Alphavirus Entry and Membrane Fusion


Abstract
:1. Introduction
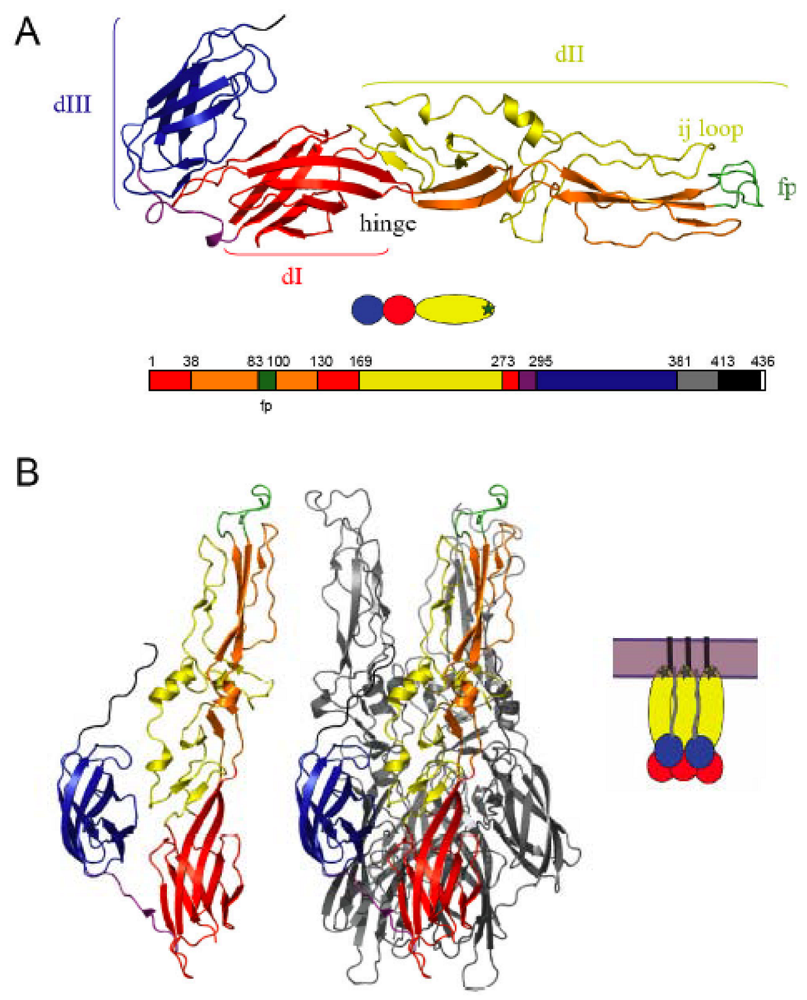
2. Alphavirus architecture and the structure of the fusion machinery
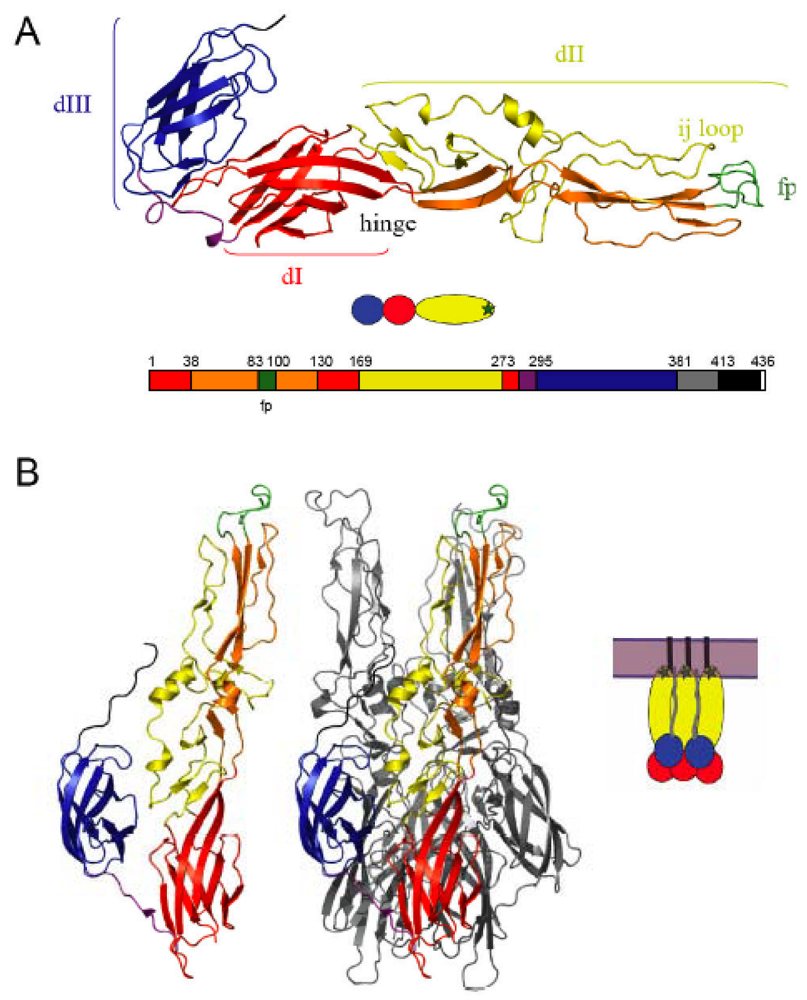
3. The alphavirus entry pathway
3.1. Receptors and attachment factors
3.2. Receptor-mediated endocytosis of alphaviruses

{kind=link}
{kind=link}
| Observation | Selected References | |
|---|---|---|
| Morphological and biochemical observation of endocytic uptake | [39,43] | |
| Infection from within endosomes | [44] | |
| Infection/fusion inhibited by dominant-negative inhibitors of endocytosis: | dynamin | [45,46][47][48] |
| eps15 | ||
| rab-5 | ||
| Infection/fusion inhibited by weak bases (e.g. NH4Cl, chloroquine) | [39,44,49] | |
| Infection/fusion inhibited by ionophores (e.g. monensin) | [50,51] | |
| Infection/fusion inhibited by vacuolar proton pump inhibitors (e.g. bafilomycin, concanamycin) | [46,49,52] | |
| Specific low pH-dependence of pseudotype infection | [53] | |
| Low pH-dependent cell-cell fusion | [54-57] | |
| Low pH-dependent virus fusion with liposomes | [58-60] | |
| Low pH-dependent fusion pore formation | [56,61] | |
| Mutations block both membrane fusion in vitro and virus infection in vivo | [62-64] | |
| Exogenous DIII blocks both fusion and infection | [65] | |
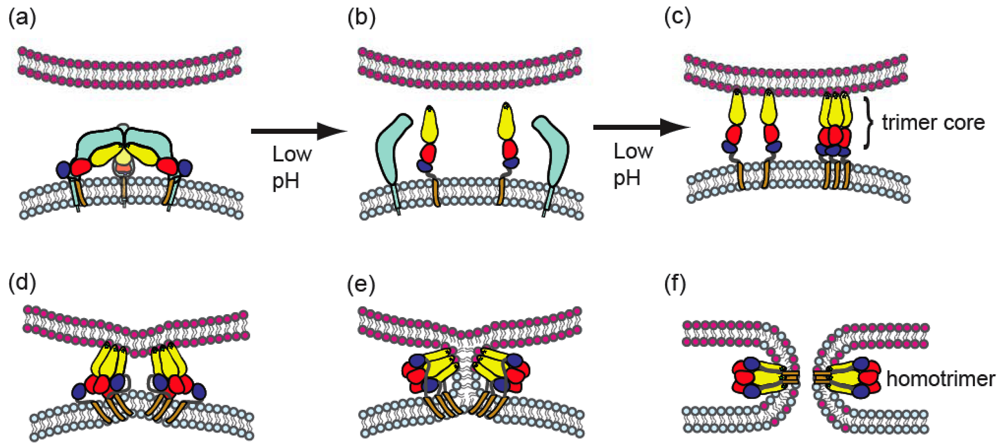
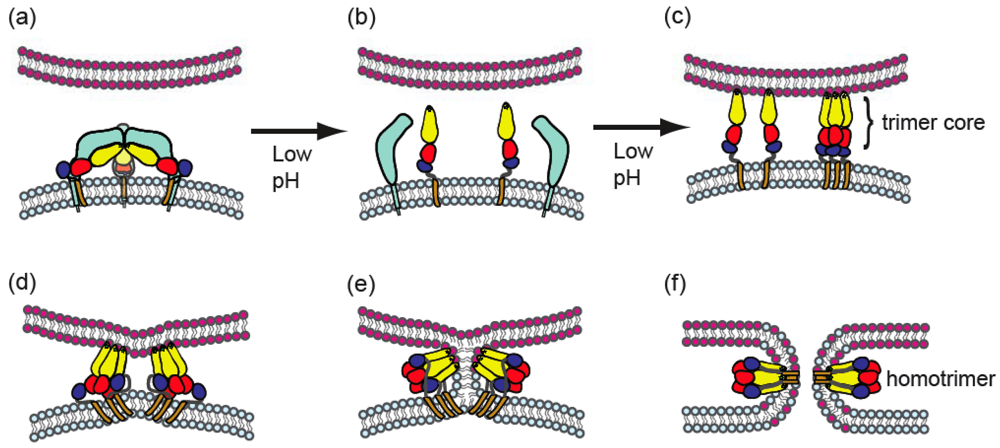
