Applications of the FIV Model to Study HIV Pathogenesis



1
Department of Veterinary Pathobiology, Oklahoma State University, Stillwater, OK 74078, USA
2
Department of Microbiology, Immunology, and Pathology, Colorado State University, Fort Collins, CO 80523, USA
3
Department of Veterinary Pathobiology, University of Missouri, Columbia, MO 65201, USA
4
Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA 92037, USA
*
Author to whom correspondence should be addressed.
Viruses 2018, 10(4), 206; https://doi.org/10.3390/v10040206
Submission received: 31 March 2018
/
Revised: 17 April 2018
/
Accepted: 17 April 2018
/
Published: 20 April 2018
(This article belongs to the Special Issue Nonprimate Lentivirus)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Feline immunodeficiency virus (FIV) is a naturally-occurring retrovirus that infects domestic and non-domestic feline species, producing progressive immune depletion that results in an acquired immunodeficiency syndrome (AIDS). Much has been learned about FIV since it was first described in 1987, particularly in regard to its application as a model to study the closely related lentivirus, human immunodeficiency virus (HIV). In particular, FIV and HIV share remarkable structure and sequence organization, utilize parallel modes of receptor-mediated entry, and result in a similar spectrum of immunodeficiency-related diseases due to analogous modes of immune dysfunction. This review summarizes current knowledge of FIV infection kinetics and the mechanisms of immune dysfunction in relation to opportunistic disease, specifically in regard to studying HIV pathogenesis. Furthermore, we present data that highlight changes in the oral microbiota and oral immune system during FIV infection, and outline the potential for the feline model of oral AIDS manifestations to elucidate pathogenic mechanisms of HIV-induced oral disease. Finally, we discuss advances in molecular biology, vaccine development, neurologic dysfunction, and the ability to apply pharmacologic interventions and sophisticated imaging technologies to study experimental and naturally occurring FIV, which provide an excellent, but often overlooked, resource for advancing therapies and the management of HIV/AIDS.
1. Feline Immunodeficiency Virus
Feline immunodeficiency virus (FIV) is a naturally-occurring retrovirus that infects domestic and non-domestic feline species. In domestic cats, FIV produces progressive immune depletion that eventually results in an acquired immunodeficiency syndrome (AIDS) [1,2,3,4,5,6,7,8,9,10]. As a consequence, FIV infection is associated with a variety of clinical syndromes, including cachexia, anterior uveitis, chronic rhinitis, gingivostomatitis and periodontitis, encephalitis and neurologic dysfunction, and lymphoma [1,4,9,11,12,13,14,15,16,17,18,19,20,21]. The acute phase of FIV infection, lasting approximately 4–8 weeks, is characterized by a sharp increase in CD4+ T lymphocytes that are accompanied by high levels of FIV viral RNA and proviral DNA in circulation [4,8,22]. These hematologic changes are typically accompanied by mild to moderate clinical signs, which include pyrexia, lethargy, and peripheral lymphadenopathy [4,22,23]. Following a prolonged asymptomatic phase, during which the levels of circulating virus remains stable and integrated provirus establishes a reservoir of latently infected target cells, there is progressive decline of CD4+ T lymphocytes and other immunocytes, resulting in functional immunodeficiency and susceptibility to opportunistic infections [6,24,25,26].
During FIV infection, the loss of CD4+ T lymphocytes is directly attributable to a viral-induced cytopathic effect, in addition to an increase in FIV-specific CD8-mediated programmed cell death, lack of thymic regeneration, and spontaneous apoptosis in response to decreased cytokine support [10,25,27,28]. The most frequent clinical disease syndromes that are associated with FIV infection manifest as a consequence of immune dysfunction, such as oral opportunistic infection (gingivitis, stomatitis, and periodontitis), immune-mediated glomerulonephritis, chronic rhinitis, and dermatitis [15,16,19,20,29,30]. Oral opportunistic infections are prevalent in a high proportion of FIV-infected cats, and frequently present as erythematous, inflammatory lesions along the gingival margin (gingivitis), multifocal areas of necrotizing inflammation within the gingival sulcus or periodontal ligament (periodontitis), or ulcerative inflammatory lesions along the buccal mucosa, hard palate, or soft palate (stomatitis) [20,31,32,33]. Changes in the salivary/oral microbiota have been increasingly associated with FIV infection, and shifts in the proportion of opportunistic pathogens in the saliva of FIV-infected cats have been associated with the development of oral inflammatory lesions [33,34]. Similarly, FIV-infected cats frequently present with severe, necrotizing, and/or ulcerative inflammatory lesions (dermatitis) due to opportunistic infection with various bacterial, fungal, protozoal, and parasitic etiologies, including mycobacteriosis, leishmaniasis, toxoplasmosis, and dermatophytosis [16,29,35,36]. Upper respiratory disease is also a frequently finding in FIV-infected cats, and may occur in conjunction with concurrent viral, bacterial, or fungal infections [4,15,37,38].
Interestingly, FIV is also associated with the occurrence of neoplastic diseases, most frequently being demonstrated by the development of lymphoma in a large proportion of infected cats [7,39]. This association has been described in both naturally and experimentally infected animals, and predominately manifests as high-grade B-cell neoplasms that are remarkably similar to HIV-associated diffuse large B-cell lymphoma (DLBCL) (Figure 1) [7,40,41,42]. Also similar to HIV, direct viral-mediated oncogenesis that is related to proviral integration within oncogenes is an uncommon feature of FIV infection, and neoplastic transformation has been attributed to the indirect consequences of viral-induced immune dysfunction that arise in response to prolonged viral infection [7,42,43,44]. Specifically, recent studies have shown that clonal proviral integration sites are not typically detected during FIV infection and that proviral loads are lower in neoplastic tissues, indicating the neoplastic growth of cells lacking provirus [7]. Conversely, FIV and other lentiviral infections are strongly associated with polyclonal B-cell expansion, immunoglobulin production, and cytokine expression of proliferative mediators in response to immune activation and dysregulation [45,46]. It is proposed that such infection kinetics provide opportunities for somatic rearrangements that are associated with the generation of B-cell receptor diversity, or mutations in immunological cells during rapid expansion that disrupt or activate oncogenes; thus, resulting in neoplastic transformation [7,42]. However, the causal relationship of FIV and lymphoma has not been fully elucidated, and further studies are necessary to evaluate the specific role that viral infection and immune function play during tumorigenesis.
FIV-induced renal disease is also observed in both experimentally and naturally infected cats, and includes pathologic changes that include glomerulonephritis, proteinuria, protein tubular casts, and tubular microcysts, as well as diffuse interstitial inflammatory infiltrates [30,47]. Mesangial widening with glomerular and interstitial amyloidosis is also observed in kidneys of FIV-infected cats, and when evaluated in the context of another frequent finding during FIV infection, hypergammaglobulinemia, indicate the potential for immune complex deposition to occur within the glomerulus as a result of chronic antigenic stimulation and immune activation [30,48,49].
Neurologic disease is an important manifestation of FIV infection, and affected cats may present with either central nervous system (CNS) or peripheral nervous system (PNS) involvement [14,17,18,50,51]. In the PNS, FIV induces significantly increased the numbers of CD3+ T cells and macrophages in dorsal root ganglia, and infected cats exhibit pronounced changes in epidermal nerve fiber densities [50,52]. FIV enters the CNS during the acute stages of infection and it is present within the brain and cerebral spinal fluid [14,17,53]. The primary neuropathogenic effect of FIV infection within the CNS manifests as infiltration and accumulation of perivascular lymphocytes and macrophages (encephalitis), activation of microglial cells and astrocytes (gliosis), and occasional neuronal loss with myelin degeneration [14,17,18,53,54]. This infiltration of inflammatory cells and the consequences that are associated with immune activation within the CNS frequently results in clinically apparent neurologic deficits and a gradual decline in CNS function, functionally manifesting as abnormal stereotypic motor behaviors, anisocoria, increased aggression, prolonged latencies in brainstem evoked potentials, delayed righting and pupillary reflexes, decreased nerve conduction velocities, and deficits in cognitive-motor functions [55,56,57,58].
2. FIV as a Molecular Analogue to HIV
FIV is a member of the Lentivirus genus within the Retroviridae family, and much has been learned about FIV since it was first described in 1987, particularly in regard to its application as a model to study the closely related lentivirus, human immunodeficiency virus (HIV) [8,9,10,59,60]. The FIV virion is approximately 100 nm in diameter, spherical, and contains two identical strands of positive-sense RNA in its 9400-base genome, which is tightly associated with the nucleocapsid protein (NC, p7) and a t-RNAlys bound to each RNA molecule, which serves as a primer for negative strand transcription [25,59,60,61]. This protein complex, along with viral enzymes that are involved with replication and maturation (protease, reverse transcriptase, integrase, and dUTPase), are enclosed within a core of capsid protein (CA, p24), and are surrounded by a shell of matrix protein (MA, p14) [25,59,60]. Viral envelope glycoproteins (gp) are embedded within an outer lipid bilayer surrounding the matrix coat, and include the surface (SU, gp95) and transmembrane (TM, gp40) subunits, which are cleaved from a 130–150 kDa membrane-bound precursor protein, glycosylated, and non-covalently anchored within the envelope in a trimeric form [25,59,60,62].
The genomic structure of FIV consists of three primary open reading frames (ORFs), gag, pol, and env, which are flanked by two long-terminal repeats (LTR) and are accompanied by numerous small ORFs containing regulatory and accessory genes, such as vif, rev, and orfA (Figure 2). FIV gag encodes the Gag polyprotein, which is cleaved by the protease to form the three mature proteins (MA, CA, and NC) and is necessary to achieve formation of mature virus particles [59,63,64]. Pol polyprotein, which is the primary product of the FIV pol gene, contains four important enzymes that are involved in virus replication and maturation: protease, reverse transcriptase (RT), integrase (IN), and dUTPase (DU) [59]. Viral protease (PR) facilitates the cleavage of Gag and Pol polyproteins into functional enzymatic or structural proteins; DU catalyzes the hydrolysis of dUTP to dUMP in effort to minimize misincorporation of potentially mutagenic dUTP into host DNA [59,65,66]; FIV RT is an RNA-dependent DNA polymerase involved in the reverse transcription of viral genomic RNA into a double-stranded copy of proviral DNA (cDNA). Once synthesized, cDNA is integrated into the host genome by a mature IN containing three functional domains: an N-terminal domain, a central catalytic core, and a C-terminal domain [67,68,69]. The FIV Env polyprotein, which is a 130–150 kDa product of the env gene, is glycosylated and trimmed within the Golgi apparatus, and proteolytically cleaved into two mature, glycosylated proteins prior to virion budding at the cell surface: SU (gp95) and TM (gp40), both of which play critical roles in virion attachment and entry into target cells [59,60].
The structural and sequence organization of FIV is very similar to HIV, which is also a member of the lentivirus genus [59]. HIV is morphologically characterized by a spherical virion that is roughly 120nm in diameter, and contains a diploid genome that is composed of two copies of single stranded, positive-sense RNA that is packaged with nucleocapsid (p7) and accessory proteins (protease, reverse transcriptase, integrase) [70]. Like FIV, the ribonucleoprotein complex at the heart of the HIV virion is contained within a dense core of Capsid protein (CA, p24) and is surrounded by a spherical shell of Matrix protein (MA, p17) [70]. Mature Env glycoproteins, SU (gp120), and TM (gp 41), are anchored within the external lipid bilayer, and play a significant role in cell entry through binding to host cell receptors.
FIV requires an initial interaction with a primary binding receptor for infection, and binds to host cells through a high-affinity interaction of the envelope SU protein (gp95) with the CD134 surface molecule present on CD4+ lymphocytes and monocytes/macrophages [71,72,73,74,75]. This interaction induces a conformational change in the SU protein, which then exposes a cryptic epitope in the V3 loop of Env; the binding site that is necessary for binding with the entry (co-) receptor CXCR4 [26,74,75]. Binding of the V3 loop exposes the serpentine region of TM (gp40), which results in the formation of a hairpin structure that allows the fusion with the cell membrane and subsequent cell entry [26,75,76]. However, as infection progresses, the production of neutralizing antibodies by the host increases the need for FIV to escape selective pressures. New viral variants arise that exhibit a decreased dependence on CD134 and an increased ability to infect cells that express CXCR4 with limited CD134 expression, such as naïve B cells, macrophages, and CD8+ T cells [2,3,60,77,78,79,80]. This expanded cell tropism results in an increase in the number of target cells that are susceptible to infection, which subsequently causes immunodepletion and clinical manifestations that are associated with AIDS-induced disease.
HIV also requires an initial interaction with a primary binding receptor for infection, and utilizes analogous modes of receptor-mediated entry as FIV utilizing chemokine co-receptors [81,82,83]. However, in lieu of CD134, HIV utilizes CD4 as the primary binding receptor and CCR5 as its primary entry receptor, although HIV is also able to utilize CXCR4 [81,82]. Much like FIV, HIV binds to CD4+ target cells through a high-affinity interaction with the CD4 receptor that induces a conformational change in the envelope glycoprotein gp120, subsequently exposing the binding sites necessary for chemokine co-receptor binding (CXCR4 or CCR5) and subsequent fusion with the cell membrane. In HIV infection, the expression of CCR5 or CXCR4 chemokine receptors on target cells is the primary determinant of cell tropism, with CCR5-mediated infection of CD4+ T cells predominating early in infection [84,85,86]. However, over the course of infection, the preference of HIV for CCR5 co-receptor usage changes to CXCR4 in up to half of infected individuals, and these CXCR4-utilizing strains exhibit a broader tropism for different T-cell subpopulations [84,87]. Furthermore, differences in CD4 and CCR5 expression levels can affect the CCR5-mediated infection of macrophages, resulting in a shift in cell tropism that is similar to what is observed during FIV infection [84,86,87].
The HIV genome encodes three primary polyproteins, Gag, Pol, and Env, as well as the regulatory protein, Rev, and accessory protein, Vif—all of which exhibit similar functions to FIV [59,60,70]. However, in addition to these, HIV also contains genes that encode additional accessory proteins that are involved in viral maturation, replication, and survival [70]. These accessory proteins include: Tat (p16/p14), a viral transcriptional activator; Vpr (p10–15), a promoter of nuclear localization and inhibitor of cell division (cell cycle arrest at G2/M); Vpu (p16); a promotor of extracellular release of viral particles; Nef (p27–25), a downregulator of CD4 and MHC I expression; Vpx (p12–16), a Vpr homolog present in HIV-2 (absent in HIV-1); and, Tev (p28), a tripartite tat-env-rev protein [70].
