1. Introduction
Aedes aegypti (
Ae. aegypti) is an endemic mosquito vector distributed worldwide, particularly in tropical and subtropical environments [
1]. This distribution enables the primary transmission of multiple viruses to humans, including the dengue virus (DENV), Chikungunya virus (CHIKV), and Zika virus (ZIKV) [
2,
3]. Based on cartographic approaches, more than 100 dengue-endemic countries have been identified, with 390 million people infected annually [
4]. DENV, which consists of four distinct serotypes, can cause a range of disease symptoms, from a self-limited febrile illness known as dengue fever (DF) to severe conditions such as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) [
5,
6]. The binding and entry of DENV into host cells is mediated by multiple receptors, followed by the internalization of virus particles and the release of viral RNA into the cytoplasm through endosomal acidification. Subsequently, the positive-sense RNA genome is transcribed and translated into a large, single polyprotein, which is then cleaved by host and viral proteases. The structure of DENV consists of three structural proteins, the capsid protein C, the membrane protein precursor M (prM) (which matures into the membrane protein M after virus particle maturation), and the envelope protein E, as well as seven nonstructural proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5) [
7].
E-20-monooxygenase (E20MO) is the product of the gene
shade (
shd) locus (cytochrome p450, E20MO) [
8]. As first reported in Drosophila, E20MO converts ecdysone (E) into 20-hydroxyecdysone (20E) [
8,
9], and it is required for oogenesis [
8]. Additionally, conversion by E20MO has also been described in other species, including the cotton bollworm [
10], blackback land crab [
11], and small brown planthopper
Laodelphax striatellus [
12], which influences development through 20E. In
Anopheles gambiae, anti-Plasmodium immunity is promoted via a 20E agonist, which enhances innate immune responses related to bacterial and malarial parasite survival [
13,
14]. E20MO also plays a role in regulating the sexually dimorphic metamorphosis of
Ericerus pela [
15]. Acetamiprid and phoxim resistances in melon aphids and silkworms [
16,
17] are also attributed to the effect of E20MO. In
Aedes aegypti, the expression of E20MO is mainly observed in the ovaries, gut, and abdominal wall [
18]. The trade-off between immunity and reproduction in
Aedes aegypti may be governed by 20E [
19]. In summary, most studies on 20E, rather than E20MO, are associated with development, insecticide resistance, and immunity against bacteria or malaria parasites. However, there are few studies on the involvement of E20MO and the shade locus in arbovirus infection or innate immunity.
The E20MO gene was screened according to the transcriptome of Aag2 cells infected with DENV2. The results showed that the E20MO gene was upregulated after DENV2 infection in Aag2 cells [
20]. E20MO knockout (KO) Aag2 cells were constructed to investigate the effect and mechanism of the E20MO gene in response to DENV2 in Aag2 cells. To compare and evaluate the viral load (VL) between the KO and wild-type (WT) cells, extracellular viral RNA from particles was quantified by RT-qPCR following the replication and assembly of viral particles. The results showed that E20MO KO cells resulted in an increase in extracellular DENV2 copies, which suggests that the E20MO gene may be closely related to dengue virus replication in Aag2 cells. The results from this study improve our understanding of the complex biological interactions between mosquitoes and arboviruses.
2. Materials and Methods
2.1. Virus and Cells
The DENV2 Guangdong strain was provided by the Guangdong Provincial Centers for Disease Control and Prevention in China [
21].
Ae. aegypti Aag2 cells were cultured in Schneider’s Drosophila medium (SDM, Gibco, Penrose, NZ, AUK) supplemented with 10% fetal bovine serum (FBS, Gibco, Penrose, NZ, AUK) at 28 °C with 5% CO
2. To amplify DENV2, C6/36 cells were cultured in RPMI medium 1640 (RPMI 1640, Gibco, Penrose, NZ, AUK) containing 10% FBS at 28 °C and 5% CO