3.3. Low pH-triggered fusion in endosomes
4. Alphavirus membrane fusion
4.1. General properties of alphavirus-membrane fusion
4.2. Lipid dependence
4.3. Conformational changes during fusion
| Observation | Selected References |
|---|---|
| Timing during virus uptake in vivo | [51,100] |
| In vivo formation requires endosomal acidification | [100] |
| Block in homotrimer formation in fusion-defective E1 mutants G91D, D188K | [62,64] |
| Timing during virus fusion in vitro | [58,101] |
| Correlation with pH-dependence of virus fusion | [35,74] |
| Exogenous DIII blocks E1 hairpin formation and fusion | [65] |
4.4. E1 ectodomain studies
4.5. E1 homotrimer structure
4.6. Properties of the alphavirus fusion pore
5. Molecular analysis of the alphavirus membrane fusion reaction
Regulatory role of p62/E2
| Virus used for selection | Glycoproteins | References | ||
|---|---|---|---|---|
| E3 | E2 | E1 | ||
| wt/p62 (SFV) | N7D, N77D, A121E, H170Y,L221Q, R244G, R244K, R250G | V11A, T159A | [140,141] | |
| SHQL(SFV) | H64R | Q4R, R244I, R244K | [139] | |
| TRSB-N(SIN) | C25R | D82G, H169L, P191T, T198M, E216G, N239H | [35,137] | |
| VEE deletion | L243N | F253S | [135] | |
| SIN E2/RRV 6k+E1(chimera) | D72N, S118N, K131E, I150L, V237F, L243S, D248Y, I380S | S310F, F399S, Q411L, I423L, C433R | [133,142,143] | |
E1 fusion loop and target membrane interaction.
