Upregulation of the Chemokine Receptor CCR2B in Epstein‒Barr Virus-Positive Burkitt Lymphoma Cell Lines with the Latency III Program

 ,
,  and
and 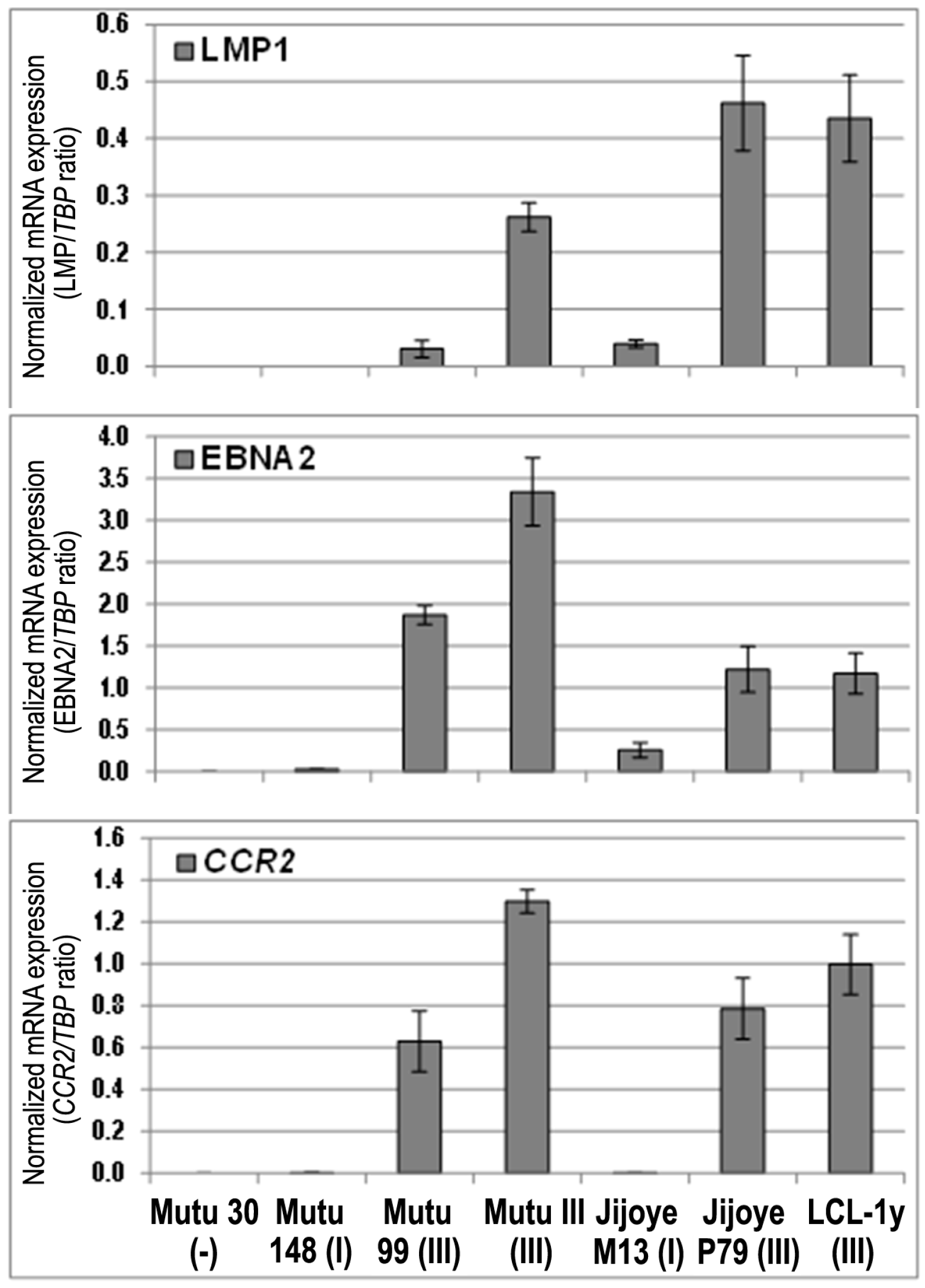{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Detection of the CCR2 and EBV Gene Expression Using Real-Time RT-PCR
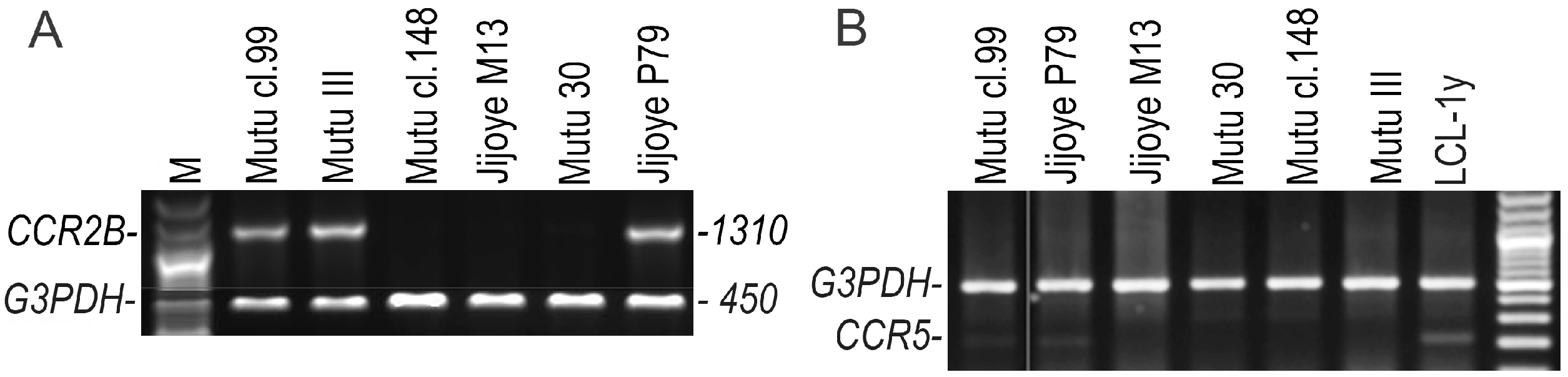
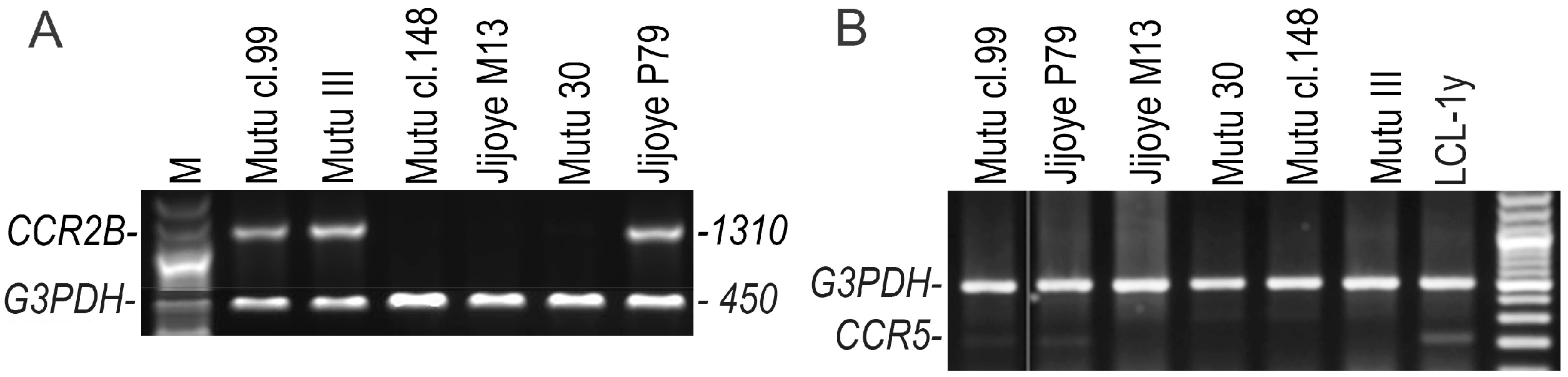
2.3. Detection of the CCR3, CCR5, CCR2A, and CCR2B Open Reading Frame (ORF) Transcripts Using RT-Duplex-PCR and Sequencing
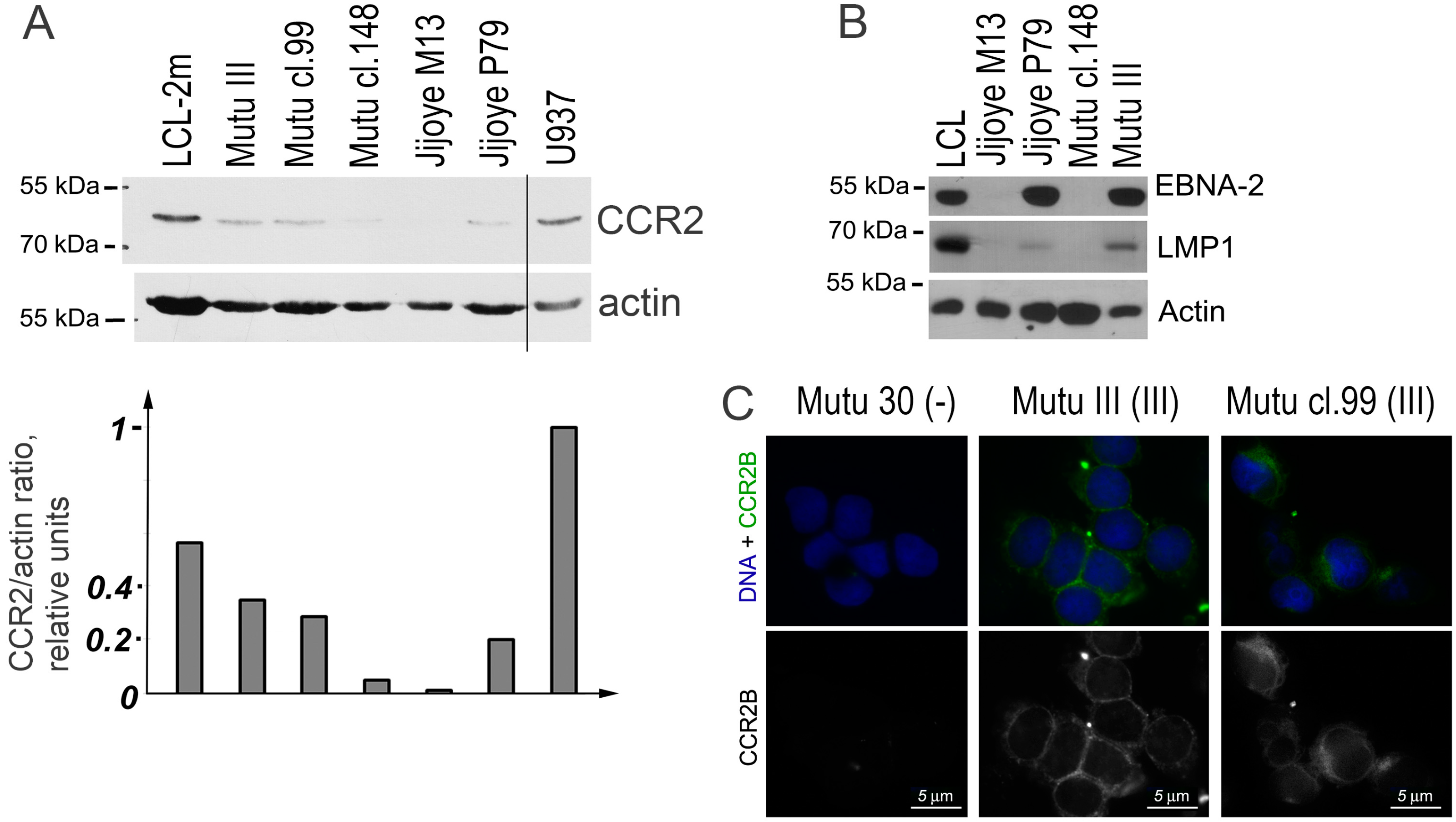
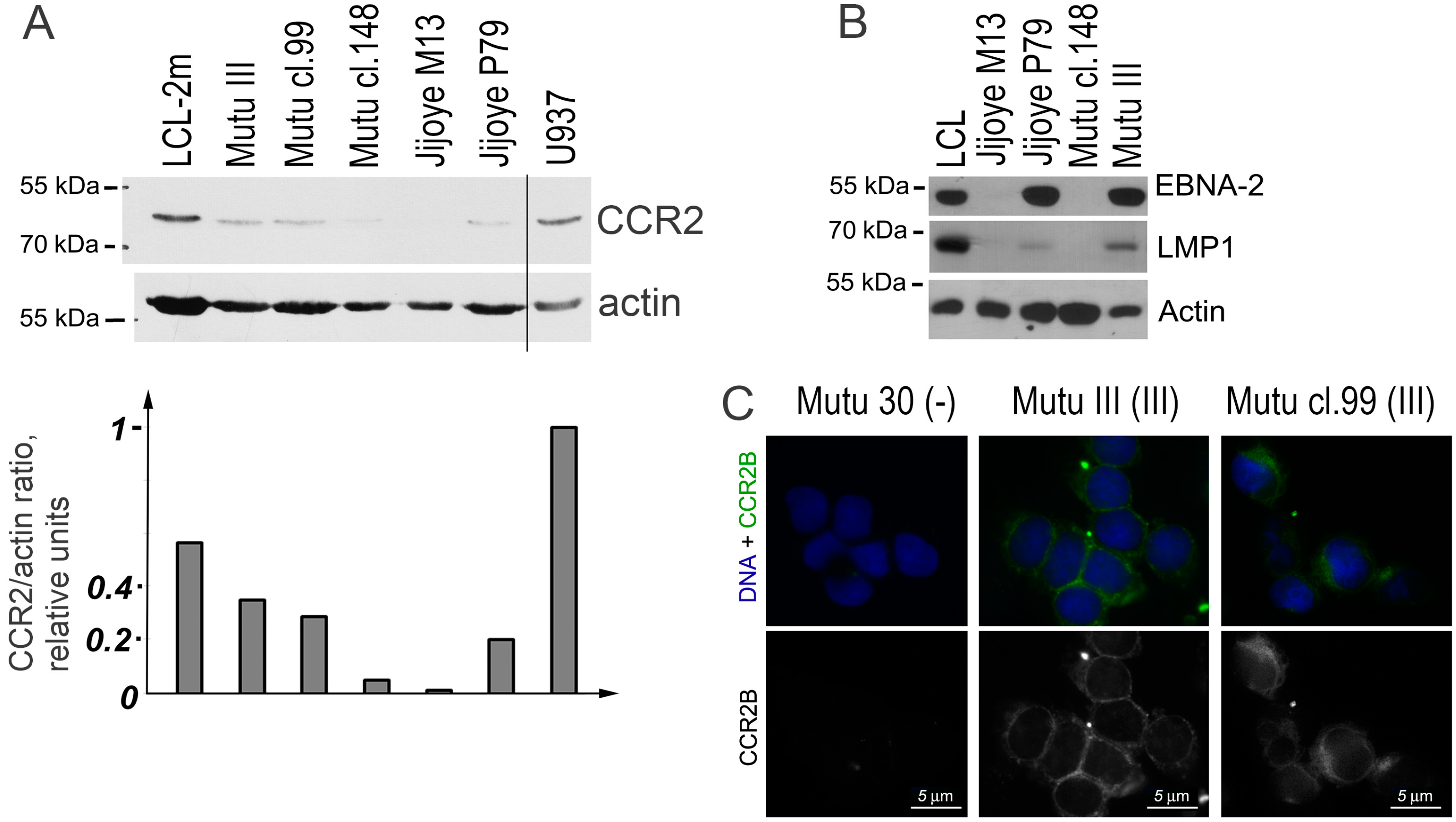
2.4. Detection of CCR2 Using Western Blot Analysis
2.5. Detection of CCR2B Using Fluorescent Immunostaining Analysis
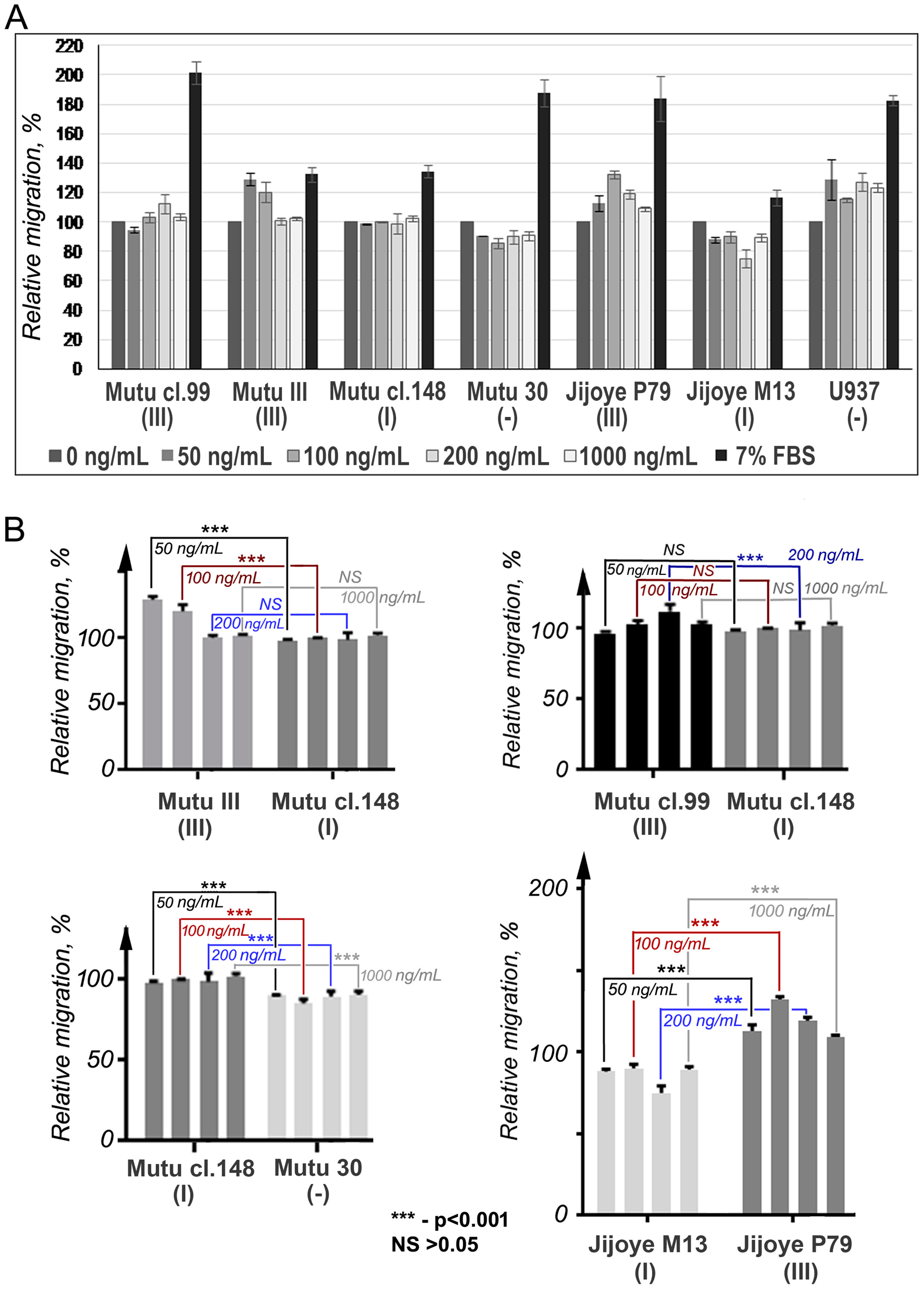
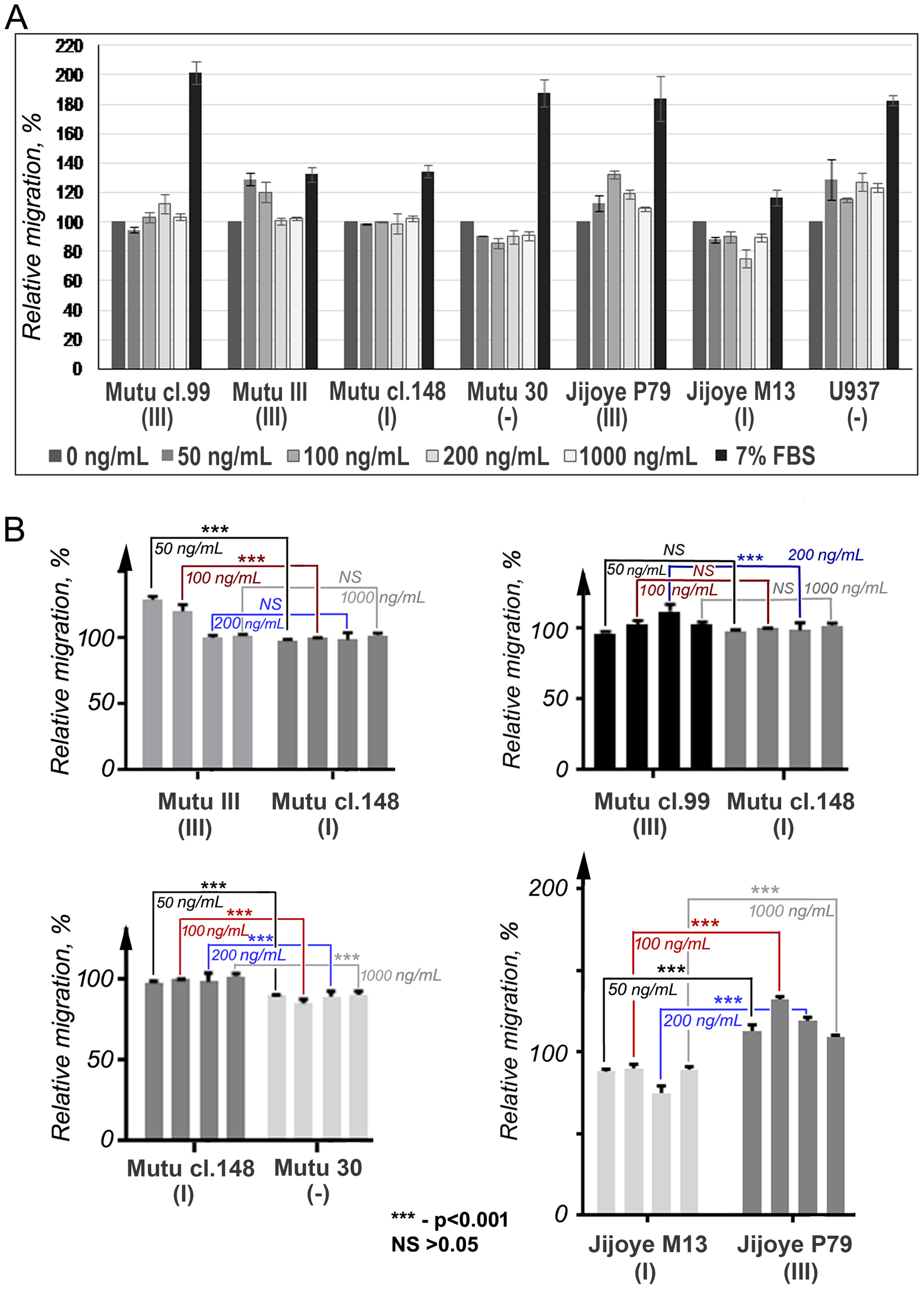
2.6. Cell Migration Assay
3. Results
4. Discussion
Author contributions
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Bornkamm, G.W. Epstein-Barr virus and the pathogenesis of Burkitt’s lymphoma: More questions than answers. Int. J. Cancer 2009, 124, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Klein, E.; Kashuba, E. Interaction of Epstein-Barr virus (EBV) with human B-lymphocytes. Biochem. Biophys. Res. Commun. 