
Parametric Studies of Titania-Supported Gold-Catalyzed Oxidation of Carbon Monoxide


1
Department of Chemical and Environmental Engineering, University of Nottingham Malaysia, Jalan Broga, 43500 Selangor, Malaysia
2
Department of Chemical Engineering and Biotechnology, National Taipei University of Technology, No. 1 Zhongxiao East Road, Section 3, Da’an District, Taipei City 106, Taiwan
*
Author to whom correspondence should be addressed.
Materials 2017, 10(7), 756; https://doi.org/10.3390/ma10070756
Submission received: 7 March 2017
/
Revised: 21 June 2017
/
Accepted: 28 June 2017
/
Published: 5 July 2017
(This article belongs to the Section Advanced Materials Characterization)
Abstract
:This paper remarks the general correlations of the shape and crystallinity of titanium dioxide (TiO2) support on gold deposition and carbon monoxide (CO) oxidation. It was found that due to the larger rutile TiO2 particles and thus the pore volume, the deposited gold particles tended to agglomerate, resulting in smaller catalyst surface area and limited gold loading, whilst anatase TiO2 enabled better gold deposition. Those properties directly related to gold particle size and thus the number of low coordinated atoms play dominant roles in enhancing CO oxidation activity. Gold deposited on anatase spheroidal TiO2 at photo-deposition wavelength of 410 nm for 5 min resulted in the highest CO oxidation activity of 0.0617 mmol CO/s.gAu (89.5% conversion) due to the comparatively highest catalyst surface area (114.4 m2/g), smallest gold particle size (2.8 nm), highest gold loading (7.2%), and highest Au0 content (68 mg/g catalyst). CO oxidation activity was also found to be directly proportional to the Au0 content. Based on diffuse reflectance infrared Fourier transform spectroscopy, we postulate that anatase TiO2-supported Au undergoes rapid direct oxidation whilst CO oxidation on rutile TiO2-supported Au could be inhibited by co-adsorption of oxygen.
1. Introduction
The first publication on the oxidation of carbon monoxide (CO) using gold (Au) as catalyst was published by Bone and Andrew [1] in 1925. In the 1970s, Bone and Sermon [2] deposited gold on different supports—magnesium oxide, aluminum oxide and silicon oxide using thermal decomposition method, and demonstrated the hydrogenation of olefins at 100 °C. Later on, Huber et al. [3,4] showed that CO oxidation occurred using a matrix mixture consisting of oxygen, CO and Au, at temperature ranging from −263 °C (10 K) to −233 °C (40 K). Until 1987, Haruta et al. [5,6] used co-precipitation and precipitation method to deposit Au on metal oxide and showed that at room temperature of 24 °C, the oxidation activity was considerably high, and that approximately 5% of Au deposited on α-iron oxide showed the best oxidation activity. Since then, development of Au catalyst has become a popular topic and has been utilized in various applications, for instance, pollution control [7,8,9,10], and fuel cell [11,12,13].
Over the years, a large number of studies were carried out to improve the catalytic activity of Au nano-particles; the dominant factors can be divided into four categories [14]: type and morphology of support [15,16,17], the Au particle size [18], the nature of the active sites [19,20], and the preparation method including the pre-treatment [21]. The optimum Au particle size was suggested to be 3.5 nm [22], or less than 5 nm [5]. It was found that when the Au particle gets smaller, the number of edge or kink atoms increases, thus increasing the surface area/periphery of interaction with the support [23,24]. The amount of low-coordinated sites in Au was also shown to be a governing factor affecting the adsorption of CO on Au surfaces [25,26,27]. Cho [28] explained that the high activity of small metallic Au particles could be attributed to the small metal clusters displaying non-metallic behavior (quantum size effects), the presence of high densities of low coordinated atoms, excess electronic charge, and Au-support interactions (active perimeter sites). Unlike Haruta [29], Park and Lee [30] showed that higher calcination temperature increased the amount of Au0, but it was the presence of Au3+ that had enhanced CO oxidation activity. Various studies showed that the morphology and type of support play an important role in catalytic performance, for instance spindle-shaped Au/α-iron oxide was more superior than rhombohedral shape [31], and cerium oxide (CeO2)-titanium (TiO2) nanorods as support endowed the Au/CeO2-TiO2 catalyst with much higher activity than the CeO2-TiO2 nanoparticles [32]. Lopez et al. [27], on the other hand, concluded that the Au particle size was the determining factor affecting the catalyst’s activity (~1/d3 scaling law for the activity, with d being the particle diameter) instead of the support. Till now, due to the contradictory results, the support type and the nature of active sites remain to be the active research areas. In view of the lack of a generalization especially for TiO2, in this study, Au catalysts with different particle size and loading were synthesized by varying the photo-deposition condition, using anatase and rutile TiO2 as the supports, and taking into consideration the effect of the shape of support—cubic and spheroidal. This study aims to provide a general correlation relating CO oxidation activity with Au particle size (and specific surface area), Au loading, and Au3+/Au0 content, for Au-TiO2 synthesized using sol–gel and light emitting diode (LED) photo-deposition method.
2. Results
2.1. TiO2 Supports and Gold Deposition
Figure 1a,b shows that the as-prepared spheroidal TiO2 particles were a mixture of both spherical and rectangular shapes, with sizes of 20~35 nm; the anatase cubic TiO2 on the other hand, appears to be cubic in shape due to the use of tetrabutylammonium hydroxide (TBAH) agent as template, with sizes of 5–10 nm. For rutile TiO2 as shown in Figure 1c,d, the less evident boundaries are probably due to crystal rearrangement at the boundary layers. The sizes of the as-prepared rutile TiO2 were in the range of 40–60 nm, about twice the sizes of anatase TiO2, due to the transformation of two or more of the anatase TiO2 particles under higher calcination temperature which favors particle growth.
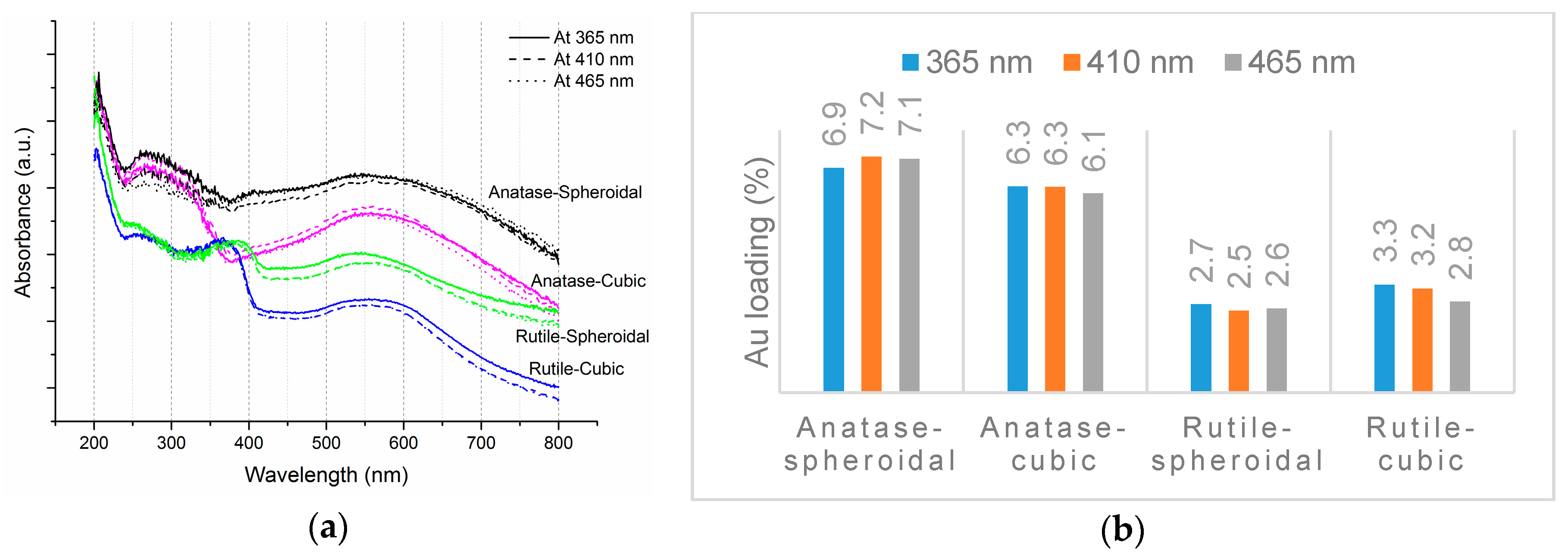
As shown in Figure 2a, the three characteristic Eg Raman modes are at 144 cm−1 (Eg(1)), 197 cm−1 (Eg(2)) and 639 cm−1 (Eg(3)), as well as two B1g Raman modes at 399 cm−1 (B1g(1)) and 519 cm−1 (B1(g), A1(g)), thus confirming the anatase TiO2 structures. Rutile TiO2 on the other hand are confirmed by the indication of four characteristic modes at 143 cm−1 (B1g), 246 cm−1, 443 cm−1 (Eg), and 609 cm−1 (A1g). The shapes of TiO2 had no significant impact on its phases or crystallinities. Figure 2b shows the UV-Visible (UV-VIS) spectra of anatase and rutile TiO2. In accordance to the transmission electron microscopy (TEM) images (Figure 1), the UV-VIS profile of rutile TiO2 was red-shifted, due to the larger particle size. The respective absorption thresholds of anatase spheroidal, anatase cubic, rutile spheroidal and rutile cubic are 408, 393, 422, 430 nm, corresponding to band-gap energies of 3.14, 3.26, 3.03, 2.98 nm.