The FIV genome contains one regulatory gene (rev) and two accessory genes (vif and orfA). FIV rev encodes Rev, which is a nucleolar polyprotein that binds to the Rev Response Element (RRE) to allow for export of partially spliced and unspliced viral RNA transcripts out of the nucleus with the help of the nuclear export protein, exportin-1 [59,60,88]. The FIV Vif protein is crucial to FIV replication and is involved in the counteraction of host defense mechanisms, such as APOBEC3, which is a cellular protein that exerts an antiviral effect by deamination of cytosine to uracil during viral replication, resulting in the degradation of synthesized minus-strand DNA [59,60,89]. FIV Vif counteracts APOBEC3 by targeting the host protein to the E3 ubiquitin ligase complex, which is subsequently degraded by the proteasome [59,60,89].
The FIV OrfA protein is encoded by the accessory gene orfA (Figure 2), and was originally considered to be a transactivator of transcription due to a role in increasing the net translation of proteins that were expressed from genes under the transcriptional control of the FIV LTR. The localization of the orfA gene in the viral genome also roughly coincides with the location of the gene encoding the HIV transactivator, Tat [90]. However, studies have failed to show an increase in transcription directed by OrfA, and there is no trans-activation response (TAR) element, as acted on by HIV Tat. Thus, an increase in net protein translation that is facilitated by OrfA must be by other means and may be involved in late steps of virion formation and the early steps of virus infectivity, although the precise role of OrfA is still undetermined [59,60,91,92,93,94]. OrfA localizes in the nucleus and causes cell cycle arrest at G2 in infected cells, reminiscent of effects that are caused by the Vpr protein in HIV-1. Also, OrfA has been shown to downregulate expression of the viral receptor for FIV (CD134) on the surface of cells, as well as E2 ubiquitin-conjugating enzymes and an ubiquitin-protein ligase [60,90,95], which is similar to the effects that are ascribed to the Nef protein on CD4 downregulation during HIV-1 infection. These potential functions of OrfA may have implications that aid in viral dissemination by preventing surface interactions with budding virions, and limit the degradation of viral proteins by host cell ubiquitin ligase mechanisms.
In 1988, Miller [96] made the observation that there was also potential to encode a peptide product from an RNA transcribed from the minus strand of the provirus. Since then, there have been a number of reports providing evidence for predicted RNA and protein products from the minus strand in HIV-1 [97,98,99,100,101,102,103,104,105,106], SIV [107], FIV [108], and in the deltaretrovirus, BLV [109]. In FIV, there are several potential short open reading frames that may be translated from a negative strand message. However, the major potential reading frame in the negative strand of both FIV and HIV, antisense protein (ASP), coincides with the Env coding region in the plus strand RNA, in the region underlying the Rev Responsive Element (RRE) encoded on the plus strand (Figure 2). A recombinant protein that has been transcribed and translated from the ASP open reading frame has been used to screen both naturally and experimentally FIV infected cats for antibodies to the protein and a small percentage (<10%) do show some level of positivity (manuscript in preparation) (Figure 2). Furthermore, knocking out the putative start codon for ASP resulted in a dramatic reduction in viral protein production, suggesting a critical role in the virus life cycle. Immunohistochemistry shows a non-nuclear localization of the protein, which is suggestive of some post-transcription event. Further studies will be required to define the role of ASP, but it may contribute to the ability of the virus to replicate by counteracting some innate anti-viral response in the cell.
3. FIV as a Model to Study HIV Pathogenesis
3.1. Immune Dysfunction
The primary immunodeficiency of FIV, which is a gradual and progressive decline in CD4+ T lymphocytes, is a hallmark feature of both natural and experimental infection, and the most obvious fundamental feature to parallel HIV infection. During both FIV and HIV infection, CD4+ lymphocyte numbers decline over an extended asymptomatic phase, and are associated with an increase in activated CD8+ lymphocytes that have antiviral activity [110,111,112,113]. The net effect of this event is a decrease in the ratio of CD4+ cells to CD8+ cells (CD4:CD8), and is used as a clinical indicator of immunosuppression in both FIV and HIV infected patients [112,113,114]. Additionally, several studies have shown that FIV induces defects in immune function that are similar to HIV, such as a decreased proliferation response of T lymphocytes in response to mitogens, a deficit in the humoral immune response, and the dysregulation of cytokine expression [10,24,59].
Large granular lymphocytes (LGLs) are a lymphoid subset comprising 10–15% of peripheral mononuclear blood cells (PBMCs) (Figure 3), and consist of either CD3− NK cells or CD3+ T-cells that mediate antibody-dependent cytotoxicity [115,116,117,118]. Analysis of LGL populations during HIV infection have been hampered by the low percentage of these cells in circulation, and has typically only been reported in association with neoplasia [118,119,120]. However, recent studies have shown that LGLs are detectable and are elevated during HIV infection in humans, and may represent viral-suppressive CD8+ T cells [118,121]. Interestingly, studies in FIV-infected cats have determined that similar elevations in LGL phenotypes may represent polyclonal T-cells with viral suppressive properties, as indicated by increased interferon-γ (IFN-γ) expression and decreased PBMC proviral loads in correlation with LGL lymphocytosis [118,122].
Conversely, recent studies have shown that CD4+ CD25+ T regulatory (Treg) cells are responsible for the inhibition of CD8+ IFN-γ production during both FIV infection [123] and HIV infection [124], highlighting the potential mechanisms by which these viruses exhibit an immunosuppressive effect on the CD8+ immune response. Furthermore, additional studies have shown that FIV directly infects and activates CD4+ CD25+ Treg cells, which are then able to suppress CD4+ CD25− T helper (Th) cells [125]. While this relationship and the potential mechanisms of Treg cell activation during HIV infection is still unclear, such comparative studies in FIV may offer potential to help our understanding of CD8+ T cell function in HIV infection.
3.2. Neurologic Dysfunction
Previous studies have shown that both FIV and HIV enter the central nervous system (CNS) at acute stages of infection, either via the trafficking of infected monocytes and lymphocytes, or by the penetration of free virus across the blood-brain or blood-CSF barriers [17,126,127,128,129,130,131]. Once present in the CNS, both FIV and HIV infection spread to microglia and astrocytes, which then serve as a reservoir for latent viral persistence [13,17,130,131,132]. Although multinucleated giant cells are rarely observed in the CNS during FIV infection, the fundamental neuropathologic finding of encephalitis is well-documented in both HIV and FIV infected patients, and the resultant proliferation and activation of these cells (gliosis) is associated with neurodegenerative processes, such as myelin degradation and neuronal injury/loss [14,17,51,54,133]. Thus, the clinical manifestations that are associated with neuropathology of FIV are likewise observed in HIV infection, and because of this, FIV has been repeatedly used as a model to investigate the pathogenesis of dementia and cognitive-motor processing deficits in AIDS patients. In vitro models of FIV have been useful to expand our understanding of role of calcium dysregulation and neural dysfunction during lentiviral infection, and have provided a unique system for the development neuroprotective treatments, such as neurotrophin ligands, which prevent the delayed accumulation of intracellular calcium and the decreased cytoskeletal damage of neuronal dendrites [17,134]. Furthermore, because of the low natural prevalence and the slow clinical course that is associated with lentiviral-induced neurologic dysfunction, experimental in vivo studies have been developed in the FIV model, which accelerate neuropathogenesis (neonatal inoculation, inoculation with neurovirulent strains, direct intracranial inoculation), allowing for increased opportunity to evaluate the viral kinetics of CNS infection, neurovirulence determinants, and the potential for novel treatments that are designed to decrease neurocognitive defects during HIV infection [53,57,134,135].
The use of neurovirulent strains of FIV has also allowed for the investigation of neuropathogenic effects on the peripheral nervous system (PNS) as a model of HIV distal symmetric polyneuropathy (DSP), demonstrating the rapid onset of peripheral neuropathy in FIV infected cats with axonal injury, macrophage activation, and detection of the virus within the nerve [50,136]. Indeed, FIV infection results in pathological events in the PNS that are very similar to HIV, including increased numbers of CD3+ T lymphocytes and activated macrophages in skin and dorsal root ganglia (DRGs) that are associated with increased expression of the pro-inflammatory cytokines, as well as changes in epidermal nerve fiber densities, which is indicative of axonal and myelin degeneration [50,52]. FIV has also been useful in the evaluation of the neurotoxicity of antiretroviral toxic neuropathy (ATN), due to mitochondrial dysfunction that is associated with nucleoside analogue reverse transcriptase (NRTI) inhibitor treatment. Thus, FIV has the potential to expand our understanding of the role of the immunopathology and progression of neuropathy in FIV-infected cats.
SIV models of neuropathogenesis have been used to study HIV-associated neurologic dysfunction (HAND), and have resulted in the elucidation of many mechanisms of neuroAIDS development, such as acute CNS infection and the importance of monocyte/macrophage activation in driving CNS lesions [137,138,139,140]. Recently, the SIV model of neuroAIDS has been adapted to study peripheral neuropathy, and significant advances have been made that implicate macrophages within dorsal root and trigeminal ganglia as a source of viral maintenance, in addition to their role in neuronal loss and neuronophagia [141,142]. These findings are coupled with additional studies that have defined impaired mitochondrial function in distal axons, which are more pronounced in ART-treated animals, indicating the potential for antiretroviral-mediated mitochondrial toxicity [143]. However, the SIV model of HAND is most commonly employed in rhesus macaques using SIV strains that arose via nosocomial infections or a lab adaptation of African monkey strains [144]. SIV neurologic disease is therefore chiefly manifested as a rapid progression to AIDS with the hallmarks of CNS inflammation that amplify pathology when compared to HIV-infected humans [139,140]. Furthermore, NHP studies are also limited by increased zoonotic risk to researchers, high cost associated with animal care and housing, the low number of animals available for research, and the potential for co-infection with a wide array of other pathogens, including rhesus rhadinovirus (RRV), lymphocryptovirus (LCV), simian cytomegalovirus (CMV), simian foamy virus (SFV), simian virus 40 (SV40), and rhesus papillomavirus (RhPV) [145,146].
In mechanistic studies of HIV-associated neurologic dysfunction, the interaction of CXCR4 with viral envelope has been shown to enhance neuronal apoptosis via Ca2+-regulating systems and NMDA receptors (NMDARs) in the synaptic membrane [147,148,149,150,151,152,153]. This neurotoxic pathway is known to involve Ca2+ influx through NMDARs, nitric oxide (NO) production, and subsequent activation cGMP-dependent protein kinase II, however, the precise cellular mechanisms by which this occurs are unknown and are difficult to assess in chronically infected human patients [154,155,156,157,158,159]. Because FIV binds to CXCR4 on the neuronal membrane in a similar non-infectious interaction as HIV, the feline model may provide answers, particularly in regard to the viral envelope-receptor interaction and synaptic activity-mediated neurotoxicity in HAND [160,161]. Given these similarities (and limitations of the SIV model), FIV represents an adjunct lentiviral model that can accurately recapitulate neuroAIDS progression in HIV-infected humans for applications, such as evaluation of ART-induced neurotoxicity, neurofibrillary tangle development, and calcium homeostasis during viral infection [14,17].
3.3. Vaccine Development
Considerable effort has been directed at the development of an anti-HIV vaccine strategy that can produce protective immunity in humans, and this effort has been paralleled in regard to FIV. A commercially available, whole inactivated virus vaccine containing two FIV subtypes (Fel-O-Vax FIV®) is currently licensed for use in the United States, and various reports have described virus neutralization and cellular immunity in a significant proportion of study animals [162,163,164]. However, the efficacy of this vaccine is still under debate, as recent studies and field evaluations have reported that the vaccine does not confer immunity against certain FIV strains (i.e., FIVGL8), and that the neutralizing antibody response and the protective rate may be low in certain cat populations (i.e., protection is not conferred to certain virulent recombinant strains of FIV) [165,166,167,168]. Other attempts at FIV vaccine development have either failed to induce protective immunity against FIV infection, or have resulted in an increased susceptibility to infection via antibody-dependent enhancement or general immune activation [169,170,171,172,173,174].
The development of an anti-HIV vaccine has been impeded by a wide variety of similar complications, such as lack of efficacy or unanticipated side effects, as well as increased susceptibility to infection via the analogous mechanisms of FIV vaccine enhancement (antibody-dependent viral enhancement or general immune activation) [175,176,177,178,179,180,181]. Indeed, vaccine-induced enhancement of viral infection has been previously reported in a large number of HIV studies [182,183,184,185], and it has been shown to occur via antibody-dependent or antibody-independent mechanisms of complement activation [186,187,188,189,190,191,192,193], as well as an increase in general immune activation and/or the expansion of lymphoid target cells [194,195,196,197,198]; features that have also been observed in FIV studies [169,170,171,172,173,174]. However, despite these setbacks in lentiviral vaccine development, there are many similarities in the disease course of HIV and FIV infection, and the use of the FIV model to circumvent these roadblocks may have great potential to provide a translational model for the development of novel immunotherapies to protect from HIV infection in humans.
Traditionally, non-human primate (NHP) models have been at the forefront of anti-HIV vaccine development due to the similarities of SIV and HIV, and have revealed several promising vaccine targets, such as nef-deleted SIV (which protects from wild-type SIV infection) and broad neutralizing antibodies utilizing chimeric SHIVs that express the HIV-1 envelope glycoprotein [199,200,201,202]. However, the successful outcome of these methods to prevent HIV infection in humans has been significantly impeded by various causes, such as restrictions on the use of live-attenuated HIV-1 in humans, as well as difficulty in producing a sufficiently efficacious neutralizing antibody response by vaccination [200]. Alternatively, various humanized mouse models have played a vital role in elucidating key aspects of the immune response to HIV, primarily through the use of generally immunocompromised mice that were engrafted with reconstituted human immune system tissues, such as human fetal thymus and liver (scid-hu-Thy/Liv), or peripheral blood lymphocytes (scid-hu-PBL) [203]. These models have been used for key studies in HIV immunopathogenesis, including mechanisms of CD4+ T-cells loss, antiretroviral therapy response, and passive immunization with monoclonal antibodies to HIV envelope protein (and testing of Env-based vaccines) [146,203,204,205,206,207]. However, because only certain parts of the human immune system can be reconstituted in humanized mouse models, interactions between the introduced human cells and the murine immune system cannot be evaluated in these hosts, nor the effects of HIV infection in non-hematopoietic tissues [203]. Although FIV lacks certain molecular similarities to HIV, it induces similar immunopathologies in its natural host, and therefore represents an important yet underutilized animal model for fully evaluating the immune response during natural lentiviral infection. Furthermore, the availability of a commercially-available vaccine in cats with efficacy against at least a subset of FIV may provide important clues to improving the efficacy of anti-HIV vaccines, and the elucidation of the mechanisms that are associated with vaccination failure in analogous FIV and HIV models of immunotherapy may provide key insights into improving the efficacy of lentiviral vaccines.
