Cyclophilin Inhibitors as a Novel HCV Therapy

Abstract
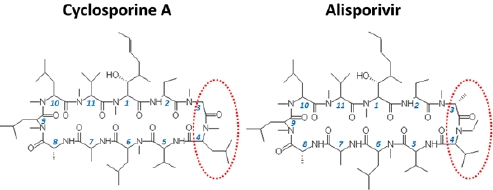
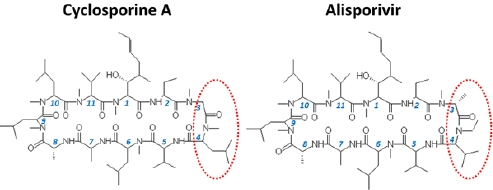
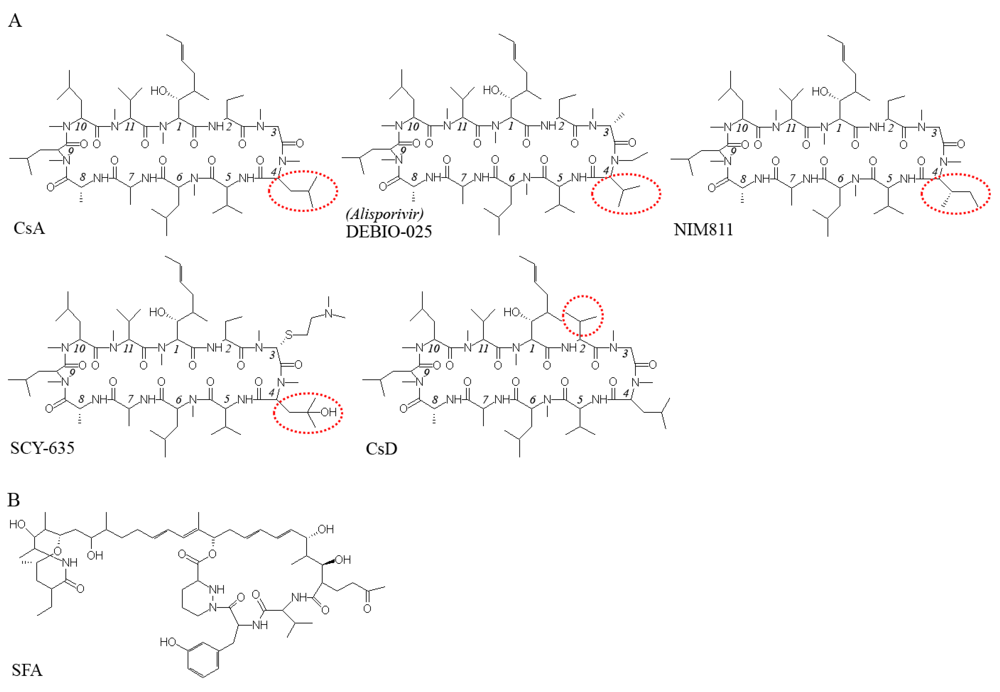
:
{kind=link}
{kind=link}
1. Introduction
2. Cyclosporine A and HCV infection
3. Cyclophilin A as an essential HCV cofactor
4. Inhibition of HCV replication by cyclophilin inhibitors
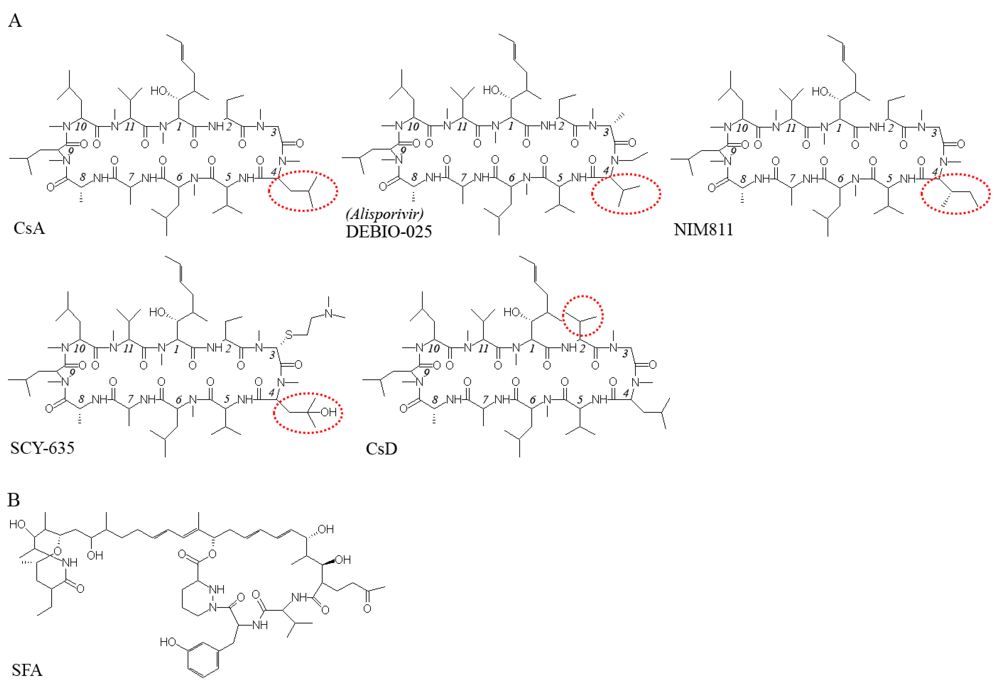
5. Viral resistance to CPIs
6. Potential mechanism of resistance to CPIs
7. Summary
Acknowledgments
References
- Kuritzkes, D.R. HIV-1 entry inhibitors: an overview. Curr. Opin. HIV AIDS 2009, 4, 82–87. [Google Scholar] [CrossRef]
- Liu, J.; Farmer Jr., J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes . Cell 1991, 66, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Teraoka, S.; Mishiro, S.; Ebihara, K.; Sanaka, T.; Yamaguchi, Y.; Nakajima, I.; Kawai, T.; Yagisawa, T.; Honda, H.; Fuchinoue, S.; et al. Effect of cyclosporine on proliferation of non-A, non-B hepatitis virus . Transplant Proc. 1988, 20, 868–876. [Google Scholar] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Hijikata, M.; Hosaka, M.; Yamaji, M.; Shimotohno, K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 2003, 38, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sakamoto, N.; Enomoto, N.; Tanabe, Y.; Kanazawa, N.; Koyama, T.; Kurosaki, M.; Maekawa, S.; Yamashiro, T.; Chen, C.H.; Itsui, Y.; Kakinuma, S.; Watanabe, M. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 2004, 313, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Hilgard, P.; Kahraman, A.; Lehmann, N.; Seltmann, C.; Beckebaum, S.; Ross, R.S.; Baba, H.A.; Malago, M.; Broelsch, C.E.; Gerken, G. Cyclosporine versus tacrolimus in patients with HCV infection after liver transplantation: effects on virus replication and recurrent hepatitis. World J. Gastroenterol. 2006, 12, 697–702. [Google Scholar] [PubMed]
- Martin, P.; Busuttil, R.W.; Goldstein, R.M.; Crippin, J.S.; Klintmalm, G.B.; Fitzsimmons, W.E.; Uleman, C. Impact of tacrolimus versus cyclosporine in hepatitis C virus-infected liver transplant recipients on recurrent hepatitis: a prospective, randomized trial. Liver Transpl. 2004, 10, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Guitard, J.; Sandres-Saune, K.; Kamar, N.; Ribes, D.; Faguer, S.; Esposito, L.; Lavit, M.; Muscari, F.; Peron, J.M.; Lavayssiere, L.; Durand, D.; Rostaing, L. Hepatitis C virus viral load after conversion from tacrolimus to cyclosporine in liver transplant patients: a pilot study. Transplant Proc. 2007, 39, 2603–2605. [Google Scholar] [CrossRef] [PubMed]
- Rayhill, S.C.; Barbeito, R.; Katz, D.; Voigt, M.; Labrecque, D.; Kirby, P.; Miller, R.; Stolpen, A.; Wu, Y.; Schmidt, W. A cyclosporine-based immunosuppressive regimen may be better than tacrolimus for long-term liver allograft survival in recipients transplanted for hepatitis C. Transplant Proc. 2006, 38, 3625–3628. [Google Scholar] [CrossRef] [PubMed]
