef]
- Safar, J.G.; Geschwind, M.D.; Deering, C.; Didorenko, S.; Sattavat, M.; Sanchez, H.; Serban, A.; Vey, M.; Baron, H.; Giles, K.; et al. Diagnosis of human prion disease. Proc.Natl.Acad.Sci.U.S.A 2005, 102, 3501–3506. [Google Scholar]
- Sajnani, G.; Silva, C.J.; Ramos, A.; Pastrana, M.A.; Onisko, B.C.; Erickson, M.L.; Antaki, E.M.; Dynin, I.; Vazquez-Fernandez, E.; Sigurdson, C.J.; et al. PK-sensitive PrP is infectious and shares basic structural features with PK-resistant PrP. PLoS.Pathog. 2012, 8, e1002547. [Google Scholar]
- Westergard, L.; Turnbaugh, J.A.; Harris, D.A. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J.Biol.Chem. 2011, 286, 44234–44242. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yusa, S.-i.; Oliveira-Martins, J.B.; Sugita-Konishi, Y.; Kikuchi, Y. Cellular Prion Protein: From Physiology to Pathology. Viruses 2012, 4, 3109-3131. https://doi.org/10.3390/v4113109
Yusa S-i, Oliveira-Martins JB, Sugita-Konishi Y, Kikuchi Y. Cellular Prion Protein: From Physiology to Pathology. Viruses. 2012; 4(11):3109-3131. https://doi.org/10.3390/v4113109
Chicago/Turabian StyleYusa, Sei-ichi, José B. Oliveira-Martins, Yoshiko Sugita-Konishi, and Yutaka Kikuchi. 2012. "Cellular Prion Protein: From Physiology to Pathology" Viruses 4, no. 11: 3109-3131. https://doi.org/10.3390/v4113109
APA StyleYusa, S.-i., Oliveira-Martins, J. B., Sugita-Konishi, Y., & Kikuchi, Y. (2012). Cellular Prion Protein: From Physiology to Pathology. Viruses, 4(11), 3109-3131. https://doi.org/10.3390/v4113109