Coronavirus Genomics and Bioinformatics Analysis



Abstract
:1. Introduction
2. Genomics
2.1. ORF1ab

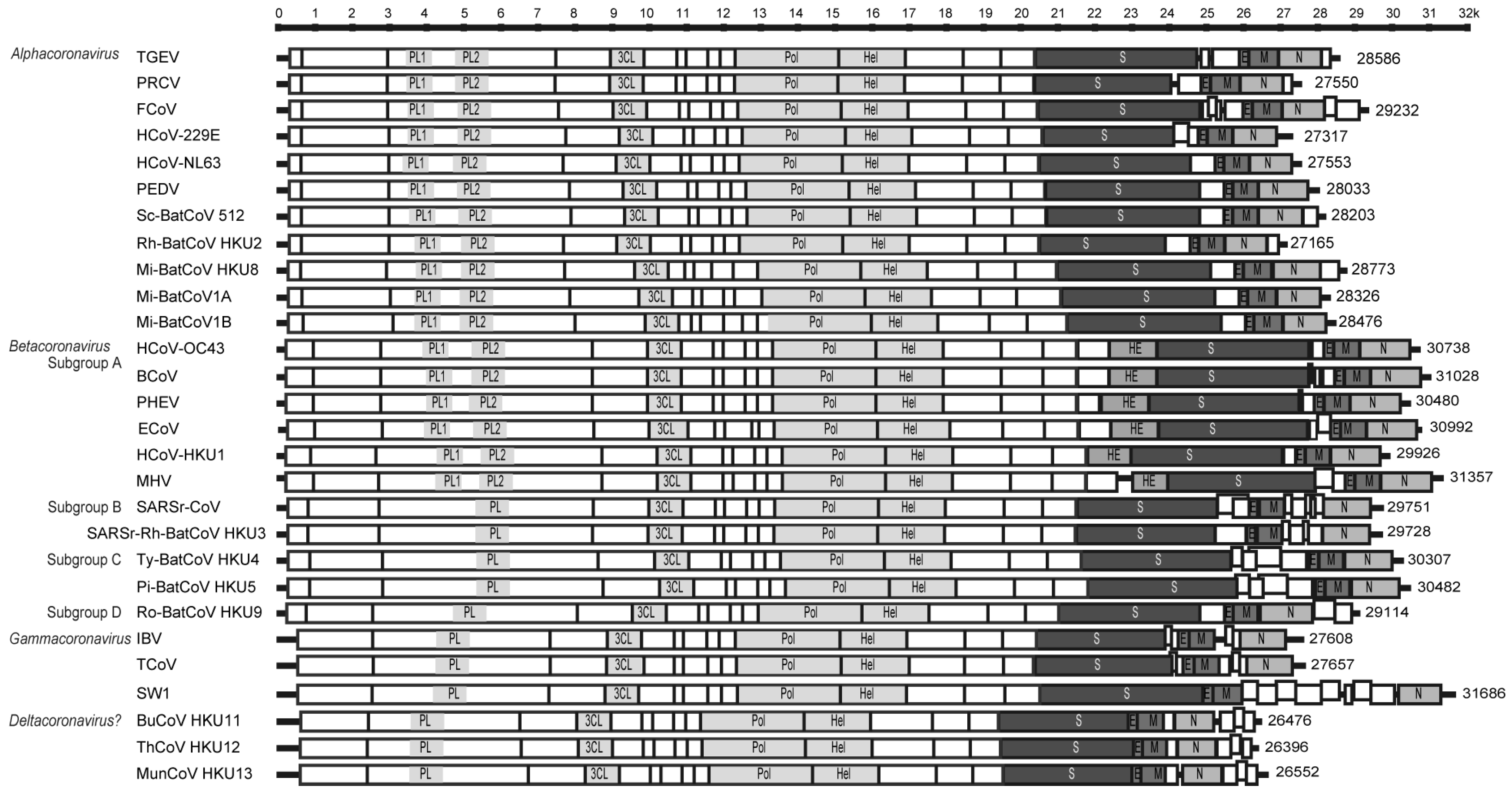{kind=link}
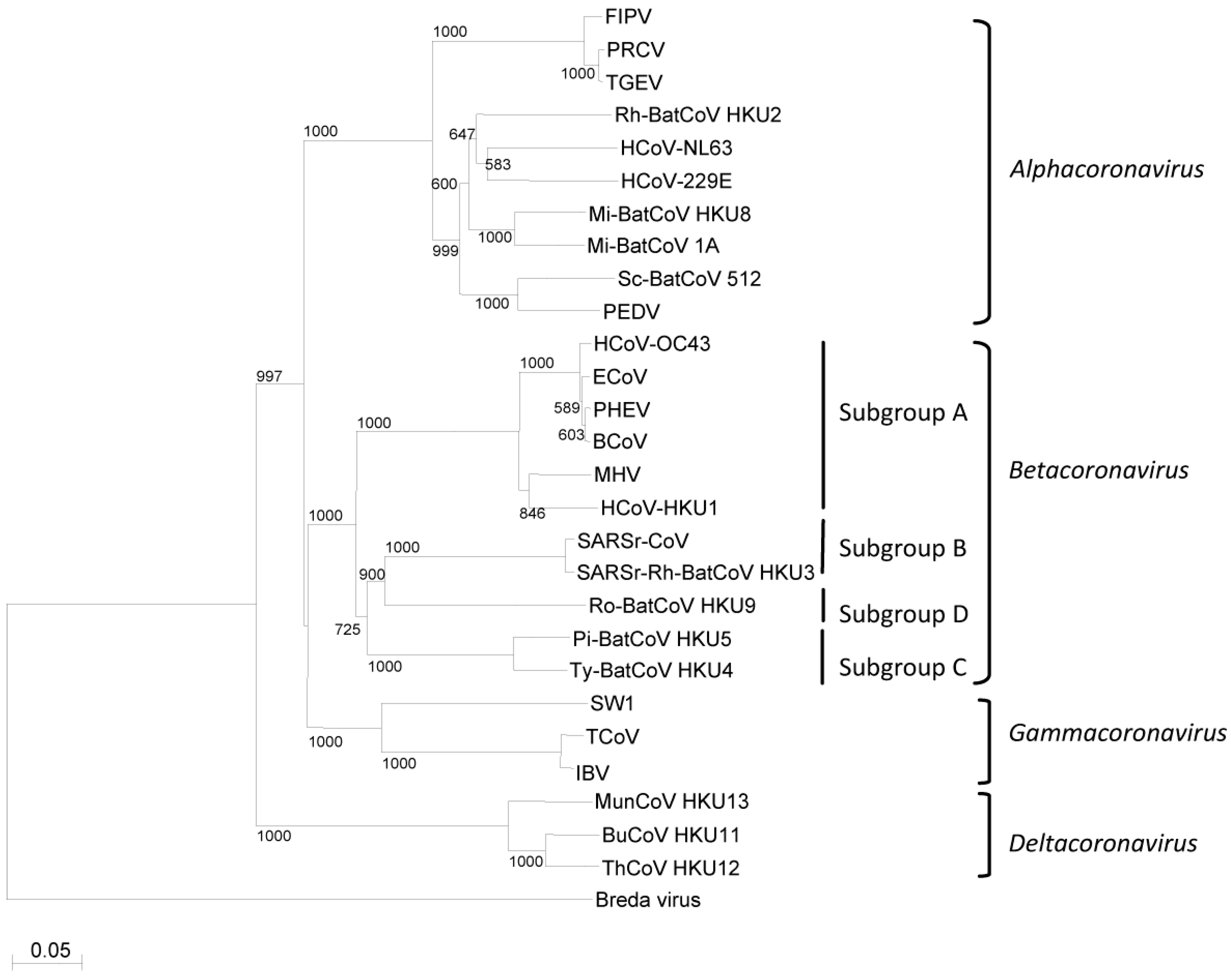{kind=link}
| Viruses | Hosts | G+C contents | Transcription regulatory sequences | No. of nsp in ORF1ab | No. of papain-like proteases in ORF1ab | No. of small ORFs between ORF1ab and N | Presence of conserved S cleavage site | No. of small ORFs downstream to N | |
| Alphacoronavirus | |||||||||
| Transmissible gastroenteritis virus | Pigs | 0.38 | CUAAAC | 16 | 2 | 2 | N | 1 | |
| Porcine respiratory coronavirus | Pigs | 0.37 | CUAAAC | 16 | 2 | 1 | N | 1 | |
| Feline coronavirus | Cats | 0.39 | CUAAAC | 16 | 2 | 4 | N | 2 | |
| Human coronavirus 229E | Humans | 0.38 | CUAAAC | 16 | 2 | 2 | N | - | |
| Human coronavirus NL63 | Humans | 0.34 | CUAAAC | 16 | 2 | 1 | N | - | |
| Porcine epidemic diarrhea virus | Pigs | 0.42 | CUAAAC | 16 | 2 | 1 | N | - | |
| Scotophilus bat coronavirus 512 | Lesser Asiatic yellow house bats | 0.40 | CUAAAC | 16 | 2 | 1 | N | 1 | |
| Rhinolophus bat coronavirus HKU2 | Chinese horseshoe bats | 0.39 | CUAAAC | 16 | 2 | 1 | N | 1 | |
| Miniopterus bat coronavirus HKU8 | Bent-winged bats | 0.42 | CUAAAC | 16 | 2 | 1 | N | 1 | |
| Miniopterus bat coronavirus 1A | Bent-winged bats | 0.38 | CUAAAC | 16 | 2 | 1 | N | - | |
