Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials

 ,
, {kind=link}
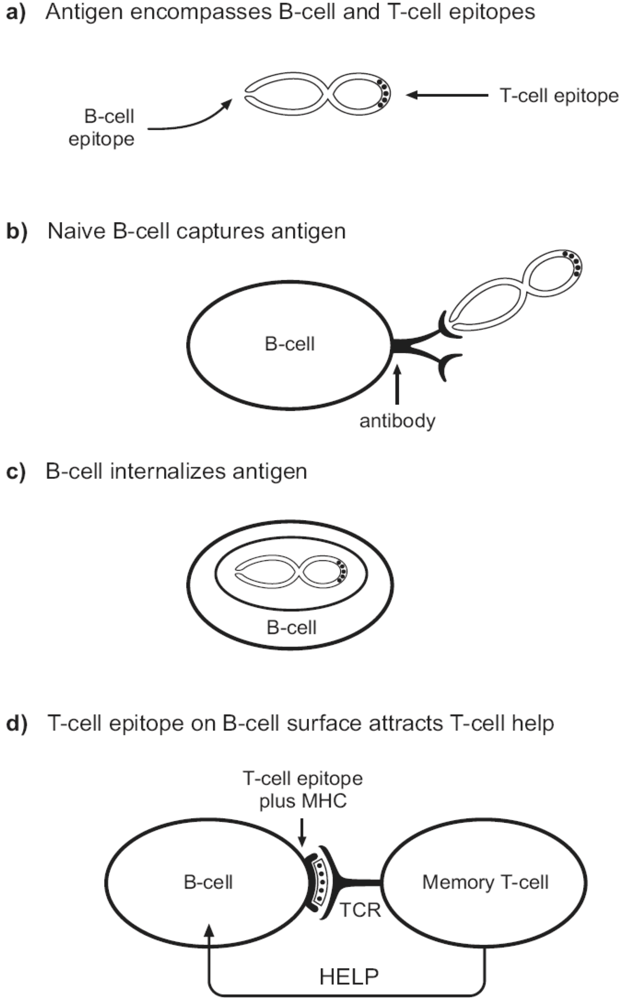
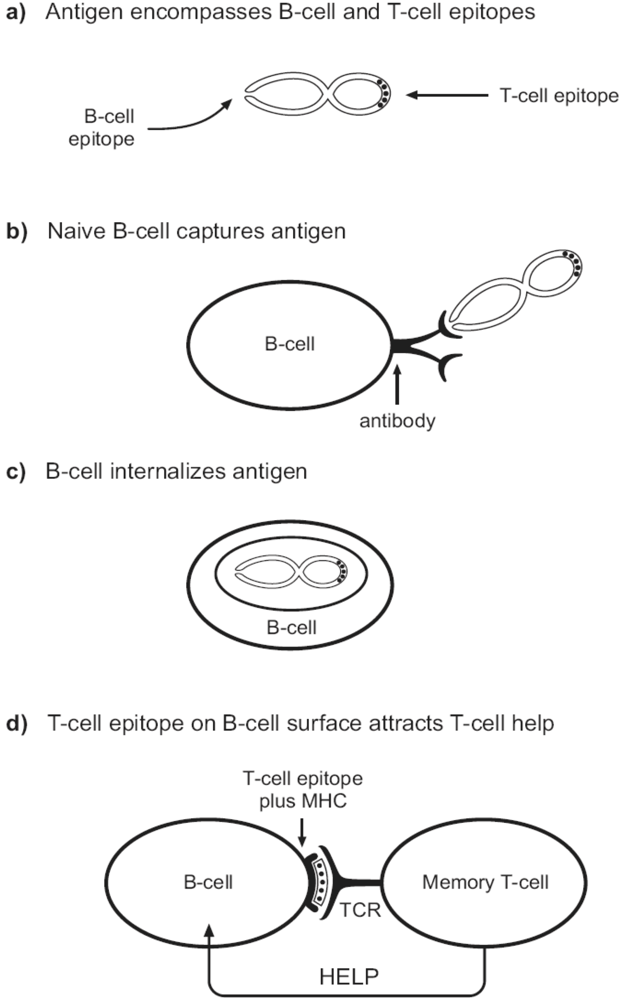{kind=link}
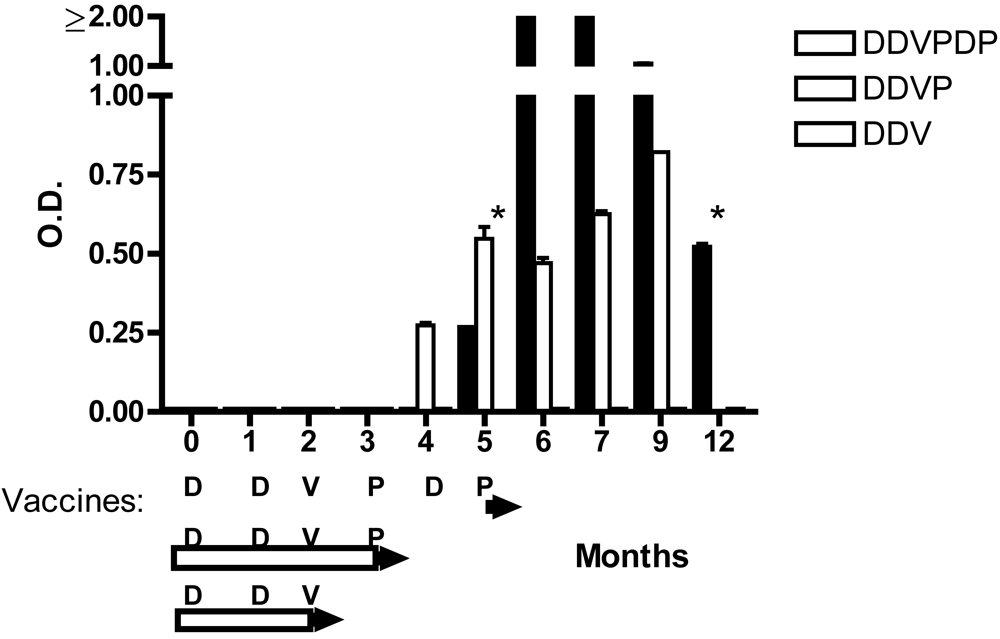{kind=link}
Abstract
:1. Introduction
2. The obstacles presented by HIV-1
2.1. Antigen diversity
2.1.1. Conserved determinants within HIV-1 as targets for vaccine design
2.1.2. Harnessing diverse lymphocytes to target a diverse pathogen
2.2. The chronic nature of HIV-1 infection
3. HIV-1 vaccine development: non recombinant vaccine strategies
3.1. Killed vaccines
3.2. Attenuated Virus Vaccines
4. Recombinant vaccine strategies
4.1. Introduction of recombinant vectors
4.2. HIV-1 gene modifications
5. Prime-boost vaccine regimens
5.1. Prime-boost with one or two delivery vehicles
5.2. Three or more vectors in prime-boost protocols
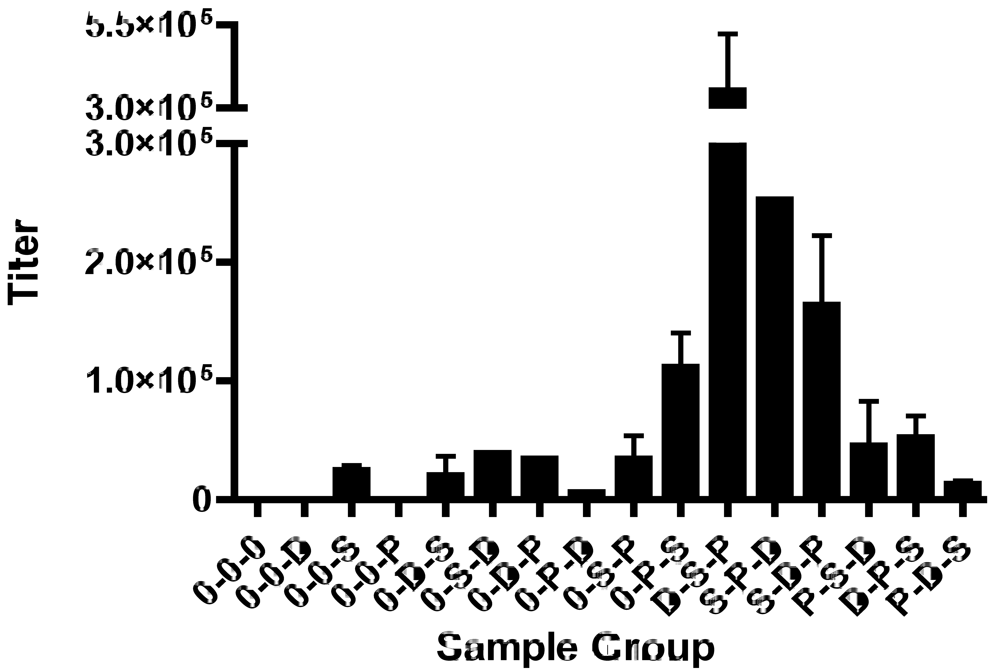
5.3. Complex factors impact prime-boost vaccine outcomes
6. Changing antigens during a prime-boost regimen
6.1. Introducing variant antigens in prime-boost vaccine protocols: lessons from the influenza virus vaccine field and original antigenic sin
6.3. HIV and OAS
7. Changing both vectors and antigens during heterologous prime-boost immunizations in HIV-1 vaccine clinical trials
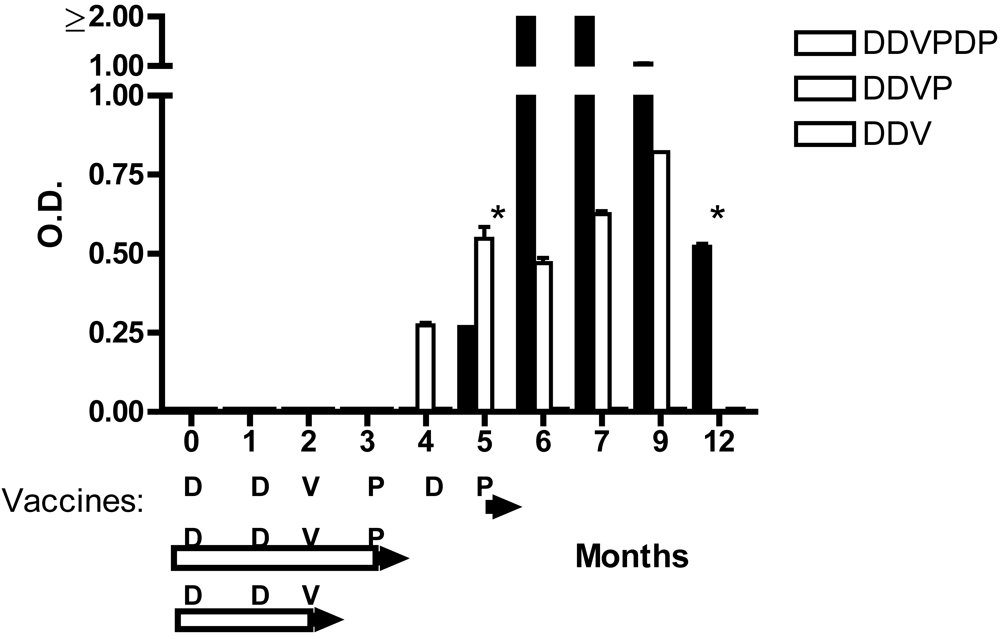
8. Conclusion
Acknowledgments
References
- Hall, H.I.; Song, R.; Rhodes, P.; Prejean, J.; An, Q.; Lee, L.M.; Karon, J.; Brookmeyer, R.; Kaplan, E.H.; McKenna, M.T.; Janssen, R.S. Estimation of HIV incidence in the United States. JAMA 2008, 300, 520–529. [Google Scholar] [CrossRef] [PubMed]
- HIV prevalence estimates--United States, 2006. MMWR Morb Mortal Wkly Rep 2008, 57, 1073–1076. [PubMed]
- McNeil Jr., DG. For first time, AIDS Vaccine shows some success. The New York Times 2009. [Google Scholar]
- Cohen, J. AIDS vaccines. HIV dodges one-two punch. Science 2004, 305, 1545–1547. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Desrosiers, R.C.; Doms, R.W.; Feinberg, M.B.; Gallo, R.C.; Hahn, B.; Hoxie, J.A.; Hunter, E.; Korber, B.; Landay, A.; Lederman, M.M.; Lieberman, J.; McCune, J.M.; Moore, J.P.; Nathanson, N.; Picker, L.; Richman, D.; Rinaldo, C.; Stevenson, M.; Watkins, D.I.; Wolinsky, S.M.; Zack, J.A. Public health. A sound rationale needed for phase III HIV-1 vaccine trials. Science 2004, 303, 316. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.; Franchini, G.; Girard, M.P.; Gotch, F.; Kaleebu, P.; Marthas, M.L.; McChesney, M.B.; McCullough, R.; Mhalu, F.; Salmon-Ceron, D.; Sekaly, R.P.; Van, R.K.; Verrier, B.; Wahren, B.; Weissenbacher, M. Support for the RV144 HIV vaccine trial. Science 2004, 305, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Robb, M.L. Failure of the Merck HIV vaccine: an uncertain step forward. Lancet 2008, 372, 1857–1858. [Google Scholar] [CrossRef] [PubMed]
- Nitayaphan, S.; Pitisuttithum, P.; Karnasuta, C.; Eamsila, C.; de, S.M.; Morgan, P.; Polonis, V.; Benenson, M.; vanCott, T.; Ratto-Kim, S.; Kim, J.; Thapinta, D.; Garner, R.; Bussaratid, V.; Singharaj, P.; el-Habib, R.; Gurunathan, S.; Heyward, W.; Birx, D.; McNeil, J.; Brown, A.E. Safety and immunogenicity of an HIV subtype B and E prime-boost vaccine combination in HIV-negative Thai adults. J. Infect. Dis. 2004, 190, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [PubMed]
- Kuiken, C.; Korber, B.; Shafer, R.W. HIV sequence databases. AIDS Rev. 2003, 5, 52–61. [Google Scholar] [PubMed]
- Gaschen, B.; Taylor, J.; Yusim, K.; Foley, B.; Gao, F.; Lang, D.; Novitsky, V.; Haynes, B.; Hahn, B.H.; Bhattacharya, T.; Korber, B. Diversity considerations in HIV-1 vaccine selection. Science 2002, 296, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Lockey, T.D.; Hurwitz, J.L. Size-heterogeneous sequences mark hot spots for asparagine, serine, and threonine insertions in HIV type 1 envelope. Aids Res. Hum. Retroviruses 1998, 14, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Kunstman, J.; Glowczwskie, J.; Kunstman, K.J.; Egan, M.A.; Peyerl, F.W.; Santra, S.; Kuroda, M.J.; Schmitz, J.E.; Beaudry, K.; Krivulka, G.R.; Lifton, M.A.; Gorgone, D.A.; Wolinsky, S.M.; Letvin, N.L. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J. Virol. 2003, 77, 7367–7375. [Google Scholar] [CrossRef] [PubMed]