2010, 96, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Klein, G. Burkitt lymphoma—A stalking horse for cancer research? Semin. Cancer Biol. 2009, 19, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Li, L.; Young, K.H. EBV-driven B-cell lymphoproliferative disorders: From biology, classification and differential diagnosis to clinical management. Exp. Mol. Med. 2015, 47, e132. [Google Scholar] [CrossRef] [PubMed]
- Shanon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. B. 2017, 372, 20160271. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Nagy, N.; Rasul, A.E. EBV genome carrying B lymphocytes that express the nuclear protein EBNA-2 but not LMP-1: Type IIb latency. Oncoimmunology 2013, 2, e23035. [Google Scholar] [CrossRef] [PubMed]
- Niller, H.H.; Szenthe, K.; Minarovits, J. Epstein-Barr virus-host cell interactions: An epigenetic dialog? Front. Genet. 2014, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. To be or not IIb: A multi-step process for Epstein-Barr virus latency establishment and consequences for B cell tumorigenesis. PLoS Pathol. 2015, 11, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.; Robertson, E.S. Epstein-Barr virus latency: Current and future. Curr. Opin. Virol. 2015, 14, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.L.; Arnold-Harris, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The Interplay between Epstein Barr Virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr virus LMP1-Mediated oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Rowe, D.T.; Gregory, C.D.; Young, L.S.; Farrell, P.J.; Rupani, H.; Rickinson, A.B. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt’s lymphoma cells. EMBO J. 1987, 6, 2743–2751. [Google Scholar] [PubMed]
- Gregory, C.D.; Kirchgens, C.; Edwards, C.F.; Young, L.S.; Rowe, M.; Forster, A.; Rabbitts, T.H.; Rickinson, A.B. Epstein-Barr virus-transformed human precursor B cell lines: Altered growth phenotype of lines with germ-line or rearranged but nonexpressed heavy chain genes. Eur. J. Immunol. 1987, 17, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.D.; Rowe, M.; Rickinson, A.B. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt’s lymphoma cell line. J. Gen. Virol. 1990, 71, 1481–1495. [Google Scholar] [CrossRef] [PubMed]
- Rincon, J.; Prieto, J.; Patarroyo, M. Expression of integrins and other adhesion molecules in Epstein-Barr virus-transformed B lymphoblastoid cells and Burkitt’s lymphoma cells. Int. J. Cancer 1992, 51, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Fujisawa, R.; Izawa, D.; Hieshima, K.; Takada, K.; Yoshie, O. Human B cells immortalized with Epstein-Barr virus upregulate CCR6 and CCR10 and downregulate CXCR4 and CXCR5. J. Virol. 2002, 76, 3072–3077. [Google Scholar] [CrossRef] [PubMed]
- Ehlin-Henriksson, B.; Liang, W.; Cagigi, A.; Mowafi, F.; Klein, G.; Nilsson, A. Changes in chemokines and chemokine receptor expression on tonsillar B cells upon Epstein-Barr virus infection. Immunology 2009, 127, 549–557. [Google Scholar] [CrossRef] [PubMed]
- White, G.E.; Iqbal, A.J.; Greaves, D.R. CC Chemokine receptors and chronic Inflammation—Therapeutic Opportunities and Pharmacological Challenges. Pharmacol. Rev. 2013, 65, 47–89. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Rott, A.; Butcher, E.C. Leukocyte chemoattractant receptors in human disease pathogenesis. Annu. Path. Mech. Dis. 2015, 10, 51–81. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.J.; Hayward, J.A.; Huang, C.; Huma, Z.E.; Sanchez, J. Mechanisms of Regulation of the Chemokine-Receptor Network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef] [PubMed]
- The IUPHAR/BPS Guide to PHARMACOLOGY Database. Available online: http://www.guidetopharmacology.org/ (accessed on 4 April 2018).
- Kholodnyuk, I.; Szeles, A.; Yang, Y.; Klein, G.; Imreh, S. Inactivation of the human Fragile Histidine Triad gene at 3p14.2 in monochromosomal human/mouse microcell hybrid-derived severe combined immunodeficient mouse tumors. Cancer Res. 2000, 60, 7119–7125. [Google Scholar] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine 2017, 98, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kholodnyuk, I.; Rudevica, Z.; Leonciks, A.; Ehlin-Henriksson, B.; Kashuba, E. Expression of the chemokine receptors CCR1 and CCR2B