Lithium Hydrazinidoborane Ammoniate LiN2H3BH3·0.25NH3, a Derivative of Hydrazine Borane
by
 and
and
Salem Ould-Amara
1, 
Dominique Granier
2,
Rodica Chiriac
3,
François Toche
3,
Pascal G. Yot
2 and
and
Umit B. Demirci
1,* 
1
IEM (Institut Europeen des Membranes), UMR5635 (CNRS, ENSCM, UM), Universite de Montpellier, Place Eugene Bataillon, CC047, F-34095 Montpellier, France
2
ICGM (Institut Charles Gerhardt Montpellier), UMR 5253 (CNRS UM ENSCM), Université de Montpellier, CC 15005, Place Eugène Bataillon, F-34095 Montpellier cedex 05, France
3
Univ Lyon, Université Claude Bernard Lyon 1, Laboratoire des Multimatériaux et Interfaces UMR CNRS 5615, LMI, F-69622 Villeurbanne, France
*
Author to whom correspondence should be addressed.
Materials 2017, 10(7), 750; https://doi.org/10.3390/ma10070750
Submission received: 14 June 2017
/
Revised: 27 June 2017
/
Accepted: 29 June 2017
/
Published: 5 July 2017
(This article belongs to the Section Energy Materials)
Abstract
:Boron- and nitrogen-based materials have shown to be attractive for solid-state chemical hydrogen storage owing to gravimetric hydrogen densities higher than 10 wt% H. Herein, we report a new derivative of hydrazine borane N2H4BH3, namely lithium hydrazinidoborane ammoniate LiN2H3BH3·0.25NH3. It is easily obtained in ambient conditions by ball-milling N2H4BH3 and lithium amide LiNH2 taken in equimolar amounts. Both compounds react without loss of any H atoms. The molecular and crystallographic structures of our new compound have been confirmed by NMR/FTIR spectroscopy and powder X-ray diffraction. The complexation of the entity LiN2H3BH3 by some NH3 has been also established by thermogravimetric and calorimetric analyses. In our conditions, LiN2H3BH3·0.25NH3 has been shown to be able to release H2 at temperatures lower than the parent N2H4BH3 or the counterpart LiN2H3BH3. It also liberates non-negligible amounts of NH3 at temperatures lower than 100 °C. This is actually quite detrimental for chemical H storage, but alternatively LiN2H3BH3·0.25NH3 might be seen as a potential NH3 carrier.
1. Introduction
Hydrogen storage is one of the major obstacles restricting the development of an “economy of hydrogen energy”. Within the past decade, many studies have been done to find new solutions, and particular attention has been paid to chemical H storage [1], and interestingly there has been new interest in old molecules and materials [2]. An example of this is hydrazine borane N2H4BH3. It was discovered in the 1960s and the first applied study presented it as an energetic material [3]. It was then barely investigated until recent years. In 2009, N2H4BH3 was re-discovered owing to its high gravimetric hydrogen density of 15.4 wt%. However, it was considered as being unsuitable for solid-state chemical H storage because of hazardous dehydrogenation properties [4]. Indeed, when heated it releases large amounts of hydrazine N2H4 together with H2, and generates a shock-sensitive solid residue above 300 °C [5]. It is worth mentioning that hydrogen storage via hydrazine borane and any other B–N–H compounds (e.g., ammonia borane NH3BH3) is irreversible. Upon thermolytic dehydrogenation, they transform into a polymeric residue of complex composition and they cannot be hydrogenated directly under H2 pressure. The only way to regenerate the starting materials is chemical recycling [1,2].
Yet, N2H4BH3 is still of interest in the field because it can be modified by reaction with an alkali hydride MH (M+ = Li+, Na+, K+) resulting in the formation of an alkali hydrazinidoborane derivative, MN2H3BH3 [6]:
N2H4BH3 (s) + MH (s) → MN2H3BH3 (s) + H2 (g)
Lithium hydrazinidoborane LiN2H3BH3 is synthesized by ball-milling and depending on the milling conditions, two polymorphs form. The α phase (monoclinic, s.g. (space group) P21/c), already reported [7], is the high-temperature phase. It can be synthesized directly [7]. Otherwise it can be formed from the β phase (orthorhombic, s.g. Pbca) at about 95 °C [8]. The latter phase is the low-temperature phase. Lithium hydrazinidoborane is thermally less stable than the parent N2H4BH3 as a result of the substitution of one Hδ+ of the N2H4 moiety by Li+ and of the subsequent lengthening and concomitant weakening of the Hδ+···Hδ− interactions (which refer to the so-called dihydrogen bonding). For example, the β phase is able to dehydrogenate from 40 °C and release 7.8 wt% of almost pure H2 up to 144 °C. Lithium hydrazinidoborane is thus more suitable for solid-state chemical H storage than the parent N2H4BH3. This is also the case for the sodium derivative NaN2H3BH3 [9]. It starts to liberate H2 below 60 °C, and over the range 60–100 °C it loses 6 wt% of almost pure H2. The interesting feature with NaN2H3BH3 is that synthesis by ball-milling has to be performed below −30 °C because of the high reactivity of sodium hydride NaH with N2H4BH3. A reaction enthalpy of −27,756.7 J mol−1 was determined by Calvet calorimetry at 25 °C [10]. The reaction is highly exothermic, especially when compared to the enthalpy of the reaction between LiH and N2H4BH3 at the same temperature (−62.8 J mol−1). In fact, the bigger the alkali cation is, the more exothermic the reactivity of the alkali hydride. The enthalpy of reaction of KH with N2H4BH3 was measured as being −70,247.2 J mol−1. Accordingly, the safest way to get potassium hydrazinidoborane KN2H3BH3 is by wet synthesis (i.e., suspension in tetrahydrofuran) in an autoclave reactor [11]. All of these alkali hydrazinidoboranes have shown better and safer dehydrogenation properties than the parent hydrazine borane. To our knowledge, there is no other derivative of N2H4BH3. Attempts in synthesizing the hydrazinidoboranes of magnesium, calcium, and aluminum M(N2H3BH3)n (Mn+ = Mg2+, Ca2+, Al3+) by ball-milling have failed. Composites MHn-N2H4BH3 have been found to form where the hydrides act as destabilizers of the borane [10].
The substitution of one Hδ+ of the N2H4 moiety of N2H4BH3 by M+ negatively affects the gravimetric hydrogen density of the as-formed material because of the loss of 1 equivalent of H2. For instance, the gravimetric hydrogen density drops from 15.4 wt% for N2H4BH3 to 11.7, 8.9, and 7.2 wt% for LiN2H3BH3, NaN2H3BH3, and KN2H3BH3, respectively. The decrease might be compensated by using an amide MNH2 instead of a hydride MH, only if the NH2 group of the amide interacts by trapping one Hδ+ of the N2H4 moiety of hydrazine borane. Consequently, the formation of a new compound may be expected with a formula resembling MN2H3BH3·xNH3. Such an approach has shown to be successful for synthesizing metal amidoborane ammoniates like Mg(NH2BH3)·NH3 and Ca(NH2BH3)·NH3 where all of the hydrogen atoms of the precursors were kept in the final material [12,13]. We therefore explored the possibility of synthesizing novel derivatives of N2H4BH3. Lithium amide LiNH2 was selected owing to the lightness of Li+. Another reason for this choice is that LiH is less reactive than NaH or KH, and thus is safer. Similar behavior was expected for LiNH2. Then, by ball-milling N2H4BH3 and LiNH2, the formation of lithium hydrazinidoborane ammoniate LiN2H3BH3·xNH3 was expected; it has a higher gravimetric hydrogen density (13.2 wt% for x = 1; 12.1 for x = 0.25) than LiN2H3BH3 (11.6 wt%). This is discussed hereafter.
2. Results
2.1. Molecular Structure
Ball-milling of LiNH2 and N2H4BH3 in our conditions resulted in the formation of a pasty solid. More details are given in the section dedicated to the experimental conditions. The pasty aspect is otherwise discussed in the discussion section. Hereafter, the precursors N2H4BH3 and LiNH2 will be denoted 1 and 2 respectively, while the ball-milling product will be identified as 3.