2. BHK-21 cells were used for plaque formation to determine DENV2 titers.
2.2. Design and Synthesis of sgRNA Targets
To investigate the role of the E20MO gene in DENV2 replication in Aag2 cells, we employed the CRISPR/Cas9 system to create an E20MO knockout (KO) cell line. In vitro, a ribonucleoprotein (RNP) complex was formed by chemically synthesizing sgRNA and Cas9 proteins, thus enhancing sgRNA stability while reducing off-target effects [
22,
23]. The DNA sequence of the
Aedes aegypti E20MO gene (gene no. 110677901) was obtained from the NCBI database (
https://www.ncbi.nlm.nih.gov/ (accessed on 18 June 2021)) and accessed through the Benching website (
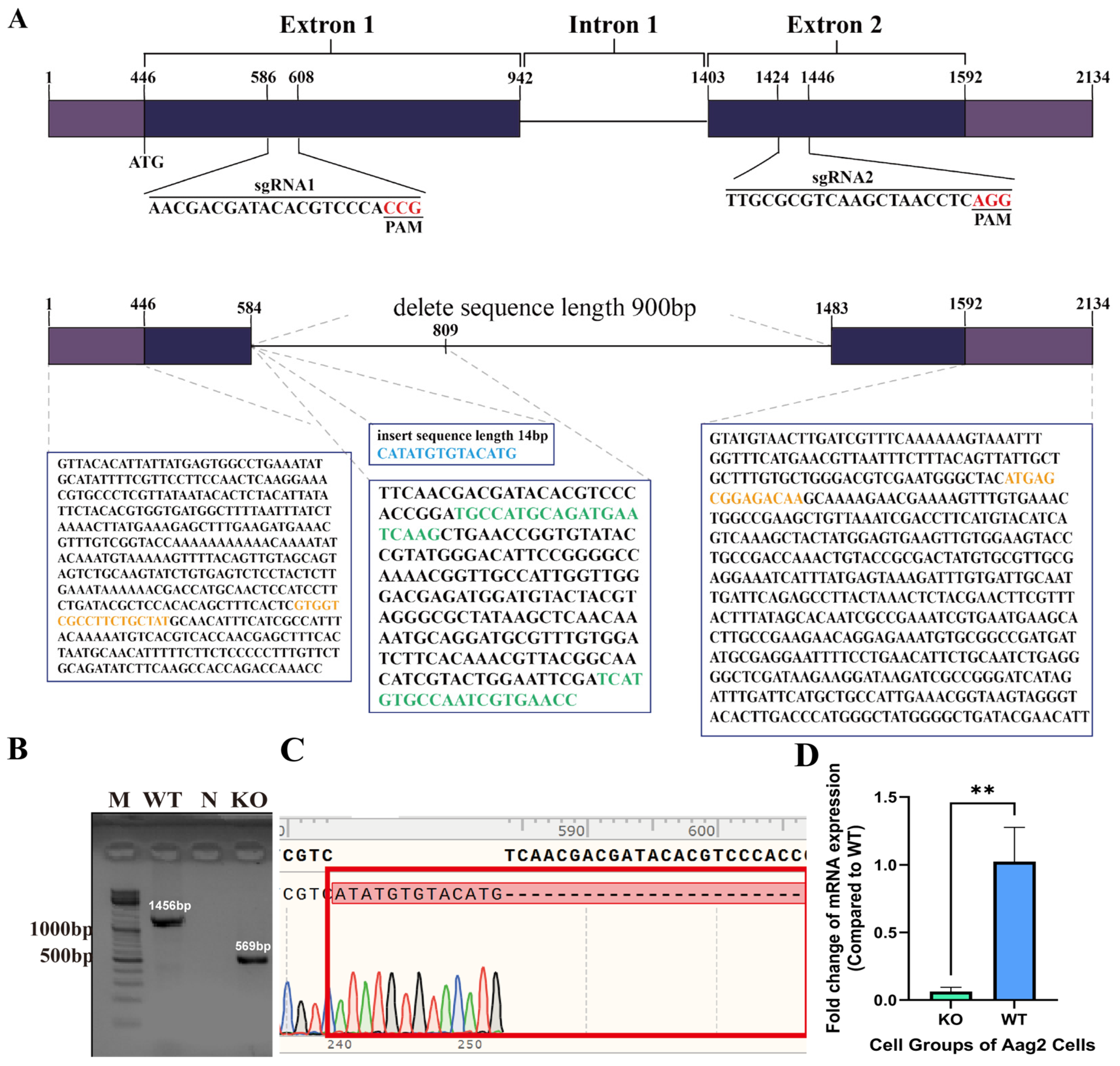
https://benching.com/ (accessed on 18 June 2021)). The target sequence for sgRNA1 was 5′-AACGACGATACACGTCCCACCGG-3′, and that for sgRNA2 was 5′-TTGCGCGTCAAGCTAACCTCAGG-3′. The sgRNA sequences were synthesized by GenScript Biotech.