3.4. HIV-Induced Oral Disease
Oral manifestations of HIV are exhibited through various disease syndromes, such as Oral Candidiasis (OC, “thrush”), Linear Gingival Erythema (LGE), Necrotizing Ulcerative Gingivitis (NUG), and Necrotizing Ulcerative Periodontitis (NUP) [208,209,210]. Despite the success of combinational antiretroviral therapy (cART) in diminishing HIV viral replication and prolonging immune function, lesions that are associated with systemic and local immune activation and opportunistic oral infections persist in HIV-infected patients [208,211,212,213]. Previous studies have demonstrated that CD4+ T-cells are rapidly and severely depleted from the intestinal mucosa following HIV infection due to the direct effects of targeted virus infection and virus-induced Fas-mediated apoptosis, resulting in a loss of mucosal integrity and a reduced capacity to control potential pathogens at mucosal surfaces—thereby triggering local and systemic pro-inflammatory responses [214,215,216,217]. Based upon the analogous microenvironments of the oral and gastrointestinal mucosa, the same effects of viral-induced immunosuppression is predicted to occur in the oral cavity, resulting in a chronic cycle of immune stimulation, leukocyte recruitment, and target cell infection that produces HIV-induced oral disease lesions [208,218].
The FIV model is particularly well suited for studies of HIV-associated oral disease, as it not only parallels HIV in its structural, biochemical, and immunological properties, but it is also the only naturally occurring lentivirus to predictably induce oral lesions in its natural host, the domestic cat [1,4,9,10,31,32]. Non-human primate (NHP) models of HIV do not reliably cause oral disease and are limited by zoonotic risk to researchers, high cost associated with animal care and housing, and low number of animals that are available for research, while humanized mouse models of HIV lack both the prevalence of oral lesions and the presence of tonsillar structures that are similar to humans [146,219,220,221]. In contrast, FIV oral manifestations are common in naturally and experimentally-infected cats [20,32,33], and the range of lesions seen include gingivitis, periodontitis, and feline chronic gingivostomatitis [32], with striking similarities to LGE, NUG, and NUP lesions noted in untreated HIV patients [1,4,111,208,222,223,224,225]. Furthermore, opportunistic infections that were detected in HIV-positive individuals are paralleled in feline oral disease syndromes [35,226,227,228,229,230,231,232,233,234,235], and feline tonsillar tissues (palatine, pharyngeal, and lingual tonsils) are analogous to those in humans [220]. Coupled with recent advances in new generation cART protocols that have potential for adaption in cat studies [236,237,238,239,240], the domestic cat model of FIV presents an easily manipulated animal model to evaluate the drivers of immune dysfunction and microbial dyscrasias during HIV infection using a controlled in vivo study design.
Thus, in order to assess in vivo mechanisms contributing to oral disease during lentiviral infection, we collected saliva from the sublingual area and ventral cheek pouches from juvenile SPF cats (12–14 month-old) and examined samples by 16S rRNA metagenomics analysis to detect differences in the oral microbiota of naïve and age-matched cats that were infected with FIV (PPR strain) of eight months duration (n = 5/group). FIVPPR is a relatively apathogenic strain of FIV that typically results in mild self-limiting gingivitis and/or periodontitis during acute infection [241], and animals did not have overt, visual signs of clinical periodontitis at the time of sampling. FIV-infected and naïve SPF animals were maintained on a similar diet, and similar anatomic regions were swabbed from all of the animals at the same time of day. DNA was extracted [242], and amplicon sequencing was performed using illumina MiSeq to generate paired-end 2 × 250 bp sequences of the hyper-variable region 4 (V4) of the 16S rDNA. Data were normalized using cumulative sum scaling [243], and used to construct a nonmetric multidimensional scaling three-dimensional (3D) plot (Figure 4A).
Significant differences were detected in the oral microbiota composition of FIV-infected cats relative to naïve animals (Figure 4A). Normalized data were tested using the Zero Inflated Gaussian model implemented in the R package metagenomeSeq [244] to identify the putative OTUs driving differences between FIV+ and FIV− cats. Significant log-fold change in abundance in 12 genera was noted between groups at the 0.1 level of significance after correction for multiple testing (Figure 4B). One FIV-positive cat developed moderate to severe erythematous gingivitis during the course of infection and saliva was collected and analyzed as described above. Upon analysis of saliva, this individual demonstrated a dramatically altered microbiome population with >95% operational taxonomic units (OTUs), corresponding to the genus Moraxellaceae as compared to the FIV+ cats with no lesions and the FIV− cats (Figure 4C).
Collectively, these results demonstrate that similar to HIV, FIV infection of domestic cats is associated with oral microbiota dysbiosis and a marked loss of microbial diversity during lentiviral-associated periodontitis. The persistence of HIV infection and periodontitis in patients on cART indicates that ancillary treatments that are specifically directed at restoring the normal oral microbiota in conjunction with cART may improve HIV periodontal progression and decrease systemic immune activation [229,245,246,247]. Feline dental disease is currently managed by comprehensive dental treatment consisting of hand and ultrasonic scaling, identical to techniques that are used in humans [248,249]. Probiotic supplementation has been successful in early studies as an adjuvant for treating periodontitis in people, and similar commercial oral probiotics products are available for the management of feline oral conditions [250,251,252]. Thus, the application of comprehensive dental cleaning with probiotic treatments in the feline model has the potential to assess the impact of local therapy for restoring oral homeostasis during lentiviral infection, and may increase our understanding of the progression and/or resolution of FIV-induced oral lesions and oral microbiome in the presence and absence of cART.
4. Conclusions
Our understanding of FIV infection of cats has progressed remarkably over the last three decades, yet much remains to be learned from this widespread lentiviral infection. Correspondingly, many aspects of HIV pathogenesis and the mechanisms of immune dysfunction are still poorly understood. Most notably, the complete elimination of HIV from the host and the restoration of immune function has not yet been achieved, nor has the means to provide protective immunity from infection. In regard to the future of HIV research, a precise understanding of the mechanisms for immunodeficiency, especially in the face of co-infections, viral-associated disease, and in the presence and absence of antiretroviral therapy will be necessary for the development of restorative or immuno-protective therapies and prophylaxis.
While being genetically divergent, FIV shares remarkable overlap with HIV in regard to molecular biology and function. Coupled with the flexibility of working with a small animal model, FIV represents a useful system to assess the in vivo aspects of lentiviral pathogenesis. As noted above, comparative pathogenesis of lentiviral immune dysfunction, neurologic, and oral disease in the feline model could aid in an understanding of HIV AIDS. Further, the successful deployment of an FIV vaccine provides great opportunities for the evaluation of lentiviral prophylaxis leading to sterilizing immunity.
The application of investigations in the molecular biology and function of genetic elements is another area that affords great potential to understand the mechanisms of lentiviral infection via the FIV model. For example, contemporary studies in FIV have recently used the 3D structure of FIV reverse transcriptase to uncover the mechanistic basis of viral resistance to non-nucleoside inhibitor drugs [253]. These studies are now uncovering crucial elements in RT structure that can be used as a template for the development of novel compounds that target conventional sites of drugs resistance, providing increased efficacy against drug-resistant strains of HIV [253].
Finally, the FIV model holds significant potential as a tractable vehicle to assess the efficacy of novel anti-retroviral therapies. Recent studies employing a progressive cART regimen that is composed of nucleoside reverse transcriptase inhibitors (emtricitabine, tenofovir) and integrase inhibitors (dolutegravir) have demonstrated the significant efficacy in FIV studies in vitro [236,237,238,239,240], and immuno-restorative therapies employing recombinant feline interferon omega (rFeIFN-ω) have resulted in the improvement of clinical symptoms in FIV-associated oral disease and feline chronic gingivostomatitis [254,255,256]. IFN-ω has also been reported to be a potent inhibitor of HIV infection in vitro, but in vivo therapeutic potential in human patients has not been evaluated [257]. Because IFN-ω exerts strong immunomodulatory effects by stimulating Natural Killer cell activity, enhancing the expression of MHC-I, and inhibiting lymphocyte proliferation, testing outcomes of IFN-ω therapy on FIV-associated disease may therefore elucidate anti-inflammatory mechanisms and offer significant potential for adoption as an agent to treat HIV-associated diseases [258].
Improvements in molecular technology and available diagnostic analyses for domestic cats, as well as the ability to apply pharmacologic interventions and sophisticated imaging technologies to the study of experimental and naturally occurring FIV provide an excellent, but often overlooked resource for advancing the therapies and management of HIV/AIDS.
Acknowledgments
Work in this article is supported by National Institute of Allergy and Infectious Diseases and the National Institute of Dental and Craniofacial Research of the National Institutes of Health under award numbers R01AI25825 and F32DE026679-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Special thanks Wendy Sprague for the large granular lymphocyte (LGL) image used in Figure 3.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siebelink, K.H.; Chu, I.-H.; Rimmelzwaan, G.F.; Weijer, K.; van Herwijnen, R.; Knell, P.; Egberink, H.F.; Bosch, M.L.; Osterhaus, A.D. Feline immunodeficiency virus (FIV) infection in the cat as a model for HIV infection in man: Fiv-induced impairment of immune function. AIDS Res. Hum. Retrovir. 1990, 6, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Dean, G.A.; Himathongkham, S.; Sparger, E.E. Differential cell tropism of feline immunodeficiency virus molecular clones in vivo. J. Virol. 1999, 73, 2596–2603. [Google Scholar] [PubMed]
- English, R.V.; Johnson, C.M.; Gebhard, D.H.; Tompkins, M.B. In vivo lymphocyte tropism of feline immunodeficiency virus. J. Virol. 1993, 67, 5175–5186. [Google Scholar] [PubMed]
- Pedersen, N.; Yamamoto, J.K.; Ishida, T.; Hansen, H. Feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 1989, 21, 111–129. [Google Scholar] [CrossRef]
- Torten, M.; Franchini, M.; Barlough, J.E.; George, J.W.; Mozes, E.; Lutz, H.; Pedersen, N.C. Progressive immune dysfunction in cats experimentally infected with feline immunodeficiency virus. J. Virol. 1991, 65, 2225–2230. [Google Scholar] [PubMed]
- Hosie, M.J.; Addie, D.; Belák, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Lutz, H.; Marsilio, F. Feline immunodeficiency: Abcd guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Magden, E.; Miller, C.; MacMillan, M.; Bielefeldt-Ohmann, H.; Avery, A.; Quackenbush, S.L.; VandeWoude, S. Acute virulent infection with feline immunodeficiency virus (FIV) results in lymphomagenesis via an indirect mechanism. Virology 2013, 436, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Ho, E.W.; Brown, M.L.; Yamamoto, J.K. Isolation of a t-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science 1987, 235, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.H.; Lin, Y.-C.; Fink, E.; Grant, C.K. Feline immunodeficiency virus (FIV) as a model for study of lentivirus infections: Parallels with HIV. Curr. HIV Res. 2010, 8, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, M.; Dean, G.A. Transmission and immunopathogenesis of fiv in cats as a model for HIV. Curr. HIV Res. 2003, 1, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Bęczkowski, P.M.; Litster, A.; Lin, T.L.; Mellor, D.J.; Willett, B.J.; Hosie, M.J. Contrasting clinical outcomes in two cohorts of cats naturally infected with feline immunodeficiency virus (FIV). Vet. Microbiol. 2015, 176, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Colitz, C.M. Feline uveitis: Diagnosis and treatment. Clin. Tech. Small Anim. Pract. 2005, 20, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.W.; Poss, M.L.; Hoover, E.A. Feline immunodeficiency virus: A neurotropic lentivirus. J. Acquir. Immune Defic. Syndr. 1990, 3, 658–668. [Google Scholar] [PubMed]
- Fletcher, N.F.; Meeker, R.B.; Hudson, L.C.; Callanan, J.J. The neuropathogenesis of feline immunodeficiency virus infection: Barriers to overcome. Vet. J. 2011, 188, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Hopper, C.; Sparkes, A.; Gruffydd-Jones, T.; Crispin, S.; Muir, P.; Harbour, D.; Stokes, C. Clinical and laboratory findings in cats infected with feline immunodeficiency virus. Vet. Rec. 1989, 125, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lappin, M. Opportunistic infections associated with retroviral infections in cats. Semin. Vet. Med. Surg. Small Anim. 1995, 10, 244–250. [Google Scholar] [PubMed]