| Miniopterus bat coronavirus 1B | Bent-winged bats | 0.39 | CUAAAC | 16 | 2 | 1 | N | - | |
| Betacoronavirus Subgroup A | |||||||||
| Human coronavirus OC43 | Humans | 0.37 | CUAAAC | 16 | 2 | 1 | Y | - | |
| Bovine coronavirus | Cows | 0.37 | CUAAAC | 16 | 2 | 3 | Y | - | |
| Porcine hemagglutinating encephalomyelitis virus | Pigs | 0.37 | CUAAAC | 16 | 2 | 2 | Y | - | |
| Equine coronavirus | Horses | 0.37 | CUAAAC | 16 | 2 | 2 | Y | - | |
| Human coronavirus HKU1 | Humans | 0.32 | CUAAAC | 16 | 2 | 1 | Y | - | |
| Mouse hepatitis virus | Mice | 0.42 | CUAAAC | 16 | 2 | 2 | Y | - | |
| Subgroup B | |||||||||
| Human SARS related coronavirus | Humans | 0.41 | ACGAAC | 16 | 1 | 7 | N | - | |
| SARS-related Rhinolophus bat coronavirus HKU3 | Chinese horseshoe bats | 0.41 | ACGAAC | 16 | 1 | 5 | N | - | |
| Subgroup C | |||||||||
| Tylonycteris bat coronavirus HKU4 | Lesser bamboo bats | 0.38 | ACGAAC | 16 | 1 | 4 | N | - | |
| Pipistrellus bat coronavirus HKU5 | Japanese pipistrelle bats | 0.43 | ACGAAC | 16 | 1 | 4 | N | - | |
| Subgroup D | |||||||||
| Rousettus bat coronavirus HKU9 | Leschenault's rousette bats | 0.41 | ACGAAC | 16 | 1 | 1 | N | 2 | |
| Gammacoronavirus | |||||||||
| Infectious bronchitis virus | Chickens | 0.38 | CUUAACAA | 15 | 1 | 4 | Y | - | |