2.3. Construction of E20MO KO Aag2 Cells
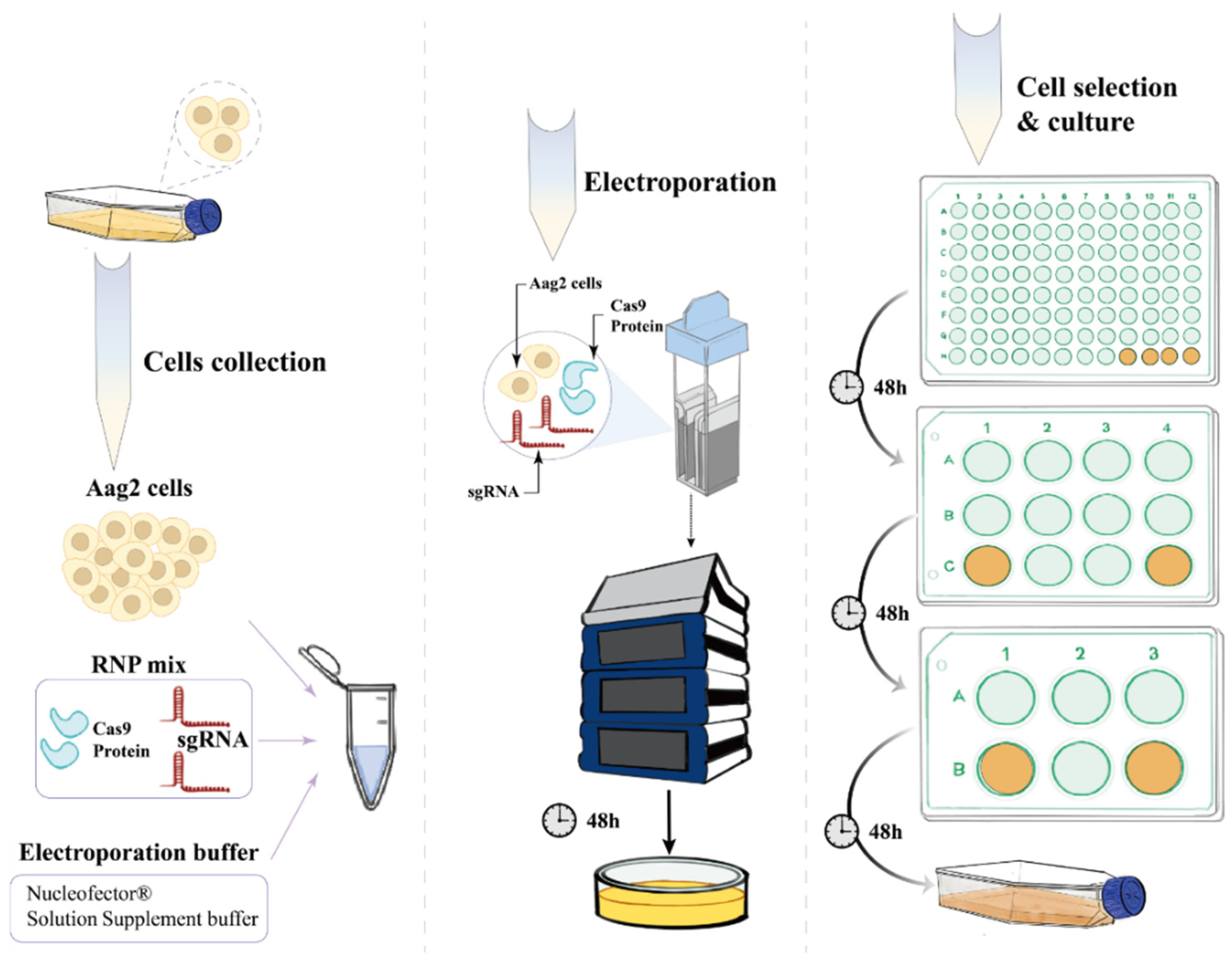
Chemical synthetic sgRNA and Cas9 proteins (632678, TAKARA, Osaka, Japan) were delivered into cells through electrical transfer. Ribonucleoprotein (RNP) complexes were formed by combining sgRNA and Cas9 proteins in vitro. RNP complex 1 (20 µL) was prepared by combining 8 µL of Guide-it™ Recombinant Cas9 (10 µg/µL) with 12 µL of sgRNA1 solution (0.1 nM/µL in water). Similarly, RNP complex 2 (20 µL) was prepared by mixing 8 µL of Guide-it™ Recombinant Cas9 (10 µg/µL) with 12 µL of sgRNA2 solution (0.1 nM/µL in water). After individual mixing for 10 min, the two RNP complexes were combined and further mixed for 5–10 min to obtain a final RNP complex of 40 µL. The wild-type Aag2 cells were washed once with PBS, and the cell pellet was collected for electroporation, with a cell number range of 5 × 104 to 1 × 106. Preparation of the electroporation buffer (total volume 40 µL) involved mixing 32.8 µL of P3 Primary Cell Nucleofector® Solution with 7.2 µL. Both the P3 Primary Cell Nucleofector® Solution and Supplement 1 were obtained from the Primary Cell 4D-Nucleofector™ X Kit S (V4XP-3032, Lonza, Tokyo, Japan). A mixture of two sgRNAs and Cas9 proteins was introduced into wild-type Aag2 cells using an electroporation system, and the cells were cultured for 6 days for the primary screening of mutants. The collected Aag2 cells were then resuspended in a mixture of RNP complexes and an electrotransfer buffer (Lonza) before being transferred to an electrotransfer cup. The electrotransfer apparatus (Lonza) was run using the K562 program for five minutes. After electrotransformation, the cells were resuspended in SDM medium containing 10% fetal bovine serum (FBS) and incubated at 28 °C. Subsequently, the cells were incubated using the K562 program for five minutes. When the cells reached 80–90% confluency in the culture dish, they were visually inspected under a microscope, and individual cells with a well-defined morphology, intact and clear edges, moderate size, and homogeneous transparency were selected to transfer into a 96-well plate. Subsequently, we sequentially transferred the cells from the 96-well plate to the 12-well plate and 6-well plate for amplification culture, aiming to obtain the final gene knockout cells.