- Meeker, R.B.; Hudson, L. Feline immunodeficiency virus neuropathogenesis: A model for HIV-induced cns inflammation and neurodegeneration. Vet. Sci. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Bielefeldt-Ohmann, H.; MacMillan, M.; Huitron-Resendiz, S.; Henriksen, S.; Elder, J.; VandeWoude, S. Strain-specific viral distribution and neuropathology of feline immunodeficiency virus. Vet. Immunol. Immunopathol. 2011, 143, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Boegler, K.; Carver, S.; MacMillan, M.; Bielefeldt-Ohmann, H.; VandeWoude, S. Pathogenesis of oral fiv infection. PLoS ONE 2017, 12, e0185138. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, A.P.; Franti, C.E.; Madewell, B.R.; Pedersen, N.C. Chronic oral infections of cats and their relationship to persistent oral carriage of feline calici-, immunodeficiency, or leukemia viruses. Vet. Immunol. Immunopathol. 1991, 29, 1–14. [Google Scholar] [CrossRef]
- Yamamoto, J.; Hansen, H.; Ho, E.; Morishita, T.; Okuda, T.; Sawa, T.; Nakamura, R.; Pedersen, N. Epidemiologic and clinical aspects of feline immunodeficiency virus infection in cats from the continental united states and canada and possible mode of transmission. J. Am. Vet. Med. Assoc. 1989, 194, 213–220. [Google Scholar] [PubMed]
- Pedersen, N. Feline immunodeficiency virus infection. In Animal Models in AIDS: International TNO Meeting, Maastricht, The Netherlands, 23–26 October 1989; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1990; pp. 165–183. [Google Scholar]
- Del Fierro, G.; Meers, J.; Thomas, J.; Chadwick, B.; Park, H.; Robinson, W. Quantification of lymphadenopathy in experimentally induced feline immunodeficiency virus infection in domestic cats. Vet. Immunol. Immunopathol. 1995, 46, 3–12. [Google Scholar] [CrossRef]
- Bendinelli, M.; Pistello, M.; Lombardi, S.; Poli, A.; Garzelli, C.; Matteucci, D.; Ceccherini-Nelli, L.; Malvaldi, G.; Tozzini, F. Feline immunodeficiency virus: An interesting model for aids studies and an important cat pathogen. Clin. Microbiol. Rev. 1995, 8, 87–112. [Google Scholar] [PubMed]
- Lecollinet, S.; Richardson, J. Vaccination against the feline immunodeficiency virus: The road not taken. Comp. Immunol. Microbiol. Infect. Dis. 2008, 31, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Taniwaki, S.A.; Figueiredo, A.S.; Araujo, J.P., Jr. Virus–host interaction in feline immunodeficiency virus (FIV) infection. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.A.; Willett, B.J.; Gault, E.A.; Jarrett, O. A longitudinal study of feline immunodeficiency virus-specific cytotoxic t lymphocytes in experimentally infected cats, using antigen-specific induction. J. Virol. 1996, 70, 6199–6206. [Google Scholar] [PubMed]
- Guiot, A.-L.; Rigal, D.; Chappuis, G. Spontaneous programmed cell death (pcd) process of lymphocytes of fiv-infected cats: Cellular targets and modulation. Vet. Immunol. Immunopathol. 1997, 58, 93–106. [Google Scholar] [CrossRef]
- Hughes, M.; Ball, N.; Love, D.; Canfield, P.; Wigney, D.; Dawson, D.; Davis, P.; Malik, R. Disseminated mycobacterium genavense infection in a fiv-positive cat. J. Feline Med. Surg. 1999, 1, 23–29. [Google Scholar] [CrossRef]
- Poli, A.; Tozon, N.; Guidi, G.; Pistello, M. Renal alterations in feline immunodeficiency virus (FIV)-infected cats: A natural model of lentivirus-induced renal disease changes. Viruses 2012, 4, 1372–1389. [Google Scholar] [CrossRef] [PubMed]
- Diehl, L.J.; Mathiason-Dubard, C.K.; O’Neil, L.L.; Obert, L.A.; Hoover, E.A. Induction of accelerated feline immunodeficiency virus disease by acute-phase virus passage. J. Virol. 1995, 69, 6149–6157. [Google Scholar] [PubMed]
- Kornya, M.R.; Little, S.E.; Scherk, M.A.; Sears, W.C.; Bienzle, D. Association between oral health status and retrovirus test results in cats. J. Am. Vet. Med. Assoc. 2014, 245, 916–922. [Google Scholar] [CrossRef] [PubMed]
- De Rozières, S.; Mathiason, C.K.; Rolston, M.R.; Chatterji, U.; Hoover, E.A.; Elder, J.H. Characterization of a highly pathogenic molecular clone of feline immunodeficiency virus clade C. J. Virol. 2004, 78, 8971–8982. [Google Scholar] [CrossRef] [PubMed]
- Weese, S.J.; Nichols, J.; Jalali, M.; Litster, A. The oral and conjunctival microbiotas in cats with and without feline immunodeficiency virus infection. Vet. Res. 2015, 46, 21. [Google Scholar] [CrossRef] [PubMed]
- Mancianti, F.; Giannelli, C.; Bendinelli, M.; Poli, A. Mycological findings in feline immunodeficiency virus-infected cats. J. Med. Vet. Mycol. 1992, 30, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.G. Leishmaniosis of companion animals in europe: An update. Vet. Parasitol. 2015, 208, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Sparkes, A.; Hopper, C.; Millard, W.; Gruffydd-Jones, T.; Harbour, D. Feline immunodeficiency virus infection clinicopathologic findings in 90 naturally occurring cases. J. Vet. Intern. Med. 1993, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K. Feline immunodeficiency virus infection: An overview. Vet. J. 1998, 155, 123–137. [Google Scholar] [CrossRef]
- Callanan, J.; Jones, B.; Irvine, J.; Willett, B.; McCandlish, I.; Jarrett, O. Histologic classification and immunophenotype of lymphosarcomas in cats with naturally and experimentally acquired feline immunodeficiency virus infections. Vet. Pathol. 1996, 33, 264–272. [Google Scholar] [CrossRef] [PubMed]
- English, R.; Nelson, P.; Johnson, C.M.; Nasisse, M.; Tompkins, W.A.; Tompkins, M.B. Development of clinical disease in cats experimentally infected with feline immunodeficiency virus. J. Infect. Dis. 1994, 170, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Gabor, L.; Love, D.; Malik, R.; Canfield, P. Feline immunodeficiency virus status of australian cats with lymphosarcoma. Aust. Vet. J. 2001, 79, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J. Viral causes of feline lymphoma: Retroviruses and beyond. Vet. J. 2014, 201, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Cho, K.-W.; Nishigaki, K.; Momoi, Y.; Nishimura, Y.; Mizuno, T.; Goto, Y.; Watari, T.; Tsujimoto, H.; Hasegawa, A. Molecular characteristics of malignant lymphomas in cats naturally infected with feline immunodeficiency virus. Vet. Immunol. Immunopathol. 1997, 57, 153–167. [Google Scholar] [CrossRef]
- Shiramizu, B.; Herndier, B.G.; McGrath, M.S. Identification of a common clonal human immunodeficiency virus integration site in human immunodeficiency virus-associated lymphomas. Cancer Res. 1994, 54, 2069–2072. [Google Scholar] [PubMed]
- Beatty, J.; Lawrence, C.; Callanan, J.; Grant, C.; Gault, E.; Neil, J.; Jarrett, O. Feline immunodeficiency virus (FIV)-associated lymphoma: A potential role for immune dysfunction in tumourigenesis. Vet. Immunol. Immunopathol. 1998, 65, 309–322. [Google Scholar] [CrossRef]
- Yamamoto, H.; Umemura, T.; Inoshima, Y.; Nakamura, M.; Adachi, I.; Miyazawa, T.; Mikami, T. Immunological and histological disorders in cats experimentally infected with feline immunodeficiency virus subtype b (TM2 strain). Vet. Microbiol. 1997, 57, 313–324. [Google Scholar] [CrossRef]
- Poli, A.; Abramo, F.; Taccini, E.; Guidi, G.; Barsotti, E.; Bendinelli, M.; Malvaldi, G. Renal involvement in feline immunodeficiency virus infection: A clinicopathological study. Nephron 1993, 64, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Takemura, N.; Sako, T.; Koyama, H.; Motoyoshi, S.; Inada, Y. Serum concentration of circulating immune complexes in cats infected with feline immunodeficiency virus detected by immune adherence hemagglutination method. J. Vet. Med. Sci. 1997, 59, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Falcone, M.; Bigalli, L.; Massi, C.; Hofmann-Lehmann, R.; Lombardi, S.; Bendinelli, M.; Lutz, H. Circulating immune complexes and analysis of renal immune deposits in feline immunodeficiency virus-infected cats. Clin. Exp. Immunol. 1995, 101, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H.; Miller, A.D. Animal models of HIV peripheral neuropathy. Future Virol. 2014, 9, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; March, P.A.; Buck, W.R.; Mathes, L.E. The feline model of neuroaids: Understanding the progression towards aids dementia. J. Psychopharmacol. 2000, 14, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Antony, J.; Liu, S.; Martinez, J.A.; Giuliani, F.; Zochodne, D.; Power, C. CD8+ lymphocyte-mediated injury of dorsal root ganglion neurons during lentivirus infection: CD154-dependent cell contact neurotoxicity. J. Neurosci. 2006, 26, 3396–3403. [Google Scholar] [CrossRef] [PubMed]
- Power, C.; Buist, R.; Johnston, J.; Del Bigio, M.; Ni, W.; Dawood, M.; Peeling, J. Neurovirulence in feline immunodeficiency virus-infected neonatal cats is viral strain specific and dependent on systemic immune suppression. J. Virol. 1998, 72, 9109–9115. [Google Scholar] [PubMed]
- Abramo, F.; BO, S.; Canese, M.G.; Poli, A. Regional distribution of lesions in the central nervous system of cats infected with feline immunodeficiency virus. AIDS Res. Hum. Retrovir. 1995, 11, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Steigerwald, E.S.; Sarter, M.; March, P.; Podell, M. Effects of feline immunodeficiency virus on cognition and behavioral function in cats. J. Acquir. Immune Defic. Syndr. 1999, 20, 411–419. [Google Scholar] [CrossRef]
- Maingat, F.; Vivithanaporn, P.; Zhu, Y.; Taylor, A.; Baker, G.; Pearson, K.; Power, C. Neurobehavioral performance in feline immunodeficiency virus infection: Integrated analysis of viral burden, neuroinflammation, and neuronal injury in cortex. J. Neurosci. 2009, 29, 8429–8437. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.; Prospero-Garcia, O.; Wheeler, D.; Wagaman, P.; Lerner, D.; Fox, H.; Whalen, L.; Bloom, F.; Elder, J.; Henriksen, S. Neurologic dysfunctions caused by a molecular clone of feline immunodeficiency virus, FIV-PPR. J. Neurovirol. 1996, 2, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.J.; Hayes, K.A.; Buck, W.R.; Podell, M.; Mathes, L.E. Neurophysiologic and immunologic abnormalities associated with feline immunodeficiency virus molecular clone FIV-PPR DNA inoculation. J. Acquir. Immune Defic. Syndr. 1999 2000, 23, 8–16. [Google Scholar] [CrossRef]
- Sparger, E.E. Fiv as a model for HIV: An overview. In In Vivo Models of HIV Disease and Control; Springer: Berlin, Germany, 2006; pp. 149–237. [Google Scholar]
- Kenyon, J.C.; Lever, A.M. The molecular biology of feline immunodeficiency virus (FIV). Viruses 2011, 3, 2192–2213. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.; Dombrowski, J.; O’Connor, T.; Montelaro, R.C.; Tonelli, Q.; Lawrence, K.; Seymour, C.; Goodness, J.; Pedersen, N.C.; Andersen, P.R. Biochemical and immunological characterization of the major structural proteins of feline immunodeficiency virus. J Gen. Virol. 1990, 71, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-Y.; Fink, E.; Hong, Y.; Wang, C.; Grant, C.K.; Elder, J.H. Fine definition of the cxcr4-binding region on the v3 loop of feline immunodeficiency virus surface glycoprotein. PLoS ONE 2010, 5, e10689. [Google Scholar] [CrossRef] [PubMed]
- Egberink, H.F.; Ederveen, J.; Montelaro, R.C.; Pedersen, N.C.; Horzinek, M.C.; Koolen, M.J. Intracellular proteins of feline immunodeficiency virus and their antigenic relationship with equine infectious anaemia virus proteins. J. Gen. Virol. 1990, 71, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.; Schnölzer, M.; Hasselkus-Light, C.; Henson, M.; Lerner, D.; Phillips, T.; Wagaman, P.; Kent, S. Identification of proteolytic processing sites within the gag and pol polyproteins of feline immunodeficiency virus. J. Virol. 1993, 67, 1869–1876. [Google Scholar] [PubMed]
- Von der Helm, K. Retroviral proteases: Structure, function and inhibition-from a non-anticipated viral enzyme to the target of a most promising HIV therapy. Biol. Chem. Hoppe Seyler 1996, 377, 765–774. [Google Scholar]
- Gadsden, M.H.; McIntosh, E.; Game, J.C.; Wilson, P.J.; Haynes, R. Dutp pyrophosphatase is an essential enzyme in saccharomyces cerevisiae. EMBO J. 1993, 12, 4425–4431. [Google Scholar] [PubMed]
- Khan, E.; Mack, J.P.; Katz, R.A.; Kulkosky, J.; Skalka, A.M. Retroviral integrase domains: DNA binding and the recognition of ltr sequences. Nucleic Acids Res. 1991, 19, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.; van der Linden, K.H.; Plasterk, R. Activities of the feline immunodeficiency virus integrase protein produced in escherichia coli. J. Virol. 1994, 68, 1468–1474. [Google Scholar] [PubMed]
- North, T.W.; Cronn, R.C.; Remington, K.M.; Tandberg, R.T.; Judd, R.C. Characterization of reverse transcriptase from feline immunodeficiency virus. J. Biol. Chem. 1990, 265, 5121–5128. [Google Scholar] [PubMed]
- Foley, B.T.; Leitner, T.K.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B.T.M. HIV Sequence Compendium 2015; Los Alamos National Lab.(LANL): Los Alamos, NM, USA, 2015. [Google Scholar]
- De Parseval, A.; Elder, J.H. Binding of recombinant feline immunodeficiency virus surface glycoprotein to feline cells: Role of cxcr4, cell-surface heparans, and an unidentified non-cxcr4 receptor. J. Virol. 2001, 75, 4528–4539. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A.; Ngo, S.; Sun, P.; Elder, J.H. Factors that increase the effective concentration of cxcr4 dictate feline immunodeficiency virus tropism and kinetics of replication. J. Virol. 2004, 78, 9132–9143. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A.; Chatterji, U.; Sun, P.; Elder, J.H. Feline immunodeficiency virus targets activated cd4+ t cells by using CD134 as a binding receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 13044–13049. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A.; Grant, C.K.; Sastry, K.J.; Elder, J.H. Sequential CD134-CXCR4 interactions in feline immunodeficiency virus (FIV): Soluble CD134 activates FIV Env for CXCR4-dependent entry and reveals a cryptic neutralization epitope. J. Virol. 2006, 80, 3088–3091. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, M.; White, R.L.; de Parseval, A.; Sastry, K.J.; Morris, G.; Grant, C.K.; Elder, J.H. Mapping of the cxcr4 binding site within variable region 3 of the feline immunodeficiency virus surface glycoprotein. J. Virol. 2008, 82, 9134–9142. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Fuller, F.J.; Tompkins, W.A. Mechanism of feline immunodeficiency virus envelope glycoprotein-mediated fusion. Virology 2004, 321, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Dean, G.A.; Reubel, G.H.; Moore, P.F.; Pedersen, N.C. Proviral burden and infection kinetics of feline immunodeficiency virus in lymphocyte subsets of blood and lymph node. J. Virol. 1996, 70, 5165–5169. [Google Scholar] [PubMed]