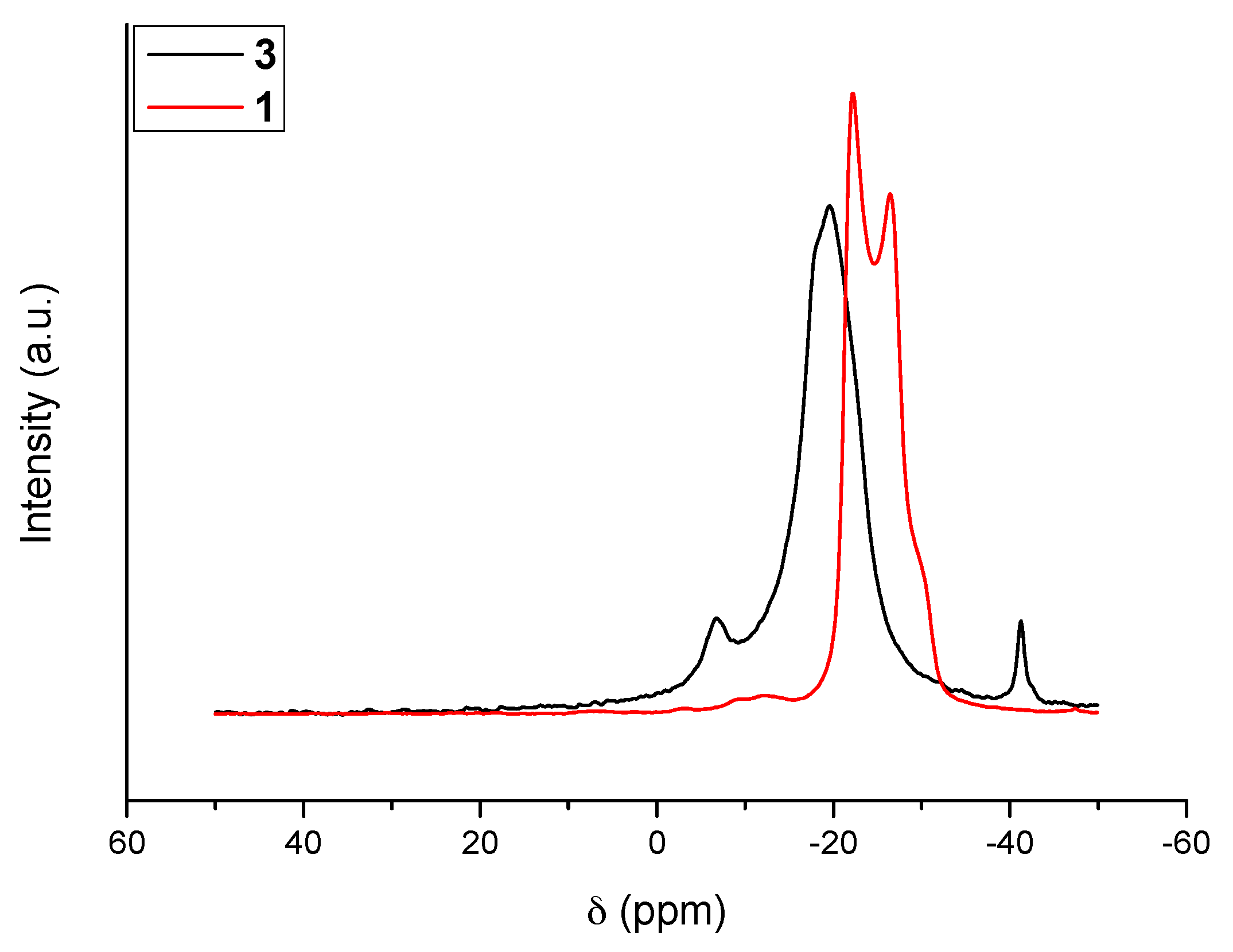
The 11B MAS NMR spectra of 1 and 3 are shown in Figure 1. With the former (i.e., N2H4BH3), the signal is typical of a quadrupolar coupling due to anisotropy around the boron atom because of strong intermolecular Hδ+···Hδ− interactions. In contrast, the new compound 3 shows a resonance of high intensity at δ −19.6 ppm; the signal is otherwise broad. Such features are indicative of isotropy around the boron atom of the NBH3 environment, change of the environment of the N–H bonds, and lengthening of the intermolecular Hδ+···Hδ− interactions. Similar observations were reported for LiN2H3BH3 as well as for NaN2H3BH3 [6,7,8,9]. There are also two other signals. The first one is centered at δ −41.3 ppm. It is typical of a BH4 environment [14]. The second one is centered at δ −6.7 ppm. It may be ascribed to N2BH2 and N3BH environments [15]. The presence of such N2BH2 and N3BH environments might indicate some decomposition of the starting borane, but the concomitant apparition of the BH4 environment does not support such an assumption. In the open literature dedicated to B–N–H compounds, the presence of both signals is generally ascribed to the formation of an ionic dimer [16]: for example, with ammonia borane NH3BH3, a similar 11B MAS NMR spectrum can be collected when part of the borane transforms to the ionic dimer diammoniate of diborane of formula [NH3BH2NH3+][BH4−]. We therefore believe that in our conditions a little part of N2H4BH3 dimerized into an ionic intermediate of formula [N2H4BH2N2H4+][BH4−] (dihydrazinoboronium borohydride or dihydrazinate of diborane):
2N2H4BH3 (s) → [N2H4BH2N2H4+][BH4−] (s)
Such a compound shows both of the N2BH2 and BH4 environments, while the BH3 environment comes from the new solid 3.
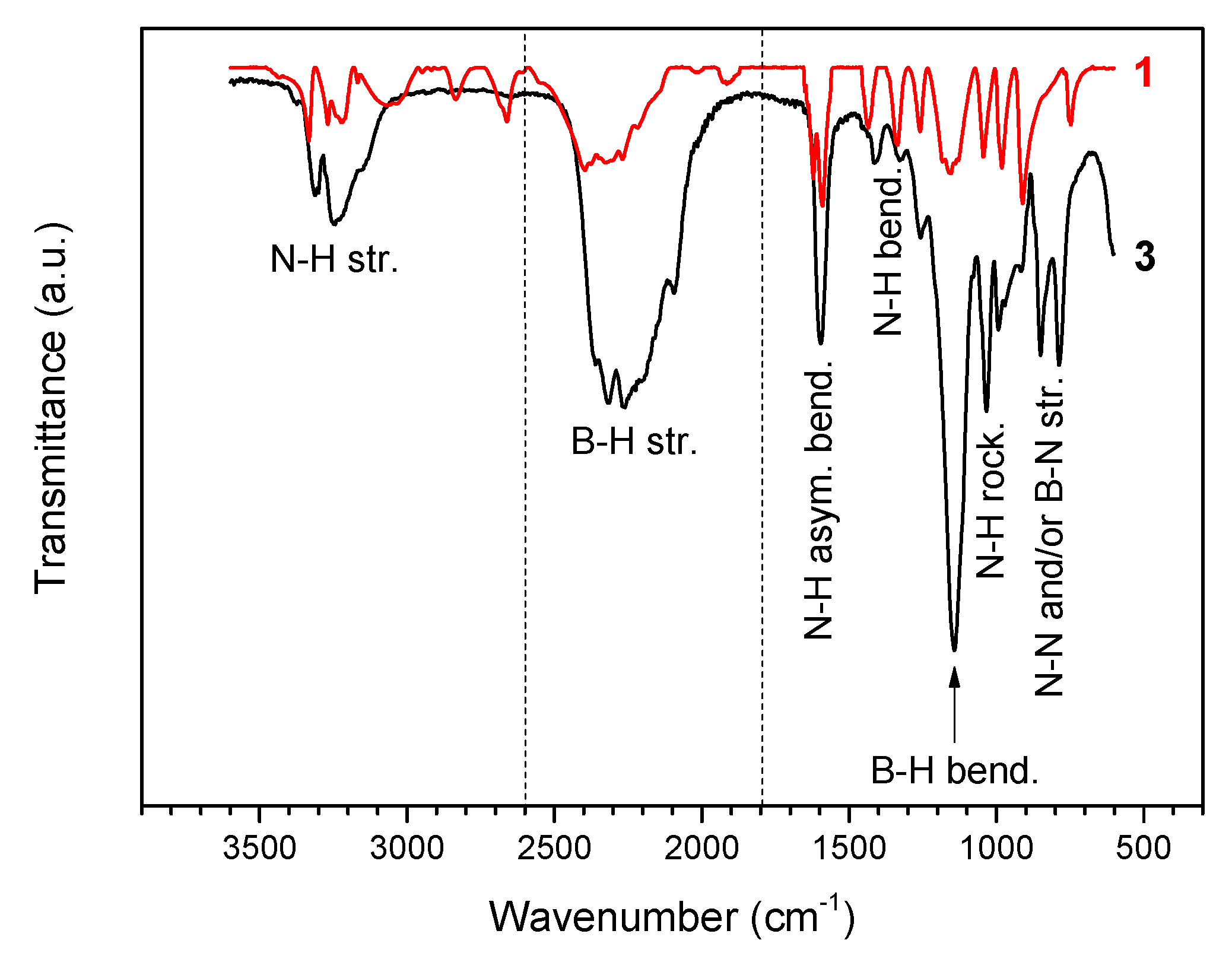
The FTIR spectrum of 3 (Figure 2) is different from that of 1. It displays a less complex fingerprint in the N–H stretching region (3450–2950 cm−1) and broadened N–H bending bands (1700–1300 cm−1), thereby suggesting that the intermolecular Hδ+···Hδ− interactions are less important and that the Hδ+···Hδ− network is weaker. There are more bands in the B–H stretching region (2600–1800 cm−1) owing to interactions of Li+ and N of 2 with Hδ− of BH3 groups. The two small bands at 1913 and 2017 cm−1 observed on the spectrum of N2H4BH3 are indicative of strong Hδ+···Hδ− interactions [5]. They cannot be seen for 3, confirming a weakened Hδ+···Hδ− network. It may be reasonably concluded that 2 has induced electronic modification in 1. In fact, the FTIR spectrum of 3 resembles that of LiN2H4BH3 reported elsewhere and similar conclusions were made for this derivative [8]. The N–H stretching vibration at 3375 cm−1 is in the range of the degenerate stretching N–H mode of ammonia, thereby suggesting weakly bound NH3 molecules [17].