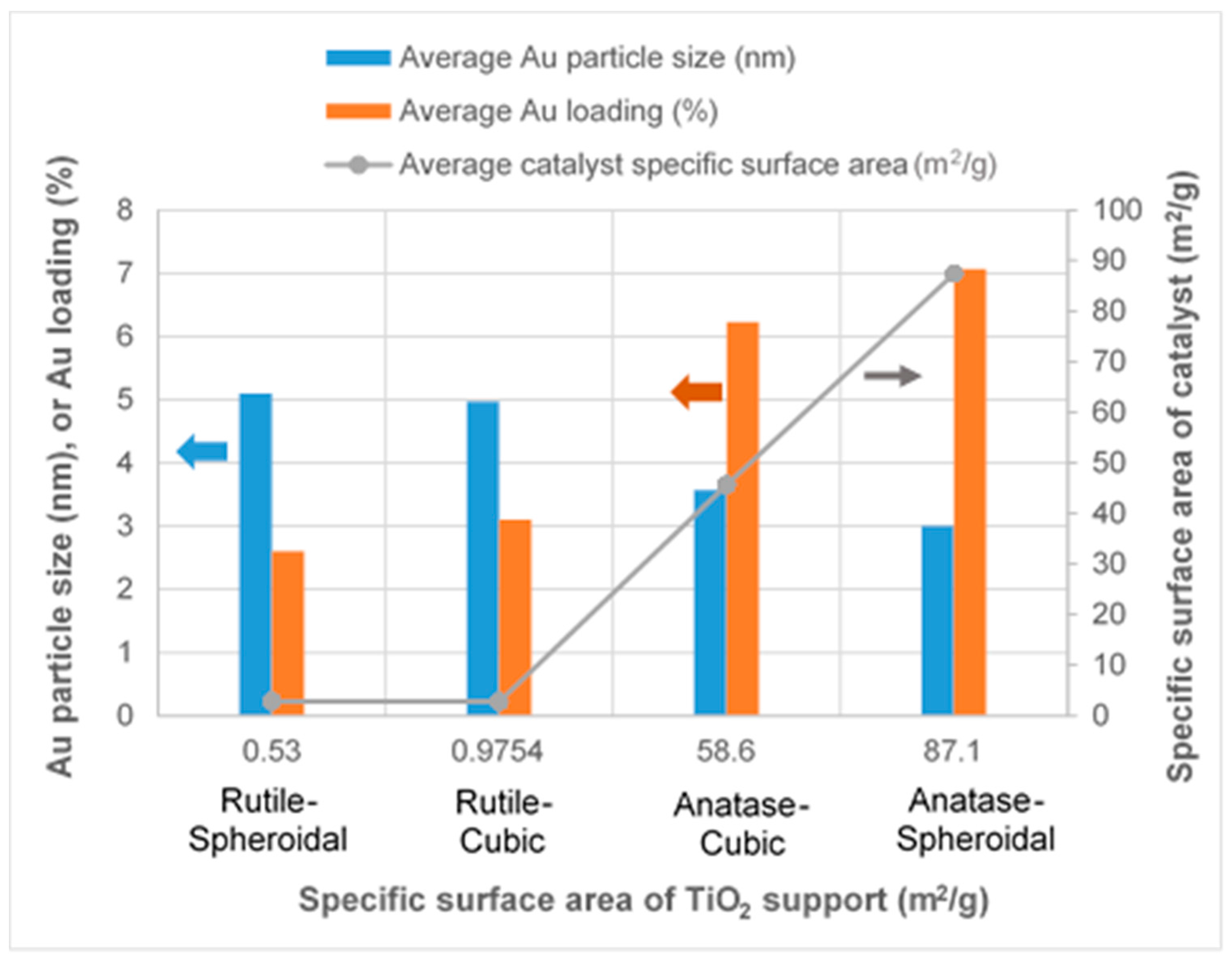
As revealed by TEM images in Figure 3a,b, Au was evenly distributed on the anatase TiO2. Under illumination wavelengths of 365, 410, and 465 nm, the average sizes of the Au particles deposited on anatase spheroidal TiO2 were 3.2, 2.8 and 3.0 nm respectively; and those on anatase cubic TiO2 were 3.5, 3.6, 3.6 nm respectively. Au deposited on the rutile TiO2 supports, on the other hand, appeared to be uneven and agglomerating. The average Au particle sizes under illumination wavelengths of 365, 410 and 465 nm were respectively 4.9, 5.3 and 5.1 nm when deposited on rutile spheroidal TiO2, and 4.8, 4.9 and 5.2 nm when deposited on rutile cubic TiO2. As shown in Figure 4a, UV-VIS spectra display the presence of a broad peak at 550 nm for all Au catalysts, which could be ascribed to the local surface plasmon resonance (LSPR) [33,34] as a signature of the presence of Au0 [35,36]. Comparing the UV-VIS spectra of anatase and rutile TiO2 supports, anatase TiO2 resulted in higher LSPR peaks compared to rutile TiO2. As verified by electron probe micro-analysis (EPMA) in Figure 4b, the higher LSPR peak could be induced by the higher Au content. It is postulated that the higher gold loading and better gold dispersion on anatase TiO2 than rutile TiO2 could be due to the difference in the surface and textural properties [37], in which, as compiled in Table 1 and Figure 5, supports with higher surface areas and bigger pore volumes such as anatase TiO2 having specific surface areas of 58.6–87 m2/g, allowed better gold deposition and dispersion (smaller Au particle size and higher Au loading), in contrast to rutile TiO2 having much smaller specific surface areas in the range of 0.53–0.98 m2/g.
2.2. CO Oxidation
Table 1 compiles the results of X-ray photoelectron spectroscopy (XPS), Brunauer, Emmett and Teller (BET), CO oxidation activity and CO conversion results of different catalysts. Among all, anatase spheroidal TiO2 support having highest specific surface area (87.1 m2/g) enabled highest gold loading (7.2%) and catalyst specific surface area (114.4 m2/g) which then enabled comparatively highest CO oxidation activity (61.7 × 10−3 mmol CO/s.gAu). As shown in Figure 6, the as-prepared Au-TiO2 catalysts contained only two gold states—Au0 and Au3+. The binding energies of Au0 (4f5/2), Au0 (4f7/2), Au3+ (4f5/2) and Au3+ (4f7/2) were respectively 86.8, 83.3, 88.4 and 84.8 eV.