- Willett, B.J.; Hosie, M.J. Chemokine receptors and co-stimulatory molecules: Unravelling feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 2008, 123, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Hosie, M.J.; Pajek, D.; Samman, A.; Willett, B.J. Feline immunodeficiency virus (FIV) neutralization: A review. Viruses 2011, 3, 1870–1890. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Hosie, M.J. The virus–receptor interaction in the replication of feline immunodeficiency virus (FIV). Curr. Opin. in Virol. 2013, 3, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Ann. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Doms, R.W. Chemokine receptors and HIV entry. AIDS 2001, 15, S34–S35. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.H.; Sundstrom, M.; de Rozieres, S.; de Parseval, A.; Grant, C.K.; Lin, Y.-C. Molecular mechanisms of fiv infection. Vet. Immunol. Immunopathol. 2008, 123, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E. How HIV changes its tropism: Evolution and adaptation? Curr. Opin. HIV AIDS 2009, 4, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Duenas-Decamp, M.J.; Sullivan, W.M.; Brown, R.; Ankghuambom, C.; Luzuriaga, K.; Robinson, J.; Burton, D.R.; Bell, J.; Simmonds, P. Variation in HIV-1 R5 macrophage-tropism correlates with sensitivity to reagents that block envelope: CD4 interactions but not with sensitivity to other entry inhibitors. Retrovirology 2008, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Clapham, P.R.; McKnight, A. HIV-1 receptors and cell tropism. Br. Med. Bull. 2001, 58, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Regoes, R.R.; Bonhoeffer, S. The HIV coreceptor switch: A population dynamical perspective. Trends Microbiol. 2005, 13, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Tomonaga, K.; Mikami, T. Molecular biology of the feline immunodeficiency virus auxiliary genes. J. Gen. Virol. 1996, 77, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- LaRue, R.S.; Lengyel, J.; Jónsson, S.R.; Andrésdóttir, V.; Harris, R.S. Lentiviral vif degrades the apobec3z3/apobec3h protein of its mammalian host and is capable of cross-species activity. J. Virol. 2010, 84, 8193–8201. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, M.; Chatterji, U.; Schaffer, L.; de Rozières, S.; Elder, J.H. Feline immunodeficiency virus orfa alters gene expression of splicing factors and proteasome-ubiquitination proteins. Virology 2008, 371, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Gemeniano, M.C.; Sawai, E.T.; Leutenegger, C.M.; Sparger, E.E. Feline immunodeficiency virus orf-a is required for virus particle formation and virus infectivity. J. Virol. 2003, 77, 8819–8830. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.; De Parseval, A.; Lerner, D.; Neil, J.; Thompson, F.; Elder, J. Influence of ORF2 on host cell tropism of feline immunodeficiency virus. Virology 1996, 215, 10–16. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A.; Elder, J.H. Demonstration that Orf2 encodes the feline immunodeficiency virus transactivating (TAT) protein and characterization of a unique gene product with partial rev activity. J. Virol. 1999, 73, 608–617. [Google Scholar] [PubMed]
- Gemeniano, M.C.; Sawai, E.T.; Sparger, E.E. Feline immunodeficiency virus Orf-A localizes to the nucleus and induces cell cycle arrest. Virology 2004, 325, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Fink, E.; Hu, Q.-Y.; Kiosses, W.B.; Elder, J.H. Orfa downregulates feline immunodeficiency virus primary receptor cd134 on the host cell surface and is important in viral infection. J. Virol. 2010, 84, 7225–7232. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H. Human immunodeficiency virus may encode a novel protein on the genomic DNA plus strand. Science 1988, 239, 1420–1422. [Google Scholar] [CrossRef] [PubMed]
- Briquet, S.; Vaquero, C. Immunolocalization studies of an antisense protein in HIV-1-infected cells and viral particles. Virology 2002, 292, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Etkin, A.F. Plus strand of the HIV provirus DNA is expressed at early stages of infection. AIDS Res. Hum. Retrovir. 1990, 6, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. HIV-1-encoded antisense rna suppresses viral replication for a prolonged period. Retrovirology 2012, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Laverdure, S.; Gross, A.; Arpin-Andre, C.; Clerc, I.; Beaumelle, B.; Barbeau, B.; Mesnard, J.M. HIV-1 antisense transcription is preferentially activated in primary monocyte-derived cells. J. Virol. 2012, 86, 13785–13789. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.B.; Ambrus, J.L., Jr.; Krawczyk, K.A.; Sharma, S.; Brooks, S.; Hsiao, C.B.; Schwartz, S.A. Human immunodeficiency virus-type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology 2006, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.L.; Vahey, M.T.; d’Arcy, L.; Ehrenberg, P.K.; Mosca, J.D.; Rappaport, J.; Redfield, R.R. Negative-strand rna transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by tat. J. Virol. 1994, 68, 979–987. [Google Scholar] [PubMed]
- Torresilla, C.; Do Carmo, S.; Larocque, É.; Douceron, E.; Mesnard, J.-M.; Mahieux, R.; Barbeau, B. The antisense protein of HTLV-2 positively modulates HIV-1 replication. Retrovirology 2014, 11, P118. [Google Scholar] [CrossRef]
- Torresilla, C.; Larocque, E.; Landry, S.; Halin, M.; Coulombe, Y.; Masson, J.Y.; Mesnard, J.M.; Barbeau, B. Detection of the HIV-1 minus-strand-encoded antisense protein and its association with autophagy. J. Virol. 2013, 87, 5089–5105. [Google Scholar] [CrossRef] [PubMed]
- Vanhee-Brossollet, C.; Thoreau, H.; Serpente, N.; D’Auriol, L.; Levy, J.P.; Vaquero, C. A natural antisense rna derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology 1995, 206, 196–202. [Google Scholar] [CrossRef]
- Cassan, E.; Arigon-Chifolleau, A.M.; Mesnard, J.M.; Gross, A.; Gascuel, O. Concomitant emergence of the antisense protein gene of HIV-1 and of the pandemic. Proc. Natl. Acad. Sci. USA 2016, 113, 11537–11542. [Google Scholar] [CrossRef] [PubMed]
- Briquet, S.; Richardson, J.; Vanhee-Brossollet, C.; Vaquero, C. Natural antisense transcripts are detected in different cell lines and tissues of cats infected with feline immunodeficiency virus. Gene 2001, 267, 157–164. [Google Scholar] [CrossRef]
- Durkin, K.; Rosewick, N.; Artesi, M.; Hahaut, V.; Griebel, P.; Arsic, N.; Burny, A.; Georges, M.; Van den Broeke, A. Characterization of novel bovine leukemia virus (BLV) antisense transcripts by deep sequencing reveals constitutive expression in tumors and transcriptional interaction with viral micrornas. Retrovirology 2016, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.M. The dynamics of CD4+ t-cell depletion in HIV disease. Nature 2001, 410, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Obert, L.A.; Hoover, E.A. Early pathogenesis of transmucosal feline immunodeficiency virus infection. J. Virol. 2002, 76, 6311–6322. [Google Scholar] [CrossRef] [PubMed]
- Obert, L.; Hoover, E. Relationship of lymphoid lesions to disease course in mucosal feline immunodeficiency virus type c infection. Vet. Pathol. 2000, 37, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.C.; Dean, G.A.; Pedersen, N.C.; Moore, P.F. Immunopathologic changes in the thymus during the acute stage of experimentally induced feline immunodeficiency virus infection in juvenile cats. J. Virol. 1997, 71, 8632–8641. [Google Scholar] [PubMed]
- Serrano-Villar, S.; Sainz, T.; Lee, S.A.; Hunt, P.W.; Sinclair, E.; Shacklett, B.L.; Ferre, A.L.; Hayes, T.L.; Somsouk, M.; Hsue, P.Y. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-aids morbidity and mortality. PLoS Pathog. 2014, 10, e1004078. [Google Scholar] [CrossRef] [PubMed]
- Loughran, T.J. Clonal diseases of large granular lymphocytes [see comments]. Blood 1993, 82, 1–14. [Google Scholar] [PubMed]
- Phillips, J.; Lanier, L. Lectin-dependent and anti-CD3 induced cytotoxicity are preferentially mediated by peripheral blood cytotoxic T lymphocytes expressing Leu-7 antigen. J. Immunol. 1986, 136, 1579–1585. [Google Scholar] [PubMed]
- Schmidt, R.E.; Murray, C.; Daley, J.F.; Schlossman, S.; Ritz, J. A subset of natural killer cells in peripheral blood displays a mature T cell phenotype. J. Exp. Med. 1986, 164, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Sprague, W.S.; Apetrei, C.; Avery, A.C.; Peskind, R.L.; Vandewoude, S. Large granular lymphocytes are universally increased in human, macaque, and feline lentiviral infection. Vet. Immunol. Immunopathol. 2015, 167, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Alekshun, T.J.; Sokol, L. Diseases of large granular lymphocytes. Cancer Control 2007, 14, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Boveri, E.; Riboni, R.; Antico, P.; Malacrida, A.; Pastorini, A. CD3+ T large granular lymphocyte leukaemia in a HIV+, HCV+, HBV+ patient. Virchows Arch. 2009, 454, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.R.; Cavenagh, J.D.; Milne, T.; Howe, D.; Wilkes, S.J.; Sinnott, P.; Forster, G.E.; Helbert, M. Benign monoclonal expansion of cd8+ lymphocytes in HIV infection. J. Clin. Pathol. 2000, 53, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Sprague, W.; TerWee, J.; VandeWoude, S. Temporal association of large granular lymphocytosis, neutropenia, proviral load, and fasl mrna in cats with acute feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 2010, 134, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Fogle, J.E.; Mexas, A.M.; Tompkins, W.A.; Tompkins, M.B. CD4+ CD25+ T regulatory cells inhibit CD8+ IFN-γ production during acute and chronic fiv infection utilizing a membrane TGF-β-dependent mechanism. AIDS Res. Hum. Retrovir. 2010, 26, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Horak, R.; Sion, M.; Riggin, L.; McNally, J.; Lin, Y.; Jackson, R.; O’Shea, A.; Roby, G.; Kovacs, C. CD25+ regulatory T cells isolated from HIV-infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV-specific CD8+ t cells in vitro. AIDS Res. Hum. Retrovir. 2007, 23, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Fogle, J.E.; Tompkins, M.B. Infection with feline immunodeficiency virus directly activates CD4+ CD25+ T regulatory cells. J. Virol. 2013, 87, 9373–9378. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J. Neurovirol. 2004, 10, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Zenger, E.; Tiffany-Castiglioni, E.; Collisson, E.W. Cellular mechanisms of feline immunodeficiency virus (FIV)-induced neuropathogenesis. Front. Biosci. 1997, 2, d527–d537. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.; Bexiga, M.; Brayden, D.; Brankin, B.; Willett, B.; Hosie, M.; Jacque, J.M.; Callanan, J. Lymphocyte migration through the blood–brain barrier (BBB) in feline immunodeficiency virus infection is significantly influenced by the pre-existence of virus and tumour necrosis factor (TNF)-α within the central nervous system (CNS): Studies using an in vitro feline bbb model. Neuropathol. Appl. Neurobiol. 2009, 35, 592–602. [Google Scholar] [PubMed]
- Hudson, L.; Bragg, D.; Tompkins, M.; Meeker, R. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. 2005, 1058, 148–160. [Google Scholar] [CrossRef] [PubMed]
- González-Scarano, F.; Martín-García, J. The neuropathogenesis of aids. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, R.; Schwinn, A.; Narayan, O.; Zink, C.; Kreth, H.; Roggendorf, W.; Dörries, R.; Schwender, S.; Imrich, H.; Ter Meulen, V. Human immunodeficiency virus infection in microglia: Correlation between cells infected in the brain and cells cultured from infectious brain tissue. Ann. Neurol. 1992, 31, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Maeda, K.; Tohya, Y.; Furuya, T.; Miyazawa, T.; Horimoto, T.; Norimine, J.; Kai, C.; Mikami, T. Replicative difference in early-passage feline brain cells among feline immunodeficiency virus isolates. Arch. Virol. 1992, 125, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Abramo, F.; Iorio, C.D.; Cantile, C.; Carli, M.A.; Pollera, C.; Vago, L.; Tosoni, A.; Costanzi, G. Neuropathology in cats experimentally infected wit feline immunodeficiency virus: A morphological, immunocytochemical and morphometric study. J. Neurovirol. 1997, 3, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Poulton, W.; Feng, W.-H.; Hudson, L.; Longo, F.M. Suppression of immunodeficiency virus-associated neural damage by the P75 neurotrophin receptor ligand, LM11A-31, in an in vitro feline model. J. Neuroimmune Pharmacol. 2012, 7, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Hudson, L.C.; Tompkins, M.B.; Vahlenkamp, T.W.; Meeker, R.B. Compartmentalization and evolution of feline immunodeficiency virus between the central nervous system and periphery following intracerebroventricular or systemic inoculation. J. Neurovirol. 2006, 12, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.M.; Hoke, A.; Zhu, Y.; Johnston, J.B.; van Marle, G.; Silva, C.; Zochodne, D.W.; Power, C. Peripheral neuropathy in lentivirus infection: Evidence of inflammation and axonal injury. Aids 2004, 18, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H.; Lackner, A.; Williams, K.C. Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol. Rev. 2013, 254, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Burdo, T.H. Monocyte mobilization, activation markers, and unique macrophage populations in the brain: Observations from siv infected monkeys are informative with regard to pathogenic mechanisms of HIV infection in humans. J. Neuroimmune Pharmacol. 2012, 7, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Lackner, A.; Mallard, J. Non-human primate models of siv infection and cns neuropathology. Curr. Opin. Virol. 2016, 19, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Hickey, W.F. Central nervous system damage, monocytes and macrophages, and neurological disorders in aids. Ann. Rev. Neurosci. 2002, 25, 537–562. [Google Scholar] [CrossRef] [PubMed]