- Firpi, R.J.; Zhu, H.; Morelli, G.; Abdelmalek, M.F.; Soldevila-Pico, C.; Machicao, V.I.; Cabrera, R.; Reed, A.I.; Liu, C.; Nelson, D.R. Cyclosporine suppresses hepatitis C virus in vitro and increases the chance of a sustained virological response after liver transplantation. Liver Transpl. 2006, 12, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Zervos, X.A.; Weppler, D.; Fragulidis, G.P.; Torres, M.B.; Nery, J.R.; Khan, M.F.; Pinna, A.D.; Kato, T.; Miller, J.; Reddy, K.R.; Tzakis, A.G. Comparison of tacrolimus with neoral as primary immunosuppression in hepatitis C patients after liver transplantation. Transplant Proc. 1998, 30, 1405–1406. [Google Scholar] [CrossRef] [PubMed]
- Firpi, R.J.; Soldevila-Pico, C.; Morelli, G.G.; Cabrera, R.; Levy, C.; Clark, V.C.; Suman, A.; Michaels, A.; Chen, C.; Nelson, D.R. The use of cyclosporine for recurrent hepatitis C after liver transplant: a randomized pilot study. Dig. Dis. Sci. 2010, 55, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Sekiyama, K.; Yamada, M.; Watanabe, T.; Yasuda, H.; Yoshiba, M. Combined interferon alpha2b and cyclosporin A in the treatment of chronic hepatitis C: controlled trial. J. Gastroenterol. 2003, 38, 567–572. [Google Scholar] [PubMed]
- Inoue, K.; Yoshiba, M. Interferon combined with cyclosporine treatment as an effective countermeasure against hepatitis C virus recurrence in liver transplant patients with end-stage hepatitis C virus related disease. Transplant Proc. 2005, 37, 1233–1234. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Umehara, T.; Ruegg, U.T.; Yasui, F.; Watanabe, T.; Yasuda, H.; Dumont, J.M.; Scalfaro, P.; Yoshiba, M.; Kohara, M. Evaluation of a cyclophilin inhibitor in hepatitis C virus-infected chimeric mice in vivo. Hepatology 2007, 45, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Dumont, J.M.; Vuagniaux, G.; Neyts, J. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitors. Antimicrob. Agents Chemother. 2009, 53, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Feinman, S.V.; Jablkowski, M.; Horban, A.; Kryczka, W.; Pawlowska, M.; Heathcote, J.E.; Mazzella, G.; Vandelli, C.; Nicolas-Metral, V.; Grosgurin, P.; Liz, J.S.; Scalfaro, P.; Porchet, H.; Crabbe, R. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 2009, 49, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Boerner, J.E.; TiongYip, C.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef] [PubMed]
- Mathy, J.E.; Ma, S.; Compton, T.; Lin, K. Combinations of cyclophilin inhibitor NIM811 with hepatitis C Virus NS3-4A Protease or NS5B polymerase inhibitors enhance antiviral activity and suppress the emergence of resistance. Antimicrob. Agents Chemother. 2008, 52, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Scorneaux, B.; Huang, Z.; Murray, M.G.; Wring, S.; Smitley, C.; Harris, R.; Erdmann, F.; Fischer, G.; Ribeill, Y. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 2010, 54, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 2005, 19, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sakamoto, N.; Tanabe, Y.; Koyama, T.; Itsui, Y.; Takeda, Y.; Chen, C.H.; Kakinuma, S.; Oooka, S.; Maekawa, S.; Enomoto, N.; Watanabe, M. Suppression of hepatitis C virus replication by cyclosporin a is mediated by blockade of cyclophilins. Gastroenterology 2005, 129, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin a is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics . PLoS Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009, 284, 16998–17005. [Google Scholar] [CrossRef] [PubMed]