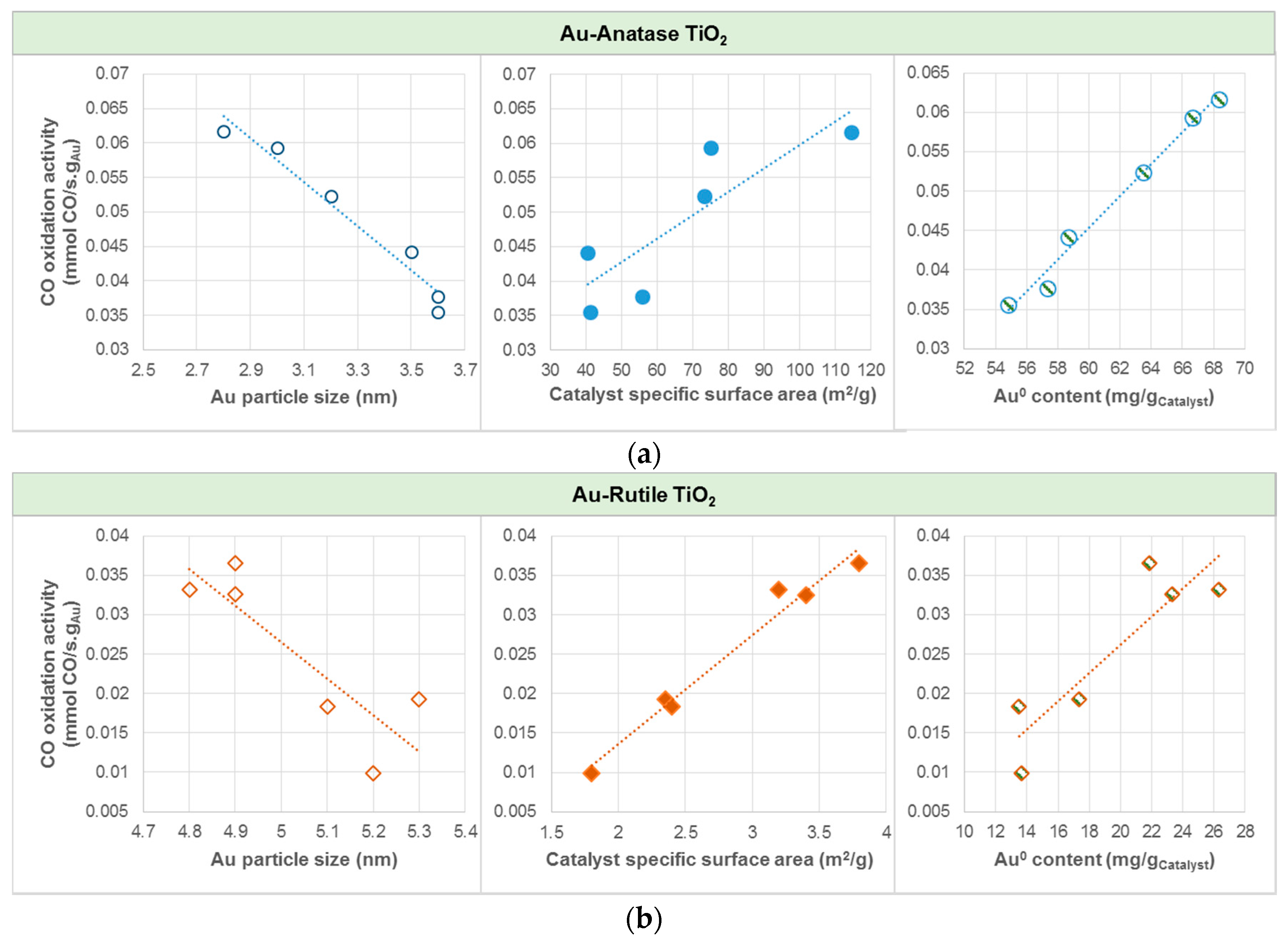
In average, gold catalyst supported on anatase was found to yield about three to six times higher CO oxidation activities than gold catalyst supported on rutile. Among the Au catalysts studied, AS(410) yielded the highest CO oxidation activity, corresponding to CO conversion of 89.5%. The plots of CO oxidation activity in Figure 7 show that, for both Au-anatase TiO2 and Au-rutile TiO2, higher oxidation activities could be attributed to the relatively smaller Au particle size and thus higher catalyst specific surface area, as well as a higher Au0 content.
It is apparent that Au particle size is a dominant factor affecting the catalyst surface area and Au loading. Smaller Au particles brings upon higher Au loading and thus higher catalyst specific area. Similar to the observations by [27], a property directly related to the size of the Au particles serves the vital role in the exceptional catalytic activity of Au catalyst, while other effects due to the support may have considerably smaller effect. It is thus postulated that small gold clusters in small support particles (more atoms having lower coordination number) induce stronger chemisorption [23,27,38], due to well-established varying electron density steps on model surfaces [39] and increase of the fraction of edge atoms [23,40].
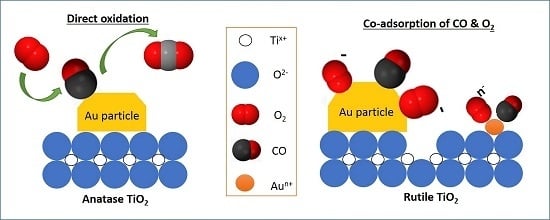
As displayed in Figure 7, CO oxidation activity was found to be directly proportional to the Au0 content. Within the range of investigation, the higher the amount of Au0, the higher the CO oxidation activity. Catalyst with Au0 content of 68 mg/gCatalyst (AS(410)) resulted in CO oxidation activity of 61.7 × 10−3 mmol CO/s.gAu (89.5% conversion); whilst that with Au0 content of 14 mg/gCatalyst (RC(465)) resulted in CO oxidation activity of 9.9 × 10−3 mmol CO/s.gAu (5.6% conversion). Hodge et al. [41] nevertheless showed that the optimum Au3+/Au0 ratio was 1.5 for Au/iron-oxide catalyzed CO oxidation, and therefore generalizations remain an issue if it is to obtain for all catalysts, as much of the literature available are confined to specific types of catalyst. CO oxidations on anatase and cubic TiO2-supported Au catalysts in our case thus do not follow the Bond and Thompson model [7], which suggests that there is a need of a certain amount of Au3+ to serve as “chemical glue” binding the support with the gold particles for increased oxidation activity. Instead, as recognized by the correlation plots in Figure 6, the presented results are in good agreement with [42], which demonstrated that the CO conversion rate of Au particle (very close to pure Au0) was 100 times greater Au atom, i.e., oxidic Au, normalized to the number of Au atoms on the surface, for Au-ceria catalyzed CO oxidation at room temperature. Despite the important roles of those properties related to Au particle size, the difference in the CO oxidation of the anatase and rutile TiO2-supported Au catalysts can be explained using the DRIFTS analysis results.
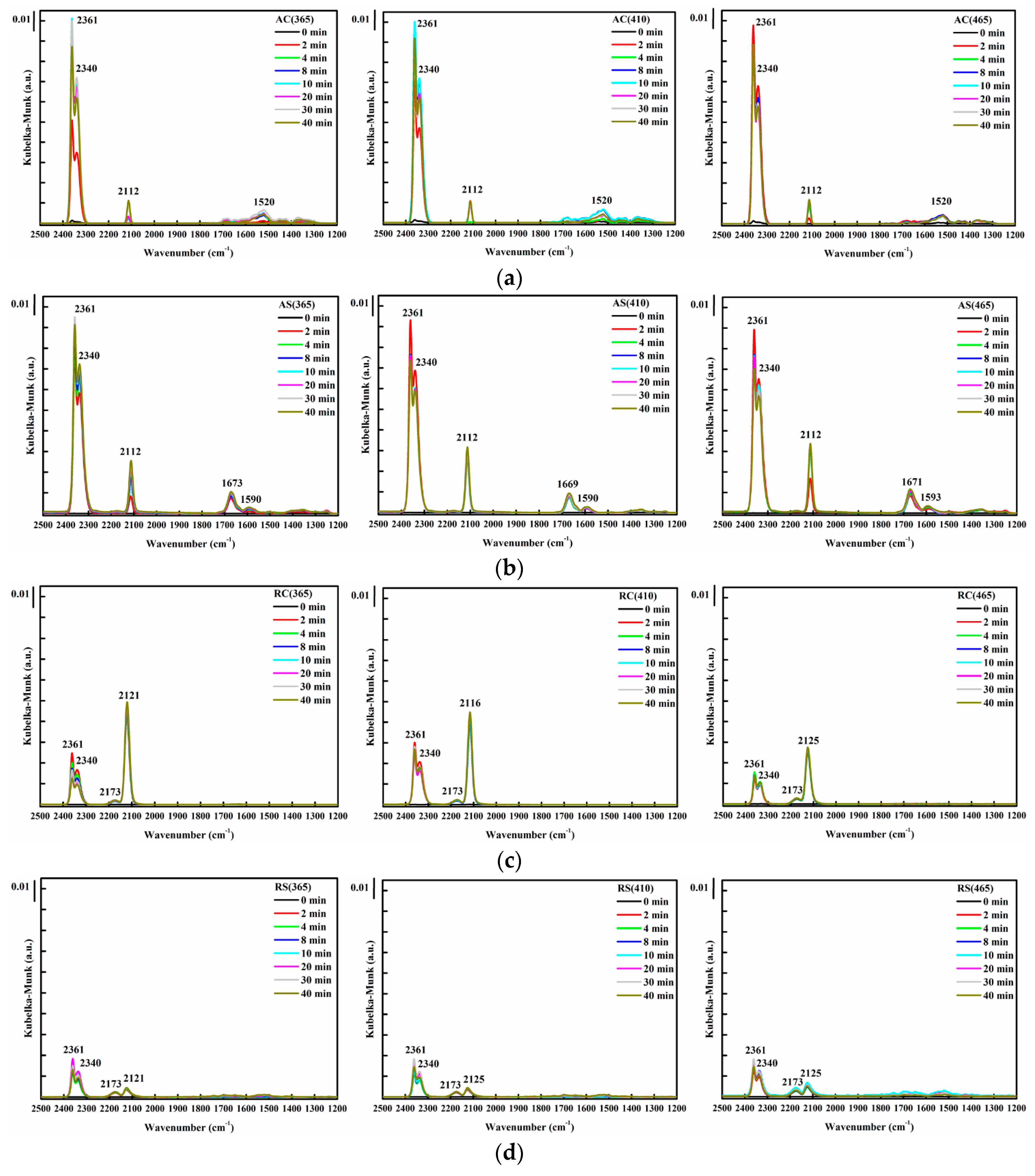
As seen in Figure 8a,b, for both cubic and spheroidal anatase TiO2-supported Au, approximately two minutes after feeding with CO, absorption peaks at 2112, 2340 and 2361 cm−1 appeared simultaneously. The former indicates adsorption of CO on Au0 [38,43], or more precisely, CO chemisorbed on the steps of small metallic Au particles [44], and the latter doublet indicates the formation of CO2 due to CO oxidation [44]. The byproduct formed was mainly monodentate carbonates (1520 cm−1) [45], bidentate carbonates (1669 cm−1) [46], and formates (1590 cm−1) [47]. The DRIFTS spectra of CO oxidation on rutile TiO2-supported Au catalysts on the other hand, revealed the noteworthy difference than those on anatase TiO2-supported Au catalysts. As shown in Figure 8c, two minutes after feeding with CO, unlike Au supported on anatase TiO2, no absorption peak at 2112 cm−1 was observed (which could also be overlapped); instead, the absorption peak at 2121 cm−1 appeared for RC(365) and RS(365); 2116 cm−1 for RC(410); and 2125 cm−1 for RC(465), RS(410) and RS(465), with the CO2 signals relatively smaller than anatase TiO2-supported Au catalysts.