2.4. E20MO KO Cell Detection at the DNA Level
After the cells reached a certain confluence, DNA was extracted from the cells (Insect DNA Extraction Kit from Shanghai Enlighten Biotech, Shanghai, China). The extracted DNA was then amplified through polymerase chain reaction (PCR) using the following specific primer sequences: forward primer (5′-GTGGTCGCCTTCTGCTAT-3′) and reverse primer (5′-TTGCTTGTCTCCGCTCAT-3′). Subsequently, the amplified products were analyzed using agarose gel electrophoresis and subjected to sequencing. For the PCR system, the following components were included: 2 × EasyTaq® PCR SuperMix from TransGen Biotech (Beijing, China) at 12.5 μL, 10 μM forward primer at 0.5 μL, 10 μM reverse primer at 0.5 μL, DNA template at 2 μL, and nuclease-free water to bring the total reaction volume to 25 μL. PCR was carried out as follows: initial denaturation at 94 °C for 10 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, extension at 72 °C for 1 min 15 s, and a final extension at 72 °C for 10 min. PCR products were analyzed by agarose gel electrophoresis, and selected PCR products were sent to the Tianyi Huiyuan Company (Beijing, China) for sequencing.
2.5. qRT-PCR Analysis of E20MO Expression in Ae. aegypti and Aag2 Cells
RNA was extracted from KO Aag2 cells and WT Aag2 cells (including DENV2-infected and noninfected cells) with TRIzol, and TransScript® Green One-Step qRT-PCR SuperMix (TransGen Biotech, Beijing, China) was used for quantitative reverse transcription PCR (qRT-PCR) with RNA as the template. The forward primer 5′-TGCCATGCAGATGAATCAAGC-3′ and reverse primer (5′-GGTTCACGATTGGCACATGA-3′) were used as the qRT-PCR primer sequences. The reaction system consisted of 2 × PerfectStartTM Green One-Step qPCR SuperMix (10 μL), TransScript® Green One-Step RT/RI Enzyme Mix (0.4 μL), forward primer (10 μM) (0.4 μL), reverse primer (10 μM) (0.4 μL), passive reference dye (50×) (0.4 μL), RNA template (2 μL), and nucleic acid-free water (to 20 μL). The reaction procedure was as follows: 45 °C for 5 min and 40 cycles of 94 °C for 30 s, 94 °C for 5 s, and 60 °C for 30 s.
2.6. Viral Infection of KO and WT Aag2 Cells
Two milliliters of KO and WT Aag2 cell suspension (5% FBS) (1 × 106 cells/mL) were seeded into 12-well plates. After 30 min of incubation, DENV2 was diluted to the appropriate ratio in SDM medium containing 5% FBS to obtain a multiplicity of infection (MOI) of 0.1. Next, 100 μL of the diluted virus was added to each cell plate, and the cells were cultured in an incubator at 28 °C with 5% CO2. One milliliter of WT and DWT cell groups (1 × 105 cells/mL) were seeded instead, and MOI was adjusted to 0.01. After infection, first, 10 μL cell suspension was mixed with trypan blue solution at a ratio of 1:1; then, the 10 μL mixture was used to determine the cell concentration. After the centrifugation of the cell suspension, 100 μL of the cell supernatant was taken daily and stored at −80 °C until the 7th day post-infection (dpi). The experiment was designed in two groups, with four biological replicates: the KO group (KO Aag2 cells + DENV2) and the WT group (WT Aag2 cells + DENV2).