- Ciesek, S.; Steinmann, E.; Wedemeyer, H.; Manns, M.P.; Neyts, J.; Tautz, N.; Madan, V.; Bartenschlager, R.; von Hahn, T.; Pietschmann, T. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 2009, 50, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ikeda, M.; Ariumi, Y.; Dansako, H.; Wakita, T.; Kato, N. HCV genotype 1b chimeric replicon with NS5B of JFH-1 exhibited resistance to cyclosporine A. Arch. Virol. 2009, 154, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Braaten, D.; Luban, J. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.M.; Zydowsky, L.D.; Liu, J.; Walsh, C.T. Crystal structure of recombinant human T-cell cyclophilin A at 2.5 A resolution . Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 9483–9487. [Google Scholar] [CrossRef] [PubMed]
- Mikol, V.; Kallen, J.; Walkinshaw, M.D. X-ray structure of a cyclophilin B/cyclosporin complex: comparison with cyclophilin A and delineation of its calcineurin-binding domain. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 5183–5186. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Zhao, Y.; Luo, F.; Weissman, I.; Friedman, J. Crystal structure of murine cyclophilin C complexed with immunosuppressive drug cyclosporin A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 11850–11854. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.; Poole, D.S.; Hoover, S.; Middleton, R.; Andrei, A.C.; Gerstner, J.; Striker, R. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology 2007, 46, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Heck, J.A.; Meng, X.; Frick, D.N. Cyclophilin B stimulates RNA synthesis by the HCV RNA dependent RNA polymerase. Biochem. Pharmacol. 2009, 77, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Robida, J.M.; Nelson, H.B.; Liu, Z.; Tang, H. Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J. Virol. 2007, 81, 5829–5840. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [PubMed]
- Takahashi, N.; Hayano, T.; Suzuki, M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 1989, 337, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Zydowsky, L.D.; Etzkorn, F.A.; Chang, H.Y.; Ferguson, S.B.; Stolz, L.A.; Ho, S.I.; Walsh, C.T. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992, 1, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, F.; Robotham, J.M.; Tang, H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 2009, 83, 6554–6565. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Hanoulle, X.; Badillo, A.; Wieruszeski, J.M.; Verdegem, D.; Landrieu, I.; Bartenschlager, R.; Penin, F.; Lippens, G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 2009, 284, 13589–13601. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.; Ansari, I.U.; Striker, R. cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A . PLoS One 2010, 5, e9815. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Lim, P.; Bobardt, M.D.; Wieland, S.; Cordek, D.G.; Vuagniaux, G.; Chisari, F.; Cameron, C.E.; Targett-Adams, P.; Parkinson, T.; Gallay, P.A. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors . J. Hepatol. 2010. [Google Scholar]