It was found that, the higher the 2112 cm−1 absorption peak in the DRIFTS spectra, the higher the CO oxidation activity, signaling the critical role of Au0. The reaction pathway of the CO oxidation on anatase TiO2-supported Au catalysts could be the rapid direct oxidation of CO at the surface of the metallic Au particles as suggested in [26,48]. On the other hand, the appearance of 2173 cm−1 absorption peak in all rutile TiO2-supported Au catalysts indicates CO adsorption on isolated carbonyl peroxo species (Auδ+-CO-O2δ−) [44] due to the higher amount of Au3+. This peak, however, did not seem to aid CO oxidation activity. Due to changes in the amount of charges, the 2112 cm−1 which was observed in anatase TiO2-supported Au blue-shifted to 2116 cm−1, signaling the CO-Ausδ+-Oδ− surface species [44,48]. The appearance of the absorption peak at 2121 or 2125 cm−1 in most rutile TiO2-supported Au catalysts signifies that electron transfer occurred from Au to the adsorbed oxygen molecules, i.e., CO was adsorbed on metallic Au interacting with a superoxidic or peroxidic oxygen molecular species [44], or the presence of CO-Auδ+ [49]. The low CO oxidation activity of rutile TiO2-supported Au catalyst was due to co-adsorption of oxygen (CO adsorption on pre-O2-adsorbed Au) which, to some extent, inhibited its CO oxidation activity [44,48,50].
3. Materials and Methods
Fourteen milliliters of titanium (IV) isopropoxide (TTIP, Sigma-Aldrich, 99%, Taipei City, Taiwan, R.O.C.) were diluted with 25 mL distilled water for 30 min hydrolysis on a magnetic stirrer. The solution was then washed with distilled water in a centrifuge to remove contaminants and excess reactants before forming white precipitate. The precipitate was calcined at 400 °C for 5 h to obtain anatase spheroidal TiO2 particles. To obtain anatase cubic TiO2 particles, the white precipitate was mixed with 0.33 M TBAH (Honeywell Fluka, Shanghai, China) (40% in water) and stirred for 20 h before being poured to a Teflon autoclave for 12 h hydrothermal treatment at 200 °C. After that, the product was washed with ethanol and then calcined at 400 °C for 5 h. The same approach was carried out but both at calcination temperature of 1000 °C for 2 h to obtain rutile spheroidal and cubic TiO2 particles.
For catalyst impregnation in the above support, 0.42 g of the above TiO2 was mixed with 25 mL distilled water and 8.5 × 10−4 M gold (III) chloride trihydrate (HAuCl4·3H2O, Sigma-Aldrich, 99%, Taipei City, Taiwan, R.O.C.). It was then adjusted to pH 9.5 using 1 M ammonium hydroxide (NH4OH, Sigma-Aldrich, 99%, Taipei City, Taiwan, R.O.C.) (28% in water). For photo-deposition of Au on TiO2, LED light tube of a fixed wavelength was placed on top of the beaker containing the solution. Three irradiation wavelengths were investigated—365, 410 and 465 nm (Thorlabs, Taipei City, Taiwan R.O.C.)—all with irradiation time of 5 min. After irradiation, the solution was washed with DI water in a centrifuge to remove solvents, the precipitate was then air-dried before being calcined at 300 °C.
The samples were analyzed with transmission electron microscopy (Hitachi H-7100, Taipei City, Taiwan R.O.C.) for displaying the morphology and size; Raman Spectrometer (DongWoo DM500i, Gyeonggi-do, Korea) and X-ray diffraction (Panalytical X’Pert PRO, Almelo, the Netherlands (with CuKα (λ = 1.5405 Å) radiation in the 2θ range from 20 to 70° at accelerating voltage of 45 kV and 40 mA) for determining the phase and crystallinity; UV-Visible spectroscopy (JASCO V-630, Gross-Umstadt, Gremany) for obtaining the band-gap energy. Electron probe micro-analysis (JEOL JXA-8200, Tokyo, Japan) for determining the Au loading; X-ray photoelectron spectroscopy (Kratos Axis Ultra DLD, Manchester, UK) for determining the elemental atomic ratios; and Physisorption BET (Micromeritics ASAP 2020, Norcross, GA, USA) for analyzing the surface area. In order to evaluate the CO oxidation efficiency, Fourier transform infrared spectroscopy (Perkin Elmer GX2000, Taipei City, Taiwan R.O.C.) (scanned in the region of 4000–1000 cm−1 at the resolution of 4 cm−1) coupled with DRIFTS sampling accessory (Harrick, Praying Mantis DRA, New York, NY, USA) was utilized to examine the CO oxidation activity using a catalyst loading of 60 mg, initial CO concentration of 2% in air, operating temperature at 20 °C, and a flowrate of 20 standard cubic centimeter per minute (SCCM). The gas outlet was connected to gas chromatography (Thermo GC Ultra Carboxen-1010, New York, NY, USA) for determining the outlet concentration. The CO oxidation activity was computed as the Change in CO concentration/(Reaction time × Catalyst weight). Au catalysts deposited on anatase cubic and anatase spheroidal TiO2 are denoted respectively as AC(X) and AS(X); and Au deposited on rutile cubic and rutile spheroidalTiO2 are denoted respectively as RC(X) and RS(X), with X being the photo-deposition wavelength used.
4. Conclusions
Au was photo-deposited on anatase cubic, anatase spheroidal, rutile cubic, and rutile spheroidal TiO2 at various illumination wavelengths—365, 410 and 465 nm for 5 min—and examined for CO oxidation activity at room temperature. In contrast to anatase TiO2, due to larger pore volume and thus larger specific surface area, Au loading on rutile TiO2 was limited and the Au particles tended to agglomerate, thus affecting the particle size and amount of Au deposited. The average Au particle sizes on anatase cubic, anatase spheroidal, rutile cubic, and rutile spheroidal were respectively 3.6, 3.0, 5.0 and 5.1 nm, with Au loading respectively of 6.2%, 7.1%, 3.1% and 2.6%. XPS results revealed the presence of Au3+ and Au0. The highest CO oxidation activity (0.0617 mmol CO/s.gAu, corresponding to CO conversion of 89.5%) was obtained from anatase spheroidal TiO2 (410)-supported Au, with Au size 2.8 nm, Au loading 7.2%, specific surface area 114.4 m2/g, and Au0 content 68 mg/gCatalyst. Based on the results, we conclude that the surface and textural properties of the support, the contribution of gold states, and the nature of the metal/support interface (which accounts for the Au particle size, and Au loading) play critical roles in CO oxidation. We also postulate that Au deposited on anatase and rutile TiO2 supports result in different reaction schemes, in which a rapid direct oxidation takes place on the former, and co-adsorption of oxygen is likely to happen on the latter and, to some extent, inhibits the CO oxidation activity.