2.7. Plasmid for E20MO Synthesis and Extraction
The PSL1180polyUBdsRED plasmid, which can express red fluorescent protein, was designed by Snapgene software 6.0.2 using dual-enzyme digestion [
24] to avoid generating scrambled and reverse ligation. Enhanced green fluorescent protein (EGFP) and green fluorescent protein-puromycin sites were added to the original vector, and the plasmid was constructed by replacing the EGFP fragment with the CDS fragment of the target gene. After the carrier was designed, it was synthesized by Bio-engineering (Shanghai) Co. Ltd. (Shanghai, China). Ten microliters of plasmid containing each gene was added to 100 mL of liquid LB medium, followed by incubation at a constant temperature (37 °C 200 R/min) for 12 h. Then, 30 mL of plasmid was centrifuged at 8000 R/min for 3 min, and the supernatant (medium) was removed; this enrichment process was repeated four times. The plasmids were extracted by an endotoxin-free plasmid extraction kit and then stored at −20 °C (Lipopolysaccharide-free Plasmid Extraction Kit DP117, Tiangen biochemical technology., Beijing, Co., Ltd., Beijing, China).
2.8. Plasmid Containing E20MO/EGFP Replenishment and Viral Infection of Cells
One milliliter of Aag2 cell suspension (5% FBS) (1 × 106 cells/mL) was seeded into 12-well plates. After 2 days of incubation, plasmids encoding E20MO and EGFP were transfected into KO cells using FuGENE Transfection Reagent (FuGENE®, Promega, Madison, WI, USA). After 24 h and after observing the cells under a fluorescence microscope, some cells were used to measure the relative gene expression of E20MO. The rest of the cells were utilized for viral cell infection. DENV2 was diluted to the appropriate concentration in SDM medium containing 5% FBS to obtain an MOI of 0.1. Subsequently, 100 μL of the diluted virus was added to each cell plate, and the cells were incubated in an incubator at 28 °C with 5% CO2. One hundred microliters of the cell supernatant were collected on the 2nd, 4th, and 6th dpi and stored at −80 °C. The experiment was designed with four groups, each having three biological replicates: the KOE group (KO Aag2 cells + EGFP plasmid + DENV2), KOC group (KO Aag2 cells + E20MO plasmid + DENV2), KO group (KO Aag2 cells + DENV2), and WT group (WT Aag2 cells + DENV2).
2.9. Analysis of Extracellular Viral Copies in the Cell Supernatant
DENV2 RNA was detected using a DENV2 non-nucleic acid detection kit from Beijing Merab Medical Technology. The reaction mixture consisted of 2× DENV2 amplification solution (10 μL), DENV2 primer and probe mixture (1 μL), reverse transcription PCR (RT-PCR) enzyme mixture (1 μL), cell supernatant (4 μL), and nuclease-free water to bring the reaction volume to 20 μL. The reaction conditions were as follows: 40 cycles of DENV2 amplification with the following temperature profile: 50 °C for 10 min and 95 °C for 10 min, followed by denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 30 s. The primers used were as follows: forward primer (5′-AATTAGAGCAGATCTGATGAA-3′), reverse primer (5′-AGCATTCCAAGTGAGAATCTCTTTGT-3′), and DENV2 primer and probe (5′-AGCATTCCAAGTGAGAATCTCTTTGTCA-3′).
2.10. Statistical Methods
All data are presented as the means ± standard deviations (SDs). The data were processed using Excel 2019 for analysis, and figures were created using GraphPad Prism 8.0 software. Statistical analysis was conducted using SPSS software 23.0.0.0. For data with a normal distribution and homogeneity of variance, unpaired t-tests were employed. In data for which the homogeneity of variance assumption was violated, the unpaired t-test with Welch’s correction was applied. For data with a nonnormal distribution, Mann–Whitney and Kolmogorov–Smirnov tests were utilized. For comparing multiple samples that followed a normal distribution, one-way ANOVA followed by Dunnett’s method was employed. In contrast, when comparing multiple samples with a nonnormal distribution, the Kruskal–Wallis test was used, followed by Dunnett’s post hoc analysis.
4. Discussion
Dengue fever is considered the most prevalent and fastest spreading viral disease in the population and is mediated by mosquitoes [
25]. At present, it is a major public health issue that endangers human life and health and hinders social and economic development.
Aedes aegypti, as the main vector of dengue virus (DENV) transmission, has promoted the outbreak of dengue fever in tropical and subtropical regions [
4]. Therefore, controlling the transmission vector of the dengue virus and cutting off the transmission route are the main measures to prevent and control the spread of dengue fever.