- Laast, V.A.; Pardo, C.A.; Tarwater, P.M.; Queen, S.E.; Reinhart, T.A.; Ghosh, M.; Adams, R.J.; Zink, M.C.; Mankowski, J.L. Pathogenesis of simian immunodeficiency virus-induced alterations in macaque trigeminal ganglia. J. Neuropathol. Exp. Neurol. 2007, 66, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Laast, V.A.; Shim, B.; Johanek, L.M.; Dorsey, J.L.; Hauer, P.E.; Tarwater, P.M.; Adams, R.J.; Pardo, C.A.; McArthur, J.C.; Ringkamp, M. Macrophage-mediated dorsal root ganglion damage precedes altered nerve conduction in siv-infected macaques. Am. J. Pathol. 2011, 179, 2337–2345. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, H.C.; Chen, W.; Borzan, J.; Mankowski, J.L.; Höke, A. Mitochondrial dysfunction in distal axons contributes to human immunodeficiency virus sensory neuropathy. Ann Neurol. 2011, 69, 100–110. [Google Scholar] [CrossRef] [PubMed]
- VandeWoude, S.; Apetrei, C. Going wild: Lessons from naturally occurring T-lymphotropic lentiviruses. Clin. Microbiol. Rev. 2006, 19, 728–762. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.V.; Lin, K.-C.; Mansfield, K.; Wachtman, L.M. Specific pathogen-free status alters immunophenotype in rhesus macaques: Implications for the study of simian immunodeficiency virus. AIDS Res. Hum. Retrovir. 2011, 27, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Denton, P.W.; Garcia, J.V. Humanized mouse models of HIV infection. AIDS Rev. 2011, 13, 135–148. [Google Scholar] [PubMed]
- Zheng, J.; Ghorpade, A.; Niemann, D.; Cotter, R.L.; Thylin, M.R.; Epstein, L.; Swartz, J.M.; Shepard, R.B.; Liu, X.; Nukuna, A.; et al. Lymphotropic virions affect chemokine receptor-mediated neural signaling and apoptosis: Implications for human immunodeficiency virus type 1-associated dementia. J. Virol. 1999, 73, 8256–8267. [Google Scholar] [PubMed]
- Hesselgesser, J.; Taub, D.; Baskar, P.; Greenberg, M.; Hoxie, J.; Kolson, D.L.; Horuk, R. Neuronal apoptosis induced by HIV-1 GP120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr. Biol. CB 1998, 8, 595–598. [Google Scholar] [CrossRef]
- Zheng, J.; Thylin, M.R.; Ghorpade, A.; Xiong, H.; Persidsky, Y.; Cotter, R.; Niemann, D.; Che, M.; Zeng, Y.C.; Gelbard, H.A.; et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J. Neuroimmunol. 1999, 98, 185–200. [Google Scholar] [CrossRef]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Bushell, T.J.; Gray, P.W.; Miller, R.J. Chemokines regulate hippocampal neuronal signaling and GP120 neurotoxicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14500–14505. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Sucher, N.J.; Kaiser, P.K.; Dreyer, E.B. Synergistic effects of HIV coat protein and nmda receptor-mediated neurotoxicity. Neuron 1991, 7, 111–118. [Google Scholar] [CrossRef]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins tat and GP120. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. 2), S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Ballester, L.Y.; Capo-Velez, C.M.; Garcia-Beltran, W.F.; Ramos, F.M.; Vazquez-Rosa, E.; Rios, R.; Mercado, J.R.; Melendez, R.I.; Lasalde-Dominicci, J.A. Up-regulation of the neuronal nicotinic receptor alpha7 by HIV glycoprotein 120: Potential implications for HIV-associated neurocognitive disorder. J. Biol. Chem. 2012, 287, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate-linked enhancement of CGMP levels in the cerebellum. Proc. Natl. Acad. Sci. USA 1989, 86, 9030–9033. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Garthwaite, G.; Palmer, R.M.; Moncada, S. Nmda receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur. J. Pharmacol. 1989, 172, 413–416. [Google Scholar] [CrossRef]
- Kornau, H.C.; Seeburg, P.H.; Kennedy, M.B. Interaction of ion channels and receptors with PDZ domain proteins. Curr. Opin. Neurobiol. 1997, 7, 368–373. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef] [PubMed]
- Rameau, G.A.; Tukey, D.S.; Garcin-Hosfield, E.D.; Titcombe, R.F.; Misra, C.; Khatri, L.; Getzoff, E.D.; Ziff, E.B. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the nmda receptor regulates ampa receptor trafficking and neuronal cell death. J. Neurosci. 2007, 27, 3445–3455. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S. Nitric oxide signaling specificity—The heart of the problem. J. Cell Sci. 2003, 116, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bragg, D.C.; Meeker, R.B.; Duff, B.A.; English, R.V.; Tompkins, M.B. Neurotoxicity of FIV and FIV envelope protein in feline cortical cultures. Brain Res. 1999, 816, 431–437. [Google Scholar] [CrossRef]
- Brenneman, D.E.; Westbrook, G.L.; Fitzgerald, S.P.; Ennist, D.L.; Elkins, K.L.; Ruff, M.R.; Pert, C.B. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature 1988, 335, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.K.; Pu, R.; Sato, E.; Hohdatsu, T. Feline immunodeficiency virus pathogenesis and development of a dual-subtype feline-immunodeficiency-virus vaccine. AIDS 2007, 21, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Uhl, E.; Heaton-Jones, T.; Pu, R.; Yamamoto, J. Fiv vaccine development and its importance to veterinary and human medicine: A review: FIV vaccine 2002 update and review. Vet. Immunol. Immunopathol. 2002, 90, 113–132. [Google Scholar] [CrossRef]
- Pu, R.; Coleman, J.; Coisman, J.; Sato, E.; Tanabe, T.; Arai, M.; Yamamoto, J.K. Dual-subtype FIV vaccine (Fel-O-Vax® FIV) protection against a heterologous subtype B FIV isolate. J. Feline Med. Surg. 2005, 7, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Bęczkowski, P.M.; Harris, M.; Techakriengkrai, N.; Beatty, J.A.; Willett, B.J.; Hosie, M.J. Neutralising antibody response in domestic cats immunised with a commercial feline immunodeficiency virus (FIV) vaccine. Vaccine 2015, 33, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Westman, M.; Malik, R.; Hall, E.; Harris, M.; Norris, J. The protective rate of the feline immunodeficiency virus vaccine: An australian field study. Vaccine 2016, 34, 4752–4758. [Google Scholar] [CrossRef] [PubMed]
- Dunham, S.P.; Bruce, J.; Klein, D.; Flynn, J.N.; Golder, M.C.; MacDonald, S.; Jarrett, O.; Neil, J.C. Prime-boost vaccination using DNA and whole inactivated virus vaccines provides limited protection against virulent feline immunodeficiency virus. Vaccine 2006, 24, 7095–7108. [Google Scholar] [CrossRef] [PubMed]
- Dunham, S.; Bruce, J.; MacKay, S.; Golder, M.; Jarrett, O.; Neil, J. Limited efficacy of an inactivated feline immunodeficiency virus vaccine. Vet. Rec. 2006, 158, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Hosie, M.J.; Osborne, R.; Reid, G.; Neil, J.C.; Jarrett, O. Enhancement after feline immunodeficiency virus vaccination. Vet. Immunol. Immunopathol. 1992, 35, 191–197. [Google Scholar] [CrossRef]
- Lombardi, S.; Garzelli, C.; Pistello, M.; Massi, C.; Matteucci, D.; Baldinotti, F.; Cammarota, G.; Da Prato, L.; Bandecchi, P.; Tozzini, F. A neutralizing antibody-inducing peptide of the V3 domain of feline immunodeficiency virus envelope glycoprotein does not induce protective immunity. J. Virol. 1994, 68, 8374–8379. [Google Scholar] [PubMed]
- Siebelink, K.; Tijhaar, E.; Huisman, R.C.; Huisman, W.; De Ronde, A.; Darby, I.H.; Francis, M.J.; Rimmelzwaan, G.F.; Osterhaus, A. Enhancement of feline immunodeficiency virus infection after immunization with envelope glycoprotein subunit vaccines. J. Virol. 1995, 69, 3704–3711. [Google Scholar] [PubMed]
- Richardson, J.; Moraillon, A.; Baud, S.; Cuisinier, A.; Sonigo, P.; Pancino, G. Enhancement of feline immunodeficiency virus (FIV) infection after DNA vaccination with the fiv envelope. J. Virol. 1997, 71, 9640–9649. [Google Scholar] [PubMed]
- Karlas, J.A.; Siebelink, K.; Peer, M.A.V.; Huisman, W.; Cuisinier, A.M.; Rimmelzwaan, G.F.; Osterhaus, A. Vaccination with experimental feline immunodeficiency virus vaccines, based on autologous infected cells, elicits enhancement of homologous challenge infection. J Gen. Virol. 1999, 80, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Giannecchini, S.; Isola, P.; Sichi, O.; Matteucci, D.; Pistello, M.; Zaccaro, L.; Del Mauro, D.; Bendinelli, M. Aids vaccination studies using an ex vivo feline immunodeficiency virus model: Failure to protect and possible enhancement of challenge infection by four cell-based vaccines prepared with autologous lymphoblasts. J. Virol. 2002, 76, 6882–6892. [Google Scholar] [CrossRef] [PubMed]
- Lun, W.-H.; Takeda, A.; Nakamura, H.; Kano, M.; Mori, K.; Sata, T.; Nagai, Y.; Matano, T. Loss of virus-specific CD4+ T cells with increases in viral loads in the chronic phase after vaccine-based partial control of primary simian immunodeficiency virus replication in macaques. J. Gen. Virol. 2004, 85, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Mueller, Y.M.; Do, D.H.; Altork, S.R.; Artlett, C.M.; Gracely, E.J.; Katsetos, C.D.; Legido, A.; Villinger, F.; Altman, J.D.; Brown, C.R. Il-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger siv-specific CD8+ t cell responses. J. Immunol. 2008, 180, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.E.; Montefiori, D.; Mitchell, W. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet 1988, 331, 790–794. [Google Scholar] [CrossRef]
- Staprans, S.I.; Hamilton, B.L.; Follansbee, S.E.; Elbeik, T.; Barbosa, P.; Grant, R.M.; Feinberg, M.B. Activation of virus replication after vaccination of HIV-1-infected individuals. J. Exp. Med. 1995, 182, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Villinger, F.; Rowe, T.; Parekh, B.S.; Green, T.A.; Mayne, A.E.; Grimm, B.; McClure, H.M.; Lackner, A.A.; Dailey, P.J.; Ansari, A.A. Chronic immune stimulation accelerates siv-induced disease progression. J. Med. Primatol. 2001, 30, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Staprans, S.I.; Barry, A.P.; Silvestri, G.; Safrit, J.T.; Kozyr, N.; Sumpter, B.; Nguyen, H.; McClure, H.; Montefiori, D.; Cohen, J.I. Enhanced siv replication and accelerated progression to aids in macaques primed to mount a cd4 t cell response to the siv envelope protein. Proc. Natl. Acad. Sci. USA 2004, 101, 13026–13031. [Google Scholar] [CrossRef] [PubMed]
- Huisman, W.; Martina, B.; Rimmelzwaan, G.; Gruters, R.; Osterhaus, A. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C. Role of complement and FC receptors in the pathogenesis of HIV-1 infection. In Immunopathogenesis of HIV Infection; Springer: Berlin, Germany, 1997; pp. 119–138. [Google Scholar]
- Müller-Eberhard, H.J. Molecular organization and function of the complement system. Ann. Rev. Biochem. 1988, 57, 321–347. [Google Scholar] [CrossRef] [PubMed]
- Willey, S.; Aasa-Chapman, M.M.; O’Farrell, S.; Pellegrino, P.; Williams, I.; Weiss, R.A.; Neil, S.J. Extensive complement-dependent enhancement of HIV-1 by autologous non-neutralising antibodies at early stages of infection. Retrovirology 2011, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Szabó, J.; Prohászka, Z.; Tóth, F.D.; Gyuris, Á.; Segesdi, J.; Bánhegyi, D.; Ujhelyi, E.; Minárovits, J.; Füst, G. Strong correlation between the complement-mediated antibody-dependent enhancement of HIV-1 infection and plasma viral load. AIDS 1999, 13, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.E.; Kawamura, T.; Lake, D.; Masuho, Y.; Mitchell, W.; Hersh, E.M. Antibodies to the primary immunodominant domain of human immunodeficiency virus type 1 (HIV-1) glycoprotein gp41 enhance HIV-1 infection in vitro. J. Virol. 1990, 64, 5301–5305. [Google Scholar] [PubMed]
- Robinson, W.; Gorny, M.; Xu, J.; Mitchell, W.; Zolla-Pazner, S. Two immunodominant domains of GP41 bind antibodies which enhance human immunodeficiency virus type 1 infection in vitro. J. Virol. 1991, 65, 4169–4176. [Google Scholar] [PubMed]
- Montefiori, D.C.; Robinson, W.E.; Mitchell, W.M. Antibody-independent, complement-mediated enhancement of HIV-1 infection by mannosidase i and ii inhibitors. Antivir. Res. 1989, 11, 137–146. [Google Scholar] [CrossRef]
- Boyer, V.; Desgranges, C.; Trabaud, M.; Fischer, E.; Kazatchkine, M. Complement mediates human immunodeficiency virus type 1 infection of a human t cell line in a CD4- and antibody-independent fashion. J. Exp. Med. 1991, 173, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C.; Stewart, K.; Ahearn, J.M.; Zhou, J. Complement-mediated binding of naturally glycosylated and glycosylation-modified human immunodeficiency virus type 1 to human CR2 (CD21). J. Virol. 1993, 67, 2699–2706. [Google Scholar] [PubMed]
- Reisinger, E.C.; Vogetseder, W.; Berzow, D.; Köfler, D.; Bitterlich, G.; Lehr, H.A.; Wachter, H.; Dierich, M.P. Complement-mediated enhancement of HIV-1 infection of the monoblastoid cell line U937. AIDS 1990, 4, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Sölder, B.; Schulz, T.; Hengster, P.; Löwer, J.; Larcher, C.; Bitterlich, G.; Kurth, R.; Wachter, H.; Dierich, M. HIV and HIV-infected cells differentially activate the human complement system independent of antibody. Immunol. Lett. 1989, 22, 135–145. [Google Scholar] [CrossRef]
- Spear, G.T.; Jiang, H.; Sullivan, B.L.; Gewürz, H.; Landay, A.L.; Lint, T.F. Direct binding of complement component C1q to human immunodeficiency virus (HIV) and human T lymphotrophic virus-I (HTLV-I) coinfected cells. AIDS Res. Hum. Retrovir. 1991, 7, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.; Broche, S.; Baud, S.; Leste-Lasserre, T.; Féménia, F.; Levy, D.; Moraillon, A.; Pancino, G.; Sonigo, P. Lymphoid activation: A confounding factor in aids vaccine development? J. Gen. Virol. 2002, 83, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M.; Greenwell-Wild, T.; Peng, G.; Hale-Donze, H.; Orenstein, J.M. Co-infection with opportunistic pathogens promotes human immunodeficiency virus type 1 infection in macrophages. J. Infect. Dis. 1999, 179, S457–S460. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Orenstein, J.M. Immune stimulation and HIV-1 viral replication. J. Leukoc. Biol. 1997, 62, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-C.; Spouge, J.L.; Conley, S.R.; Tsai, W.-P.; Merges, M.J.; Nara, P.L. Human plasma enhances the infectivity of primary human immunodeficiency virus type 1 isolates in peripheral blood mononuclear cells and monocyte-derived macrophages. J. Virol. 1995, 69, 6054–6062. [Google Scholar] [PubMed]