| Turkey coronavirus | Turkeys | 0.38 | CUUAACAA | 15 | 1 | 5 | Y | - | |
| Beluga whale coronavirus | Beluga whales | 0.39 | AAACA | 15 | 1 | 8 | N | - | |
| Deltacoronavirus | |||||||||
| Bulbul coronavirus HKU11 | Chinese bulbuls | 0.39 | ACACCA | 15 | 1 | 1 | N | 3 | |
| Thrush coronavirus HKU12 | Gray-backed thrushes | 0.38 | ACACCA | 15 | 1 | 1 | N | 3 | |
| Munia coronavirus HKU13 | White-rumped munias | 0.43 | ACACCA | 15 | 1 | 1 | N | 3 | |

2.2. Haemagglutinin esterase
2.3. Spike
2.4. Envelope and membrane
2.5. Nucleocapsid
2.6. Other small ORFS
3. Phylogeny
4. Evolutionary rate and divergence
5. Recombination analysis
6. Codon usage bias
7. Database
8. Concluding remarks
Acknowledgments
References and Notes
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lai, K.K.; Huang, Y.; Lee, P.; Luk, G.S.; Dyrting, K.C.; Chan, K.H.; Yuen, K.Y. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 2009, 83, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Wang, M.; Lau, S.K.; Xu, H.; Poon, R.W.; Guo, R.; Wong, B.H.; Gao, K.; Tsoi, H.W.; Huang, Y.; Li, K.S.; Lam, C.S.; Chan, K.H.; Zheng, B.J.; Yuen, K.Y. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- ICTV Virus Taxonomy: 2009 Release. Available online: http://ictvonline.org/virusTaxonomy.asp?version=2009 (accessed on 1 August 2010).