The main function of E20MO is to catalyze the generation of 20E. Most studies on the E20MO gene have been conducted in organisms such as cotton bollworms, black-backed land crabs, gray planthoppers, melon aphids, and silkworms [
10,
11,
12,
16,
17]. Discussions have focused on 20E, which has been shown to have functions pertaining to growth, development, and insecticide resistance. However, there is relatively little research on the relationship between the E20MO gene and DENV2 infection in mosquitoes. Notably, in a previous study, we found the E20MO gene through screening transcriptome data of
Aedes aegypti Aag2 cells infected with DENV2 [
20]. This study focused on exploring the effect of the E20MO gene on DENV2 replication in Aag2 cells.
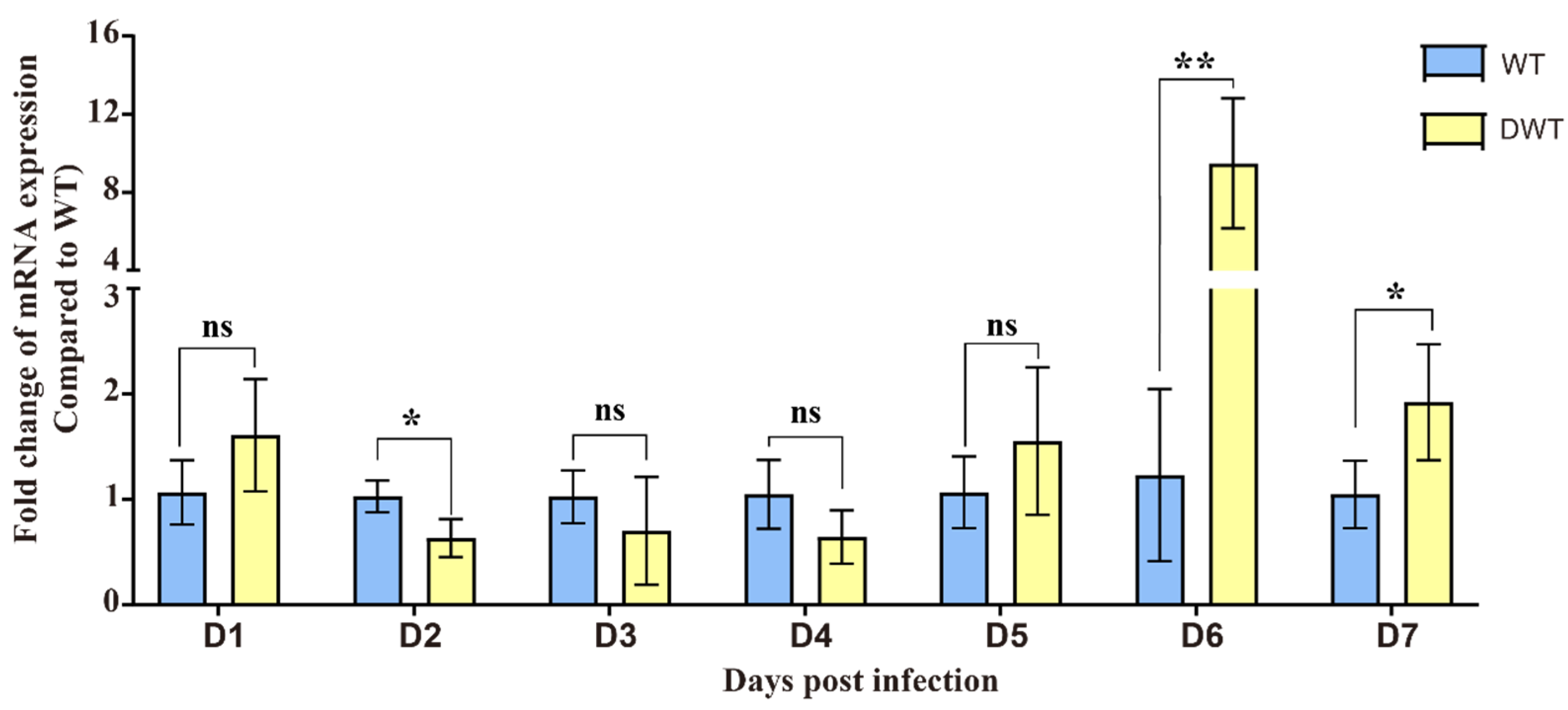
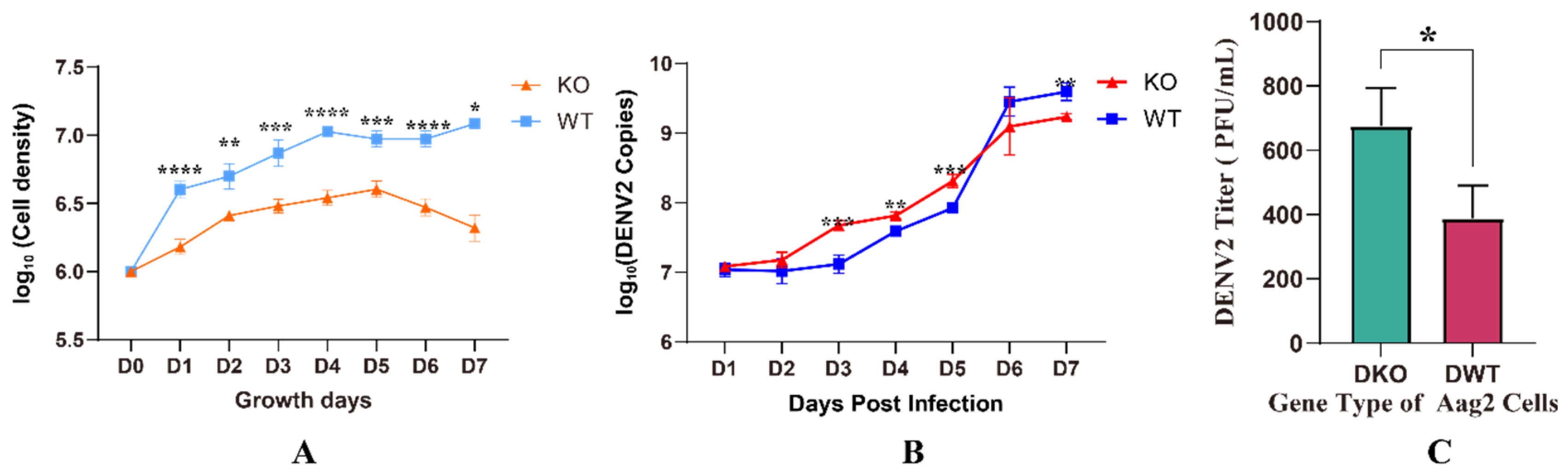
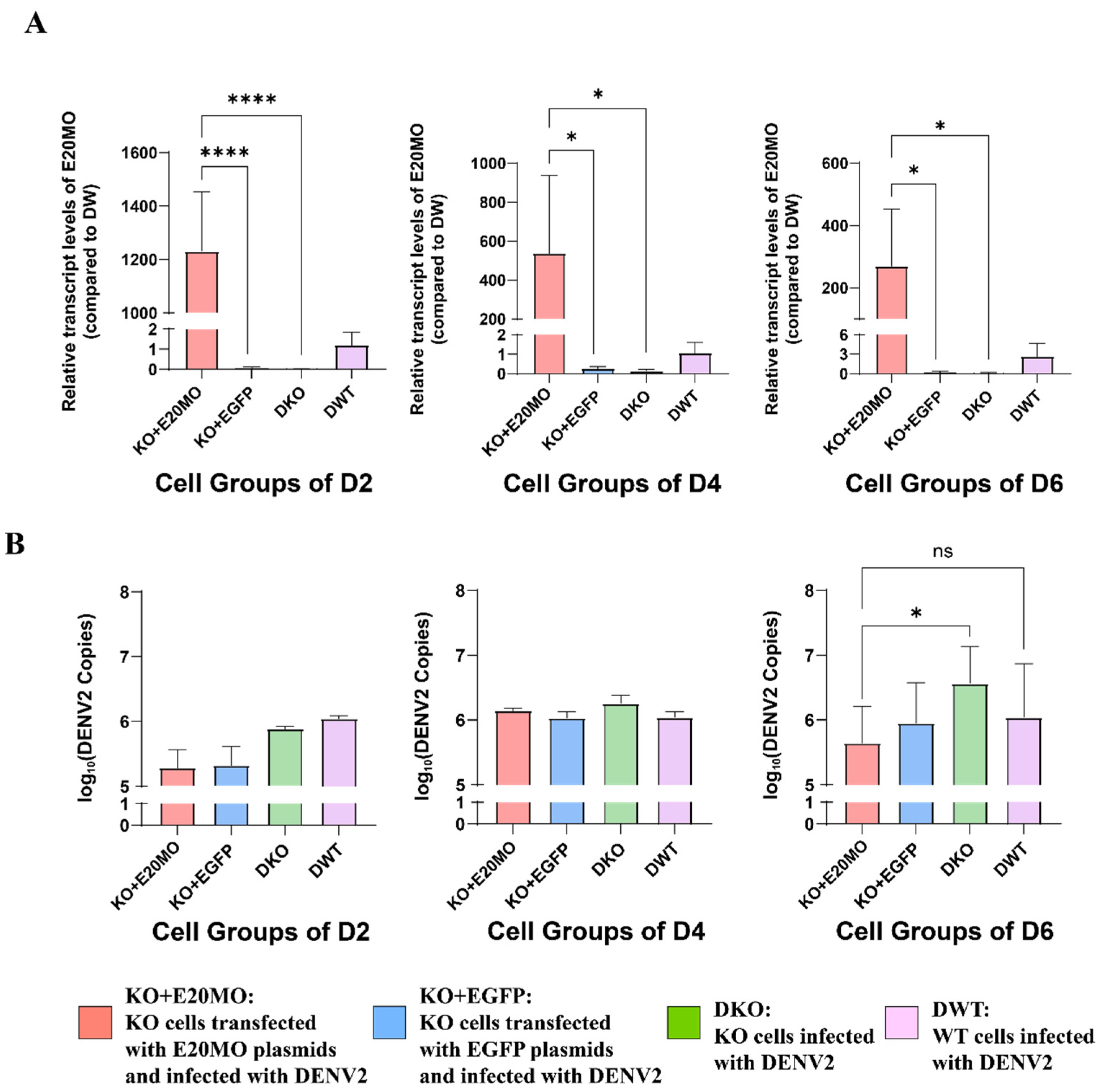
After exposure to exogenous DENV2, there was no significant change in the expression of the E20MO gene in the early stages, but at 6 dpi, the E20MO gene was significantly upregulated. This suggests that the expression of the E20MO gene may be related to DENV2 infection. Even though the relative expression of the E20MO gene did not differ significantly from 2 dpi to 5 dpi, it may have played a role in DENV2 infection, and differences in mRNA could be detected on day 6. After successfully constructing an E20MO KO cell line, a significant difference in cell density was observed between KO and WT cells, which may be attributed to the compromised cellular health in Aag2 cells following the knockout of the E20MO gene. Following the infection of the cell line with the virus, there was an ascending trend in the DENV2 viral