- Wrin, T.; Crawford, L.; Sawyer, L.; Weber, P.; Sheppard, H.W.; Hanson, C.V. Neutralizing antibody responses to autologous and heterologous isolates of human immunodeficiency virus. J. Acquir. Immune Defic. Syndr. 1994, 7, 211–219. [Google Scholar] [PubMed]
- Siciliano, S.J.; Kuhmann, S.E.; Weng, Y.; Madani, N.; Springer, M.S.; Lineberger, J.E.; Danzeisen, R.; Miller, M.D.; Kavanaugh, M.P.; DeMartino, J.A.; Kabat, D. A critical site in the core of the CCR5 chemokine receptor required for binding and infectivity of human immunodeficiency virus type 1. J. Biol. Chem. 1999, 274, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, G.J.; Davide, J.P.; Weinhold, K.; Waterbury, J.A.; Profy, A.T.; LEwis, J.A.; Lanlois, A.J.; Dreesman, G.R.; Boswell, R.N.; Shadduck, P.; Holley, L.H.; Karplus, M.; Bolognesi, D.P.; Matthews, T.J.; Emini, E.A.; Putney, S.D. Conserved sequence and structural elements in the HIV-1 principal neutralizing determinant. Science 1990, 249, 932–935. [Google Scholar] [PubMed]
- White-Scharf, M.E.; Potts, B.J.; Smith, L.M.; Sokolowski, K.A.; Rusche, J.R.; Silver, S. Broadly neutralizing monoclonal antibodies to the V3 region of HIV-1 can be elicited by peptide immunization. Virology 1993, 192, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Pinter, A.; Honnen, W.J.; Kayman, S.C.; Trochev, O.; Wu, Z. Potent neutralization of primary HIV-1 isolates by antibodies directed against epitopes present in the V1/V2 domain of HIV-1 gp120. Vaccine 1998, 16, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Kostyu, D.D.; Cox, J.; Dawson, D.V.; Flores, J.; Weinhold, K.J.; Osmanov, S. Identification of highly conserved and broadly cross-reactive HIV type 1 cytotoxic T lymphocyte epitopes as candidate immunogens for inclusion in Mycobacterium bovis BCG-vectored HIV vaccines. Aids Res. Hum. Retroviruses 2000, 16, 1433–1443. [Google Scholar] [PubMed]
- Chow, Y.H.; Wei, O.L.; Phogat, S.; Sidorov, I.A.; Fouts, T.R.; Broder, C.C.; Dimitrov, D.S. Conserved structures exposed in HIV-1 envelope glycoproteins stabilized by flexible linkers as potent entry inhibitors and potential immunogens. Biochemistry 2002, 41, 7176–7182. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; Lehrman, J.K.; Priddy, F.H.; Olsen, O.A.; Frey, S.M.; Hammond, P.W.; Miiro, G.; Serwanga, J.; Pozniak, A.; McPhee, D.; Manigart, O.; Mwananyanda, L.; Karita, E.; Inwoley, A.; Jaoko, W.; Dehovitz, J.; Bekker, L.G.; Pitisuttithum, P.; Paris, R.; Allen, S.; Kaminsky, S.; Zamb, T.; Moyle, M.; Koff, W.C.; Poignard, P.; Burton, D.R. Broad and Potent Neutralizing Antibodies from an African Donor Reveal a New HIV-1 Vaccine Target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, I.K.; Stamatatos, L.; Kan, E.; Vajdy, M.; Lian, Y.; Hilt, S.; Martin, L.; Vita, C.; Zhu, P.; Roux, K.H.; Vojtech, L.; Montefiori, C.; Donnelly, J.; Ulmer, J.B.; Barnett, S.W. Purification, characterization, and immunogenicity of a soluble trimeric envelope protein containing a partial deletion of the V2 loop derived from SF162, an R5-tropic human immunodeficiency virus type 1 isolate. J. Virol. 2003, 77, 11244–11259. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Pancera, M.; Kwong, P.D.; Kolchinsky, P.; Grundner, C.; Wang, L.; Hendrickson, W.A.; Sodroski, J.; Wyatt, R. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology 2003, 313, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.K.; Kong, W.P.; Wu, B.Y.; Yang, Z.Y.; Friborg, J.; Ling, X.; King, S.R.; Montefiori, D.C.; Nabel, G.J. Modifications of the human immunodeficiency virus envelope glycoprotein enhance immunogenicity for genetic immunization. J. Virol. 2002, 76, 5357–5368. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, R.A.; Follis, K.E.; Trahey, M.; Scarborough, J.D.; Littman, D.R.; Nunberg, J.H. Fusion-competent vaccines: broad neutralization of primary isolates of HIV (Retraction: Science2002, 296, 1025). Science 1999, 283, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Fleming, J.; St Clair, E.W.; Katinger, H.; Stiegler, G.; Kunert, R.; Robinson, J.; Scearce, R.M.; Plonk, K.; Staats, H.F.; Ortel, T.L.; Liao, H.X.; Alam, S.M. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 2005, 308, 1906–1908. [Google Scholar] [CrossRef] [PubMed]
- Murphy K, Travers P. Garland Science: New York, NY, USA, 2008.