Characterization of the E1 homotrimer
Cholesterol dependence
Role of the E1 ij loop
Roles of the E1 stem and transmembrane domains
In vitro reconstitution of trimerization
6. Future directions
Acknowledgments
References
- Marsh, M.; Helenius, A. Virus entry: open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J. Viruses and endosome membrane dynamics. Curr. Opin. Cell Biol. 2009, 21, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Viral entry. Curr. Topics Microbiol. Immunol. 2005, 285, 1–23. [Google Scholar]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Kuhn, R.J. Togaviridae: The Viruses and Their Replication. In Fields Virology; Knipe, D.M., Ed.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Mancini, E.J.; Clarke, M.; Gowen, B.E.; Rutten, T.; Fuller, S.D. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki forest virus. Mol. Cell 2000, 5, 255–266. [Google Scholar] [CrossRef]
- Zhang, W.; Mukhopadhyay, S.; Pletnev, S.V.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Placement of the structural proteins in sindbis virus. J. Virol. 2002, 76, 11645–11658. [Google Scholar] [CrossRef] [PubMed]
- Roussel, A.; Lescar, J.; Vaney, M.-C.; Wengler, G.; Wengler, G.; Rey, F.A. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 2006, 14, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Zhang, W.; Gabler, S.; Chipman, P.R.; Strauss, E.G.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure 2006, 14, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Rey, F.A. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Vaney, M.-C.; Roussel, A.; Vigouroux, A.; Reilly, B.; Lepault, J.; Kielian, M.; Rey, F.A. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 2004, 427, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2A resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 2005, 79, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, W.; Ogata, S.; Clements, D.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Conformational changes of the flavivirus E glycoprotein. Structure (Camb) 2004, 12, 1607–1618. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Nelson, C.A.; Chen, B.R.; Diamond, M.S.; Fremont, D.H. Crystal structure of the West Nile virus envelope glycoprotein. J. Virol. 2006, 80, 11467–11474. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Kar, K.; Anthony, K.; Gould, L.H.; Ledizet, M.; Fikrig, E.; Marasco, W.A.; Koski, R.A.; Modis, Y. Crystal structure of west nile virus envelope glycoprotein reveals viral surface epitopes. J. Virol. 2006, 80, 11000–11008. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M.; Rey, F.A. Virus membrane fusion proteins: more than one way to make a hairpin. Nat.Rev.Micro. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- Sanchez-San Martin, C.; Liu, C.Y.; Kielian, M. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends in Microbiol. 2009, 17, 514–521. [Google Scholar] [CrossRef]
- Kielian, M. Membrane fusion and the alphavirus life cycle. Adv. Virus Res. 1995, 45, 113–151. [Google Scholar]
- Kielian, M.; Helenius, A. Entry of Alphaviruses. In The Togaviridae and Flaviviridae; Schlesinger, S., Schlesinger, M.J. Eds.; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- La Linn, M.; Eble, J.A.; Lubken, C.; Slade, R.W.; Heino, J.; Davies, J.; Suhrbier, A. An arthritogenic alphavirus uses the alpha1beta1 integrin collagen receptor. Virol. 2005, 336, 229–239. [Google Scholar] [CrossRef]
- Klimstra, W.B.; Nangle, E.M.; Smith, M.S.; Yurochko, A.D.; Ryman, K.D. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J. Virol. 2003, 77, 12022–12032. [Google Scholar] [CrossRef] [PubMed]
- Klimstra, W.B.; Ryman, K.D.; Johnston, R.E. Adaptation of sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 1998, 72, 7357–7366. [Google Scholar] [PubMed]
- Byrnes, A.P.; Griffin, D.E. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 1998, 72, 7349–7356. [Google Scholar] [PubMed]
- Jan, J.T.; Byrnes, A.P.; Griffin, D.E. Characterization of a Chinese hamster ovary cell line developed by retroviral insertional mutagenesis that is resistant to Sindbis virus infection. J. Virol. 1999, 73, 4919–4924. [Google Scholar] [PubMed]
- Heil, M.L.; Albee, A.; Strauss, J.H.; Kuhn, R.J. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 2001, 75, 6303–6309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Heil, M.; Kuhn, R.J.; Baker, T.S. Heparin binding sites on Ross River virus revealed by electron cryo-microscopy. Virol. 2005, 332, 511–518. [Google Scholar] [CrossRef]
- Byrnes, A.P.; Griffin, D.E. Large-plaque mutants of sindbis virus show reduced binding to heparan sulfate, heightened viremia, and slower clearance from the circulation. J. Virol. 2000, 74, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.A.; Klimstra, W.B.; Johnston, R.E. Mutations in the E2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virol. 2000, 276, 93–103. [Google Scholar] [CrossRef]
- Wang, E.; Brault, A.C.; Powers, A.M.; Kang, W.; Weaver, S.C. Glycosaminoglycan binding properties of natural venezuelan equine encephalitis virus isolates. J. Virol. 2003, 77, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Klimstra, W.B.; Heidner, H.W.; Johnston, R.E. The furin protease cleavage recognition sequence of sindbis virus PE2 can mediate virion attachment to cell surface heparan sulfate. J. Virol. 1999, 73, 6299–6306. [Google Scholar] [PubMed]