load, with significant differences compared to the DENV2 viral load in the control group. Although the viral load in WT cells was higher than that in KO cells at 6 and 7 dpi, this result may be due to the poor growth and development of KO cells and the damage caused by the virus, resulting in a significantly lower cell density than that of WT cells, thus leading to a decrease in viral load. This result indicates that after the loss of the gene, the decrease in E20MO in Aag2 cells leads to an increase in the DENV2 viral load, suggesting that the E20MO gene may affect DENV2 replication in Aag2 cells. To further validate this function, we transferred plasmids containing the E20MO gene into Aag2 cells, which was confirmed by a fluorescence microscope and qRT-PCR, and infected the cells with DENV2. For cells that were successfully transformed with the plasmid, the viral load was significantly higher in KO cells than in cells transformed with the E20MO gene plasmids. This may be due to the overexpression of the E20MO gene in the Aag2 cells, which hindered the replication of DENV2 within the cell. This further suggests that the E20MO gene may hinder DENV2 replication.
Previous investigations conducted with Drosophila [
26,
27,
28] have demonstrated that 20E signaling plays a role in regulating the synthesis of antimicrobial proteins (AMPs) [
26,
27]. This regulation is achieved through both PGRP-LC-dependent and PGRP-LC-independent mechanisms, mediated by the ecdysone receptor and ultraspiracle heterodimer [
27]. Additional evidence from Drosophila suggests that 20E also enhances the activation of hemocytes, resulting in increased mobility, responsiveness to injury, and phagocytic activity [