- Gerhard, W.; Yewdell, J.; Frankel, M.E.; Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 1981, 290, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Mouquet, H.; Feldhahn, N.; Seaman, M.S.; Velinzon, K.; Pietzsch, J.; Ott, R.G.; Anthony, R.M.; Zebroski, H.; Hurley, A.; Phogat, A.; Chakrabarti, B.; Li, Y.; Connors, M.; Pereyra, F.; Walker, B.D.; Wardemann, H.; Ho, D.; Wyatt, R.T.; Mascola, J.R.; Ravetch, J.V.; Nussenzweig, M.C. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 2009, 458, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Clark, H.F.; Offit, P.A.; Plotkin, S.A.; Heaton, P.M. The new pentavalent rotavirus vaccine composed of bovine (strain WC3) -human rotavirus reassortants. Pediatr. Infect. Dis. J. 2006, 25, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Krogstad, P.; Cherry, J.D. Quadrivalent human vaccine - a call to action and for additional research. Pediatr. Res. 2007, 62, 527. [Google Scholar] [CrossRef] [PubMed]
- Biagini, R.E.; Schlottmann, S.A.; Sammons, D.L.; Smith, J.P.; Snawder, J.C.; Striley, C.A.; MacKenzie, B.A.; Weissman, D.N. Method for simultaneous measurement of antibodies to 23 pneumococcal capsular polysaccharides. Clin. Diagn. Lab. Immunol. 2003, 10, 744–750. [Google Scholar] [PubMed]
- Vesikari, T.; Wysocki, J.; Chevallier, B.; Karvonen, A.; Czajka, H.; Arsene, J.P.; Lommel, P.; Dieussaert, I.; Schuerman, L. Immunogenicity of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) compared to the licensed 7vCRM vaccine. Pediatr. Infect. Dis. J. 2009, 28, S66–S76. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, S. Prevention of varicella and zoster by live attenuated VZV vaccine. Front Biosci. 2008, 13, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Varicella-related deaths--United States, January 2003-June 2004. MMWR Morb. Mortal Wkly. Rep. 2005, 54, 272–274. [PubMed]
- Bjorklund, A.; Aschan, J.; Labopin, M.; Remberger, M.; Ringden, O.; Winiarski, J.; Ljungman, P. Risk factors for fatal infectious complications developing late after allogeneic stem cell transplantation. Bone Marrow Transpl. 2007, 40, 1055–1062. [Google Scholar] [CrossRef]
- Murphey-Corb, M.; Martin, L.N.; vison-Fairburn, B.; Montelaro, R.C.; Miller, M.; West, M.; Ohkawa, S.; Baskin, G.B.; Zhang, J.Y.; Putney, S.D. A formalin-inactivated whole SIV vaccine confers protection in macaques. Science 1989, 246, 1293–1297. [Google Scholar] [PubMed]
- Carmichael, A.J.; Sissons, J.G. Vaccines against HIV. QJM 1995, 88, 77–79. [Google Scholar] [PubMed]
- Stott, E.J. Anti-cell antibody in macaques. Nature 1991, 353, 393. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, H.W. Inactivated- or Killed-Virus HIV/AIDS Vaccines. Curr. Drug Targets Infect. Disord. 2005, 5, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.D.; Kirchhoff, F.; Czajak, S.C.; Sehgal, P.K.; Desrosiers, R.C. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 1992, 258, 1938–1941. [Google Scholar] [PubMed]
- Hofmann-Lehmann, R.; Vlasak, J.; Williams, A.L.; Chenine, A.L.; McClure, H.M.; Anderson, D.C.; O'Neil, S.; Ruprecht, R.M. Live attenuated, nef-deleted SIV is pathogenic in most adult macaques after prolonged observation. AIDS 2003, 17, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Stahl-Hennig, C.; Dittmer, U.; Nisslein, T.; Pekrun, K.; Petry, H.; Jurkiewicz, E.; Fuchs, D.; Wachter, H.; Rud, E.W.; Hunsmann, G. Attenuated SIV imparts immunity to challenge with pathogenic spleen-derived SIV but cannot prevent repair of the nef deletion. Immunol. Lett. 1996, 51, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Whatmore, A.M.; Cook, N.; Hall, G.A.; Sharpe, S.; Rud, E.W.; Cranage, M.P. Repair and evolution of nef in vivo modulates simian immunodeficiency virus virulence. J. Virol. 1995, 69, 5117–5123. [Google Scholar] [PubMed]
- Baba, T.W.; Jeong, Y.S.; Pennick, D.; Bronson, R.; Greene, M.F.; Ruprecht, R.M. Pathogenicity of live, attenuated SIV after mucosal infection of neonatal macaques. Science 1995, 267, 1820–1825. [Google Scholar] [PubMed]
- Wyand, M.S.; Manson, K.H.; Garcia-Moll, M.; Montefiori, D.; Desrosiers, R.C. Vaccine protection by a triple deletion mutant of simian immunodeficiency virus. J. Virol. 1996, 70, 3724–3733. [Google Scholar] [PubMed]
- Sealy, R.; Zhan, X.; Lockey, T.D.; Martin, L.; Blanchard, J.; Traina-Dorge, V.; Hurwitz, J.L. SHIV infection protects against heterologous pathogenic SHIV challenge in macaques: a gold-standard for HIV-1 vaccine development? Curr. HIV Res. 2009, 7, 497–503. [Google Scholar] [PubMed]
- Cranage, M.P.; Whatmore, A.M.; Sharpe, S.A.; Cook, N.; Polyanskaya, N.; Leech, S.; Smith, J.D.; Rud, E.W.; Dennis, M.J.; Hall, G.A. Macaques infected with live attenuated SIVmac are protected against superinfection via the rectal mucosa. Virology 1997, 229, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Stahl-Hennig, C.; Dittmer, U.; Nisslein, T.; Petry, H.; Jurkiewicz, E.; Fuchs, D.; Wachter, H.; Matz-Rensing, K.; Kuhn, E.M.; Kaup, F.J.; Rud, E.W.; Hunsmann, G. Rapid development of vaccine protection in macaques by live-attenuated simian immunodeficiency virus. J. Gen. Virol. 1996, 77, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Stephens, E.B.; Joag, S.V.; Atkinson, B.; Sahni, M.; Li, Z.; Foresman, L.; Adany, I.; Narayan, O. Infected macaques that controlled replication of SIVmac or nonpathogenic SHIV developed sterilizing resistance against pathogenic SHIV(KU-1). Virology 1997, 234, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, T.; Hardison, R.C.; Lacy, E.; Lauer, J.; O'Connell, C.; Quon, D.; Sim, G.K.; Efstratiadis, A. The isolation of structural genes from libraries of eucaryotic DNA. Cell 1978, 15, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M.; Hogness, D.S. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Proc. Natl. Acad. Sci. USA 1975, 72, 3961–3965. [Google Scholar] [CrossRef]
- Shubeita, H.E.; Sambrook, J.F.; McCormick, A.M. Molecular cloning and analysis of functional cDNA and genomic clones encoding bovine cellular retinoic acid-binding protein. Proc. Natl. Acad. Sci. USA 1987, 84, 5645–5649. [Google Scholar] [CrossRef]
- Breton, M.; Zhao, C.; Ouellette, M.; Tremblay, M.J.; Papadopoulou, B. A recombinant non-pathogenic Leishmania vaccine expressing human immunodeficiency virus 1 (HIV-1) Gag elicits cell-mediated immunity in mice and decreases HIV-1 replication in human tonsillar tissue following exposure to HIV-1 infection. J. Gen. Virol. 2007, 88, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Farmer, J.L.; Hampton, R.G.; Boots, E. Flow cytometric assays for monitoring production of recombinant HIV-1 gp160 in insect cells infected with a baculovirus expression vector. J. Virol. Meth. 1989, 26, 279–290. [Google Scholar] [CrossRef]