- Smit, J.M.; Klimstra, W.B.; Ryman, K.D.; Bittman, R.; Johnston, R.E.; Wilschut, J. PE2 cleavage mutants of Sindbis virus: correlation between viral infectivity and pH-dependent membrane fusion activation of the spike heterodimer. J. Virol. 2001, 75, 11196–11204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fugere, M.; Day, R.; Kielian, M. Furin processing and proteolytic activation of Semliki Forest virus. J. Virol. 2003, 77, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, H.; Mitchell, D.A.; Drickamer, K.; Weis, W.I. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 2001, 294, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Robbins, P.W. Regulation of asparagine-linked oligosaccharide processing. J. Biol. Chem. 1984, 259, 2375–2382. [Google Scholar] [PubMed]
- Helenius, A.; Kartenbeck, J.; Simons, K.; Fries, E. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 1980, 84, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Helenius, A. How viruses enter animal cells. Science 2004, 304, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Garoff, H.; Wilschut, J.; Liljeström, P.; Wahlberg, J.M.; Bron, R.; Suomalainen, M.; Smyth, J.; Salminen, A.; Barth, B.U.; Zhao, H. Assembly and entry mechanisms of Semliki Forest virus. Arch .Virol. 1994, 9, 329–338. [Google Scholar]
- Sieczkarski, S.B.; Whittaker, G.R. Dissecting virus entry via endocytosis. J. Gen. Virol. 2002, 83, 1535–1545. [Google Scholar] [PubMed]
- Marsh, M.; Helenius, A. Adsorptive endocytosis of Semliki Forest virus. J. Mol. Biol. 1980, 142, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Marsh, M.; White, J. Inhibition of Semliki Forest virus penetration by lysosomotropic weak bases. J. Gen. Virol. 1982, 58, 47–61. [Google Scholar] [CrossRef] [PubMed]
- DeTulleo, L.; Kirchhausen, T. The clathrin endocytic pathway in viral infection. EMBO J. 1998, 17, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Earp, L.J.; Delos, S.E.; Netter, R.C.; Bates, P.; White, J.M. The avian retrovirus avian sarcoma/leukosis virus subtype A reaches the lipid mixing stage of fusion at neutral pH. J. Virol. 2003, 77, 3058–3066. [Google Scholar] [CrossRef] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 2003, 4, 333–343. [Google Scholar] [CrossRef]
- Glomb-Reinmund, S.; Kielian, M. The role of low pH and disulfide shuffling in the entry and fusion of Semliki Forest virus and Sindbis virus. Virol. 1998, 248, 372–381. [Google Scholar] [CrossRef]
- Marsh, M.; Wellsteed, J.; Kern, H.; Harms, E.; Helenius, A. Monensin inhibits Semliki Forest virus penetration into culture cells. Proc. Natl. Acad. Sci. USA 1982, 79, 5297–5301. [Google Scholar] [CrossRef]
- Kielian, M.C.; Marsh, M.; Helenius, A. Kinetics of endosome acidification detected by mutant and wild-type Semliki Forest virus. EMBO J. 1986, 5, 3103–3109. [Google Scholar] [PubMed]
- Irurzun, A.; Nieva, J.L.; Carrasco, L. Entry of Semliki Forest virus into cells: Effects of concanamycin A and nigericin on viral membrane fusion and infection. Virol. 1997, 227, 488–492. [Google Scholar] [CrossRef]
- Sharkey, C.M.; North, C.L.; Kuhn, R.J.; Sanders, D.A. Ross River virus glycoprotein-pseudotyped retroviruses and stable cell lines for their production. J. Virol. 2001, 75, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Matlin, K.; Helenius, A. Cell fusion by Semliki Forest, influenza and Vesicular Stomatitis viruses. J. Cell Biol. 1981, 89, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.; Flaviano, A.; Kohler, U.; Koblet, H. Fusion of Semliki Forest virus infected Aedes albopictus cells at low pH is a fusion from within. Arch. Virol. 1986, 89, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Lanzrein, M.; Käsermann, N.; Weingart, R.; Kempf, C. Early events of Semliki Forest Virus-induced cell-cell fusion. Virol. 1993, 196, 541–547. [Google Scholar] [CrossRef]
- Zaitseva, E.; Mittal, A.; Griffin, D.E.; Chernomordik, L.V. Class II fusion protein of alphaviruses drives membrane fusion through the same pathway as class I proteins. J. Cell Biol. 2005, 169, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Bron, R.; Wahlberg, J.M.; Garoff, H.; Wilschut, J. Membrane fusion of Semliki Forest virus in a model system: Correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 1993, 12, 693–701. [Google Scholar] [PubMed]
- Smit, J.M.; Bittman, R.; Wilschut, J. Low-pH-dependent fusion of sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J. Virol. 1999, 73, 8476–8484. [Google Scholar] [PubMed]
- White, J.; Helenius, A. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc. Natl. Acad. Sci. USA 1980, 77, 3273–3277. [Google Scholar] [CrossRef]
- Samsonov, A.V.; Chatterjee, P.K.; Razinkov, V.I.; Eng, C.H.; Kielian, M.; Cohen, F.S. Effects of membrane potential and sphingolipid structures on fusion of Semliki Forest virus. J. Virol. 2002, 76, 12691–12702. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M.; Klimjack, M.R.; Ghosh, S.; Duffus, W.A. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 1996, 134, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Chanel-Vos, C.; Kielian, M. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J. Virol. 2004, 78, 13543–13552. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Kielian, M. E1 mutants identify a critical region in the trimer interface of the Semliki Forest virus fusion protein. J. Virol. 2009, 83, 11298–11306. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Kielian, M. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J. Cell Biol. 2005, 171, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Doxsey, S.J.; Brodsky, F.M.; Blank, G.S.; Helenius, A. Inhibition of endocytosis by anti-clathrin antibodies. Cell 1987, 50, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.L.; Fuchs, R.; Kielian, M.; Helenius, A.; Mellman, I. Acidification of endosome subpopulations in wild-type Chinese hamster ovary cells and temperature-sensitive acidification-defective mutants. J. Cell Biol. 1989, 108, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Bolzau, E.; Helenius, A. Penetration of Semliki Forest virus from acidic prelysomal vacuoles. Cell 1983, 32, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, T.M.; Moore, A.C.; Kolokoltsov, A.A.; Davey, R.A. Venezuelan equine encephalitis virus infection of mosquito cells requires acidification as well as mosquito homologs of the endocytic proteins Rab5 and Rab7. Virol. 2007, 369, 78–91. [Google Scholar] [CrossRef]