28,
29]. This indicates that Aag2 cells may regulate the immune mechanism through the E20MO gene, thereby inhibiting the replication and spread of the dengue virus within cells.
Furthermore, Rebekah A. Reynolds et al. found that the hormone 20E plays a role in preparing mosquito innate immunity against bacteria and malaria parasites, partly due to the activation of cellular immune pathways, and possibly involves a series of humoral factors, such as antimicrobial peptide CEC3, CLIP serine proteases, and lysozymes, making mosquitoes more resistant to pathogen infection [
13]. Therefore, 20E can enhance the immune system response and reduce susceptibility to Plasmodium infection. We speculate that after DENV2 infection, Aag2 cells activate the pathway that leads to the expression of the E20MO gene and, subsequently, the activation of certain immune pathways, thereby hindering the replication of DENV2 within the cell. Additionally, the E20MO gene was originally found to be highly expressed in the midgut of larvae of
Drosophila [
8] and most specifically expressed in the midgut of female
Aedes aegypti mosquitoes [
18]. A study has suggested that the determinant of mosquito infection was identified as the DII region of the E protein of DNEV, and changing the DII region significantly enhanced
Aedes aegypti midgut infection [
30]. Therefore, the E20MO gene may influence the recognition or interaction of the host organism with the E protein of DENV2, thereby leading to an increased susceptibility of Aag2 cells to DENV2 infection upon the knockout of the E20MO gene. These hypotheses need to be tested through experiments.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