- Hanke, T.; Barnfield, C.; Wee, E.G.; Agren, L.; Samuel, R.V.; Larke, N.; Liljestrom, P. Construction and immunogenicity in a prime-boost regimen of a Semliki Forest virus-vectored experimental HIV clade A vaccine. J. Gen. Virol. 2003, 84, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Berglund, P.; Quesada-Rolander, M.; Putkonen, P.; Biberfeld, G.; Thorstensson, R.; Liljestrom, P. Outcome of immunization of cynomolgus monkeys with recombinant Semliki Forest virus encoding human immunodeficiency virus type 1 envelope protein and challenge with a high dose of SHIV-4 virus. Aids Res. Hum. Retroviruses 1997, 13, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; West, A.; Reap, E.; MacDonald, G.; Collier, M.; Dryga, S.; Maughan, M.; Connell, M.; Walker, C.; McGrath, K.; Cecil, C.; Ping, L.H.; Frelinger, J.; Olmsted, R. Alphavirus replicon particles as candidate HIV vaccines. IUBMB Life 2002, 53, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, M.D.; Natuk, R.J.; Chengalvala, M.; Chanda, P.K.; Murthy, K.K.; Murthy, S.; Mizutani, S.; Lee, S.G.; Wade, M.S.; Bhat, B.M. Immunogenicity of recombinant adenovirus-human immunodeficiency virus vaccines in chimpanzees following intranasal administration. Aids Res. Hum. Retroviruses 1994, 10, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Xin, K.Q.; Urabe, M.; Yang, J.; Nomiyama, K.; Mizukami, H.; Hamajima, K.; Nomiyama, H.; Saito, T.; Imai, M.; Monahan, J.; Okuda, K.; Ozawa, K.; Okuda, K. A novel recombinant adeno-associated virus vaccine induces a long-term humoral immune response to human immunodeficiency virus. Hum. Gene Ther. 2001, 12, 1047–1061. [Google Scholar] [PubMed]
- Lin, J.; Calcedo, R.; Vandenberghe, L.H.; Bell, P.; Somanathan, S.; Wilson, J.M. A new genetic vaccine platform based on an adeno-associated virus isolated from a rhesus macaque. J. Virol. 2009, 83, 12738–12750. [Google Scholar] [CrossRef] [PubMed]
- Rencher, S.D.; Lockey, T.D.; Srinivas, R.V.; Owens, R.J.; Hurwitz, J.L. Eliciting HIV-1 envelope-specific antibodies with mixed vaccinia virus recombinants. Vaccine 1997, 15, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Amara, R.R.; Villinger, F.; Altman, J.D.; Lydy, S.L.; O'Neil, S.P.; Staprans, S.I.; Montefiori, D.C.; Xu, Y.; Herndon, J.G.; Wyatt, L.S.; Candido, M.A.; Kozyr, N.L.; Earl, P.L.; Smith, J.M.; Ma, H.L.; Grimm, B.D.; Hulsey, M.L.; McClure, H.M.; McNicholl, J.M.; Moss, B.; Robinson, H.L. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Vaccine 2002, 20, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Amara, R.R.; Smith, J.M.; Staprans, S.I.; Montefiori, D.C.; Villinger, F.; Altman, J.D.; O'Neil, S.P.; Kozyr, N.L.; Xu, Y.; Wyatt, L.S.; Earl, P.L.; Herndon, J.G.; McNicholl, J.M.; McClure, H.M.; Moss, B.; Robinson, H.L. Critical role for Env as well as Gag-Pol in control of a simian-human immunodeficiency virus 89.6P challenge by a DNA prime/recombinant modified vaccinia virus Ankara vaccine. J. Virol. 2002, 76, 6138–6146. [Google Scholar] [CrossRef] [PubMed]
- Clements-Mann, M.L.; Weinhold, K.; Matthews, T.J.; Graham, B.S.; Gorse, G.J.; Keefer, M.C.; McElrath, M.J.; Hsieh, R.-H.; Mestecky, J.; Zolla-Pazner, S.; Mascola, J.; Schwartz, D.; Siliciano, R.; Corey, L.; Wright, P.F.; Belshe, R.; Dolin, R.; Jackson, S.; Xu, S.; Fast, P.; Walker, M.C.; Stablein, D.; Excler, J.-L.; Tartaglia, J.; Duliege, A.-M.; Sinangil, F.; Paoletti, E.; NIAID AIDS VACCINE EVALUATION GROUP. Immune responses to human immunodeficiency virus (HIV) type 1 induced by canarypox expressing HIV-1-MN gp120, HIV-1-SF2 recombinant gp120, or both vaccines in seronegative adults. J. Infect. Dis. 1998, 177, 1246. [Google Scholar]
- Pialoux, G.; Excler, J.-L.; Rivière, Y.; Gonzalez-Canali, G.; Feuillie, V.; Coulaud, P.; Gluckman, J.-C.; Matthews, T.J.; Meignier, B.; Kieny, M.-P.; Gonnet, P.; Diaz, I.; Méric, C.; Paoletti, E.; Tartaglia, J.; Salomon, H.; Plotkin, S.; The Agis Group; L'Agence Natl de Recherche sur le Sida. A prime-boost approach to HIV preventive vaccine using a recombinant canarypox virus expressing glycoprotein 160 (MN) followed by a recombinant glycoprotein 160 (MN/LAI). Aids Res. Hum. Retroviruses 1995, 11, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.J.; Zhao, A.; Best, S.J.; Chandler, J.D.; Boyle, D.B.; Ramshaw, I.A. Enhanced T-cell immunogenicity and protective efficacy of a human immunodeficiency virus type 1 vaccine regimen consisting of consecutive priming with DNA and boosting with recombinant fowlpox virus. J. Virol. 1998, 72, 10180–10188. [Google Scholar] [PubMed]
- Stubbs, A.C.; Martin, K.S.; Coeshott, C.; Skaates, S.V.; Kuritzkes, D.R.; Bellgrau, D.; Franzusoff, A.; Duke, R.C.; Wilson, C.C. Whole recombinant yeast vaccine activates dendritic cells and elicits protective cell-mediated immunity. Nat. Med. 2001, 7, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Leiner, I.; Kerksiek, K.; Buonocore, L.; Pamer, E.; Rose, J.K. Robust recall and long-term memory T-cell responses induced by prime-boost regimens with heterologous live viral vectors expressing human immunodeficiency virus type 1 Gag and Env proteins. J. Virol. 2002, 76, 7506–7517. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, M.; Zhou, C.; Zhao, X.; Iijima, N.; Frankel, F.R. Novel vaccination protocol with two live mucosal vectors elicits strong cell-mediated immunity in the vagina and protects against vaginal virus challenge. J. Immunol. 2008, 180, 2504–2513. [Google Scholar] [PubMed]
- Humbert, M.; Rasmussen, R.A.; Ong, H.; Kaiser, F.M.; Hu, S.L.; Ruprecht, R.M. Inducing cross-clade neutralizing antibodies against HIV-1 by immunofocusing. PLoS ONE 2008, 3, e3937. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, A.; Roshorm, Y.; Lockett, L.J.; Xu, Z.Z.; Hopkins, R.; Shaw, J.; Both, G.W.; Hanke, T. Ovine atadenovirus, a novel and highly immunogenic vector in prime-boost studies of a candidate HIV-1 vaccine. Vaccine 2009, 28, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, U.D.; Larsen, M.H.; Kim, J.; Porcelli, S.A.; Jacobs, W.R.; Fennelly, G.J. Recombinant pro-apoptotic Mycobacterium tuberculosis generates CD8+ T cell responses against human immunodeficiency virus type 1 Env and M. tuberculosis in neonatal mice. Vaccine 2009, 28, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von, L.D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- de Goede, A.L.; Boers, P.H.; Dekker, L.J.; Osterhaus, A.D.; Gruters, R.A.; Rimmelzwaan, G.F. Characterization of recombinant influenza A virus as a vector for HIV-1 p17Gag. Vaccine 2009, 27, 5735–5739. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P.; Geng, Y.; Ng, H.L.; Yang, O.O.; Krogstad, P. Packaging limits and stability of HIV-1 sequences in a coxsackievirus B vector. Vaccine 2009, 27, 3992–4000. [Google Scholar] [CrossRef] [PubMed]
- Lemiale, F.; Korokhov, N. Lentiviral vectors for HIV disease prevention and treatment. Vaccine 2009, 27, 3443–3449. [Google Scholar] [CrossRef] [PubMed]
- Chin'ombe, N.; Bourn, W.R.; Williamson, A.L.; Shephard, E.G. Oral vaccination with a recombinant Salmonella vaccine vector provokes systemic HIV-1 subtype C Gag-specific CD4+ Th1 and Th2 cell immune responses in mice. Virol. J. 2009, 6, 87. [Google Scholar] [CrossRef]