Acknowledgments
The partial financial support from the Ministry of Science and Technology of the Republic of China, R.O.C. (MOST103-2119-M-027-001) is gratefully appreciated. The authors would like to thank Chun-Hao Pan for his assistance in the experiments.
Author Contributions
Siewhui Chong analyzed the data and wrote the paper; Thomas Chung-Kuang Yang conceived and designed the experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bone, W.A.; Andrew, G.W. Studies upon Catalytic Combustion. Part I. The Union of Carbon Monoxide and Oxygen in Contact with a Gold Surface. R. Soc. Lond. 1925, 109, 459–476. [Google Scholar] [CrossRef]
- Bond, G.C.; Sermon, P.A. Gold catalysts for olefin hydrogenation. Gold Bull. 1973, 6, 102–105. [Google Scholar] [CrossRef]
- Huber, H.; McIntosh, D.; Ozin, G. A metal atom model for the oxidation of carbon monoxide to carbon dioxide. The gold atom-carbon monoxide-dioxygen reaction and the gold atom-carbon dioxide reaction. Inorg. Chem. 1977, 16, 975–979. [Google Scholar] [CrossRef]
- McIntosh, D.; Ozin, G.A. Direct synthesis using gold atoms. Monodioxygen gold, Au(O2). Inorg. Chem. 1976, 15, 2869–2871. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Mellor, J.; Palazov, A.; Grigorova, B.; Greyling, J.; Reddy, K.; Letsoalo, M.; Marsh, J. The application of supported gold catalysts to automotive pollution abatement. Catal. Today 2002, 72, 145–156. [Google Scholar] [CrossRef]
- Petrov, L.A. Gold Based Catalyst for Exhaust Gas Purification. WO Patent 199851401 A1, 19 November 1998. [Google Scholar]
- Marecot, P.; Emmanuel, R. Rhone Poulenc Chimie. Low Temperature Oxidation of CO. French Patent 2771310 A1, 28 May 1999. [Google Scholar]
- Buelow, M.T.; Chin, S.W.; Hoke, J.B.; Leclerc, N.R.; Robinson, D.M. Catalyst Coatings for Pollution Control. Google Patents WO2015066272 A3, 30 October 2014. [Google Scholar]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M. FTIR study of methanol decomposition on gold catalyst for fuel cells. J. Power Sources 2003, 118, 304–310. [Google Scholar] [CrossRef]
- Tumilty, J.A.J.; Mellor, J.; Grigorova, B. Gold Catalyst for Fuel Cell. Google Patents WO2000013791 A1, 16 March 2000. [Google Scholar]
- Tabata, T.; Terada, T.; Nagata, T.; Kataoka, M.; Takahashi, H.; Mizutani, N.; Horiuchi, Y. Fuel Cell and Supported Catalyst Used Therefor. Google Patents US 20100196802 A1, 5 August 2010. [Google Scholar]
- Hussain, A. A Computational Study of Catalysis by Gold in Applications of CO Oxidation. Ph.D. Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2010. [Google Scholar]
- Chen, S.; Zhang, B.; Su, D.; Huang, W. Titania Morphology-Dependent Gold—Titania Interaction, Structure, and Catalytic Performance of Gold/Titania Catalysts. ChemCatChem 2015, 7, 3290–3298. [Google Scholar] [CrossRef]
- Cao, L.; Chen, D.; Li, W.; Caruso, R.A. Hierarchically Porous Titania Networks with Tunable Anatase: Rutile Ratios and Their Enhanced Photocatalytic Activities. ACS Appl. Mater. Interfaces 2014, 6, 13129–13137. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Yang, T.C.-K. Illumination wavelength and time dependent nano gold photo-deposition and CO oxidation. Results Phys. 2017, 7, 1167–1174. [Google Scholar] [CrossRef]
- Stevanovic, A.; Ma, S.; Yates, J.T., Jr. Effect of Gold Nanoparticles on Photoexcited Charge Carriers in Powdered TiO2—Long Range Quenching of Photoluminescence. J. Phys. Chem. C 2014, 118, 21275–21280. [Google Scholar] [CrossRef]
- Landmann, M.; Rauls, E.; Schmidt, W. The electronic structure and optical response of rutile, anatase and brookite TiO2. J. Phys. Condens. Matter 2012, 24, 195503. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.Y.; Yun, H.J.; Yu, S.; Kim, W.; Kim, N.D.; Yi, J. Effect of TiO2 crystalline phase on CO oxidation over CuO catalysts supported on TiO2. J. Mol. Catal. A Chem. 2013, 368, 72–77. [Google Scholar] [CrossRef]
- Saavedra, J.; Powell, C.; Panthi, B.; Pursell, C.J.; Chandler, B.D. CO oxidation over Au/TiO2 catalyst: pretreatment effects, catalyst deactivation, and carbonates production. J. Catal. 2013, 307, 37–47. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Stoltze, P.; Nørskov, J.K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. [Google Scholar] [CrossRef]
- Overbury, S.; Schwartz, V.; Mullins, D.R.; Yan, W.; Dai, S. Evaluation of the Au size effect: CO oxidation catalyzed by Au/TiO2. J. Catal. 2006, 241, 56–65. [Google Scholar] [CrossRef]
- Lopez, N.; Nørskov, J.K. Catalytic CO oxidation by a gold nanoparticle: A density functional study. J. Am. Chem. Soc. 2002, 124, 11262–11263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-P.; Hu, P.; Alavi, A. Catalytic role of gold in gold-based catalysts: A density functional theory study on the CO oxidation on gold. J. Am. Chem. Soc. 2002, 124, 14770–14779. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Cho, A. Connecting the dots to custom catalysts. Science 2003, 299, 1684–1685. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, J.S. Effects of Pretreatment Conditions on CO Oxidation over Supported Au Catalysts. J. Catal. 1999, 186, 1–11. [Google Scholar] [CrossRef]