- Mellman, I.; Fuchs, R.; Helenius, A. Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 1986, 55, 663–700. [Google Scholar] [PubMed]
- Marsh, M.; Bron, R. SFV infection in CHO cells: Cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 1997, 110, 95–103. [Google Scholar] [PubMed]
- Fan, D.P.; Sefton, B.N. The entry into host cells of Sindbis virus, Vesicular Stomatitis virus and Sendai virus. Cell 1978, 15, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Glomb-Reinmund, S.; Kielian, M. fus-1, a pH-shift mutant of Semliki Forest virus, acts by altering spike subunit interactions via a mutation in the E2 subunit. J. Virol. 1998, 72, 4281–4287. [Google Scholar] [PubMed]
- Abell, B.A.; Brown, D.T. Sindbis virus membrane fusion is mediated by reduction of glycoprotein disulfide bridges at the cell surface. J. Virol. 1993, 67, 5496–5501. [Google Scholar] [PubMed]
- Flynn, D.C.; Meyer, W.J.; Mackenzie, J.M.; Johnston, R.E. A conformational change in Sindbis virus glycoproteins E1 and E2 is detected at the plasma membrane as a consequence of early virus-cell interaction. J. Virol. 1990, 64, 3643–3653. [Google Scholar] [PubMed]
- Kielian, M.; Chatterjee, P.K.; Gibbons, D.L.; Lu, Y.E. Specific roles for lipids in virus fusion and exit: Examples from the alphaviruses. In Subcellular Biochemistry Vol. 34. Fusion of Biological Membranes and Related Problems . 2000, 409–455. [Google Scholar]
- Umashankar, M.; Sanchez San Martin, C.; Liao, M.; Reilly, B.; Guo, A.; Taylor, G.; Kielian, M. Differential Cholesterol Binding by Class II Fusion Proteins Determines Membrane Fusion Properties. J. Virol. 2008, 82, 9245–9253. [Google Scholar] [CrossRef] [PubMed]
- Mooney, J.J.; Dalrymple, J.M.; Alving, C.R.; Russell, P.K. Interaction of Sindbis virus with liposomal model membranes. J. Virol. 1975, 15, 225–231. [Google Scholar] [PubMed]
- Kielian, M.C.; Helenius, A. The role of cholesterol in the fusion of Semliki Forest virus with membranes. J. Virol. 1984, 52, 281–283. [Google Scholar] [PubMed]
- Dawidowicz, E.A. Dynamics of membrane lipid metabolism and turnover. Annu. Rev. Biochem. 1987, 56, 43–61. [Google Scholar] [PubMed]
- Nes, W.R.; McKean, M.L. Occurence, Physiology, and Ecology of Sterols. Biochemistry of Steroids and Other Isopentenoids 1977, 411–533. [Google Scholar]
- Silberkang, M.; Havel, C.M.; Friend, D.S.; McCarthy, B.J.; Watson, J.A. Isoprene synthesis in isolated embryonic Drosophila cells I. Sterol-deficient eukaryotic cells. J. Biol. Chem. 1983, 258, 8303–8311. [Google Scholar]
- Phalen, T.; Kielian, M. Cholesterol is required for infection by Semliki Forest virus. J. Cell Biol. 1991, 112, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, M.T.; Phalen, T.; Kielian, M. Cholesterol is required in the exit pathway of Semliki Forest virus. J. Cell Biol. 1993, 123, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Vashishtha, M.; Phalen, T.; Marquardt, M.T.; Ryu, J.S.; Ng, A.C.; Kielian, M. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 1998, 140, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.E.; Cassese, T.; Kielian, M. The cholesterol requirement for Sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 1999, 73, 4272–4278. [Google Scholar] [PubMed]
- Lu, Y.E.; Kielian, M. Semliki Forest virus budding: Assay, mechanisms and cholesterol requirement. J. Virol. 2000, 74, 7708–7719. [Google Scholar] [CrossRef] [PubMed]
- Hafer, A.; Whittlesey, R.; Brown, D.T.; Hernandez, R. Differential incorporation of cholesterol by Sindbis virus grown in mammalian or insect cells. J. Virol. 2009, 83, 9113–9121. [Google Scholar] [CrossRef] [PubMed]
- Bowers, D.F.; Abell, B.A.; Brown, D.T. Replication and tissue tropism of the alphavirus Sindbis in the mosquito Aedes albopictus. Virol. 1995, 212, 1–12. [Google Scholar] [CrossRef]
- Scott, T.W.; Weaver, S.C. Eastern equine encephalomyelitis virus: Epidemiology and evolution of mosquito transmission. Adv. Virus Res. 1989, 37, 277–328. [Google Scholar] [PubMed]
- Marquardt, M.T.; Kielian, M. Cholesterol-depleted cells that are relatively permissive for Semliki Forest virus infection. Virol. 1996, 224, 198–205. [Google Scholar] [CrossRef]
- Nieva, J.L.; Bron, R.; Corver, J.; Wilschut, J. Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO J. 1994, 13, 2797–2804. [Google Scholar] [PubMed]
- Wilschut, J.; Corver, J.; Nieva, J.L.; Bron, R.; Moesby, L.; Reddy, K.C.; Bittman, R. Fusion of Semliki Forest virus with cholesterol-containing liposomes at low pH: A specific requirement for sphingolipids. Mol. Membrane Biol. 1995, 12, 143–149. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem.. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [PubMed]
- Waarts, B.L.; Bittman, R.; Wilschut, J. Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J. Biol. Chem.. 2002, 277, 38141–38147. [Google Scholar] [CrossRef] [PubMed]