- Chege, G.K.; Thomas, R.; Shephard, E.G.; Meyers, A.; Bourn, W.; Williamson, C.; Maclean, J.; Gray, C.M.; Rybicki, E.P.; Williamson, A.L. A prime-boost immunisation regimen using recombinant BCG and Pr55(gag) virus-like particle vaccines based on HIV type 1 subtype C successfully elicits Gag-specific responses in baboons. Vaccine 2009, 27, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Sanford, H.B.; Garry, D.; Lang, S.; Klumpp, S.A.; Watanabe, D.; Bronson, R.T.; Lifson, J.D.; Rosati, M.; Pavlakis, G.N.; Felber, B.K.; Knipe, D.M.; Desrosiers, R.C. Ability of herpes simplex virus vectors to boost immune responses to DNA vectors and to protect against challenge by simian immunodeficiency virus. Virology 2007, 357, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.J.; De, R.R.; Mokhonov, V.V.; Mokhonova, E.I.; Fernandez, C.S.; Alcantara, S.; Rollman, E.; Mason, R.D.; Loh, L.; Peut, V.; Reece, J.C.; Wang, X.J.; Wilson, K.M.; Suhrbier, A.; Khromykh, A. Evaluation of recombinant Kunjin replicon SIV vaccines for protective efficacy in macaques. Virology 2008, 374, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Liniger, M.; Zuniga, A.; Morin, T.N.; Combardiere, B.; Marty, R.; Wiegand, M.; Ilter, O.; Knuchel, M.; Naim, H.Y. Recombinant measles viruses expressing single or multiple antigens of human immunodeficiency virus (HIV-1) induce cellular and humoral immune responses. Vaccine 2009, 27, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Nasar, F.; Megati, S.; Luckay, A.; Lee, M.; Udem, S.A.; Eldridge, J.H.; Egan, M.A.; Emini, E.; Clarke, D.K. Prime-Boost Vaccination with Recombinant Mumps Virus and Recombinant Vesicular Stomatitis Virus Vectors Elicits an Enhanced HIV-1 Gag-Specific Cellular Immune Response in Rhesus Macaques. J. Virol. 2009. [Google Scholar]
- Faul, E.J.; Wanjalla, C.N.; McGettigan, J.P.; Schnell, M.J. Interferon-beta expressed by a rabies virus-based HIV-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology 2008, 382, 226–238. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.S.; Klaas, M.; Gonzalez-Rabade, N.; Poage, M.; Badillo-Corona, J.A.; Zhou, F.; Karcher, D.; Bock, R.; Gray, J.C.; Dix, P.J. Plastid transformation of high-biomass tobacco variety Maryland Mammoth for production of human immunodeficiency virus type 1 (HIV-1) p24 antigen. Plant Biotechnol. J. 2008. [Google Scholar]
- Robinson, H.L.; Hunt, L.A.; Webster, R.G. Protection against a lethal influenza virus challenge by immunization with a haemagglutinin-expressing plasmid DNA. Vaccine 1993, 11, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Fynan, E.F.; Santoro, J.C.; Robinson, H. Protection of ferrets against influenza challenge with a DNA vaccine to the haemagglutinin. Vaccine 1994, 12, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Peet, N.; Delves, P.J.; de Souza, B.; Cambridge, G.; Wilkinson, L.; Klonisch, T.; Matear, P.; Vyakarnam, A.; Lund, T. The immune response to HIV gp120 induced by nucleic acid immunization (NAI). Ann. NY Acad. Sci. 1995, 772, 257–260. [Google Scholar] [CrossRef]
- Yao, Q.; Bu, Z.; Vzorov, A.; Yang, C.; Compans, R.W. Virus-like particle and DNA-based candidate AIDS vaccines. Vaccine 2003, 21, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Douglas, A.R.; Skehel, J.J.; Daniels, R.S. Analysis of murine antibody responses to baculovirus-expressed human immunodeficiency virus type 1 envelope glycoproteins. J. Gen. Virol. 1994, 75, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Tonks, M.; Carbone, F.; Ryskamp, T.; Morrow, W.J.; Hanna, N. Induction of antigen-specific class I-restricted cytotoxic T cells by soluble proteins in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 8308–8312. [Google Scholar] [CrossRef]
- Crotty, S.; Felgner, P.; Davies, H.; Glidewell, J.; Villarreal, L.; Ahmed, R. Cutting edge: long-term B cell memory in humans after smallpox vaccination. J. Immunol. 2003, 171, 4969–4973. [Google Scholar] [PubMed]
- Amanna, I.J.; Slifka, M.K.; Crotty, S. Immunity and immunological memory following smallpox vaccination. Immunol. Rev. 2006, 211, 320–337. [Google Scholar] [CrossRef] [PubMed]
- Hyland, L.; Sangster, M.; Sealy, R.; Coleclough, C. Respiratory virus infection of mice provokes a permanent humoral immune response. J. Virol. 1994, 68, 6083–6086. [Google Scholar] [PubMed]
- Barnett, S.W.; Srivastava, I.K.; Ulmer, J.B.; Donnelly, J.J.; Rappuoli, R. Development of V2-deleted trimeric envelope vaccine candidates from human immunodeficiency virus type 1 (HIV-1) subtypes B and C. Microbes Infect. 2005, 7, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.; Smith, U.R.; Gilbert, P.; McLane, M.F.; Chigwedere, P.; Williamson, C.; Ndung'u, T.; Klein, I.; Chang, S.Y.; Peter, T.; Thior, I.; Foley, B.T.; Gaolekwe, S.; Rybak, N.; Gaseitsiwe, S.; Vannberg, F.; Marlink, R.; Lee, T.H.; Essex, M. Human immunodeficiency virus type 1 subtype C molecular phylogeny: consensus sequence for an AIDS vaccine design? J. Virol. 2002, 76, 5435–5451. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Muldoon, M.; Theiler, J.; Gao, F.; Gupta, R.; Lapedes, A.; Hahn, B.H.; Wolinsky, S.; Bhattacharya, T. Timing the ancestor of the HIV-1 pandemic strains. Science 2000, 288, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, J.; Yoon, H.; Kuiken, C.; Yusim, K.; Perkins, S.; Theiler, J.; Bhattacharya, T.; Korber, B.; Fischer, W. Web-based design and evaluation of T-cell vaccine candidates. Bioinformatics 2008, 24, 1639–1640. [Google Scholar] [CrossRef] [PubMed]
- Weaver, E.A.; Lu, Z.; Camacho, Z.T.; Moukdar, F.; Liao, H.X.; Ma, B.J.; Muldoon, M.; Theiler, J.; Nabel, G.J.; Letvin, N.L.; Korber, B.T.; Hahn, B.H.; Haynes, B.F.; Gao, F. Cross-subtype T-cell immune responses induced by a human immunodeficiency virus type 1 group m consensus env immunogen. J. Virol. 2006, 80, 6745–6756. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Koenig, S.; Moss, B. Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. J. Virol. 1991, 65, 31–41. [Google Scholar] [PubMed]
- Thomson, S.A.; Jaramillo, A.B.; Shoobridge, M.; Dunstan, K.J.; Everett, B.; Ranasinghe, C.; Kent, S.J.; Gao, K.; Medveckzy, J.; Ffrench, R.A.; Ramshaw, I.A. Development of a synthetic consensus sequence scrambled antigen HIV-1 vaccine designed for global use. Vaccine 2005, 23, 4647–4657. [Google Scholar] [CrossRef] [PubMed]
- D'Costa, S.; Slobod, K.S.; Webster, R.G.; White, S.W.; Hurwitz, J.L. Structural features of HIV envelope defined by antibody escape mutant analysis. Aids Res. Hum. Retroviruses 2001, 17, 1205–1209. [Google Scholar] [PubMed]
- Sealy, R.; Chaka, W.; Surman, S.; Brown, S.A.; Cresswell, P.; Hurwitz, J.L. Target peptide sequence within infectious human immunodeficiency virus type 1 does not ensure envelope-specific T-helper cell reactivation: influences of cysteine protease and gamma interferon-induced thiol reductase activities. Clin. Vaccine Immunol. 2008, 15, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Arunachalam, B.; Phan, U.T.; Dong, C.; Garrett, W.S.; Cannon, K.S.; Alfonso, C.; Karlsson, L.; Flavell, R.A.; Cresswell, P. Defective antigen processing in GILT-free mice. Science 2001, 294, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Sjolander, S.; Bolmstedt, A.; Akerblom, L.; Horal, P.; Olofsson, S.; Morein, B.; Sjolander, A. N-linked glycans in the CD4-binding domain of human immunodeficiency virus type 1 envelope glycoprotein gp160 are essential for the in vivo priming of T cells recognizing an epitope located in their vicinity. Virology 1996, 215, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Esparza, J.; Heyward, W.L.; Osmanov, S. HIV vaccine development: from basic research to human trials. AIDS 1996, 10, S123–S132. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.L.; Klaniecki, J.; Dykers, T.; Sridhar, P.; Travis, B.M. Neutralizing antibodies against HIV-1 BRU and SF2 isolates generated in mice immunized with recombinant vaccinia virus expressing HIV-1 (BRU) envelope glycoproteins and boosted with homologous gp160. Aids Res. Hum. Retroviruses 1991, 7, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-L.; Abrams, K.; Barber, G.N.; Moran, P.; Zarling, J.M.; Langlois, A.J.; Kuller, L.; Morton, W.R.; Benveniste, R.E. Protection of macaques against SIV infection by subunit vaccines of SIV envelope glycoprotein gp160. Science 1992, 255, 456–459. [Google Scholar] [PubMed]