- Wang, G.-H.; Li, W.-C.; Jia, K.-M.; Spliethoff, B.; Schüth, F.; Lu, A.-H. Shape and size controlled α-Fe2O3 nanoparticles as supports for gold-catalysts: Synthesis and influence of support shape and size on catalytic performance. Appl. Catal. A 2009, 364, 42–47. [Google Scholar] [CrossRef]
- Li, S.; Zhu, H.; Qin, Z.; Wang, G.; Zhang, Y.; Wu, Z.; Li, Z.; Chen, G.; Dong, W.; Wu, Z.; et al. Morphologic effects of nano CeO2–TiO2 on the performance of Au/CeO2–TiO2 catalysts in low-temperature CO oxidation. Appl. Catal. B 2014, 144, 498–506. [Google Scholar] [CrossRef]
- Jovic, V.; Chen, W.-T.; Sun-Waterhouse, D.; Blackford, M.G.; Idriss, H.; Waterhouse, G.I. Effect of gold loading and TiO2 support composition on the activity of Au/TiO2 photocatalysts for H2 production from ethanol–water mixtures. J. Catal. 2013, 305, 307–317. [Google Scholar] [CrossRef]
- Yang, K.; Liu, J.; Si, R.; Chen, X.; Dai, W.; Fu, X. Comparative study of Au/TiO2 and Au/Al2O3 for oxidizing CO in the presence of H2 under visible light irradiation. J. Catal. 2014, 317, 229–239. [Google Scholar] [CrossRef]
- Ramirez-Garza, R.E.; Pawelec, B.; Zepeda, T.A.; Martinez-Hernandez, A. Total CO oxidation over Fe-containing Au/HMS catalysts: Effects of gold loading and catalyst pretreatment. Catal. Today 2011, 172, 95–102. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Shin, C.-H.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]
- Ho, K.; Yeung, K. Properties of TiO2 support and the performance of Au/TiO2 catalyst for CO oxidation reaction. Gold Bull. 2007, 40, 15–30. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Gold-catalysed oxidation of carbon monoxide. Gold Bull. 2000, 33, 41–50. [Google Scholar] [CrossRef]
- Falicov, L.; Somorjai, G. Correlation between catalytic activity and bonding and coordination number of atoms and molecules on transition metal surfaces: Theory and experimental evidence. Proc. Natl. Acad. Sci. USA 1985, 82, 2207–2211. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.C.; Thompson, D.T. Catalysis by Gold. Catal. Rev. Sci. Eng. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Hodge, N.A.; Kiely, C.J.; Whyman, R.; Siddiqui, M.R.H.; Hutchings, G.J.; Pankhurst, Q.A.; Wagner, F.E.; Rajaram, R.R.; Golunski, S.E. Microstructural comparison of calcined and uncalcined gold/iron-oxide catalysts for low-temperature CO oxidation. Catal. Today 2002, 72, 133–144. [Google Scholar] [CrossRef]
- Guo, L.-W.; Du, P.-P.; Fu, X.-P.; Ma, C.; Zeng, J.; Si, R.; Huang, Y.-Y.; Jia, C.-J.; Zhang, Y.-W.; Yan, C.-H. Contributions of distinct gold species to catalytic reactivity for carbon monoxide oxidation. Nat. Commun. 2016, 7, 13481. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, M.; Lippits, M.; Nieuwenhuys, B. CO adsorption and oxidation on Au/TiO2. Catal. Lett. 1998, 56, 195–197. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Lu, P.; Akita, T.; Ichikawa, S.; Haruta, M. Au/TiO2 nanosized samples: A catalytic, TEM, and FTIR study of the effect of calcination temperature on the CO oxidation. J. Catal. 2001, 202, 256–267. [Google Scholar] [CrossRef]
- Srinivas, G.; Wright, J.; Bai, C.S.; Cook, R. Au/metal oxides for low temperature CO oxidation. Stud. Surf. Sci. Catal. 1996, 101, 427–433. [Google Scholar]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Spectroscopic observation of dual catalytic sites during oxidation of CO on a Au/TiO2 catalyst. Science 2011, 333, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bell, A.T. The mechanism of dimethyl carbonate synthesis on Cu-exchanged zeolite Y. J. Catal. 2008, 255, 153–161. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Tsubota, S.; Haruta, M. FTIR study of carbon monoxide oxidation and scrambling at room temperature over gold supported on ZnO and TiO2. 2. J. Phys. Chem. 1996, 100, 3625–3631. [Google Scholar] [CrossRef]
- Raphulu, M.; McPherson, J.; Van der Lingen, E.; Anderson, J.; Scurrell, M. Investigation of the active site and the mode of Au/TiO2 catalyst deactivation using Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS). Gold Bull. 2010, 43, 21–28. [Google Scholar] [CrossRef]
- Bollinger, M.A.; Vannice, M.A. A kinetic and DRIFTS study of low-temperature carbon monoxide oxidation over Au-TiO2 catalysts. Appl. Catal. B Environ. 1996, 8, 417–443. [Google Scholar] [CrossRef]
Figure 1.
TEM images of (a) anatase spheroidal TiO2; (b) anatase cubic TiO2; (c) rutile spheroidal TiO2; and (d) rutile cubic TiO2.
Figure 1.
TEM images of (a) anatase spheroidal TiO2; (b) anatase cubic TiO2; (c) rutile spheroidal TiO2; and (d) rutile cubic TiO2.

Figure 2.
Raman and UV-VIS spectra of anatase and rutile TiO2 in both (a) cubic, and (b) spheroidal shapes. a.u.: arbitrary unit.
Figure 2.
Raman and UV-VIS spectra of anatase and rutile TiO2 in both (a) cubic, and (b) spheroidal shapes. a.u.: arbitrary unit.
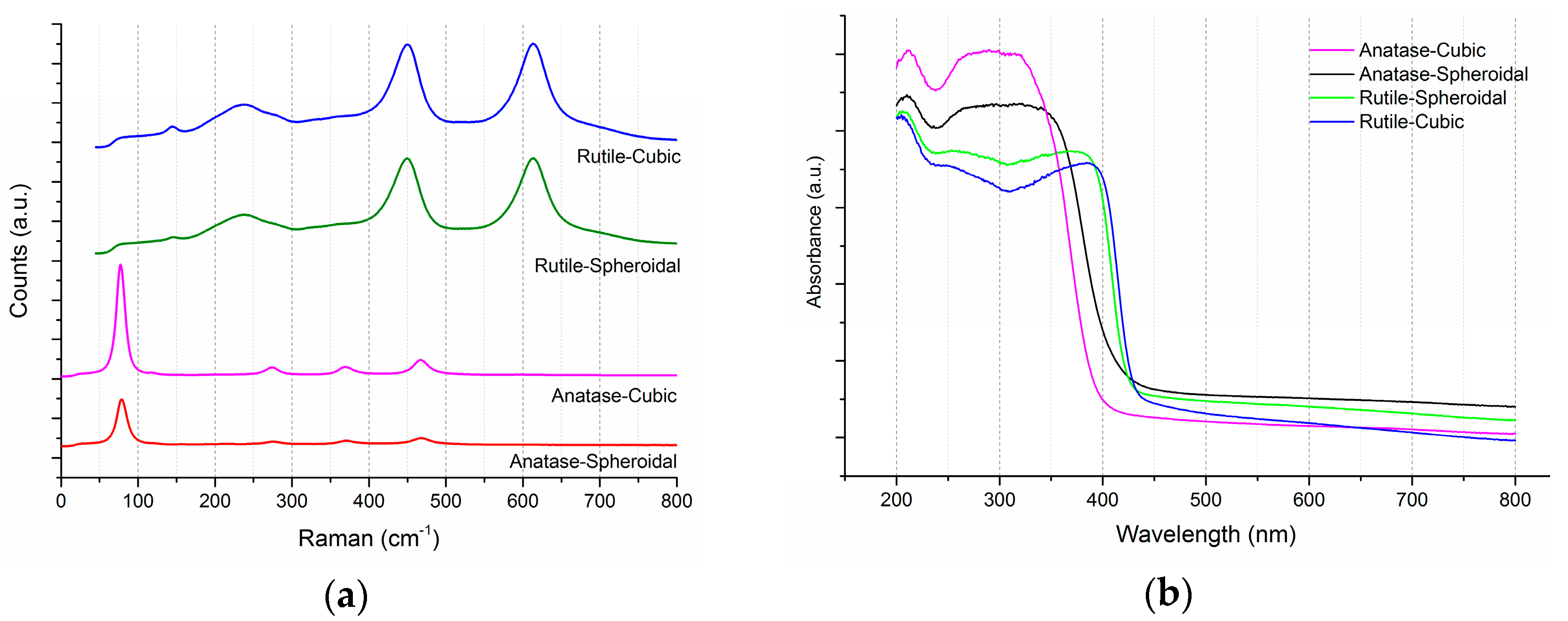
Figure 3.
TEM images of (a) Au-anatase spheroidal TiO2; (b) Au-anatase cubic TiO2; (c) Au-rutile spheroidal TiO2; and (d) Au-rutile cubic TiO2, at photo-deposition wavelength of 365 nm.
Figure 3.
TEM images of (a) Au-anatase spheroidal TiO2; (b) Au-anatase cubic TiO2; (c) Au-rutile spheroidal TiO2; and (d) Au-rutile cubic TiO2, at photo-deposition wavelength of 365 nm.

Figure 4.
(a) UV-VIS spectra and (b) EPMA results of Au-TiO2.