- Liu, S.; Chen, J.; Chen, J.; Kong, X.; Shao, Y.; Han, Z.; Feng, L.; Cai, X.; Gu, S.; Liu, M. Isolation of avian infectious bronchitis coronavirus from domestic peafowl (Pavo cristatus) and teal (Anas). J. Gen. Virol. 2005, 86, 719–725. [Google Scholar] [CrossRef]
- Tang, X.C.; Zhang, J.X.; Zhang, S.Y.; Wang, P.; Fan, X.H.; Li, L.F.; Li, G.; Dong, B.Q.; Liu, W.; Cheung, C.L.; Xu, K.M.; Song, W.J.; Vijaykrishna, D.; Poon, L.L.; Peiris, J.S.; Smith, G.J.; Chen, H.; Guan, Y. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 2006, 80, 7481–7490. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Li, K.S.; Poon, R.W.; Wong, B.H.; Tsoi, H.W.; Yip, B.C.; Huang, Y.; Chan, K.H.; Yuen, K.Y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Hartwig, N.G.; Bestebroer, T.M.; Niemeyer, B.; de Jong, J.C.; Simon, J.H.; Osterhaus, A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 6212–6216. [Google Scholar] [CrossRef]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; Poon, L.L.; Wong, S.S.; Guan, Y.; Peiris, J.S.; Yuen, K.Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14040–14045. [Google Scholar] [CrossRef]
- Chu, D.K.; Peiris, J.S.; Chen, H.; Guan, Y.; Poon, L.L. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 2008, 89, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Mihindukulasuriya, K.A.; Wu, G.; St Leger, J.; Nordhausen, R.W.; Wang, D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 2008, 82, 5084–5088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guy, J.S.; Snijder, E.J.; Denniston, D.A.; Timoney, P.J.; Balasuriya, U.B. Genomic characterization of equine coronavirus. Virology 2007, 369, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Wang, M.; Lam, C.S.; Xu, H.; Guo, R.; Chan, K.H.; Zheng, B.J.; Yuen, K.Y. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 2007, 367, 428–439. [Google Scholar] [CrossRef]
- Lai, M.M.; Perlman, S.; L., A. Coronaviridae. In Fields virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 1305–1335. [Google Scholar]
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between nonsegmented RNA genomes of murine coronaviruses. J. Virol. 1985, 56, 449–456. [Google Scholar] [CrossRef]
- Woo, P.C.; Huang, Y.; Lau, S.K.; Tsoi, H.W.; Yuen, K.Y. In silico analysis of ORF1ab in coronavirus HKU1 genome reveals a unique putative cleavage site of coronavirus HKU1 3C-like protease. Microbiol. Immunol. 2005, 49, 899–908. [Google Scholar] [CrossRef]
- Nasr, F.; Filipowicz, W. Characterization of the Saccharomyces cerevisiae cyclic nucleotide phosphodiesterase involved in the metabolism of ADP-ribose 1",2"-cyclic phosphate. Nucleic Acids Res. 2000, 28, 1676–1683. [Google Scholar] [CrossRef]
- Luytjes, W.; Bredenbeek, P.J.; Noten, A.F.; Horzinek, M.C.; Spaan, W.J. Sequence of mouse hepatitis virus A59 mRNA 2: indications for RNA recombination between coronaviruses and influenza C virus. Virology 1988, 166, 415–422. [Google Scholar] [CrossRef]
- Haijema, B.J.; Volders, H.; Rottier, P.J. Live, attenuated coronavirus vaccines through the directed deletion of group-specific genes provide protection against feline infectious peritonitis. J. Virol. 2004, 78, 3863–3871. [Google Scholar] [CrossRef]
- Olsen, C.W. A review of feline infectious peritonitis virus: molecular biology, immunopathogenesis, clinical aspects, and vaccination. Vet. Microbiol. 1993, 36, 1–37. [Google Scholar] [CrossRef]
- Tung, F.Y.; Abraham, S.; Sethna, M.; Hung, S.L.; Sethna, P.; Hogue, B.G.; Brian, D.A. The 9-kDa hydrophobic protein encoded at the 3' end of the porcine transmissible gastroenteritis coronavirus genome is membrane-associated. Virology 1992, 186, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; Butt, K.M.; Wong, K.L.; Chan, K.W.; Lim, W.; Shortridge, K.F.; Yuen, K.Y.; Peiris, J.S.; Poon, L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef]