The aforementioned spectroscopy results are indicative of the formation of a compound with the speculated molecular formula LiN2H3BH3·xNH3 (lithium hydrazinidoborane ammoniate).
2.2. Crystallography
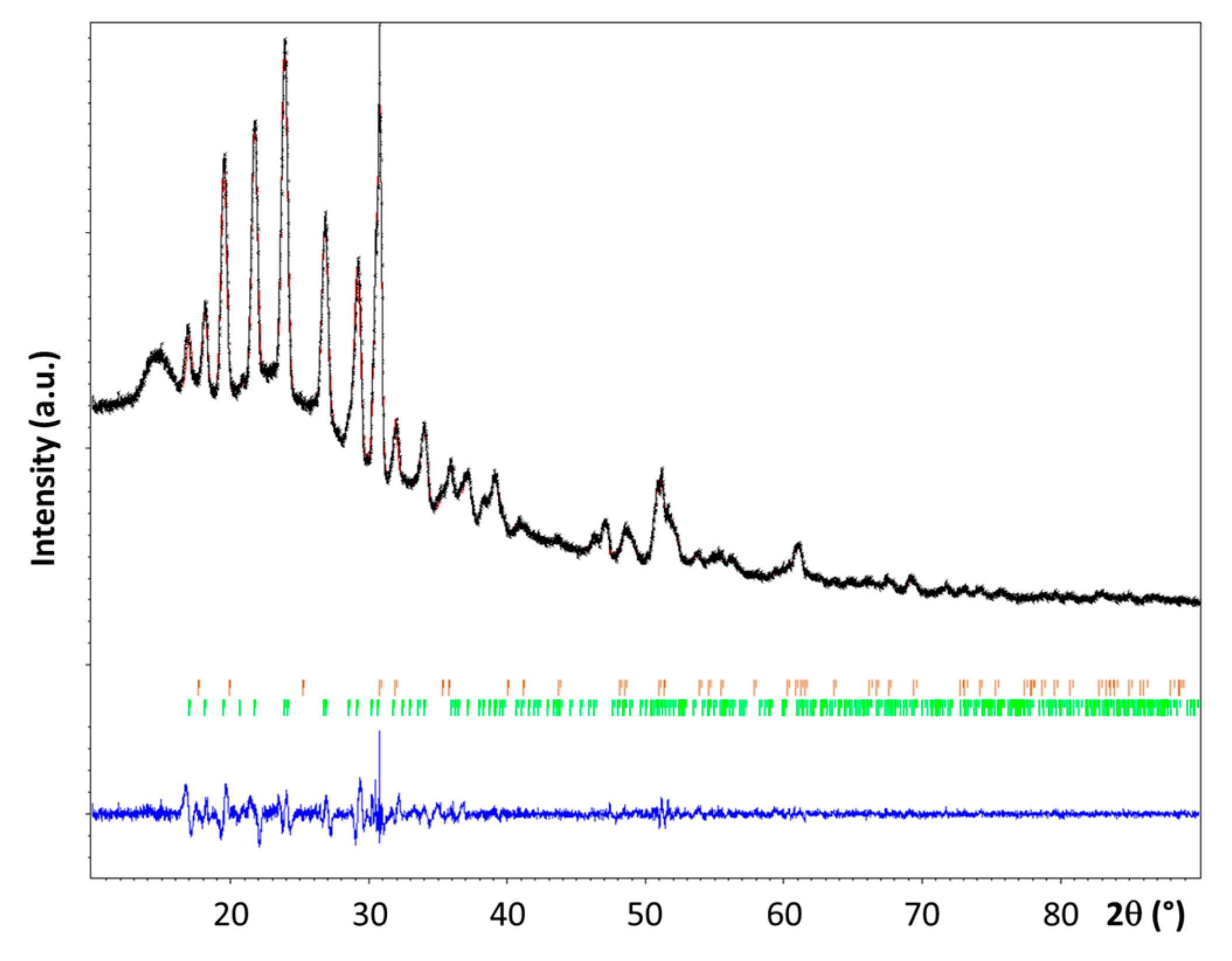
The powder XRD pattern recorded for 3 is shown in Figure 3. It was compared to the patterns of the reactants 1 [3,7,18] and 2 [19]. The presence of a new crystalline phase was confirmed, though some residual diffraction peaks coming from 2 have been also detected.
The diffraction peaks of 3 have been successfully indexed as a single phase using DICVOL06 [20] after removing the peaks supposed to be overlapped with the LiNH2 diffraction lines. The new phase 3 crystallizes in the monoclinic system, with a space group P21/n (No. 14) and Z = 4; the unit cell parameters a = 7.649(1) Å, b = 7.502(1) Å, c = 5.973(1) Å, and β = 97.81(1)° were found. The unit cell volume (~340 Å3) is larger than the one reported for α–LiN2H3BH3 (328 Å3) [7]. The structure factor of 3 and that of the α [7] and β [8] phases of LiN2H3BH3 were compared; they present strong differences. This is indicative of the formation of a new phase. The position of the Li element in the unit cell was located by Direct Methods using EXPO2014 [21]. The crystal structure was then successfully refined by the Rietveld method (Figure 3) using the Jana 2006 program [22]. The structure parameters that were obtained are presented in Table 1 and Table 2. For the refinement the lengths of the N–N and N–B bonds and the N–N–B angle were fixed to 1.50 Å, 1.58 Å, and 113.8° respectively. The refinement was carried out while taking into account the presence of 2. As a result, the relative weight amounts of 3 (LiN2H3BH3·xNH3) and 2 (LiNH2) were found to be close to be 95.4 and 4.6 wt%, respectively. Nevertheless, the refinement of the position of the x molecule of NH3 into the crystalline network has not been possible. Hence, considering the difference of volume (14 Å3) between the cell volume of 3 and that of α–LiN2H3BH3, it can be assumed the presence of one non-H atom per unit cell. This atom is proposed to be N of NH3, suggesting then the molecular formula LiN2H3BH3·0.25NH3 for 3.
2.3. Thermal Analyses and Evolving Gas Analyses
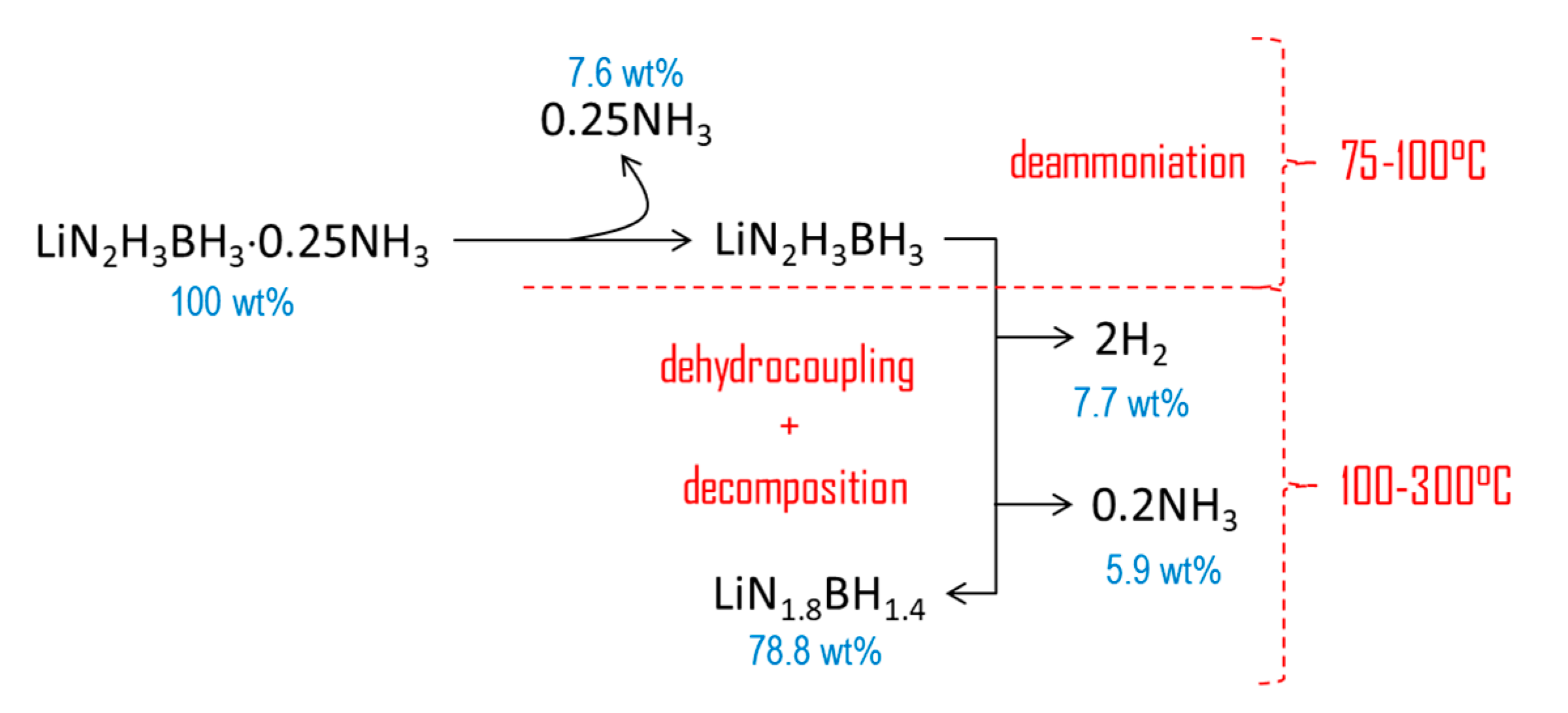
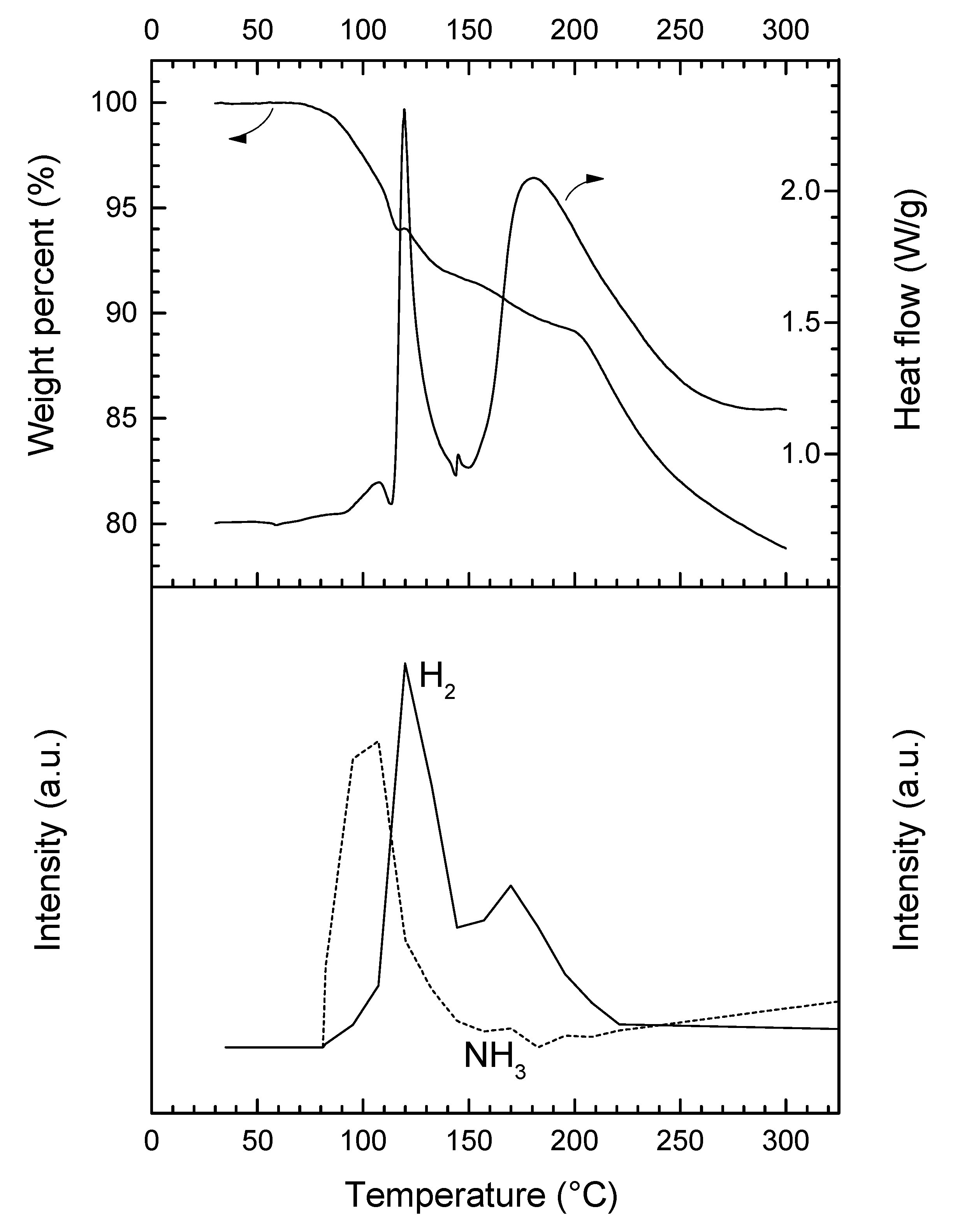