- Richmond, J.F.L.; Mustafa, F.; Lu, S.; Santoro, J.C.; Weng, J.; O'Connell, M.; Fenyo, E.M.; Hurwitz, J.L.; Montefiori, D.C.; Robinson, H.L. Screening of HIV-1 Env glycoproteins for the ability to raise neutralizing antibody using DNA immunization and recombinant vaccinia virus boosting. Virology 1997, 230, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.J.; Leong, K.H.; Ramshaw, I.A. DNA vaccination against virus infection and enhancement of antiviral immunity following consecutive immunization with DNA and viral vectors. Immunol. Cell Biol. 1997, 75, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Ramshaw, I.A.; Ramsay, A.J. The prime-boost strategy: exciting prospects for improved vaccination. Immunol. Today JID - 8008346 2000, 21, 163–165. [Google Scholar] [CrossRef]
- Estcourt, M.J.; Ramsay, A.J.; Brooks, A.; Thomson, S.A.; Medveckzy, C.J.; Ramshaw, I.A. Prime-boost immunization generates a high frequency, high-avidity CD8(+) cytotoxic T lymphocyte population. Int. Immunol. 2002, 14, 31–37. [Google Scholar] [CrossRef] [PubMed]
- McConkey, S.J.; Reece, W.H.; Moorthy, V.S.; Webster, D.; Dunachie, S.; Butcher, G.; Vuola, J.M.; Blanchard, T.J.; Gothard, P.; Watkins, K.; Hannan, C.M.; Everaere, S.; Brown, K.; Kester, K.E.; Cummings, J.; Williams, J.; Heppner, D.G.; Pathan, A.; Flanagan, K.; Arulanantham, N.; Roberts, M.T.; Roy, M.; Smith, G.L.; Schneider, J.; Peto, T.; Sinden, R.E.; Gilbert, S.C.; Hill, A.V. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 2003, 9, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Winfrey, S.; Yang, Z.Y.; Xu, L.; Rao, S.S.; Srivastava, I.; Barnett, S.W.; Nabel, G.J.; Mascola, J.R. Efficient protein boosting after plasmid DNA or recombinant adenovirus immunization with HIV-1 vaccine constructs. Vaccine 2007, 25, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Bart, P.A.; Stohr, W.; Tapia, G.; Garcia, M.; Medjitna-Rais, E.; Burnet, S.; Cellerai, C.; Erlwein, O.; Barber, T.; Moog, C.; Liljestrom, P.; Wagner, R.; Wolf, H.; Kraehenbuhl, J.P.; Esteban, M.; Heeney, J.; Frachette, M.J.; Tartaglia, J.; McCormack, S.; Babiker, A.; Weber, J.; Pantaleo, G. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J. Exp. Med. 2008, 205, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.S.; Xu, L.; Beaudry, K.; Martin, K.L.; Beddall, M.H.; Miura, A.; Sambor, A.; Chakrabarti, B.K.; Huang, Y.; Bailer, R.; Koup, R.A.; Mascola, J.R.; Nabel, G.J.; Letvin, N.L. Multiclade human immunodeficiency virus type 1 envelope immunogens elicit broad cellular and humoral immunity in rhesus monkeys. J. Virol. 2005, 79, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.A.; Chong, S.Y.; Megati, S.; Montefiori, D.C.; Rose, N.F.; Boyer, J.D.; Sidhu, M.K.; Quiroz, J.; Rosati, M.; Schadeck, E.B.; Pavlakis, G.N.; Weiner, D.B.; Rose, J.K.; Israel, Z.R.; Udem, S.A.; Eldridge, J.H. Priming with plasmid DNAs expressing interleukin-12 and simian immunodeficiency virus gag enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. Aids Res. Hum. Retroviruses 2005, 21, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Kalyanaraman, V.S.; Nair, B.C.; Whitney, S.; Keen, T.; Hocker, L.; Hudacik, L.; Rose, N.; Mboudjeka, I.; Shen, S.; Wu-Chou, T.H.; Montefiori, D.; Mascola, J.; Markham, P.; Lu, S. Immunization of rhesus macaques with a polyvalent DNA prime/protein boost human immunodeficiency virus type 1 vaccine elicits protective antibody response against simian human immunodeficiency virus of R5 phenotype. Virology 2006, 348, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Sun, Y.; Parvani, J.G.; Philippon, V.; Wyand, M.S.; Manson, K.; Gomez-Yafal, A.; Mazzara, G.; Panicali, D.; Markham, P.D.; Montefiori, D.C.; Letvin, N.L. Heterologous prime/boost immunization of rhesus monkeys by using diverse poxvirus vectors. J. Virol. 2007, 81, 8563–8570. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, M.D.; Natuk, R.; Myagkikh, M.; Kalyan, N.; Aldrich, K.; Sinangil, F.; Alipanah, S.; Murthy, S.C.; Chanda, P.K.; Nigida, S.M.; Markham, P.D.; Zolla-Pazner, S.; Steimer, K.; Wade, M.; Reitz, M.S.; Arthur, L.O.; Mizutani, S.; Davis, A.; Hung, P.P.; Gallo, R.C.; Eichberg, J.; Robert-Guroff, M. Long-term protection of chimpanzees against high-dose HIV-1 challenge induced by immunization. Nat. Med. 1997, 3, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pinczewski, J.; Gomez-Roman, V.R.; Venzon, D.; Kalyanaraman, V.S.; Markham, P.D.; Aldrich, K.; Moake, M.; Montefiori, D.C.; Lou, Y.; Pavlakis, G.N.; Robert-Guroff, M. Improved protection of rhesus macaques against intrarectal simian immunodeficiency virus SIV(mac251) challenge by a replication-competent Ad5hr-SIVenv/rev and Ad5hr-SIVgag recombinant priming/gp120 boosting regimen. J. Virol. 2003, 77, 8354–8365. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Wang, L.R.; Gomez-Roman, V.R.; vis-Warren, A.; Montefiori, D.C.; Kalyanaraman, V.S.; Venzon, D.; Zhao, J.; Kan, E.; Rowell, T.J.; Murthy, K.K.; Srivastava, I.; Barnett, S.W.; Robert-Guroff, M. Replicating rather than nonreplicating adenovirus-human immunodeficiency virus recombinant vaccines are better at eliciting potent cellular immunity and priming high-titer antibodies. J. Virol. 2005, 79, 10200–10209. [Google Scholar] [CrossRef] [PubMed]
- Patterson, L.J.; Robert-Guroff, M. Replicating adenovirus vector prime/protein boost strategies for HIV vaccine development. Expert Opin. Biol. Ther. 2008, 8, 1347–1363. [Google Scholar] [CrossRef]
- Tatsis, N.; Lasaro, M.O.; Lin, S.W.; Xiang, Z.Q.; Zhou, D.; Dimenna, L.; Li, H.; Bian, A.; Abdulla, S.; Li, Y.; Giles-Davis, W.; Engram, J.; Ratcliffe, S.J.; Silvestri, G.; Ertl, H.C.; Betts, M.R. Adenovirus vector-induced immune responses in nonhuman primates: responses to prime boost regimens. J. Immunol. 2009, 182, 6587–6599. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Sun, Y.; Korioth-Schmitz, B.; Fitzgerald, J.; Charbonneau, C.; Santos, G.; Seaman, M.S.; Ratcliffe, S.J.; Montefiori, D.C.; Nabel, G.J.; Ertl, H.C.; Letvin, N.L. Heterologous prime/boost immunizations of rhesus monkeys using chimpanzee adenovirus vectors. Vaccine 2009, 27, 5837–5845. [Google Scholar] [CrossRef] [PubMed]
- Fix A. Vaccine Briefs: Phase II prime-boost trial begins in the US. IAVI Report, The publication on AIDS Vaccine Research July-August 2009.