- Eickmann, M.; Becker, S.; Klenk, H.D.; Doerr, H.W.; Stadler, K.; Censini, S.; Guidotti, S.; Masignani, V.; Scarselli, M.; Mora, M.; Donati, C.; Han, J.H.; Song, H.C.; Abrignani, S.; Covacci, A.; Rappuoli, R. Phylogeny of the SARS coronavirus. Science 2003, 302, 1504–1505. [Google Scholar] [CrossRef]
- Sanchez, C.M.; Gebauer, F.; Sune, C.; Mendez, A.; Dopazo, J.; Enjuanes, L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 1992, 190, 92–105. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Maes, P.; Van Reeth, K.; Nauwynck, H.; Pensaert, M.; Van Ranst, M. Evolutionary history of the closely related group 2 coronaviruses: porcine hemagglutinating encephalomyelitis virus, bovine coronavirus, and human coronavirus OC43. J. Virol. 2006, 80, 7270–7274. [Google Scholar] [CrossRef]
- Song, H.D.; Tu, C.C.; Zhang, G.W.; Wang, S.Y.; Zheng, K.; Lei, L.C.; Chen, Q.X.; Gao, Y.W.; Zhou, H.Q.; Xiang, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 2430–2435. [Google Scholar] [CrossRef]
- Hon, C.C.; Lam, T.Y.; Shi, Z.L.; Drummond, A.J.; Yip, C.W.; Zeng, F.; Lam, P.Y.; Leung, F.C. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Smith, G.J.; Zhang, J.X.; Peiris, J.S.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- BEAST Home Page. http://beast.bio.ed.ac.uk/Main_Page (accessed on 1 August 2010).
- Lau, S.K.; Li, K.S.; Huang, Y.; Shek, C.T.; Tse, H.; Wang, M.; Choi, G.K.; Xu, H.; Lam, C.S.; Guo, R.; Chan, K.H.; Zheng, B.J.; Woo, P.C.; Yuen, K.Y. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Chow, K.Y.; Leung, F.C. Estimated timing of the last common ancestor of the SARS coronavirus. N. Engl. J. Med. 2003, 349, 2469–2470. [Google Scholar] [CrossRef]
- Salemi, M.; Fitch, W.M.; Ciccozzi, M.; Ruiz-Alvarez, M.J.; Rezza, G.; Lewis, M.J. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J. Virol. 2004, 78, 1602–1603. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef]
- Lai, M.M. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [CrossRef]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Nidovirus transcription: how to make sense...? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef]
- Herrewegh, A.A.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef]
- Keck, J.G.; Matsushima, G.K.; Makino, S.; Fleming, J.O.; Vannier, D.M.; Stohlman, S.A.; Lai, M.M. In vivo RNA-RNA recombination of coronavirus in mouse brain. J. Virol. 1988, 62, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Kottier, S.A.; Cavanagh, D.; Britton, P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology 1995, 213, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Lavi, E.; Haluskey, J.A.; Masters, P.S. The pathogenesis of MHV nucleocapsid gene chimeric viruses. Adv. Exp. Med. Biol. 1998, 440, 537–541. [Google Scholar]
- Motokawa, K.; Hohdatsu, T.; Aizawa, C.; Koyama, H.; Hashimoto, H. Molecular cloning and sequence determination of the peplomer protein gene of feline infectious peritonitis virus type I. Arch. Virol. 1995, 140, 469–480. [Google Scholar] [CrossRef]
- Wesseling, J.G.; Vennema, H.; Godeke, G.J.; Horzinek, M.C.; Rottier, P.J. Nucleotide sequence and expression of the spike (S) gene of canine coronavirus and comparison with the S proteins of feline and porcine coronaviruses. J. Gen. Virol. 1994, 75 (Pt 7), 1789–1794. [Google Scholar] [CrossRef]