Under heating at 5 °C min−1, 3 is stable up to 75 °C (Figure 4). Then, it decomposes according to a complex pathway. The DSC curve is characterized by two major (maximum at 119.4 and 180.2 °C) and two minor exothermic events (107.4 and 145.5 °C). This is consistent with the occurrence of several weight losses observed by TGA. Over 75 °C, 3 starts to liberate NH3, which is characterized by the first exothermic event (0.9 kJ mol−1) peaking at 107.4 °C. This is consistent with the molecular structure of an adduct like LiN2H3BH3·xNH3. The first major weight loss (8.3 wt%) that occurs over the temperature range 75–145 °C is associated with the second exothermic event (11.5 kJ mol−1) peaking at 119.4 °C. It is mainly due to the dehydrogenation of 3. There is then an exothermic signal of low heat (0.15 kJ mol−1). It is followed by the second major decomposition (12.9 wt% and 50.5 kJ mol−1) that takes place over the temperature range 145–300 °C. At 300 °C, the overall weight loss is 21.2 wt% and it cannot be rationalized with the loss of H2 only (maximum of 13.2 wt% H in 3 if x = 1; 12.1 wt% H if x = 0.25). The weight proportion of NH3 in the adduct LiN2H3BH3·xNH3 is theoretically of 24.7 wt% NH3 if x = 1, and 7.6 wt% NH3 if x = 0.25. Hence, 3 releases H2 and significant amounts of NH3.
In our conditions, 3 releases most of the NH3 during the first decomposition step. Similar trends were reported for lithium amidoborane ammoniate Li(NH2BH3)·NH3, for which the evolution of NH3 peaks at 52 °C while that of H2 shows the maximum at around 100 °C [23]. Similar observations were reported for calcium amidoborane ammoniate Ca(NH2BH3)·NH3, making the authors suggest that the adducted NH3 is weakly bound to the cation Ca2+ [13].
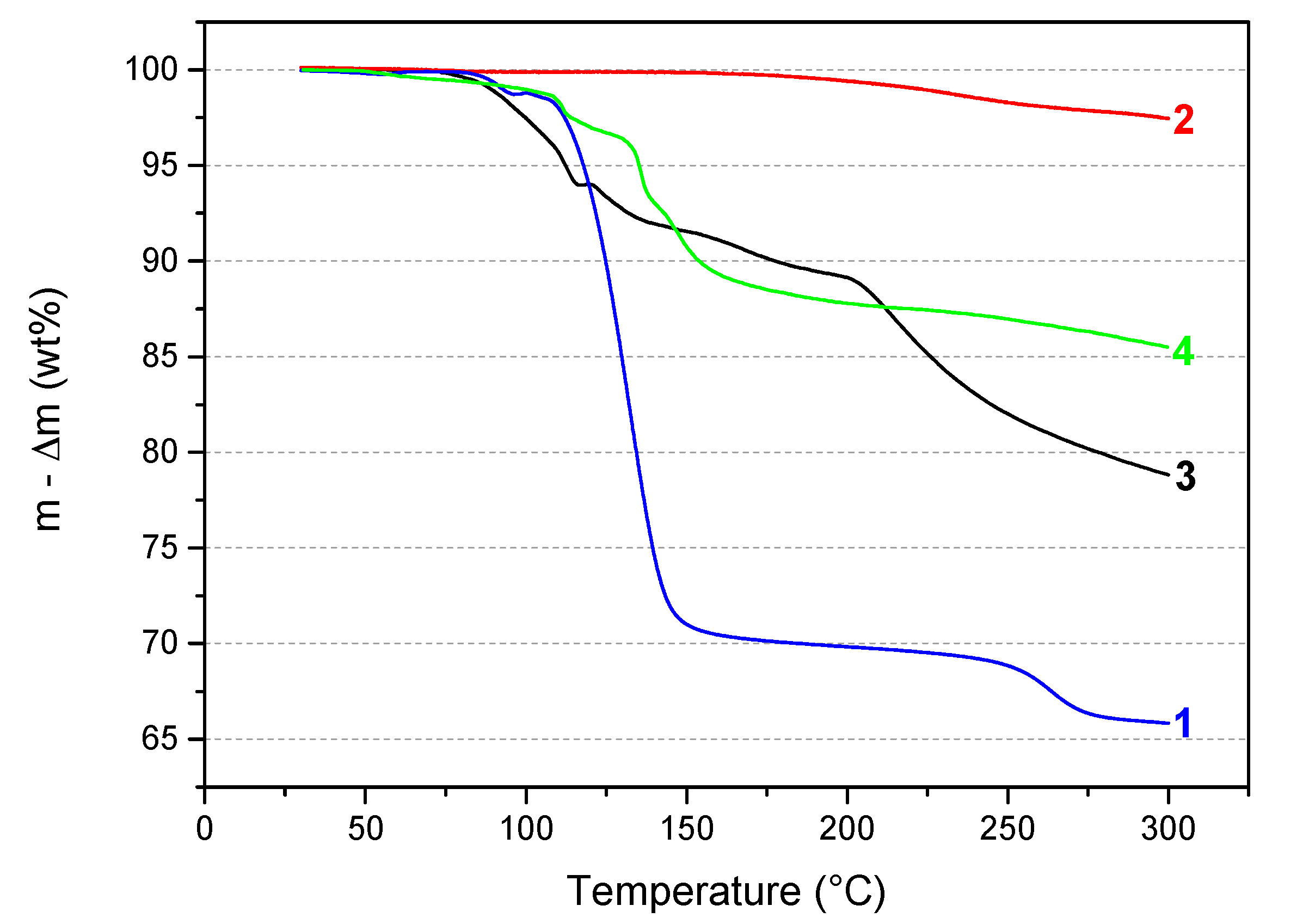
In our laboratory, TGA has been used as an efficient screening tool to evaluate the potential of any new B–N–H compound as well as to compare several of them. Accordingly, the thermal behavior of the sample 3 was first compared to that of the reactants 1 and 2. The TGA curves are shown in Figure 5. In comparison, 2 is quite stable under heating at 5 °C min−1. The weight loss of about 0.6 wt% is negligible before 200 °C and increases to 2.5 wt% up to 300 °C. With respect to 1, it has a much different TGA profile. Therefore, 3 is a compound that is different from the precursors. As mentioned in the previous paragraphs, 3 is somehow comparable to the ammonia-free derivative LiN2H3BH3 (denoted 4 in Figure 5). So, the thermal behavior of 3 was also compared to that of 4. It is worth mentioning that 4 is known to dehydrogenate according to a complex mechanism not releasing unwanted by-products like NH3 [7,8]. The thermal behaviors of these two compounds are different. 3 starts its decomposition at a lower temperature than 4 and shows a higher weight loss due to the release of NH3 molecules from both the complexation and the decomposition phenomena. In other words, the presence of NH3 in 3 leads to a thermolytic behavior which is different from that observed with 4.