- Catanzaro, A.T.; Roederer, M.; Koup, R.A.; Bailer, R.T.; Enama, M.E.; Nason, M.C.; Martin, J.E.; Rucker, S.; Andrews, C.A.; Gomez, P.L.; Mascola, J.R.; Nabel, G.J.; Graham, B.S. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine 2007, 25, 4085–4092. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L. Working towards an HIV/AIDS vaccine. Hum. Vaccin. 2009, 5, 436–438. [Google Scholar] [PubMed]
- Brave, A.; Boberg, A.; Gudmundsdotter, L.; Rollman, E.; Hallermalm, K.; Ljungberg, K.; Blomberg, P.; Stout, R.; Paulie, S.; Sandstrom, E.; Biberfeld, G.; Earl, P.; Moss, B.; Cox, J.H.; Wahren, B. A new multi-clade DNA prime/recombinant MVA boost vaccine induces broad and high levels of HIV-1-specific CD8(+) T-cell and humoral responses in mice. Mol. Ther. 2007, 15, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdotter, L.; Nilsson, C.; Brave, A.; Hejdeman, B.; Earl, P.; Moss, B.; Robb, M.; Cox, J.; Michael, N.; Marovich, M.; Biberfeld, G.; Sandstrom, E.; Wahren, B. Recombinant Modified Vaccinia Ankara (MVA) effectively boosts DNA-primed HIV-specific immune responses in humans despite pre-existing vaccinia immunity. Vaccine 2009, 27, 4468–4474. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L.; Sharma, S.; Zhao, J.; Kannanganat, S.; Lai, L.; Chennareddi, L.; Yu, T.; Montefiori, D.C.; Amara, R.R.; Wyatt, L.S.; Moss, B. Immunogenicity in macaques of the clinical product for a clade B DNA/MVA HIV vaccine: elicitation of IFN-gamma, IL-2, and TNF-alpha coproducing CD4 and CD8 T cells. Aids Res. Hum. Retroviruses 2007, 23, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Rollman, E.; Hinkula, J.; Arteaga, J.; Zuber, B.; Kjerrstrom, A.; Liu, M.; Wahren, B.; Ljungberg, K. Multi-subtype gp160 DNA immunization induces broadly neutralizing anti-HIV antibodies. Gene Ther. 2004, 11, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Rollman, E.; Brave, A.; Boberg, A.; Gudmundsdotter, L.; Engstrom, G.; Isaguliants, M.; Ljungberg, K.; Lundgren, B.; Blomberg, P.; Hinkula, J.; Hejdeman, B.; Sandstrom, E.; Liu, M.; Wahren, B. The rationale behind a vaccine based on multiple HIV antigens. Microbes Infect. 2005, 7, 1414–1423. [Google Scholar] [PubMed]
- Brave, A.; Ljungberg, K.; Boberg, A.; Rollman, E.; Isaguliants, M.; Lundgren, B.; Blomberg, P.; Hinkula, J.; Wahren, B. Multigene/multisubtype HIV-1 vaccine induces potent cellular and humoral immune responses by needle-free intradermal delivery. Mol. Ther. 2005, 12, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kennedy, J.S.; West, K.; Montefiori, D.C.; Coley, S.; Lawrence, J.; Shen, S.; Green, S.; Rothman, A.L.; Ennis, F.A.; Arthos, J.; Pal, R.; Markham, P.; Lu, S. Cross-subtype antibody and cellular immune responses induced by a polyvalent DNA prime-protein boost HIV-1 vaccine in healthy human volunteers. Vaccine 2008, 26, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Jackson, B.; West, K.; Wang, S.; Lu, S.; Kennedy, J.S.; Goepfert, P.A. Multifunctional T-cell characteristics induced by a polyvalent DNA prime/protein boost human immunodeficiency virus type 1 vaccine regimen given to healthy adults are dependent on the route and dose of administration. J. Virol. 2008, 82, 6458–6469. [Google Scholar] [CrossRef] [PubMed]
- Rencher, S.D.; Slobod, K.S.; Dawson, D.; Lockey, T.D.; Hurwitz, J.L. Does the key to a successful HIV vaccine lie among the envelope sequences of infected individuals? Aids Res. Hum. Retroviruses 1995, 11, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Slobod, K.S.; Hurwitz, J.L. Does envelope V3 peptide comprise the principal neutralizing determinant for HIV? Vaccine 1995, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, K.; Rollman, E.; Eriksson, L.; Hinkula, J.; Wahren, B. Enhanced immune responses after DNA vaccination with combined envelope genes from different HIV-1 subtypes. Virology 2002, 302, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, J.L.; Slobod, K.S.; Lockey, T.D.; Wang, S.; Chou, T.H.; Lu, S. Application of the Polyvalent Approach to HIV-1 Vaccine Development. Curr. Drug Targets Infect. Disord. 2005, 5, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.K.; Ling, X.; Yang, Z.Y.; Montefiori, D.C.; Panet, A.; Kong, W.P.; Welcher, B.; Louder, M.K.; Mascola, J.R.; Nabel, G.J. Expanded breadth of virus neutralization after immunization with a multiclade envelope HIV vaccine candidate. Vaccine 2005, 23, 3434–3445. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.-P.; Huang, Y.; Yang, Z.-Y.; Chakrabarti, B.K.; Moodie, Z.; Nabel, G.J. Immunogenicity of multiple gene and clade human immunodeficiency virus type 1 DNA vaccines. J. Virol. 2003, 77, 12764–12772. [Google Scholar] [CrossRef] [PubMed]
- Slobod, K.S.; Lockey, T.D.; Howlett, N.; Srinivas, R.V.; Rencher, S.D.; Freiden, P.J.; Doherty, P.C.; Hurwitz, J.L. Subcutaneous administration of a recombinant vaccinia virus vaccine expressing multiple envelopes of HIV-1. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Sealy, R.; Slobod, K.S.; Flynn, P.; Branum, K.; Surman, S.; Jones, B.; Freiden, P.; Lockey, T.; Howlett, N.; Hurwitz, J.L. Preclinical and clinical development of a multi-envelope, DNA-virus-protein (D-V-P) HIV-1 vaccine. Int. Rev. Immunol. 2009, 28, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Caver, T.E.; Lockey, T.D.; Srinivas, R.V.; Webster, R.G.; Hurwitz, J.L. A novel vaccine regimen utilizing DNA, vaccinia virus and protein immunizations for HIV-1 envelope presentation. Vaccine 1999, 17, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Stambas, J.; Brown, S.A.; Gutierrez, A.; Sealy, R.; Yue, W.; Jones, B.; Lockey, T.D.; Zirkel, A.; Freiden, P.; Brown, B.; Surman, S.; Coleclough, C.; Slobod, K.S.; Doherty, P.C.; Hurwitz, J.L. Long lived multi-isotype anti-HIV antibody responses following a prime-double boost immunization strategy. Vaccine 2005, 23, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, J.L.; Zhan, X.; Brown, S.A.; Bonsignori, M.; Stambas, J.; Lockey, T.D.; Sealy, R.; Surman, S.; Freiden, P.; Jones, B.; Martin, L.; Blanchard, J.; Slobod, K.S. HIV-1 vaccine development: Tackling virus diversity with a multi-envelope cocktail. Frontiers Bioscience 2008, 13, 609–620. [Google Scholar] [CrossRef]
- Slobod, K.S.; Brown, S.A.; Surman, S.; Zirkel, A.; Jones, B.; Zhan, X.; Sealy, B.; Stambas, J.; Brown, B.; Bonsignori, M.; Lockey, T.D.; Freiden, P.; Hurwitz, J.L.; Doherty, P.C.; Coleclough, C. Overcoming diversity with a multi-envelope HIV vaccine. Current Topics in Virology 2004, 4, 159–168. [Google Scholar]
- Yu, S.; Feng, X.; Shu, T.; Matano, T.; Hasegawa, M.; Wang, X.; Ma, H.; Li, H.; Li, Z.; Zeng, Y. Potent specific immune responses induced by prime-boost-boost strategies based on DNA, adenovirus, and Sendai virus vectors expressing gag gene of Chinese HIV-1 subtype B. Vaccine 2008, 26, 6124–6131. [Google Scholar] [CrossRef] [PubMed]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral immunity due to long-lived plasma cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef]
- Brown, S.A.; Hurwitz, J.L.; Zirkel, A.; Surman, S.; Takimoto, T.; Alymova, I.; Coleclough, C.; Portner, A.; Doherty, P.C.; Slobod, K.S. A recombinant Sendai virus is controlled by CD4+ effector T cells responding to a secreted human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 2007, 81, 12535–12542. [Google Scholar] [CrossRef] [PubMed]
- Matano, T.; Kano, M.; Nakamura, H.; Takeda, A.; Nagai, Y. Rapid appearance of secondary immune responses and protection from acute CD4 depletion after a highly pathogenic immunodeficiency virus challenge in macaques vaccinated with a DNA prime/Sendai virus vector boost regimen. J. Virol. 2001, 75, 11891–11896. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; McElhaugh, M.J.; Eisenlohr, L.C. Generation of CD8(+) T cell memory in response to low, high, and excessive levels of epitope. J. Immunol. 2002, 168, 4455–4461. [Google Scholar] [PubMed]
- Park, S.O.; Han, Y.W.; Aleyas, A.G.; George, J.A.; Yoon, H.A.; Lee, J.H.; Kang, H.Y.; Kang, S.H.; Eo, S.K. Low-dose antigen-experienced CD4(+) T cells display reduced clonal expansion but facilitate an effective memory pool in response to secondary exposure. Immunology 2008, 123, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Moyes, C.D.; Milne, A.; Waldon, J. Very low dose hepatitis B vaccination in the newborn: anamnestic response to booster at four years. J. Med. Virol. 1990, 30, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hellerstein, M.; McDonnel, M.; Amara, R.R.; Wyatt, L.S.; Moss, B.; Robinson, H.L. Dose-response studies for the elicitation of CD8 T cells by a DNA vaccine, used alone or as the prime for a modified vaccinia Ankara boost. Vaccine 2007, 25, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Amara, R.R.; McClure, H.M.; Patel, M.; Sharma, S.; Yi, H.; Chennareddi, L.; Herndon, J.G.; Butera, S.T.; Heneine, W.; Ellenberger, D.L.; Parekh, B.; Earl, P.L.; Wyatt, L.S.; Moss, B.; Robinson, H.L. Multiprotein HIV type 1 clade B DNA/MVA vaccine: construction, safety, and immunogenicity in Macaques. Aids Res. Hum. Retroviruses 2004, 20, 654–665. [Google Scholar] [PubMed]