- Herrewegh, A.A.; Vennema, H.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. The molecular genetics of feline coronaviruses: comparative sequence analysis of the ORF7a/7b transcription unit of different biotypes. Virology 1995, 212, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Motokawa, K.; Hohdatsu, T.; Hashimoto, H.; Koyama, H. Comparison of the amino acid sequence and phylogenetic analysis of the peplomer, integral membrane and nucleocapsid proteins of feline, canine and porcine coronaviruses. Microbiol. Immunol. 1996, 40, 425–433. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Tsoi, H.W.; Huang, Y.; Poon, R.W.; Chu, C.M.; Lee, R.A.; Luk, W.K.; Wong, G.K.; Wong, B.H.; Cheng, V.C.; Tang, B.S.; Wu, A.K.; Yung, R.W.; Chen, H.; Guan, Y.; Chan, K.H.; Yuen, K.Y. Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J. Infect. Dis. 2005, 192, 1898–1907. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Yip, C.C.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef]
- Woo, P.C.; Wong, B.H.; Huang, Y.; Lau, S.K.; Yuen, K.Y. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology 2007, 369, 431–442. [Google Scholar] [CrossRef]
- Huang, Y.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. CoVDB: a comprehensive database for comparative analysis of coronavirus genes and genomes. Nucleic Acids Res. 2008, 36, D504–511. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed]

| References | Gene | No. of SARSr-CoV strains | Estimated mean substitution rate (no. of substitutions per site per year) | Methods for estimating TMRCA | TMRCA of human/civet SARSr-CoV (95% HPD) | TMRCA of (human/civet)/ Bat Rp3 SARSr-CoV (95% HPD) | TMRCA of (human/civet/Bat Rp3 SARSr-CoV)/ SARSr-Rh-BatCoV (95% HPD) | |||
| Human | Civet | Bat Rp3 | SARSr-Rh-BatCoV | |||||||
| Zeng et al. 2003 [38] | Spike | 139 | - | Linear regression | Dec 2002 (Sep 2002, Jan 2003) | - | - | |||
| Salemi et al. 2004 [39] | ORF1ab | 10 | 4/35×10−4b | Molecular clock model | - | - | - | |||
| Zhao et al. 2004 [40] | Genome | 16 | 8-23.8×10−4 | Three strategies described by the author | Spring 2002 | - | - | |||
| Song et al. 2005 [32] | CDSsa | 3 | 5 | 2.92×10−3 | Linear regression | Nov 2002 | - | - | ||
| Vijaykrishna et al. 2007 [35] | Helicase | 3 | 3 | 1 | 5 | 2.0×10-2, 1.7×10-2c | Relaxed clock model | 1999 (1990-2003) | 1986 (1964-2002) | 1961 (1918-1995) |
| Hon et al. 2008 [33] | ORF1ab | 13 | 6 | 1 | 4 | 2.79×10-3 | Various clock models | 2002.63 (2002.14-2002.96) | 1998.51 (1993.55-2001.32) | ~1985d |
| Lau et al. 2010 [37] | ORF1ab | 8 | 8 | 1 | 15 | 2.82×10-3 | Relaxed clock model | 2001 (1999.16-2002.14) | 1995.10 (1986.53-2000.13) | 1972.39 (1935.28-1990.63) |
- a Concatenated CDS of ORF1ab, S, E, M and N.
- b The rate for all sites is 4×10-4. The rate for variable sites is 35×10-4.
- c Two numbers present the estimated rate of SARSr-Rh-BatCoV lineage and the estimated rate of human/civet/bat SARSr-CoV lineage respectively.
- d The date obtained from the figure of the reference but was not mentioned in the reference’s text.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804-1820. https://doi.org/10.3390/v2081803
Woo PCY, Huang Y, Lau SKP, Yuen K-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses. 2010; 2(8):1804-1820. https://doi.org/10.3390/v2081803
Chicago/Turabian StyleWoo, Patrick C. Y., Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen. 2010. "Coronavirus Genomics and Bioinformatics Analysis" Viruses 2, no. 8: 1804-1820. https://doi.org/10.3390/v2081803
APA StyleWoo, P. C. Y., Huang, Y., Lau, S. K. P., & Yuen, K.-Y. (2010). Coronavirus Genomics and Bioinformatics Analysis. Viruses, 2(8), 1804-1820. https://doi.org/10.3390/v2081803