is up-regulated in peripheral blood B cells upon EBV infection and in established lymphoblastoid cell lines. Virology 2017, 512, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Karpova, M.B.; Schoumans, J.; Blennow, E.; Ernberg, I.; Henter, J.I.; Smirnov, A.F.; Nordenskjöld, M.; Fadeel, B. Combined spectral karyotyping, comparative genomic hybridization, and in vitro apoptyping of a panel of Burkitt’s lymphoma-derived B cell lines reveals an unexpected complexity of chromosomal aberrations and a recurrence of specific abnormalities in chemoresistant cell lines. Int. J. Oncol. 2006, 28, 605–617. [Google Scholar] [PubMed]
- Nagy, N.; Maeda, A.; Bandobashi, K.; Kis, L.L.; Nishikawa, J.; Trivedi, P.; Faggioni, A.; Klein, G.; Klein, E. SH2D1A expression in Burkitt lymphoma cells is restricted to EBV positive group I lines and is downregulated in parallel with immunoblastic transformation. Int. J. Cancer 2002, 100, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskaja, K.; Ehlin-Henriksson, B.; Bartkova, J.; Bartek, J.; Scuderi, R.; Szekely, L.; Wiman, K.G.; Klein, G. Phenotype-related differences in the expression of D-type cyclins in human B cell-derived lines. Cell Growth Differ. 1996, 7, 1723–1732. [Google Scholar] [PubMed]
- Tierney, R.J.; Steven, N.; Young, L.S.; Rickinson, A.B. Epstein-Barr virus latency in blood mononuclear cells: Analysis of viral gene transcription during primary infection and in the carrier state. J. Virol. 1994, 68, 7374–7385. [Google Scholar] [PubMed]
- Bell, A.; Groves, K.; Kelly, G.L.; Croom-Carter, D.; Hui, E.; Chan, A.T.; Rickinson, A.B. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt’s lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol. 2006, 87, 2885–2890. [Google Scholar] [CrossRef] [PubMed]
- Kholodnyuk, I.D.; Kozireva, S.; Kost-Alimova, M.; Kashuba, V.; Klein, G.; Imreh, S. Down regulation of 3p genes, LTF, SLC38A3 and DRR1, upon growth of human chromosome 3-mouse fibrosarcoma hybrids in severe combined immunodeficiency mice. Int. J. Cancer 2006, 119, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Expression Atlas Database—EMBL-EBI. Available online: https://www.ebi.ac.uk/gxa/home (accessed on 6 April 2018).