3. Discussion and Concluding Remarks
In good agreement with our primary objective, a new hydrogen-rich B–N–H compound has been successfully synthesized by solid-state reaction (mechanosynthesis/ball-milling) of N2H4BH3 and LiNH2. The molecular analyses suggest a compound with the formula LiN2H3BH3·xNH3. This is a new type of coordination compound consisting of a metal cation Li+, a [N2H3BH3]− anionic unit, and an xNH3 ligand [24]. The crystallographic analysis allowed quantifying the x value thanks to the difference in the cell volumes of LiN2H3BH3·xNH3 and another well-described derivative—LiN2H3BH3. Indeed, x was found to be equal to 0.25 suggesting then the following reaction:
LiNH2 (s) + N2H4BH3 (s) → LiN2H3BH3·0.25NH3 (s) + 0.75NH3 (g)
Note that we also worked on a mixture of LiNH2 and N2H4BH3 (results not reported) where the mole ratio was lower than 1. We indeed considered 0.75 mole of LiNH2 and 1 mole of N2H4BH3. Like for LiN2H3BH3·0.25NH3, a paste-like solid was obtained. The as-obtained product showed NMR and FTIR spectra that were comparable to those of LiN2H3BH3·0.25NH3. With respect to the powder XRD pattern, the diffraction peaks were comparable to those observed for LiN2H3BH3·0.25NH3, but with additional peaks belonging to the excess of N2H4BH3. The TGA results were also comparable. In other words, the results and the observations were consistent with the occurrence of the reaction shown by Equation (3).
The reaction of LiNH2 and N2H4BH3 in our conditions can be interpreted as follows. The Lewis base NH2 of LiNH2 would favorably react with the acidic hydrogen Hδ+ of the N2H4 moiety of the borane resulting in NH3. In parallel, the Li+ cation would combine with the Hδ+-deficient N2H3− entity towards the formation of the entity LiN2H3BH3. It seems that a quarter of the as-formed NH3 complexes LiN2H3BH3 leading to the formation of a LiN2H3BH3·0.25NH3 like-compound. The rest of the NH3 (the 0.75 equivalent) can be evacuated under vacuum at ambient conditions.
Ammonia is known to have good affinity with the B–N–H compounds. A first example is ammonia borane. Exposed to an atmosphere of NH3, NH3BH3 is able to complex up to six molecules of NH3, resulting in the formation of a pasty solid [25]. Furthermore, NH3 is an excellent solvent of NH3BH3; the solubility is 259.7 g of NH3BH3 in 100 g of NH3 and the solvated borane shows good stability [26]. A second example is an alkali derivative of NH3BH3. It was observed that exposure of LiNH2BH3 to NH3 produces a sticky liquid containing a 1:1 molar ratio of LiNH2BH3 to NH3 [23]. Keeping in mind these reported observations, we did a simple experiment. We synthesized LiN2H4BH3 as reported in our previous report [8] and exposed it to a stream of NH3. Like for the aforementioned LiNH2BH3 [23], our sample changed to a sticky liquid/pasty material. Hence, we may conclude that the “excess” of 0.75 mole of NH3 (Equation (3)) explains the paste-like state of 3 right after ball-milling.
The new compound LiN2H3BH3·0.25NH3 was primarily synthesized for assessing its potential as chemical H storage material. Ammoniates of B–N–H compounds are of interest as, compared to their ammonia-free counterparts, they should have better dehydrogenation properties owing to the active participation of NH3 in the dehydrogenation process [13]. In our conditions, LiN2H3BH3·xNH3 has been shown to release H2 at temperatures lower than LiN2H3BH3, which is consistent with the previous remark. However, LiN2H3BH3·0.25NH3 liberates non-negligible amounts of NH3. According to Chua et al. [13], contamination of H2 by small amounts of NH3 is unavoidable, but in our conditions the amount of NH3 represents 7.6 wt% of the starting material. Under heating, the x molecules of NH3 are first released at temperatures lower than 100°C, and then the remaining solid, supposed to be mainly LiN2H3BH3, decomposes into H2 and NH3. Taking into account the overall weight loss of 21.2 wt% between 75 and 300 °C and the contents of H (12.1 wt%) and NH3 (7.6 wt%) in LiN2H3BH3·0.25NH3, there is a difference of 1.5 wt% (assuming the loss of all H atoms). This is indicative of the decomposition of LiN2H3BH3·0.25NH3 to some extent. For example, if one assumes the formation of 2 equivalents of H2 up to 300 °C (i.e., 7.7 wt%, which means the loss of 4 H atoms over a maximum of 6 in LiN2H3BH3), the proportion of NH3 stemming from the decomposition of LiN2H3BH3 would be 5.9 wt% at 300 °C. This is illustrated in Figure 6 where a mechanism of decomposition is suggested on the basis of both the present results and the results reported for LiN2H3BH3 elsewhere [8]. A polymeric product of unknown composition forms, similar to the residues recovered upon the dehydrogenation of most of the B–N–H compounds [5,6,7,8,9,10,11,12,27]. The nature of such residues is unknown yet because of their difficult characterization (amorphous to X-ray and too complex composition for FTIR and NMR spectroscopy techniques).
With respect to the aforementioned release of NH3, such a feature is unfortunately quite detrimental for the targeted application, namely chemical H storage. Indeed, the stored hydrogen is intended to be generated on demand for fueling a low-temperature fuel cell, but traces of ammonia (as low as 1 ppm) are able to severely degrade the fuel cell performance to impractical levels [28]. Given that the release of the 0.25 NH3 cannot be avoided with LiN2H3BH3·0.25NH3, its ammonia-free counterpart LiN2H3BH3 [8] seems to be more suitable for chemical H storage.
Yet, our new compound LiN2H3BH3·0.25NH3 might have potential for another application. It might be seen as a potential NH3 carrier as it is theoretically able to release 7.6 wt% of NH3 from 75 °C.
4. Materials and Methods
Hydrogen-storage grade LiNH2 was purchased from Sigma-Aldrich and used as received. Hydrazine borane N2H4BH3 was synthesized by salt metathesis according to an optimized procedure reported in details elsewhere [5]. Typically, an equimolar mixture of sodium borohydride NaBH4 (Acros Organics, Geel, Belgium) and hydrazine hemisulfate N2H4·1/2H2SO4 (Sigma-Aldrich, St Quentin Fallavier, France) in dioxane were prepared in a 250-mL three-necked round-bottom flask kept under argon flow. The mixture was allowed to react at 40 °C for 48 h. Then, the solution of N2H4BH3 was separated from insoluble Na2SO4 by filtration, the solvent was removed by extraction under vacuum at room temperature for 4 h, and the as-obtained borane was dried under dynamic vacuum at room temperature for 24 h. Both LiNH2 and N2H4BH3 were stored and handled in an argon-filled glove box (MBraun M200B, O2 < 0.1 ppm, H2O < 0.1 ppm).
The synthesis of our new compound was done as follows. In the argon-filled glove box, LiNH2 and N2H4BH3 were separately weighted (total of about 350 mg; equimolar ratio) and transferred in a 50-mL stainless steel jar. Several stainless steel balls (Ø 10 mm) of a total weight of 40 g were added and the jar was sealed to be taken out of the glove box. The mixture was ball-milled at ambient conditions by using a RETSCH PM 100 planetary ball mill: 2 min of milling at 300 rpm + 2 min of break, 10 times. In doing so, a paste-like solid was obtained. It was subjected to vacuum overnight. The paste-like state was modified, appearing less pasty. The sample was finally transferred into a vial to be kept in the argon-filled glove box. Note that these conditions were optimized in a preliminary systematic study (not reported herein). We indeed worked with different N2H4BH3/LiNH2 ratios (1:0.75 and 1:0.5), using different ball-milling conditions and a wet synthesis approach (solubilization/dispersion of the reactants in moisture-free organic solvents like tetrahydrofuran and dioxane). For consistency and clarity, only the optimized conditions are presented.