- Thibault, S.; Tardif, M.R.; Barat, C.; Tremblay, M.J. TLR2 signaling renders quiescent naive and memory CD4+ T cells more susceptible to productive infection with X4 and R5 HIV-type 1. J. Immunol. 2007, 179, 4357–4366. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.D.; Kirchhoff, F.; Czajak, S.C.; Sehgal, P.K.; Desrosiers, R.C. Protective effects of a live attenuated siv vaccine with a deletion in the nef gene. Science 1992, 258, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; Silvestri, G. Non-human primate models in aids research. Curr. Opin. HIV AIDS 2013, 8, 255–261. [Google Scholar] [PubMed]
- Hessell, A.J.; Hangartner, L.; Hunter, M.; Havenith, C.E.; Beurskens, F.J.; Bakker, J.M.; Lanigan, C.M.; Landucci, G.; Forthal, D.N.; Parren, P.W. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 2007, 449, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Mascola, J.R.; Stiegler, G.; VanCott, T.C.; Katinger, H.; Carpenter, C.B.; Hanson, C.E.; Beary, H.; Hayes, D.; Frankel, S.S.; Birx, D.L. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat. Med. 2000, 6, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Evans, D.T. Animal models for HIV/AIDs research. Nat. Rev. Microbiol. 2012, 10, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Gauduin, M.-C.; Parren, P.W.; Weir, R.; Barbas, C.F.; Burton, D.R.; Koup, R.A. Passive immunization with a human monoclonal antibody protects hu-PBL-SCID mice against challenge by primary isolates of HIV-1. Nat. Med. 1997, 3, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.; Hanley, M.B.; Moreno, M.B.; Wieder, E.; McCune, J.M. Human immunodeficiency virus-1 infection interrupts thymopoiesis and multilineage hematopoiesis in vivo. Blood 1998, 91, 2672–2678. [Google Scholar] [PubMed]
- McCune, J.M.; Namikawa, R.; Shih, C.-C.; Rabin, L.; Kaneshima, H. Suppression of HIV infection in AZT-treated SCID-HU mice. Science 1990, 247, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Safrit, J.T.; Fung, M.S.; Andrews, C.A.; Braun, D.G.; Sun, W.N.; Chang, T.W.; Koup, R.A. Hu-PBL-SCID mice can be protected from HIV-1 infection by passive transfer of monoclonal antibody to the principal neutralizing determinant of envelope GP120. AIDS 1993, 7, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Mataftsi, M.; Skoura, L.; Sakellari, D. Hiv infection and periodontal diseases: An overview of the post-HAART era. Oral Dis. 2011, 17, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 1999, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Coogan, M.M.; Greenspan, J.; Challacombe, S.J. Oral lesions in infection with human immunodeficiency virus. Bull. World Health Organ. 2005, 83, 700–706. [Google Scholar] [PubMed]
- Miziara, I.D.; Weber, R. Oral lesions as predictors of highly active antiretroviral therapy failure in brazilian HIV-infected children. J. Oral Pathol. Med. 2008, 37, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.F.; de Araújo Castro, G.F.B.; de Souza, I.P.R.; Pinheiro, M. Pediatric HIV-related oral manifestations: A five-year retrospective study. Braz. Oral Res. 2004, 18, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Vaseliu, N.; Carter, A.B.; Kline, N.E.; Kozinetz, C.; Cron, S.G.; Matusa, R.; Kline, M.W. Longitudinal study of the prevalence and prognostic implications of oral manifestations in romanian children infected with human immunodeficiency virus type 1. Pediatr. Infect. Dis. J. 2005, 24, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Funderburg, N.T.; Brenchley, J.M. Microbial translocation, immune activation, and HIV disease. Trends Microbiol. 2013, 21, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Horowitz, A.; Hurley, A.; Hogan, C.; Boden, D.; Racz, P.; Markowitz, M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 2004, 200, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Douek, D.C. Hiv disease: Fallout from a mucosal catastrophe? Nat. Immunol. 2006, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Baskin, G.; Murphey-Corb, M.; Watson, E.; Martin, L. Necropsy findings in rhesus monkeys experimentally infected with cultured simian immunodeficiency virus (SIV)/delta. Vet. Pathol. Online 1988, 25, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Casteleyn, C.; Breugelmans, S.; Simoens, P.; Van den Broeck, W. The tonsils revisited: Review of the anatomical localization and histological characteristics of the tonsils of domestic and laboratory animals. Clin. Dev. Immunol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- McClure, H.; Anderson, D.; Fultz, P.; Ansari, A.; Lockwood, E.; Brodie, A. Spectrum of disease in macaque monkeys chronically infected with SIV/SMM. Vet. Immunol. Immunopathol. 1989, 21, 13–24. [Google Scholar] [CrossRef]
- Dos Santos, L.D.C.; Castro, G.F.; de Souza, I.P.R.; Oliveira, R.H.S. Oral manifestations related to immunosuppression degree in HIV-positive children. Braz. Dent. J. 2001, 12, 135–138. [Google Scholar]
- Sparkes, A.; Caney, S.M. Feline Medicine: Self-Assessment Color Review; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Willett, B.; Flynn, N.; Hosic, M. FIV infection of the domestic cat: An animal model for aids. Immunol. Today 1997, 18, 182–189. [Google Scholar] [CrossRef]
- Yamamoto, J.K.; Sparger, E.; Ho, E.W.; Andersen, P.R.; O’connor, T.; Mandell, C.; Lowenstine, L.; Munn, R.; Pedersen, N. Pathogenesis of experimentally induced feline immunodeficiency virus infection in cats. Am. J. Vet. Res. 1988, 49, 1246–1258. [Google Scholar] [PubMed]
- Brady, L.; Walker, C.; Oxford, G.; Stewart, C.; Magnusson, I.; McArthur, W. Oral diseases, mycology and periodontal microbiology of HIV-1-infected women. Mol. Oral Microbiol. 1996, 11, 371–380. [Google Scholar] [CrossRef]
- Flaitz, C.M.; Hicks, M.J. Oral candidiasis in children with immune suppression: Clinical appearance and therapeutic considerations. ASDC J. Dent. Child. 1999, 66, 154, 161–164. [Google Scholar] [PubMed]
- Holt, S.C.; Ebersole, J.L. Porphyromonas gingivalis, treponema denticola, and tannerella forsythia: The ‘red complex’, a prototype polybacterial pathogenic consortium in periodontitis. Periodontology 2000 2005, 38, 72–122. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Saxena, D.; Chen, Z.; Liu, G.; Abrams, W.R.; Phelan, J.A.; Norman, R.G.; Fisch, G.S.; Corby, P.M.; Dewhirst, F. HIV infection and microbial diversity in saliva. J. Clin. Microbiol. 2014, 52, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Love, D.; Johnson, J.; Moore, L. Bacteroides species from the oral cavity and oral-associated diseases of cats. Vet. Microbiol. 1989, 19, 275–281. [Google Scholar] [CrossRef]
- Murray, P.A.; Grassi, M.; Winkler, J.R. The microbiology of HIV-associated periodontal lesions. J. Clin. Periodontol. 1989, 16, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Nokta, M. Oral manifestations associated with HIV infection. Curr. HIV/AIDS Rep. 2008, 5, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.M.; Love, D.N. Associations amongst three feline porphyromonas species from the gingival margin of cats during periodontal health and disease. Vet. Microbiol. 1999, 65, 195–207. [Google Scholar] [CrossRef]
- Rams, T.E.; Andriolo, M., Jr.; Feik, D.; Abel, S.N.; McGivern, T.M.; Slots, J. Microbiological study of HIV-related periodontitis. J. Periodontol. 1991, 62, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Sims, T.; Moncla, B.; Page, R. Serum antibody response to antigens of oral gram-negative bacteria by cats with plasma cell gingivitis-pharyngitis. J. Dent. Res. 1990, 69, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Vahlenkamp, T.; Egberink, H.; Hartmann, K.; Witvrouw, M.; Pannecouque, C.; Casara, P.; Nave, J.; De Clercq, E. Antiretroviral activities of acyclic nucleoside phosphonates [9-(2-phosphonylmethoxyethyl) adenine, 9-(2-phosphonylmethoxyethyl) guanine, (R)-9-(2-phosphonylmethoxypropyl) adenine, and mdl 74,968] in cell cultures and murine sarcoma virus-infected newborn nmri mice. Antimicrob. Agents Chemother. 1997, 41, 611–616. [Google Scholar] [PubMed]
- Vahlenkamp, T.W.; De Ronde, A.; Balzarini, J.; Naesens, L.; De Clercq, E.; Van Eijk, M.; Horzinek, M.C.; Egberink, H.F. (R)-9-(2-phosphonylmethoxypropyl)-2, 6-diaminopurine is a potent inhibitor of feline immunodeficiency virus infection. Antimicrob. Agents Chemother. 1995, 39, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Remington, K.M.; Preston, B.D.; Schinazi, R.F.; North, T.W. A novel point mutation at position 156 of reverse transcriptase from feline immunodeficiency virus confers resistance to the combination of (−)-β-2′, 3′-dideoxy-3′-thiacytidine and 3′-azido-3′-deoxythymidine. J. Virol. 1998, 72, 2335–2340. [Google Scholar] [PubMed]
- Hartmann, K.; Wooding, A.; Bergmann, M. Efficacy of antiviral drugs against feline immunodeficiency virus. Vet. Sci. 2015, 2, 456–476. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.M.; McCrackin, M.A.; Schinazi, R.F.; Hill, P.B.; Vahlenkamp, T.W.; Tompkins, M.B.; Hartmann, K. Antiviral efficacy of nine nucleoside reverse transcriptase inhibitors against feline immunodeficiency virus in feline peripheral blood mononuclear cells. Am. J. Vet. Res. 2014, 75, 273–281. [Google Scholar] [CrossRef] [PubMed]
- De Rozieres, S.; Thompson, J.; Sundstrom, M.; Gruber, J.; Stump, D.S.; Aymeric, P.; VandeWoude, S.; Elder, J.H. Replication properties of clade A/C chimeric feline immunodeficiency viruses and evaluation of infection kinetics in the domestic cat. J. Virol. 2008, 82, 7953–7963. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Mackie, R.S.; Harrison, T.; Shariat, B.; Kind, T.; Kehl, T.; Löchelt, M.; Boucher, C.; VandeWoude, S. Targeted enrichment for pathogen detection and characterization in three felid species. J. Clin. Microbiol. 2017, 55, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.N.; Pop, M.; Bravo, H. Metagenomeseq: Statistical analysis for sparse high-throughput sequencing. Bioconductor 2014, 1, 1–32. [Google Scholar]
- Paulson, J.N.; Pop, M.; Bravo, H. Metagenomeseq: Statistical analysis for sparse high-throughput sequencing. Bioconduct. Package 2013, 1, 1–35. [Google Scholar]
- Hunt, P.W.; Deeks, S.G.; Rodriguez, B.; Valdez, H.; Shade, S.B.; Abrams, D.I.; Kitahata, M.M.; Krone, M.; Neilands, T.B.; Brand, R.J. Continued CD4 cell count increases in HIV-infected adults experiencing 4 years of viral suppression on antiretroviral therapy. Aids 2003, 17, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Leggott, P.J. Oral manifestations of HIV infection in children. Oral Surg. Oral Med. Oral Pathol. 1992, 73, 187–192. [Google Scholar] [CrossRef]
- Heron, S.E.; Elahi, S. HIV infection and compromised mucosal immunity: Oral manifestations and systemic inflammation. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Girard, N.; Servet, E.; Biourge, V.; Hennet, P. Periodontal health status in a colony of 109 cats. J. Vet. Dent. 2009, 26, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.; Tutt, C. Periodontal disease in cats: Back to basics—With an eye on the future. J. Feline Med. Surg. 2015, 17, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Zahradnik, R.; Magnusson, I.; Walker, C.; McDonell, E.; Hillman, C.; Hillman, J. Preliminary assessment of safety and effectiveness in humans of probiora3™, a probiotic mouthwash. J. Appl. Microbiol. 2009, 107, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Seminario-Amez, M.; López-López, J.; Estrugo-Devesa, A.; Ayuso-Montero, R.; Jané-Salas, E. Probiotics and oral health: A systematic review. Med. Oral Patol. Oral Cir. Bucal 2017, 22, e282–e288. [Google Scholar] [CrossRef] [PubMed]
- Teughels, W.; Durukan, A.; Ozcelik, O.; Pauwels, M.; Quirynen, M.; Haytac, M.C. Clinical and microbiological effects of lactobacillus reuteri probiotics in the treatment of chronic periodontitis: A randomized placebo-controlled study. J. Clin. Periodontol. 2013, 40, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Galilee, M.; Alian, A. The structure of FIV reverse transcriptase and its implications for non-nucleoside inhibitor resistance. PLoS Pathog. 2018, 14, e1006849. [Google Scholar] [CrossRef] [PubMed]
- Doménech, A.; Miró, G.; Collado, V.M.; Ballesteros, N.; Sanjosé, L.; Escolar, E.; Martin, S.; Gomez-Lucia, E. Use of recombinant interferon omega in feline retrovirosis: From theory to practice. Vet. Immunol. Immunopathol. 2011, 143, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.O.; Gil, S.; Duarte, A.; McGahie, D.; Sepúlveda, N.; Niza, M.M.; Tavares, L. Evaluation of viremia, proviral load and cytokine profile in naturally feline immunodeficiency virus infected cats treated with two different protocols of recombinant feline interferon omega. Res. Vet. Sci. 2015, 99, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Mari, K.; Maynard, L.; Sanquer, A.; Lebreux, B.; Eun, H.M. Therapeutic effects of recombinant feline interferon-co on feline leukemia virus (FELV)-infected and felv/feline immunodeficiency virus (FIV)-coinfected symptomatic cats. J. Vet. Intern. Med. 2004, 18, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Künzi, M.S.; Pitha, P.M. Role of interferon-stimulated gene ISG-15 in the interferon-ω-mediated inhibition of human immunodeficiency virus replication. J. Interferon Cytokine Res. 1996, 16, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Adolf, G. Human interferon omega—A review. Mult. Scler. Houndmills Basingstoke Engl. 1995, 1, S44–S47. [Google Scholar]
Figure 1.