- Meyer, W.J.; Johnston, R.E. Structural rearrangement of infecting Sindbis virions at the cell surface: Mapping of newly accessible epitopes. J. Virol. 1993, 67, 5117–5125. [Google Scholar] [PubMed]
- Meyer, W.J.; Gidwitz, S.; Ayers, V.K.; Schoepp, R.J.; Johnston, R.E. Conformational alteration of Sindbis virion glycoproteins induced by heat, reducing agents, or low pH. J. Virol. 1992, 66, 3504–3513. [Google Scholar] [PubMed]
- Wahlberg, J.M.; Boere, W.A.M.; Garoff, H. The heterodimeric association between the membrane proteins of Semliki Forest virus changes its sensitivity to low pH during virus maturation. J. Virol. 1989, 63, 4991–4997. [Google Scholar] [PubMed]
- Wahlberg, J.M.; Garoff, H. Membrane fusion process of Semliki Forest virus I: Low pH-induced rearrangement in spike protein quaternary structure precedes virus penetration into cells. J. Cell Biol. 1992, 116, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Justman, J.; Klimjack, M.R.; Kielian, M. Role of spike protein conformational changes in fusion of Semliki Forest virus. J. Virol. 1993, 67, 7597–7607. [Google Scholar] [PubMed]
- Hammar, L.; Markarian, S.; Haag, L.; Lankinen, H.; Salmi, A.; Cheng, H.R. Prefusion rearrangements resulting in fusion peptide exposure in Semliki forest virus. J. Biol. Chem.. 2003, 278, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Ahn, A.; Liao, M.; Hammar, L.; Cheng, R.H.; Kielian, M. Multistep regulation of membrane insertion of the fusion peptide of Semliki Forest virus. J. Virol. 2004, 78, 3312–3318. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, J.M.; Bron, R.; Wilschut, J.; Garoff, H. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J. Virol. 1992, 66, 7309–7318. [Google Scholar] [PubMed]
- Kielian, M.; Helenius, A. pH-induced alterations in the fusogenic spike protein of Semliki Forest Virus. J. Cell Biol. 1985, 101, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Shome, S.G.; Kielian, M. Differential roles of two conserved glycine residues in the fusion peptide of Semliki Forest virus. Virol. 2001, 279, 146–160. [Google Scholar] [CrossRef]
- Salminen, A.; Wahlberg, J.M.; Lobigs, M.; Liljeström, P.; Garoff, H. Membrane fusion process of Semliki Forest virus II: Cleavage- dependent reorganization of the spike protein complex controls virus entry. J. Cell Biol. 1992, 116, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Corver, J.; Bron, R.; Snippe, H.; Kraaijeveld, C.; Wilschut, J. Membrane fusion activity of Semliki forest virus in a liposomal model system: Specific inhibition by Zn2+ ions. Virol. 1997, 238, 14–21. [Google Scholar] [CrossRef]
- Kielian, M.; Jungerwirth, S.; Sayad, K.U.; DeCandido, S. Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J. Virol. 1990, 64, 4614–4624. [Google Scholar] [PubMed]
- Ahn, A.; Klimjack, M.R.; Chatterjee, P.K.; Kielian, M. An epitope of the Semliki Forest virus fusion protein exposed during virus-membrane fusion. J. Virol. 1999, 73, 10029–10039. [Google Scholar] [PubMed]
- Phinney, B.S.; Blackburn, K.; Brown, D.T. The surface conformation of sindbis virus glycoproteins E1 and E2 at neutral and low pH, as determined by mass spectrometry-based mapping. J. Virol. 2000, 74, 5667–5678. [Google Scholar] [CrossRef] [PubMed]
- Klimjack, M.R.; Jeffrey, S.; Kielian, M. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 1994, 68, 6940–6946. [Google Scholar] [PubMed]
- Wengler, G.; Wengler, G.; Rey, F.A. The isolation of the ectodomain of the alphavirus E1 protein as a soluble hemagglutinin and its crystallization. Virol. 1999, 257, 472–482. [Google Scholar] [CrossRef]
- Lu, Y.E.; Eng, C.H.; Shome, S.G.; Kielian, M. In vivo generation and characterization of a soluble form of the Semliki forest virus fusion protein. J. Virol. 2001, 75, 8329–8339. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Garoff, H. Role of cell surface spikes in alphavirus budding. J. Virol. 1992, 66, 7089–7095. [Google Scholar] [PubMed]
- Sanchez-San Martin, C.; Sosa, H.; Kielian, M. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 2008, 4, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Erk, I.; Reilly, B.; Navaza, J.; Kielian, M.; Rey, F.A.; Lepault, J. Visualization of the target-membrane-inserted fusion protein of Semliki Forest virus by combined electron microscopy and crystallography. Cell 2003, 114, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Reilly, B.; Ahn, A.; Vaney, M.-C.; Vigouroux, A.; Rey, F.A.; Kielian, M. Purification and crystallization reveal two types of interactions of the fusion protein homotrimer of Semliki Forest virus. J. Virol. 2004, 787, 3514–3523. [Google Scholar] [CrossRef]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Bressanelli, S.; Stiasny, K.; Allison, S.L.; Stura, E.A.; Duquerroy, S.; Lescar, J.; Heinz, F.X.; Rey, F.A. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004, 23, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Kielian, M. Molecular dissection of the Semliki Forest virus homotrimer reveals two functionally distinct regions of the fusion protein. J. Virol. 2002, 76, 1194–1205. [Google Scholar] [PubMed]
- Markosyan, R.M.; Kielian, M.; Cohen, F.S. Fusion induced by a class II viral fusion protein, semliki forest virus E1, is dependent on the voltage of the target cell. J. Virol. 2007, 81, 11218–11225. [Google Scholar] [CrossRef] [PubMed]
- Wengler, G.; Koschinski, A.; Repp, H. During entry of alphaviruses, the E1 glycoprotein molecules probably form two separate populations that generate either a fusion pore or ion-permeable pores. J. Gen. Virol. 2004, 85, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Lanzrein, M.; Weingart, R.; Kempf, C. pH-dependent pore formation in Semliki forest virus-infected Aedes albopictus cells. Virol. 1993, 193, 296–302. [Google Scholar] [CrossRef]