The molecular structure was analyzed by Fourier transform infrared (FTIR) spectroscopy (Nicolet 710, range 3600–600 cm−1, 32 scans). The attenuated total reflection (ATR) mode (enabling sample to be examined directly) was used at ambient conditions and under air. To minimize the contact of the sample with atmospheric O2 and H2O, a vial containing a few milligrams of the borane was prepared in the glove box, taken out, and opened just before the measurement. The number of scans was fixed to 32 to avoid excessive contact with air. In such conditions, reliable spectra are generally collected.
The molecular structure was also analyzed by 11B magic-angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy (Varian VNMR400, 128.37 MHz, 20000 rpm, −10 °C). In the glove box, a few milligrams (or grains) of the sample was transferred into the NMR tube (Ø 10 mm) and dissolved by acetonitrile-d3 (Eurisotop, Gif sur Yvette, France). The sealed tube was then taken out of the glove box to be analyzed by NMR according to the standard procedures for this technique.
The crystal structure was analyzed by powder X-ray diffraction (PXRD) using a PANalytical X’PERT Pro multipurpose diffractometer (Cu-Kα1/α2 radiation, λ = 1.5418 Å, 45 kV, 30 mA). All the patterns were collected using Bragg–Brentano geometry on a spinning sample holder loaded into a glove box (Jacomex PBOX, H2O < 5 ppm, O2 < 5 ppm). The powders were protected from air and oxygen contamination using a Kapton foil.
The thermal behavior of the samples was analyzed by thermogravimetric analysis (TGA; Q500 TA Instruments; heating rate 5 °C min−1; N2 flow rate 50 mL min−1) and differential scanning calorimetry (DSC; 2920 MDSC TA Instruments, manufacturer, New Castle, DE, USA). For both techniques, our standard conditions were as follows: sample weight of 2–3 mg; aluminum crucible of 100 mL with a pinhole (Ø 670 mm); temperature range 25–300 °C; heating rate 5 °C min−1; N2 flow rate of 50 mL min−1. A micro-gas chromatograph-mass spectrometer detector (µGC-MSD; S.R.A. Instruments, Agilent Technologies, Lyon, France) was used in coupling with a TGA/DSC 2 from Mettler-Toledo for the identification and quantification of the gaseous by-products. The µGC is equipped with micro-thermal conductivity detectors and two columns: one molecular sieve column for the detection and quantification of H2 (10 m × 0.32 mm; 5 Å; carrier gas Ar; 70 °C; head column pressure fixed at 28 psi), and one poraPLOT U column for NH3 (8 m × 0.15 mm i.d.; carrier gas He; 130°C; head column pressure fixed at 30 psi).
Acknowledgments
This work has been partially funded by the French State via the ANR (Agence Nationale de la Recherche) and the program “Investissements d’Avenir” with the reference ANR-10-LABX-05-01. The work has also been partially funded by the Région Languedoc-Roussillon and the program “Chercheur(se)s d’Avenir 2013” (project C3/2013 008555). U.B.D. thanks Jean-Fabien Petit for a few of the preliminary ball-milling syntheses, Torben R. Jensen (Aarhus University) for suggesting the use of LiNH2, and Lars Jepsen (Aarhus University) for his help in XRD.
Author Contributions
S.O.A. did most of the experimental work within the frame of his PhD thesis. D.G. did the XRD analyses. R.C. and F.T. were in charge with the thermal characterizations as well as the evolved gas analyses. P.G.Y. was in charge with the crystal structure determination and the treatment of XRD data; he is the co-supervisor of S.O.A. U.B.D. is the leader of the project and the main supervisor of S.O.A.; he also analyzed the molecular data and wrote most of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sherif, S.A.; Goswami, D.Y.; Stefanakos, E.K.; Steinfeld, A. Handbook of Hydrogen Energy; CRC Press: New York, NY, USA, 2014. [Google Scholar]
- Staubitz, A.; Robertson, A.P.M.; Sloan, M.E.; Manners, I. Amine- and phosphine-borane adducts: New interest in old molecules. Chem. Rev. 2010, 110, 4023–4078. [Google Scholar] [CrossRef] [PubMed]
- Goubeau, V.J.; Ricker, E. Borinhydrazin und seine pyrolyseprodukte. Z. Anorg. Allg. Chem. 1961, 310, 123–142. [Google Scholar] [CrossRef]
- Hügle, T.; Kühnel, M.F.; Lentz, D. Hydrazine borane: A promising hydrogen storage material. J. Am. Chem. Soc. 2009, 131, 7444–7446. [Google Scholar] [CrossRef] [PubMed]
- Moury, R.; Moussa, G.; Demirci, U.B.; Hannauer, J.; Bernard, S.; Petit, E.; van der Lee, A.; Miele, P. Hydrazine borane: Synthesis, characterization, and application prospects in chemical hydrogen storage. Phys. Chem. Chem. Phys. 2012, 14, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Moury, R.; Demirci, U.B. Hydrazine borane and hydrazinidoboranes as chemical hydrogen storage materials. Energies 2015, 8, 3118–3141. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Pinkerton, F.E.; Udovic, T.J.; Yildirim, T.; Rush, J.J. Metal hydrazinoborane LiN2H3BH3 and LiN2H3BH3·2N2H4BH3: Crystal structures and high-extent dehydrogenation. Energy Environ. Sci. 2012, 5, 7531–7535. [Google Scholar] [CrossRef]
- Moury, R.; Demirci, U.B.; Ban, V.; Filinchuk, Y.; Ichikawa, T.; Zeng, L.; Goshome, K.; Miele, P. Lithium hydrazinidoborane, a polymorphic material with potential for chemical hydrogen storage. ChemSusChem 2014, 26, 3249–3255. [Google Scholar] [CrossRef]
- Moury, R.; Demirci, U.B.; Ichikawa, T.; Filinchuk, Y.; Chiriac, R.; van der Lee, A.; Miele, P. Sodium hydrazinidoborane, a chemical hydrogen storage material. ChemSusChem 2013, 6, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Pylypko, S.; Petit, J.F.; Ould-Amara, S.; Ndhili, N.; Taihei, A.; Chiriac, R.; Ichikawa, T.; Cretin, M.; Miele, P.; Demirci, U.B. Metal hydride-hydrazine borane: Towards hydrazinidoboranes or composites as hydrogen carriers. Int. J. Hydrogen Energy 2015, 40, 14875–14884. [Google Scholar] [CrossRef]
- Chua, Y.S.; Pei, Q.; Ju, X.; Zhou, W.; Udovic, T.J.; Wu, G.; Xiong, Z.; Chen, P.; Wu, H. Alkali metal hydride modification on hydrazine borane for improved dehydrogenation. J. Phys. Chem. C 2014, 118, 11244–11251. [Google Scholar] [CrossRef]
- Chua, Y.S.; Wu, G.; Xiong, Z.; Karkamkar, A.; Guo, J.; Jian, M.; Wong, M.W.; Autrey, T.; Chen, P. Synthesis, structure and dehydrogenation of magnesium amidoborane monoammoniate. Chem. Commun. 2010, 46, 5752–5754. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Wu, G.; Zhou, W.; Udovic, T.J.; Wu, G.; Xiong, Z.; Wong, M.W.; Chen, P. Monoammoniate of calcium amidoborane: Synthesis, structure, and hydrogen-storage properties. Inorg. Chem. 2012, 51, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Remhof, A.; Rentsch, D.; Züttel, A.; Giri, S.; Jena, P. A novel strategy for reversible hydrogen storage in Ca(BH4)2. Chem. Commun. 2015, 51, 11008–11011. [Google Scholar] [CrossRef] [PubMed]
- Esteruelas, M.A.; Nolis, P.; Oliván, M.; Oñate, E.; Vallribera, A.; Vélez, A. Ammonia borane dehydrogenation promoted by a pincer-square-planar rhodium(I) monohydride: A stepwise hydrogen transfer from the substrate to the catalyst. Inorg. Chem. 2016, 55, 7176–7181. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.C.; Shaw, W.J.; Linehan, J.C.; Schmid, B.; Autrey, T. In situ solid state 11B MAS-NMR studies of the thermal decomposition of ammonia borane: Mechanistic studies of the hydrogen release pathways from a solid state hydrogen storage material. Phys. Chem. Chem. Phys. 2007, 9, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, W.; Pinkerton, F.E.; Meyer, M.S.; Srinivas, G.; Yildirim, T.; Udovic, T.J.; Rush, J.J. A new family of metal borohydride ammonia borane complexes: Synthesis, structures, and hydrogen storage properties. J. Mater. Chem. 2010, 20, 6550–6556. [Google Scholar] [CrossRef]
- Yot, P.G.; Miele, P.; Demirci, U.B. In situ Synchrotron X-ray thermodiffraction of boranes. Crystals 2016, 6, 16. [Google Scholar] [CrossRef]
- Yang, J.B.; Zhou, X.D. Crystal and electronic structures of LiNH2. Appl. Phys. Lett. 2006, 88, 041914. [Google Scholar] [CrossRef]
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Petricek, V.; Dusek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Xia, G.; Yu, X.; Guo, Y.; Wu, Z.; Yang, C.; Liu, H.; Dou, S. Amminelithium amidoborane Li(NH3)NH2BH3: A new coordination compound with favorable dehydrogenation characteristics. Chem. Eur. J. 2010, 16, 3763–3769. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Wu, H.; Zhou, W.; Wang, P. A simple and efficient approach to synthesize amidoborane ammoniates: Case study for Mg(NH2BH3)2(NH3)3 with unusual coordination structure. J. Mater. Chem. 2012, 22, 13174–13179. [Google Scholar] [CrossRef]
- Gao, L.; Fang, H.; Li, Z.; Yu, X.; Fan, K. Liquefaction of solid-state BH3NH3 by gaseous NH3. Inorg. Chem. 2011, 50, 4301–4306. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.G.; Parry, R.W. The crystalline compound ammonia-borane, H3NBH3. J. Am. Chem. Soc. 1955, 77, 6084–6085. [Google Scholar] [CrossRef]
- Summerscales, O.T.; Gordon, J.C. Regeneration of ammonia borane from spent fuel materials. Dalton Trans. 2013, 42, 10075–10084. [Google Scholar] [CrossRef] [PubMed]
- Halseid, R.; Vie, P.J.S.; Tunold, R. Effect of ammonia on the performance of polymer electrolyte membrane fuel cells. J. Power Sources 2006, 154, 343–350. [Google Scholar] [CrossRef]
Figure 1.