Immunohistochemistry from a feline immunodeficiency virus (FIV)-infected cat with primary B cell lymphoma. (A) Mesenteric lymph Node; 40× and 4× (inset). Normal amounts of interlobular T lymphocytes (arrows) are present throughout the lymph node. Anti-CD3 (IHC) with DAB as chromogen and hematoxylin counterstain. (B) Mesenteric lymph Node; 40× and 4× (inset). Neoplastic B lymphocytes (arrows) multifocally expand the normal lymph node architecture. Anti-CD79a IHC with DAB as chromogen and hematoxylin counterstain.
Figure 1.
Immunohistochemistry from a feline immunodeficiency virus (FIV)-infected cat with primary B cell lymphoma. (A) Mesenteric lymph Node; 40× and 4× (inset). Normal amounts of interlobular T lymphocytes (arrows) are present throughout the lymph node. Anti-CD3 (IHC) with DAB as chromogen and hematoxylin counterstain. (B) Mesenteric lymph Node; 40× and 4× (inset). Neoplastic B lymphocytes (arrows) multifocally expand the normal lymph node architecture. Anti-CD79a IHC with DAB as chromogen and hematoxylin counterstain.
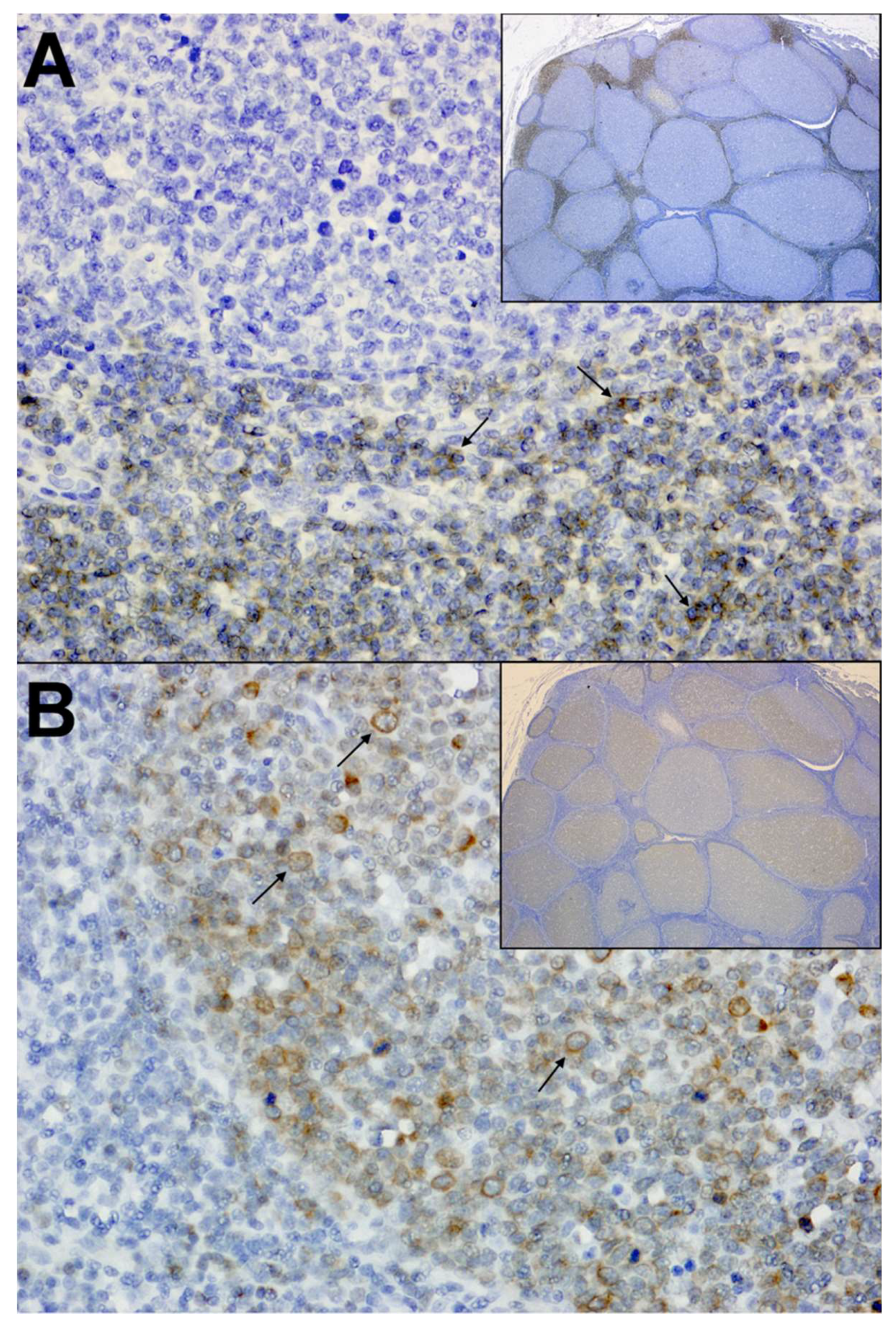
Figure 2.
Genomic organization of FIV and human immunodeficiency virus (HIV). The genomic structure of FIV consists of three primary open reading frames (ORFs), gag, pol, and env, which are flanked by two long-terminal repeats (LTR) and accompanied by numerous small ORFs containing regulatory and accessory genes such as vif and orfA. Potential short ORFs (antisense ORFs—ASP, shown in red) may be translated from a negative strand message. The genomic structure of HIV consists of three similar primary ORFs (gag, pol, and env), as well as numerous small ORFs containing regulatory and accessory genes, such as vif, vpr, vpu, tat, rev, and nef. Similar to FIV, the antisense ORF, ASP (shown in red) may be translated from a negative strand message.
Figure 2.
Genomic organization of FIV and human immunodeficiency virus (HIV). The genomic structure of FIV consists of three primary open reading frames (ORFs), gag, pol, and env, which are flanked by two long-terminal repeats (LTR) and accompanied by numerous small ORFs containing regulatory and accessory genes such as vif and orfA. Potential short ORFs (antisense ORFs—ASP, shown in red) may be translated from a negative strand message. The genomic structure of HIV consists of three similar primary ORFs (gag, pol, and env), as well as numerous small ORFs containing regulatory and accessory genes, such as vif, vpr, vpu, tat, rev, and nef. Similar to FIV, the antisense ORF, ASP (shown in red) may be translated from a negative strand message.
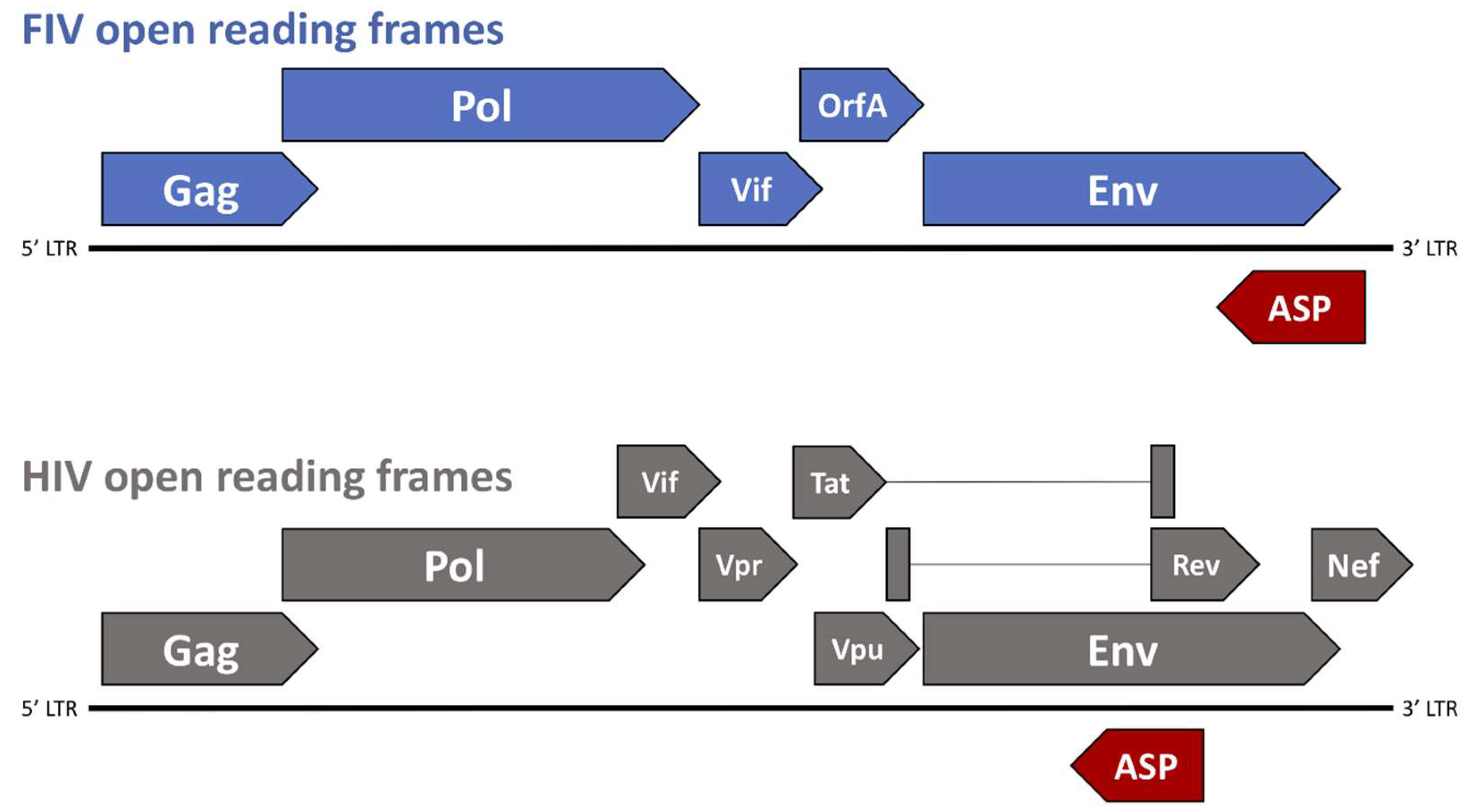
Figure 3.
Cytological morphology of a large granular lymphocyte (LGL). Recent studies have determined that elevations in LGL phenotypes during both FIV and HIV infection may represent polyclonal T-cells with viral suppressive properties. Wright-Giemsa stain, bar = 5 μm.
Figure 3.
Cytological morphology of a large granular lymphocyte (LGL). Recent studies have determined that elevations in LGL phenotypes during both FIV and HIV infection may represent polyclonal T-cells with viral suppressive properties. Wright-Giemsa stain, bar = 5 μm.

Figure 4.
Salivary microbiome alterations during FIV infection. (A) Three-dimensional (3D) Nonmetric Multidimensional Scaling (NMDS) separates clusters of FIV− and FIV+ cat microbiome samples. Ovals represent the 90% confidence ellipsoids around the centroid of the clusters (FIV+ = red; FIV− = blue). (B) Operational taxonomic units (OTUs) with significant log-fold change in abundance between FIV+ and FIV− cats at the 0.1 level of significance (after correcting for multiple testing). The list on the left shows the genera of each of these OTUs. Red indicates over representation of that OTU in the FIV+ cats. (C) FIV+ cat with clinical gingivitis/periodontitis with near monoculture of Moraxellaceae (outer circle) as compared to the mean microbial community structure of cats that are FIV + (middle circle) and cats that are FIV negative (inner circle).
Figure 4.
Salivary microbiome alterations during FIV infection. (A) Three-dimensional (3D) Nonmetric Multidimensional Scaling (NMDS) separates clusters of FIV− and FIV+ cat microbiome samples. Ovals represent the 90% confidence ellipsoids around the centroid of the clusters (FIV+ = red; FIV− = blue). (B) Operational taxonomic units (OTUs) with significant log-fold change in abundance between FIV+ and FIV− cats at the 0.1 level of significance (after correcting for multiple testing). The list on the left shows the genera of each of these OTUs. Red indicates over representation of that OTU in the FIV+ cats. (C) FIV+ cat with clinical gingivitis/periodontitis with near monoculture of Moraxellaceae (outer circle) as compared to the mean microbial community structure of cats that are FIV + (middle circle) and cats that are FIV negative (inner circle).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Miller, C.; Abdo, Z.; Ericsson, A.; Elder, J.; VandeWoude, S. Applications of the FIV Model to Study HIV Pathogenesis. Viruses 2018, 10, 206. https://doi.org/10.3390/v10040206
AMA Style
Miller C, Abdo Z, Ericsson A, Elder J, VandeWoude S. Applications of the FIV Model to Study HIV Pathogenesis. Viruses. 2018; 10(4):206. https://doi.org/10.3390/v10040206
Chicago/Turabian StyleMiller, Craig, Zaid Abdo, Aaron Ericsson, John Elder, and Sue VandeWoude. 2018. "Applications of the FIV Model to Study HIV Pathogenesis" Viruses 10, no. 4: 206. https://doi.org/10.3390/v10040206
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.