- Nyfeler, S.; Senn, K.; Kempf, C. Expression of Semliki Forest virus E1 protein in Escherichia coli. Low pH-induced pore formation. J. Biol. Chem. 2001, 276, 15453–15457. [Google Scholar] [CrossRef] [PubMed]
- Wengler, G.; Koschinski, A.; Dreyer, F. Entry of alphaviruses at the plasma membrane converts the viral surface proteins into an ion-permeable pore that can be detected by electrophysiological analyses of whole-cell membrane currents. J. Gen. Virol. 2003, 84, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Koschinski, A.; Wengler, G.; Repp, H. Rare earth ions block the ion pores generated by the class II fusion proteins of alphaviruses and allow analysis of the biological functions of these pores. J. Gen. Virol. 2005, 86, 3311–3320. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.M.; Li, G.; Schoen, P.; Corver, J.; Bittman, R.; Lin, K.C.; Wilschut, J. Fusion of alphaviruses with liposomes is a non-leaky process. FEBS Lett 2002, 521, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk-Nitschko, K.; Schlesinger, M.J. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virol. 1990, 175, 274–281. [Google Scholar] [CrossRef]
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Liljeström, P.; Lusa, S.; Huylebroeck, D.; Garoff, H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 1991, 65, 4107–4113. [Google Scholar] [PubMed]
- McInerney, G.M.; Smit, J.M.; Liljestrom, P.; Wilschut, J. Semliki Forest virus produced in the absence of the 6K protein has an altered spike structure as revealed by decreased membrane fusion capacity. Virol. 2004, 325, 200–206. [Google Scholar] [CrossRef]
- Kim, K.H.; Strauss, E.G.; Strauss, J.H. Adaptive mutations in Sindbis virus E2 and Ross River virus E1 that allow efficient budding of chimeric viruses. J. Virol. 2000, 74, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Berglund, P.; Sjoberg, M.; Garoff, H.; Atkins, G.J.; Sheahan, B.J.; Liljestrom, P. Semliki Forest virus expression system: production of conditionally infectious recombinant particles. Biotechnology (N Y) 1993, 11, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; Brown, K.W.; Greenwald, G.F.; Zajac, A.J.; Zacny, V.L.; Smith, J.F.; Johnston, R.E. Attenuated mutants of venezuelan equine encephalitis virus containing lethal mutations in the PE2 cleavage signal combined with a second-site suppressor mutation in E1. Virol. 1995, 212, 102–110. [Google Scholar] [CrossRef]
- Russell, D.L.; Dalrymple, J.M.; Johnston, R.E. Sindbis virus mutations which coordinately affect glycoprotein processing, penetration, and virulence in mice . J. Virol. 1989, 63, 16l9–1629. [Google Scholar]
- Heidner, H.W.; McKnight, K.L.; Davis, N.L.; Johnston, R.E. Lethality of PE2 incorporation into Sindbis virus can be suppressed by second-site mutations in E3 and E2. J. Virol. 1994, 68, 2683–2692. [Google Scholar] [PubMed]
- Ferlenghi, I.; Gowen, B.; Haas, F.D.; Mancini, E.J.; Garoff, H.; Sjoberg, M.; Fuller, S.D. The first step:maturation of the Semliki Forest virus spike occurs through a dramatic localized conformational change. J. Mol. Biol. 1998, 283, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Tubulekas, I.; Liljeström, P. Suppressors of cleavage-site mutations in the p62 envelope protein of Semliki Forest virus reveal dynamics in spike structure and function. J. Virol. 1998, 72, 2825–2831. [Google Scholar] [PubMed]
- Zhang, X.; Kielian, M. Mutations that promote furin-independent growth of Semliki Forest virus affect p62-E1 interactions and membrane fusion. Virol. 2004, 327, 287–296. [Google Scholar] [CrossRef]
- Zhang, X.; Kielian, M. An interaction site of the envelope proteins of Semliki Forest virus that is preserved after proteolytic activation. Virol. 2005, 337, 344–352. [Google Scholar] [CrossRef]
- Strauss, E.G.; Lenches, E.M.; Strauss, J.H. Molecular genetic evidence that the hydrophobic anchors of glycoproteins E2 and E1 interact during assembly of alphaviruses. J. Virol. 2002, 76, 10188–10194. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.S.; Strauss, E.G.; Strauss, J.H. Molecular genetic study of the interaction of Sindbis virus E2 with Ross River virus E1 for virus budding. J. Virol. 1998, 72, 1418–1423. [Google Scholar] [PubMed]
- Kielian, M.C.; Keränen, S.; Kääriäinen, L.; Helenius, A. Membrane fusion mutants of Semliki Forest virus. J. Cell Biol. 1984, 98, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Garoff, H.; Frischauf, A.-M.; Simons, K.; Lehrach, H.; Delius, H. Nucleotide sequence of cDNA coding for Semliki Forest virus membrane glycoproteins. Nature 1980, 288, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.; Gibbons, D.L.; Kielian, M. The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J. Virol. 2002, 76, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Levy-Mintz, P.; Kielian, M. Mutagenesis of the putative fusion domain of the Semliki Forest virus spike protein. J. Virol. 1991, 65, 4292–4300. [Google Scholar] [PubMed]
- Duffus, W.A.; Levy-Mintz, P.; Klimjack, M.R.; Kielian, M. Mutations in the putative fusion peptide of Semliki Forest virus affect spike protein oligomerization and virus assembly. J. Virol. 1995, 69, 2471–2479. [Google Scholar] [PubMed]