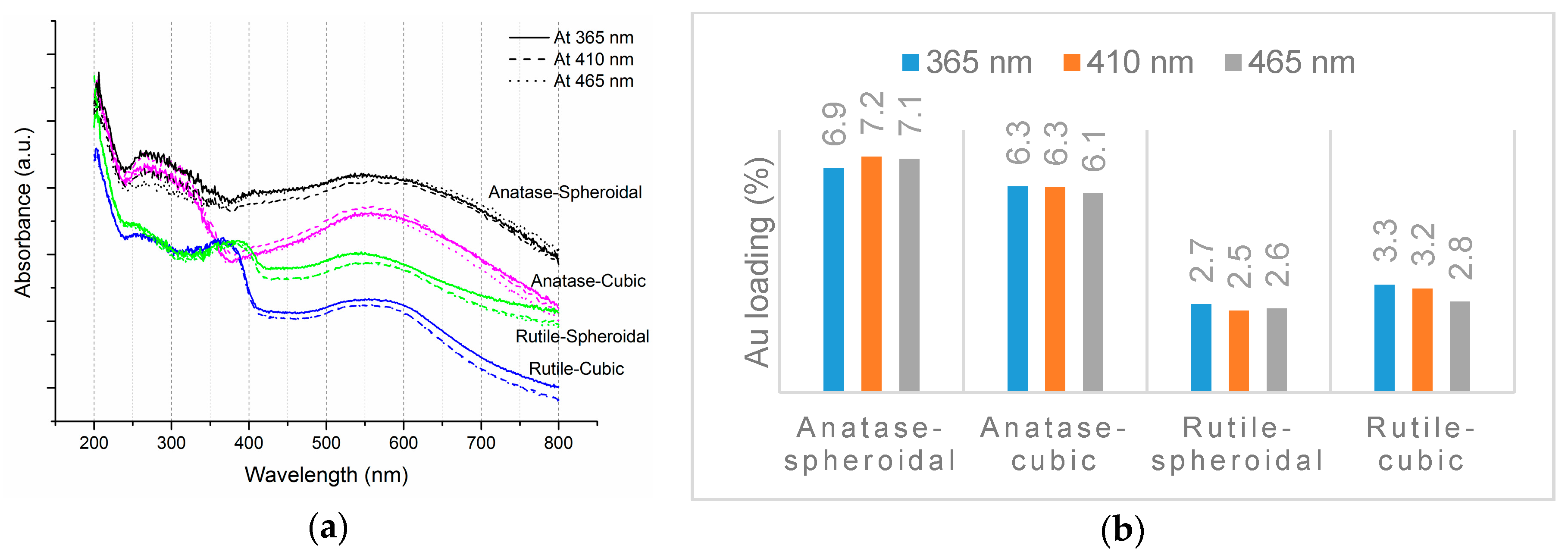
Figure 5.
Effects of different TiO2 supports on Au particle size, Au loading and catalyst specific surface area.
Figure 5.
Effects of different TiO2 supports on Au particle size, Au loading and catalyst specific surface area.
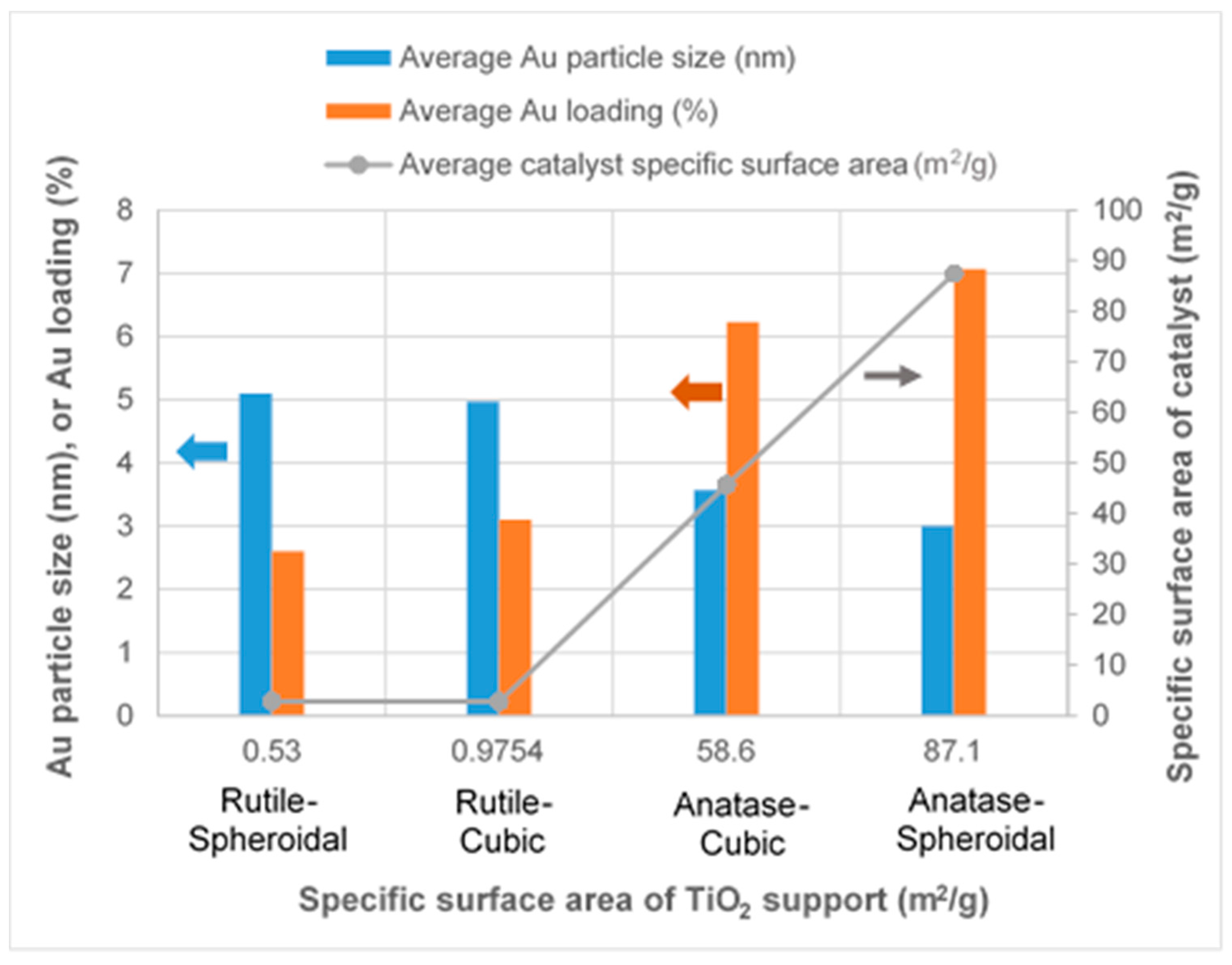
Figure 6.
XPS spectra of (a) AS(410), and (b) RS(365).
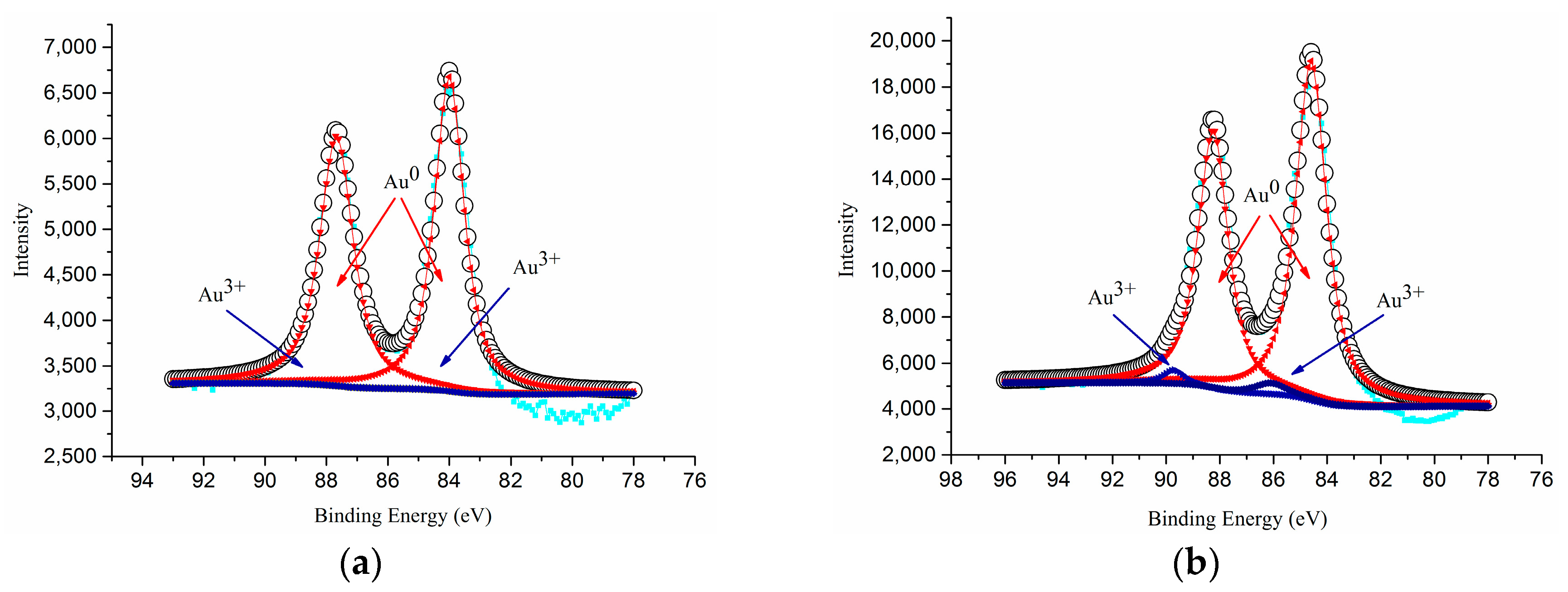
Figure 7.