11B MAS NMR spectra of 3. For comparison the spectrum of N2H4BH3 (1) is shown.
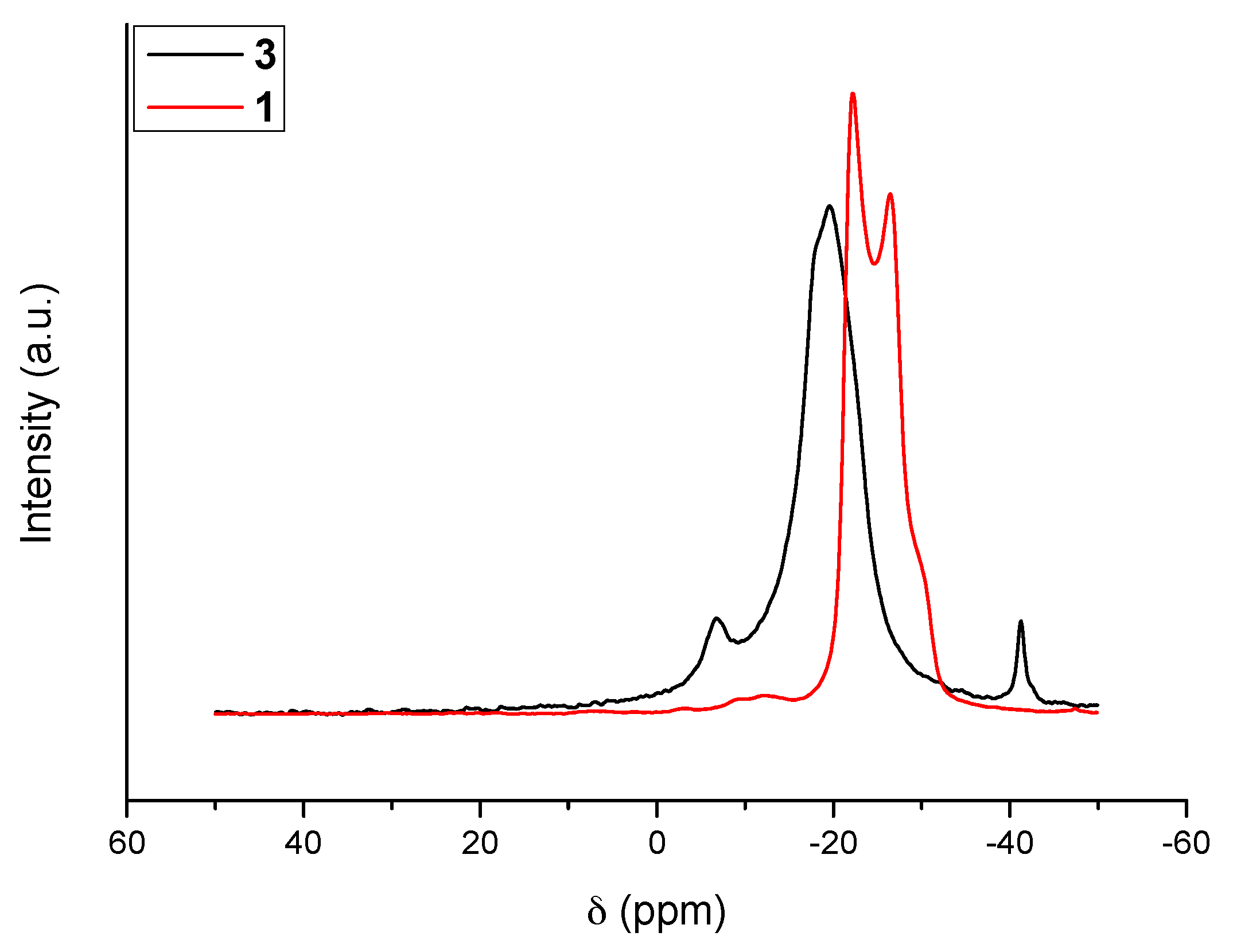
Figure 2.
FTIR spectrum of 3 and, for comparison, that of 1 (N2H4BH3). The different vibrational modes are indicated.
Figure 2.
FTIR spectrum of 3 and, for comparison, that of 1 (N2H4BH3). The different vibrational modes are indicated.
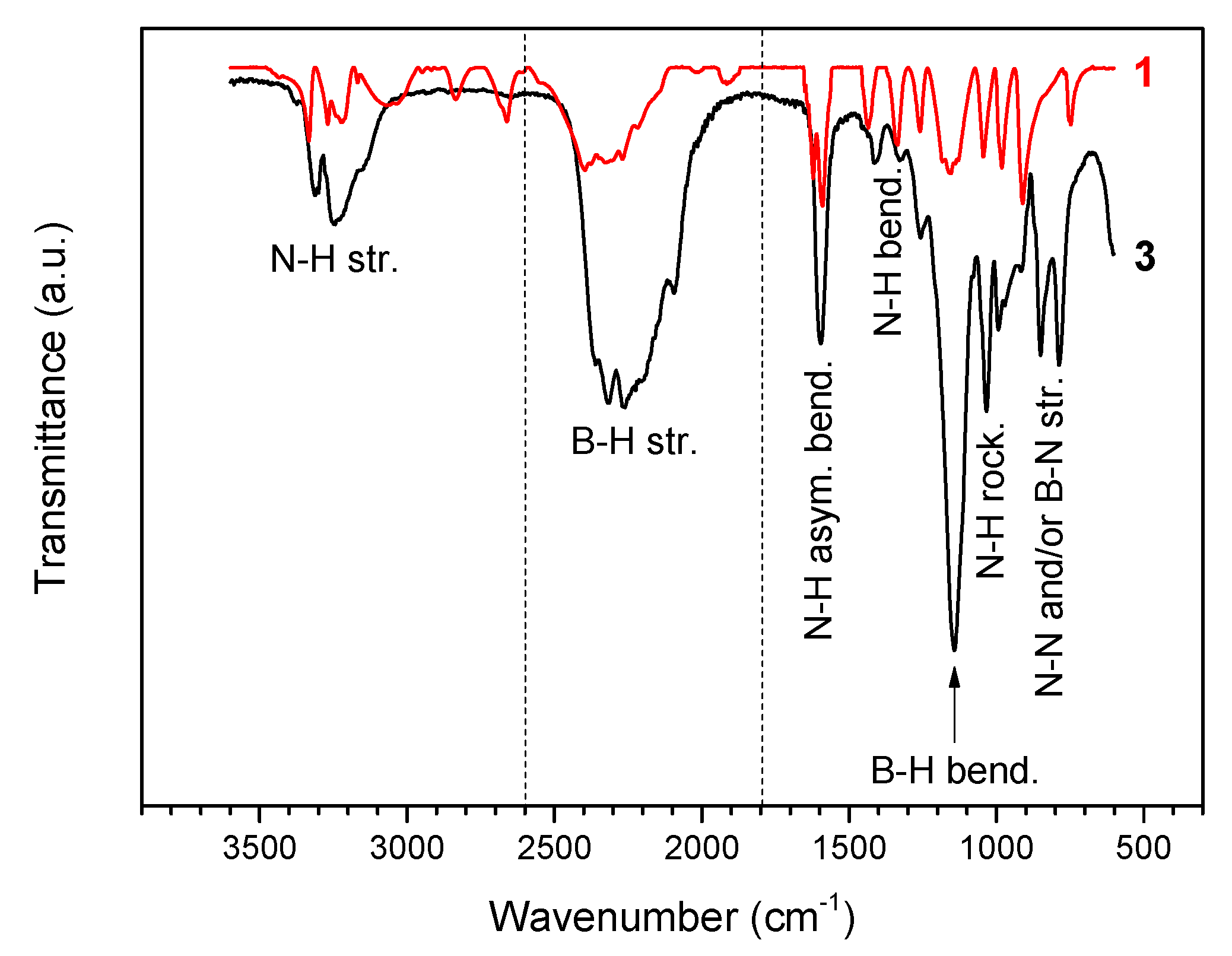
Figure 3.