- Chatterji, U.; Bobardt, M.D.; Lim, P.; Gallay, P.A. Cyclophilin A-independent recruitment of NS5A and NS5B into hepatitis C virus replication complexes. J. Gen. Virol. 2010, 91, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Watashi, K.; Murata, T.; Hishiki, T.; Hijikata, M.; Shimotohno, K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 2006, 343, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Landrieu, I.; Hanoulle, X.; Bonachera, F.; Hamel, A.; Sibille, N.; Yin, Y.; Wieruszeski, J.M.; Horvath, D.; Wei, Q.; Vuagniaux, G.; Lippens, G. Structural basis for the non-immunosuppressive character of the cyclosporin A analogue Debio 025. Biochemistry 2010, 49, 4679–4686. [Google Scholar] [CrossRef] [PubMed]
- Sadeg, N.; Pham-Huy, C.; Rucay, P.; Righenzi, S.; Halle-Pannenko, O.; Claude, J.R.; Bismuth, H.; Duc, H.T. In vitro and in vivo comparative studies on immunosuppressive properties of cyclosporines A, C, D and metabolites M1, M17 and M21. Immunopharmacol. Immunotoxicol. 1993, 15, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Horban, A.; Gallay, P.; Bobardt, M.; Selvarajah, S.; Wiercinska-Drapalo, A.; Siwak, E.; Cielniak, I.; Higersberger, J.; Kierkus, J.; Aeschlimann, C.; Grosgurin, P.; Nicolas-Metral, V.; Dumont, J.M.; Porchet, H.; Crabbe, R.; Scalfaro, P. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 2008, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.; Tyrrell, D.L.; Kneteman, N.M. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001, 7, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Rosenwirth, B.; Billich, A.; Datema, R.; Donatsch, P.; Hammerschmid, F.; Harrison, R.; Hiestand, P.; Jaksche, H.; Mayer, P.; Peichl, P.; et al. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog . Antimicrob. Agents Chemother. 1994, 38, 1763–1772. [Google Scholar] [PubMed]
- Hopkins, S.D.; Heuman, E.; Gavis, J.; Lalezari, E.; Glutzer, B.; DiMassimo, P.; Rusnak, S.; Wring, C.; Smitley, R.; Ribeill, Y. Safety, plasma, pharmacokinetics, and anti-viral activity of SCY-635 in adult patients with chronic hepatitis C virus infection . J. Hepatology 2009, 50, S36. [Google Scholar] [CrossRef]
- Jeffery, J.R. Cyclosporine analogues. Clin. Biochem. 1991, 24, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Watashi, K.; Inoue, D.; Hijikata, M.; Shimotohno, K. Identification of cellular and viral factors related to anti-hepatitis C virus activity of cyclophilin inhibitor. Cancer Sci. 2009, 100, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Puyang, X.; Poulin, D.L.; Mathy, J.E.; Anderson, L.J.; Ma, S.; Fang, Z.; Zhu, S.; Lin, K.; Fujimoto, R.; Compton, T.; Wiedmann, B. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob. Agents Chemother. 2010, 54, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Liu, J.O. Sanglifehrin A, a novel cyclophilin-binding immunosuppressant, inhibits IL-2-dependent T cell proliferation at the G1 phase of the cell cycle. J. Immunol. 2001, 166, 5611–5618. [Google Scholar] [PubMed]