Effects of Au particle size, catalyst specific surface area, and Au0 content on CO oxidation activity for (a) Au-Anatase TiO2, and (b) Au-Rutile TiO2. Note: Au0 content in mg/gCatalyst was calculated by the total amount of catalyst multiplied by the gold loading and the Au0 atomic percentage.
Figure 7.
Effects of Au particle size, catalyst specific surface area, and Au0 content on CO oxidation activity for (a) Au-Anatase TiO2, and (b) Au-Rutile TiO2. Note: Au0 content in mg/gCatalyst was calculated by the total amount of catalyst multiplied by the gold loading and the Au0 atomic percentage.
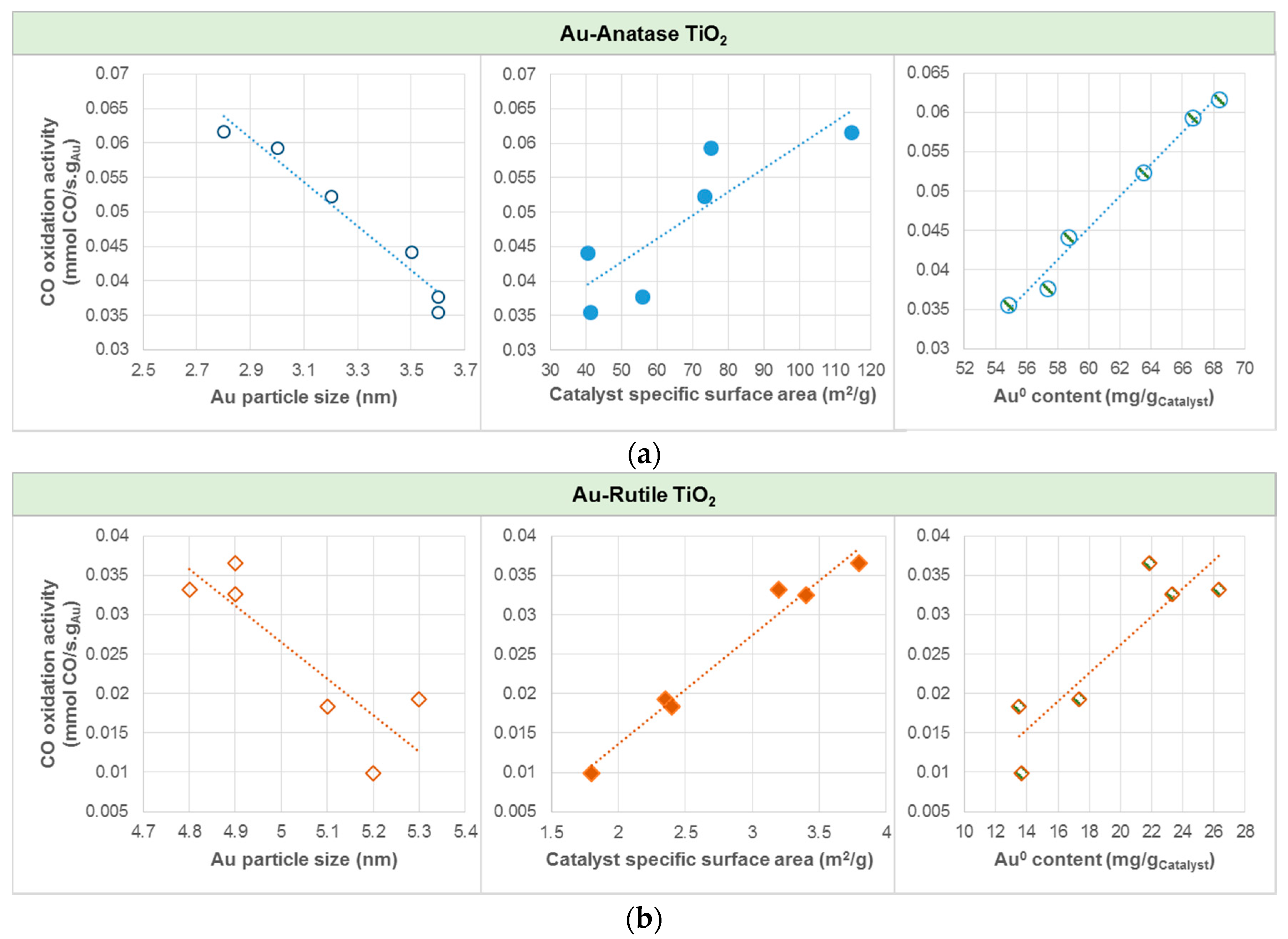
Figure 8.
DRIFTS spectra of CO oxidation using Au catalyst on various supports: (a) anatase cubic TiO2; (b) anatase spheroidal TiO2; (c) rutile cubic TiO2; and (d) rutile spheroidal TiO2, photo-deposited at different wavelengths (from left to right, 365, 410 and 465 nm).
Figure 8.
DRIFTS spectra of CO oxidation using Au catalyst on various supports: (a) anatase cubic TiO2; (b) anatase spheroidal TiO2; (c) rutile cubic TiO2; and (d) rutile spheroidal TiO2, photo-deposited at different wavelengths (from left to right, 365, 410 and 465 nm).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Au loading, Au particle size, X-ray photoelectron spectroscopy (XPS) analysis of Au0 and Au3+, Brunauer–Emmett–Teller (BET) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) results.
Table 1.
Au loading, Au particle size, X-ray photoelectron spectroscopy (XPS) analysis of Au0 and Au3+, Brunauer–Emmett–Teller (BET) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) results.
| TiO2 Support | Sample Notation | Au Loading (%) | Au Particle Size (nm) | Au0 (Atomic %) | Au3+ (Atomic %) | Au3+/Au0 Ratio | Specific Surface Area (m2/g) | Conversion (%) | CO Oxidation Activity (mmol CO/s.gAu) × 10−3 |
|---|---|---|---|---|---|---|---|---|---|
| Anatase–Spheroidal | - | - | - | - | - | - | 87.1 | - | - |
| AS(365) | 6.9 | 3.2 | 92 | 8 | 0.087 | 73.0 | 72.8 | 52.3 | |
| AS(410) | 7.2 | 2.8 | 95 | 5 | 0.052 | 114.4 | 89.5 | 61.7 | |
| AS(465) | 7.1 | 3.0 | 94 | 6 | 0.064 | 74.9 | 84.9 | 59.3 | |
| Anatase–Cubic | - | - | - | - | - | - | 58.6 | - | - |
| AC(365) | 6.3 | 3.5 | 93 | 7 | 0.075 | 40.3 | 56.1 | 44.2 | |
| AC(410) | 6.3 | 3.6 | 91 | 9 | 0.099 | 55.7 | 47.9 | 37.7 | |
| AC(465) | 6.1 | 3.6 | 90 | 10 | 0.111 | 41.0 | 43.7 | 35.5 | |
| Rutile–Spheroidal | - | - | - | - | - | - | 0.53 | - | - |
| RS(365) | 2.7 | 4.9 | 81 | 19 | 0.235 | 3.8 | 19.9 | 36.6 | |
| RS(410) | 2.5 | 5.3 | 69 | 31 | 0.449 | 2.4 | 9.7 | 19.2 | |
| RS(465) | 2.6 | 5.1 | 52 | 48 | 0.923 | 2.4 | 9.6 | 18.3 | |
| Rutile–Cubic | - | - | - | - | - | - | 0.9754 | - | - |
| RC(365) | 3.3 | 4.8 | 80 | 20 | 0.250 | 3.2 | 22.1 | 33.2 | |
| RC(410) | 3.2 | 4.9 | 73 | 27 | 0.370 | 3.4 | 21.0 | 32.6 | |
| RC(465) | 2.8 | 5.2 | 49 | 51 | 1.041 | 1.8 | 5.6 | 9.9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chong, S.; Yang, T.C.-K. Parametric Studies of Titania-Supported Gold-Catalyzed Oxidation of Carbon Monoxide. Materials 2017, 10, 756. https://doi.org/10.3390/ma10070756
AMA Style
Chong S, Yang TC-K. Parametric Studies of Titania-Supported Gold-Catalyzed Oxidation of Carbon Monoxide. Materials. 2017; 10(7):756. https://doi.org/10.3390/ma10070756
Chicago/Turabian StyleChong, Siewhui, and Thomas Chung-Kuang Yang. 2017. "Parametric Studies of Titania-Supported Gold-Catalyzed Oxidation of Carbon Monoxide" Materials 10, no. 7: 756. https://doi.org/10.3390/ma10070756
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.