- Barouch, D.H.; Liu, J.; Lynch, D.M.; O'Brien, K.L.; La, P.A.; Simmons, N.L.; Riggs, A.M.; Clark, S.; Abbink, P.; Montefiori, D.C.; Landucci, G.; Forthal, D.N.; Self, S.G.; Carville, A.; Mansfield, K.; Goudsmit, J. Protective efficacy of a single immunization of a chimeric adenovirus vector-based vaccine against simian immunodeficiency virus challenge in rhesus monkeys. J. Virol. 2009, 83, 9584–9590. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.M.; Galvani, A.P.; Bush, R.M. Ecological and immunological determinants of influenza evolution. Nature 2003, 422, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Stone, R. Swine flu outbreak. China first to vaccinate against novel H1N1 virus. Science 2009, 325, 1482–1483. [Google Scholar] [CrossRef] [PubMed]
- Fazekas de St.Groth, S.; Webster, R.G. Disquisitions on original antigenic sinI. Evidence in man. J. Exp. Med. 1966, 124, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Fazekas de St.Groth, S.; Webster, R.G. Disquisitions on original antigenic sinII. Proof in lower creatures. J. Exp. Med. 1966, 124, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Skountzou, I.; Compans, R.; Jacob, J. Original antigenic sin responses to influenza viruses. J. Immunol. 2009, 183, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.Y.; Mays, I.; Garman, L.; Helms, C.; James, J.; Air, G.M.; Capra, J.D.; Ahmed, R.; Wilson, P.C. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Gearhart, P.J.; Hurwitz, J.L.; Cebra, J.J. Successive switching of antibody isotype expressed within the lines of a B-cell clone. Proc. Natl. Acad. Sci. USA 1980, 77, 5424–5428. [Google Scholar] [CrossRef]
- Gearhart, P.J.; Sigal, N.H.; Klinman, N.R. Heterogeneity of the BALB/c antiphosphorylcholine antibody response at the precursor cell level. J. Exp. Med. 1975, 141, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Klinman DM, Higgins KW. Sequential immunizations with rgp120s from independent isolates of human immunodeficiency virus type 1 induce the preferential expansion of broadly crossreactive B cells. J.Exp.Med. 1991, 173, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Slobod, K.S.; Surman, S.; Brown, S.A.; Coleclough, C.; Hurwitz, J.L. Minor components of a multi-envelope HIV vaccine are recognized by type-specific T-helper cells. Vaccine 2004, 22, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Surman, S.; Lockey, T.D.; Slobod, K.S.; Jones, B.; Riberdy, J.M.; White, S.W.; Doherty, P.C.; Hurwitz, J.L. Localization of CD4+ T cell epitope hotspots to exposed strands of HIV envelope glycoprotein suggests structural influences on antigen processing. Proc. Natl. Acad. Sci. USA 2001, 98, 4587–4592. [Google Scholar] [CrossRef]
- Brown, S.A.; Stambas, J.; Zhan, X.; Slobod, K.S.; Coleclough, C.; Zirkel, A.; Surman, S.; White, S.W.; Doherty, P.C.; Hurwitz, J.L. Clustering of Th cell epitopes on exposed regions of HIV envelope despite defects in antibody activity. J. Immunol. 2003, 171, 4140–4148. [Google Scholar] [PubMed]
- Wang, Y.; Flesch, I.E.; Tscharke, D.C. Vaccinia virus CD8+ T-cell dominance hierarchies cannot be altered by prior immunization with individual peptides. J. Virol. 2009, 83, 9008–9012. [Google Scholar] [CrossRef] [PubMed]
- Berman, P.W.; Gray, A.M.; Wrin, T.; Vennari, J.C.; Eastman, D.J.; Nakamura, G.R.; Francis, D.P.; Gorse, G.; Schwartz, D.H. Genetic and immunologic characterization of viruses infecting MN-rgp120-vaccinated volunteers. J. Infect. Dis. 1997, 176, 384–397. [Google Scholar] [PubMed]
- Gonzales, M.J.; Delwart, E.; Rhee, S.Y.; Tsui, R.; Zolopa, A.R.; Taylor, J.; Shafer, R.W. Lack of detectable human immunodeficiency virus type 1 superinfection during 1072 person-years of observation. J. Infect. Dis. 2003, 188, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Tsui, R.; Herring, B.L.; Barbour, J.D.; Grant, R.M.; Bacchetti, P.; Kral, A.; Edli, B.R.; Delwart, E.L. Human immunodeficiency virus type 1 superinfection was not detected following 215 years of injection drug user exposure. J. Virol. 2004, 78, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Slobod, K.S.; Coleclough, C.; Bonsignori, M.; Brown, S.A.; Zhan, X.; Surman, S.; Zirkel, A.; Jones, B.G.; Sealy, R.E.; Stambas, J.; Brown, B.; Lockey, T.D.; Freiden, P.J.; Doherty, P.C.; Blanchard, J.L.; Martin, L.N.; Hurwitz, J.L. HIV Vaccine Rationale, Design and Testing. Curr. HIV Res. 2005, 3, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Martin, L.N.; Slobod, K.S.; Coleclough, C.; Lockey, T.D.; Brown, S.A.; Stambas, J.; Bonsignori, M.; Sealy, R.E.; Blanchard, J.L.; Hurwitz, J.L. Multi-envelope HIV-1 vaccine devoid of SIV components controls disease in macaques challenged with heterologous pathogenic SHIV. Vaccine 2005, 23, 5306–5320. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, J.L.; Lockey, T.D.; Jones, B.; Freiden, P.; Sealy, R.; Coleman, J.; Howlett, N.; Branum, K.; Slobod, K.S. First phase I clinical trial of an HIV-1 subtype D gp140 envelope protein vaccine: immune activity induced in all study participants. AIDS 2008, 22, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Nara, P.L.; Lin, G. HIV-1: The Confounding Variables of Virus Neutralization. Curr. Drug Targets Infect. Disord. 2005, 5, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Srivastava, I.; Gomez-Roman, V.R.; zur, M.J.; Sun, Y.; Kan, E.; Hilt, S.; Engelbrecht, S.; Himathongkham, S.; Luciw, P.A.; Otten, G.; Ulmer, J.B.; Donnelly, J.J.; Rabussay, D.; Montefiori, D.; van Rensburg, E.J.; Barnett, S.W. Evaluation of envelope vaccines derived from the South African subtype C human immunodeficiency virus type 1 TV1 strain. J. Virol. 2005, 79, 13338–13349. [Google Scholar] [CrossRef] [PubMed]
- Bures, R.; Gaitan, A.; Zhu, T.; Graziosi, C.; McGrath, K.M.; Tartaglia, J.; Caudrelier, P.; El Habib, R.; Klein, M.; Lazzarin, A.; Stablein, D.M.; Deers, M.; Corey, L.; Greenberg, M.L.; Schwartz, D.H.; Montefiori, D.C. Immunization with recombinant canarypox vectors expressing membrane-anchored glycoprotein 120 followed by glycoprotein 160 boosting fails to generate antibodies that neutralize R5 primary isolates of human immunodeficiency virus type 1. Aids Res. Hum. Retroviruses 2000, 16, 2019–2035. [Google Scholar] [PubMed]
- Hosoi, S.; Borsos, T.; Dunlop, N.; Nara, P.L. Heat-labile, complement-like factor(s) of animal sera prevent(s) HIV-1 infectivity in vitro. AIDS 1990, 3, 366–371. [Google Scholar]
- Van Rompay, K.K.; Berardi, C.J. Passive immunization of newborn rhesus macaques prevents oral simian immunodeficiency virus infection. J. Infect. Dis. 1998, 177, 1247–1259. [Google Scholar] [PubMed]
- Forthal, D.N.; Landucci, G.; Cole, K.S.; Marthas, M.; Becerra, J.C.; Van, R.K. Rhesus macaque polyclonal and monoclonal antibodies inhibit simian immunodeficiency virus in the presence of human or autologous rhesus effector cells. J. Virol. 2006, 80, 9217–9225. [Google Scholar] [CrossRef] [PubMed]
- Holl, V.; Hemmerter, S.; Burrer, R.; Schmidt, S.; Bohbot, A.; Aubertin, A.M.; Moog, C. Involvement of Fc gamma RI (CD64) in the mechanism of HIV-1 inhibition by polyclonal IgG purified from infected patients in cultured monocyte-derived macrophages. J. Immunol. 2004, 173, 6274–6283. [Google Scholar] [PubMed]
- Gomez-Roman, V.R.; Florese, R.H.; Patterson, L.J.; Peng, B.; Venzon, D.; Aldrich, K.; Robert-Guroff, M. A simplified method for the rapid fluorometric assessment of antibody-dependent cell-mediated cytotoxicity. J. Immunol. Methods 2006, 308, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, E.; Lewis, M.W.; Hansen, S.G.; Strelow, L.I.; Nelson, J.A.; Sexton, G.J.; Hanifin, J.M.; Slifka, M.K. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 2003, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.D.; Ada, G.L. Persistence of influenza virus-specific antibody-secreting cells and B-cell memory after primary murine influenza virus infection. Cell Immunol. 1987, 109, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, J.; O'Donnell, D.R.; Rohwedder, A.; Openshaw, P.J. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am. J. Respir. Crit. Care Med. 2004, 169, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.J.; Diaz, G.; Cross, R.; Doherty, P.C. Analysis of clonotype distribution and persistence for an influenza virus-specific CD8+ T cell response. Immunity 2003, 18, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Gray, D. A role for antigen in the maintenance of immunological memory. Nat Rev. Immunol. 2002, 2, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Lam, K.P.; Rajewsky, K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature 2000, 407, 636–642. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Brown, S.A.; Surman, S.L.; Sealy, R.; Jones, B.G.; Slobod, K.S.; Branum, K.; Lockey, T.D.; Howlett, N.; Freiden, P.; Flynn, P.; et al. Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials. Viruses 2010, 2, 435-467. https://doi.org/10.3390/v2020435
Brown SA, Surman SL, Sealy R, Jones BG, Slobod KS, Branum K, Lockey TD, Howlett N, Freiden P, Flynn P, et al. Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials. Viruses. 2010; 2(2):435-467. https://doi.org/10.3390/v2020435
Chicago/Turabian StyleBrown, Scott A., Sherri L. Surman, Robert Sealy, Bart G. Jones, Karen S. Slobod, Kristen Branum, Timothy D. Lockey, Nanna Howlett, Pamela Freiden, Patricia Flynn, and et al. 2010. "Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials" Viruses 2, no. 2: 435-467. https://doi.org/10.3390/v2020435
APA StyleBrown, S. A., Surman, S. L., Sealy, R., Jones, B. G., Slobod, K. S., Branum, K., Lockey, T. D., Howlett, N., Freiden, P., Flynn, P., & Hurwitz, J. L. (2010). Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials. Viruses, 2(2), 435-467. https://doi.org/10.3390/v2020435