- Zenke, G.; Strittmatter, U.; Fuchs, S.; Quesniaux, V.F.; Brinkmann, V.; Schuler, W.; Zurini, M.; Enz, A.; Billich, A.; Sanglier, J.J.; Fehr, T. Sanglifehrin A, a novel cyclophilin-binding compound showing immunosuppressive activity with a new mechanism of action. J. Immunol. 2001, 166, 7165–7171. [Google Scholar] [PubMed]
- Rong, L.; Dahari, H.; Ribeiro, R.M.; Perelson, A.S. Rapid emergence of protease inhibitor resistance in hepatitis C virus . Sci. Transl. Med. 2, 30–32.
- Yang, F.; Robotham, J.M.; Grise, H.; Frausto, S.; Madan, V.; Zayas, M.; Bartenschlager, R.; Robinson, M.; Greenstein, A.E.; Nag, A.; Logan, T.; Bienkiewicz, E.; Tang, H. A Major Determinant of Cyclophilin Dependence and Cyclosporine Susceptibility of Hepatitis C Virus Identified by a Genetic Approach . PLoS Pathog. 2010, in press. [Google Scholar]
- Moradpour, D.; Evans, M.J.; Gosert, R.; Yuan, Z.; Blum, H.E.; Goff, S.P.; Lindenbach, B.D.; Rice, C.M. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 2004, 78, 7400–7409. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.B.; Tang, H. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence-based inhibition of HCV RNA and protein expression. J. Virol. 2006, 80, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 2005, 79, 2346–2355. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Robida, J.M.; Chinnaswamy, S.; Yi, G.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kao, C.C.; Tang, H. Mutations in the hepatitis C virus polymerase that increase RNA binding can confer resistance to cyclosporine A. Hepatology 2009, 50, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O'Boyle, D.R.; Lemm, J.A.; Wang, C.; Knipe, J.O.; Chien, C.; Colonno, R.J.; Grasela, D.M.; Meanwell, N.A.; Hamann, L.G. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect . Nature 465, 96–100. [CrossRef] [PubMed]
- Ishii, N.; Watashi, K.; Hishiki, T.; Goto, K.; Inoue, D.; Hijikata, M.; Wakita, T.; Kato, N.; Shimotohno, K. Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 2006, 80, 4510–4520. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.A. Cyclophilin inhibitors. Clin. Liver Dis. 2009, 13, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Colgan, J.; Asmal, M.; Neagu, M.; Yu, B.; Schneidkraut, J.; Lee, Y.; Sokolskaja, E.; Andreotti, A.; Luban, J. Cyclophilin A regulates TCR signal strength in CD4+ T cells via a proline-directed conformational switch in Itk. Immunity 2004, 21, 189–201. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Tang, H. Cyclophilin Inhibitors as a Novel HCV Therapy. Viruses 2010, 2, 1621-1634. https://doi.org/10.3390/v2081621
Tang H. Cyclophilin Inhibitors as a Novel HCV Therapy. Viruses. 2010; 2(8):1621-1634. https://doi.org/10.3390/v2081621
Chicago/Turabian StyleTang, Hengli. 2010. "Cyclophilin Inhibitors as a Novel HCV Therapy" Viruses 2, no. 8: 1621-1634. https://doi.org/10.3390/v2081621
APA StyleTang, H. (2010). Cyclophilin Inhibitors as a Novel HCV Therapy. Viruses, 2(8), 1621-1634. https://doi.org/10.3390/v2081621