- Mellado, M.; Rodriguez-Frade, J.M.; Vila-Coro, A.J.; Fernandez, S.; Martin, D.A.; Jones, D.R.; Toran, J.L.; Martinez, A. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001, 20, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Sohy, D.; Yano, H.; de Nadai, P.; Urizar, E.; Guillabert, A.; Javitch, J.A.; Parmentier, M.; Springael, J.Y. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protein effects of “selective” antagonists. J. Biol. Chem. 2009, 284, 31270–31279. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Uguccioni, M. Modulation of Chemokine Responses: Synergy and Cooperativity. Front. Immunol. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, H.; Wang, J.; Bunjhoo, H.; Xiong, W.; Xu, Y.; Zhao, J. Relationship between CCR2-V64I polymorphism and cancer risk: A meta-analysis. Gene 2013, 524, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.X.; Zhang, X.; Li, S.; Koiiche, R.D.; Sindsceii, J.H.; Song, H. Monocyte chemotactic protein-1 and CC chemokine receptor 2 polymorphisms and prognosis of renal cell carcinoma. Tumour Biol. 2013, 34, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Narter, K.F.; Agachan, B.; Sozen, S.; Cincin, Z.B.; Isbir, T. CCR2-64I is a risk factor for development of bladder cancer. Genet. Mol. Res. 2010, 9, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.; Satoh, Y.; Hoshikawa, Y.; Sairenji, T. Detection of Epstein-Barr virus in salivas and throat washings in healthy adults and children. Microbes Infect. 2000, 2, 115–120. [Google Scholar] [CrossRef]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rowe, M. Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 2011, 7, e1001338. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Herrera, R.; Palefsky, J.M. Epstein-Barr virus transcytosis through polarized oral epithelial cells. J. Virol. 2013, 87, 8179–8194. [Google Scholar] [CrossRef] [PubMed]
- Buettner, M.; Meyer, B.; Schreck, S.; Niedobitek, G. Expression of RANTES and MCP-1 in epithelial cells is regulated via LMP1 and CD40. Int. J. Cancer 2007, 121, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.; Herrera, R.; Veluppillai, P.; Greenspan, J.; Greenspan, D.; Palefsky, J.M. Epstein-Barr virus (EBV)-infected monocytes facilitate dissemination of EBV within the oral mucosal epithelium. J. Virol. 2007, 81, 5484–5496. [Google Scholar] [CrossRef] [PubMed]
- Shimakage, M.; Kimura, M.; Yanoma, S.; Ibe, M.; Yokota, S.; Tsujino, G.; Kozuka, T.; Dezawa, T.; Tamura, S.; Ohshima, A.; et al. Expression of latent and replicative-infection genes of Epstein-Barr virus in macrophage. Arch. Virol. 1999, 144, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Savard, M.; Bélanger, C.; Tardif, M.; Gourde, P.; Flamand, L.; Gosselin, J. Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 2000, 74, 2612–2619. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozireva, S.; Rudevica, Z.; Baryshev, M.; Leonciks, A.; Kashuba, E.; Kholodnyuk, I. Upregulation of the Chemokine Receptor CCR2B in Epstein‒Barr Virus-Positive Burkitt Lymphoma Cell Lines with the Latency III Program. Viruses 2018, 10, 239. https://doi.org/10.3390/v10050239
Kozireva S, Rudevica Z, Baryshev M, Leonciks A, Kashuba E, Kholodnyuk I. Upregulation of the Chemokine Receptor CCR2B in Epstein‒Barr Virus-Positive Burkitt Lymphoma Cell Lines with the Latency III Program. Viruses. 2018; 10(5):239. https://doi.org/10.3390/v10050239
Chicago/Turabian StyleKozireva, Svetlana, Zhanna Rudevica, Mikhail Baryshev, Ainars Leonciks, Elena Kashuba, and Irina Kholodnyuk. 2018. "Upregulation of the Chemokine Receptor CCR2B in Epstein‒Barr Virus-Positive Burkitt Lymphoma Cell Lines with the Latency III Program" Viruses 10, no. 5: 239. https://doi.org/10.3390/v10050239
APA StyleKozireva, S., Rudevica, Z., Baryshev, M., Leonciks, A., Kashuba, E., & Kholodnyuk, I. (2018). Upregulation of the Chemokine Receptor CCR2B in Epstein‒Barr Virus-Positive Burkitt Lymphoma Cell Lines with the Latency III Program. Viruses, 10(5), 239. https://doi.org/10.3390/v10050239