- Gibbons, D.L.; Ahn, A.; Chatterjee, P.K.; Kielian, M. Formation and characterization of the trimeric form of the fusion protein of Semliki Forest virus. J. Virol. 2000, 74, 7772–7780. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.H.; Martin, S.R.; Wharton, S.A.; Skehel, J.J.; Bayley, P.M.; Wiley, D.C. Conformational changes in the hemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virol. 1986, 155, 484–497. [Google Scholar] [CrossRef]
- Russell, C.J.; Jardetzky, T.S.; Lamb, R.A. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001, 20, 4024–4034. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.M.; Chaudhry, C.; Kim, P.S. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc .Natl. Acad. Sci. USA 1997, 94, 14306–14313. [Google Scholar] [CrossRef]
- Stiasny, K.; Allison, S.L.; Mandl, C.W.; Heinz, F.X. Role of Metastability and Acidic pH in Membrane Fusion by Tick-Borne Encephalitis Virus. J. Virol. 2001, 75, 7392–7398. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, T.; Mueller, D.S.; Mark, A.E.; Young, P.R.; Kobe, B. The Role of Histidine Residues in Low-pH-Mediated Viral Membrane Fusion. Structure 2006, 14, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.L.; Zheng, Y.; Kielian, M. Role of conserved histidine residues in the low pH-dependence of the Semliki Forest virus fusion protein. J. Virol. 2009, 83, 4670–4677. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Eng, C.H.; Kielian, M. Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J. Virol. 2002, 76, 12712–12722. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Vashishtha, M.; Kielian, M. Biochemical consequences of a mutation that controls the cholesterol dependence of Semliki Forest virus fusion. J. Virol. 2000, 74, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.C.; Lavenir, R.; Pardigon, N.; Reynes, J.M.; Pettinelli, F.; Biscornet, L.; Diancourt, L.; Michel, S.; Duquerroy, S.; Guigon, G.; Frenkiel, M.P.; Brehin, A.C.; Cubito, N.; Despres, P.; Kunst, F.; Rey, F.A.; Zeller, H.; Brisse, S. Genome Microevolution of Chikungunya Viruses Causing the Indian Ocean Outbreak . PLoS Med. 2006, 3, e263. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, M.; Moutailler, S.; Coudrier, D.; Rousseaux, C.; Khun, H.; Huerre, M.; Thiria, J.; Dehecq, J.S.; Fontenille, D.; Schuffenecker, I.; Despres, P.; Failloux, A.B. Two Chikungunya Isolates from the Outbreak of La Reunion (Indian Ocean) Exhibit Different Patterns of Infection in the Mosquito, Aedes albopictus . PLoS ONE 2007, 2, e1168. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential . PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; McGee, C.E.; Volk, S.M.; Vanlandingham, D.L.; Weaver, S.C.; Higgs, S. Epistatic roles of E2 glycoprotein mutations in adaption of chikungunya virus to Aedes albopictus and Ae. aegypti mosquitoes . PLoS One 2009, 4, e6835. [Google Scholar] [CrossRef] [PubMed]
- Chanel-Vos, C.; Kielian, M. Second-site revertants of a Semliki Forest virus fusion-block mutation reveal the dynamics of a class II membrane fusion protein. J. Virol. 2006, 80, 6115–6122. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Kielian, M. Site-directed antibodies against the stem region reveal low pH-induced conformational changes of the Semliki Forest virus fusion protein. J. Virol. 2006, 80, 9599–9607. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Kielian, M. Functions of the stem region of the Semliki Forest virus fusion protein during virus fusion and assembly. J. Virol. 2006, 80, 11362–11369. [Google Scholar] [CrossRef] [PubMed]
- Earp, L.J.; Delos, S.E.; Park, H.E.; White, J.M. The many mechanisms of viral membrane fusion proteins. Curr. Topics Microbiol. Immunol. 2005, 285, 25–66. [Google Scholar]
- Barth, B.U.; Suomalainen, M.; Liljeström, P.; Garoff, H. Alphavirus assembly and entry: Role of the cytoplasmic tail of the E1 spike subunit. J. Virol. 1992, 66, 7560–7564. [Google Scholar] [PubMed]
- Hernandez, R.; Sinodis, C.; Horton, M.; Ferreira, D.; Yang, C.; Brown, D.T. Deletions in the transmembrane domain of a sindbis virus glycoprotein alter virus infectivity, stability, and host range. J. Virol. 2003, 77, 12710–12719. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg, M.; Garoff, H. Interactions between the transmembrane segments of the alphavirus E1 and E2 proteins play a role in virus budding and fusion. J. Virol. 2003, 77, 3441–3450. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Willis, J.H.; Sinodis, C.N.; Hernandez, R.; Brown, D.T. Single and multiple deletions in the transmembrane domain of the Sindbis virus E2 glycoprotein identify a region critical for normal virus growth. Virol. 2006, 347, 199–207. [Google Scholar] [CrossRef]
- Liao, M.; Kielian, M. The conserved glycine residues in the transmembrane domain of the Semliki Forest virus fusion protein are not required for assembly and fusion. Virol. 2005, 332, 430–437. [Google Scholar] [CrossRef]
- Paredes, A.M.; Ferreira, D.; Horton, M.; Saad, A.; Tsuruta, H.; Johnston, R.; Klimstra, W.; Ryman, K.; Hernandez, R.; Chiu, W.; Brown, D.T. Conformational changes in Sindbis virions resulting from exposure to low pH and interactions with cells suggest that cell penetration may occur at the cell surface in the absence of membrane fusion. Virol. 2004, 324, 373–386. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Kielian, M.; Chanel-Vos, C.; Liao, M. Alphavirus Entry and Membrane Fusion. Viruses 2010, 2, 796-825. https://doi.org/10.3390/v2040796
Kielian M, Chanel-Vos C, Liao M. Alphavirus Entry and Membrane Fusion. Viruses. 2010; 2(4):796-825. https://doi.org/10.3390/v2040796
Chicago/Turabian StyleKielian, Margaret, Chantal Chanel-Vos, and Maofu Liao. 2010. "Alphavirus Entry and Membrane Fusion" Viruses 2, no. 4: 796-825. https://doi.org/10.3390/v2040796