Observed (in black) and calculated (in red) powder X-ray diffraction profiles for the Rietveld refinement of the LiN2H3BH3·xNH3 phase. The bottom curve (in blue) is the difference plot on the same scale intensity and the tick marks (in green for LiN2H3BH3·xNH3 and in orange for LiNH2) are the calculated angles for the Bragg peaks in 2θ (λ = 1.5418 Å).
Figure 3.
Observed (in black) and calculated (in red) powder X-ray diffraction profiles for the Rietveld refinement of the LiN2H3BH3·xNH3 phase. The bottom curve (in blue) is the difference plot on the same scale intensity and the tick marks (in green for LiN2H3BH3·xNH3 and in orange for LiNH2) are the calculated angles for the Bragg peaks in 2θ (λ = 1.5418 Å).
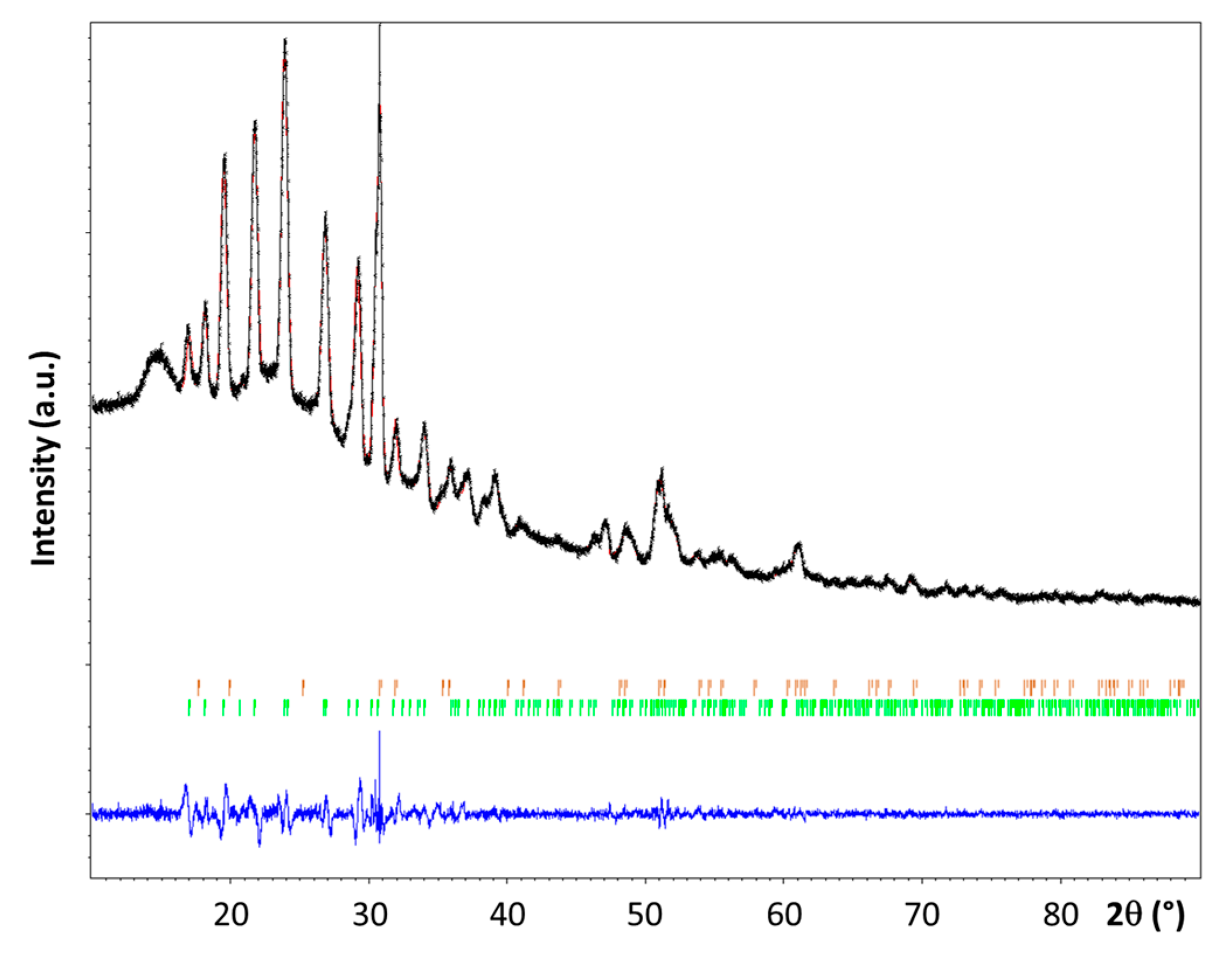
Figure 4.
TGA, DSC, and µGC-MS (H2 m/z = 2, NH3 m/z = 17) data for 3 (heating rate of 5 °C min−1).
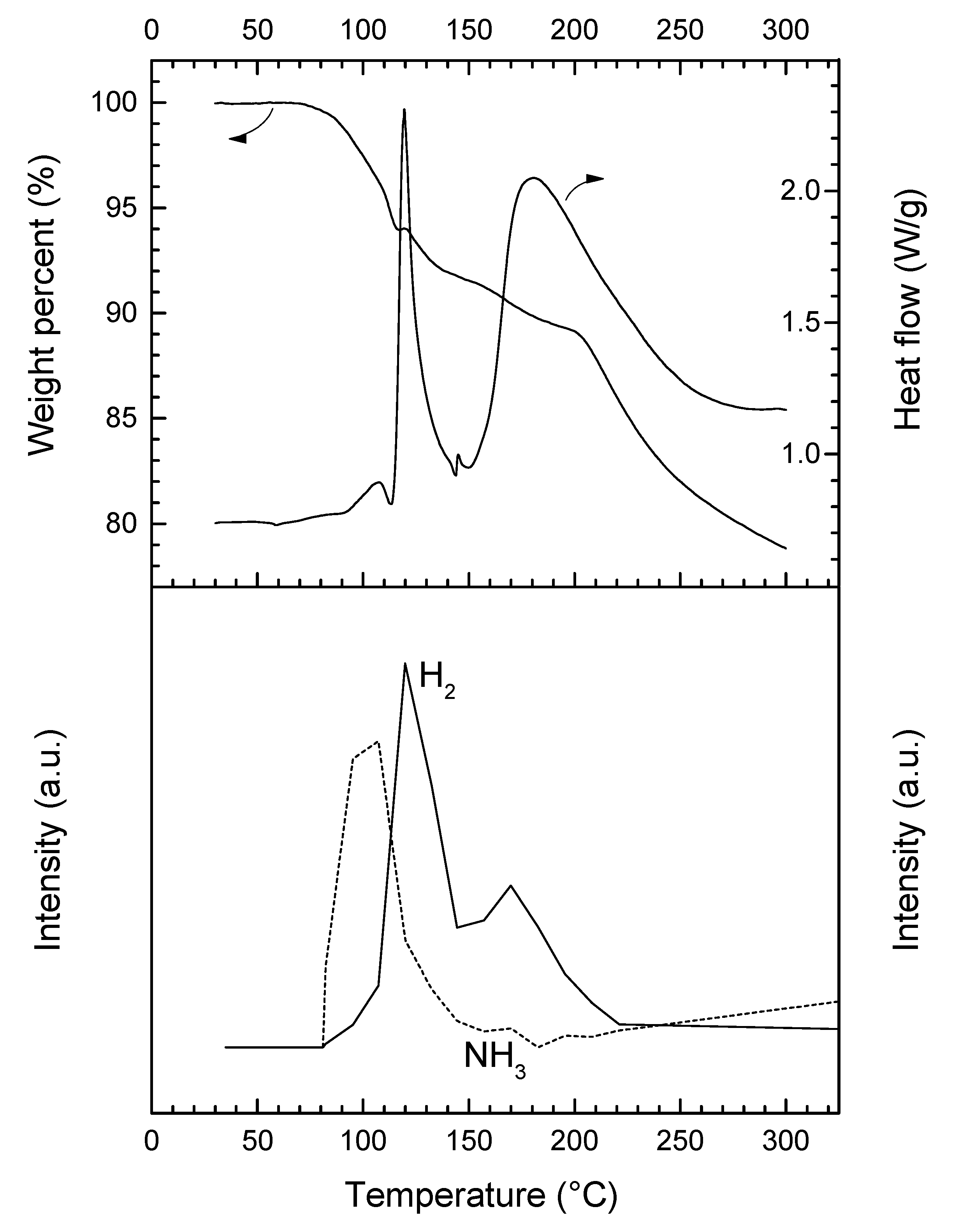
Figure 5.
Superimposition of the TGA curves of 1 (N2H4BH3; from ref. [5]), 2 (LiNH2), 3 (LiN2H3BH3·xNH3), and 4 (LiN2H3BH3; from ref. [8]).
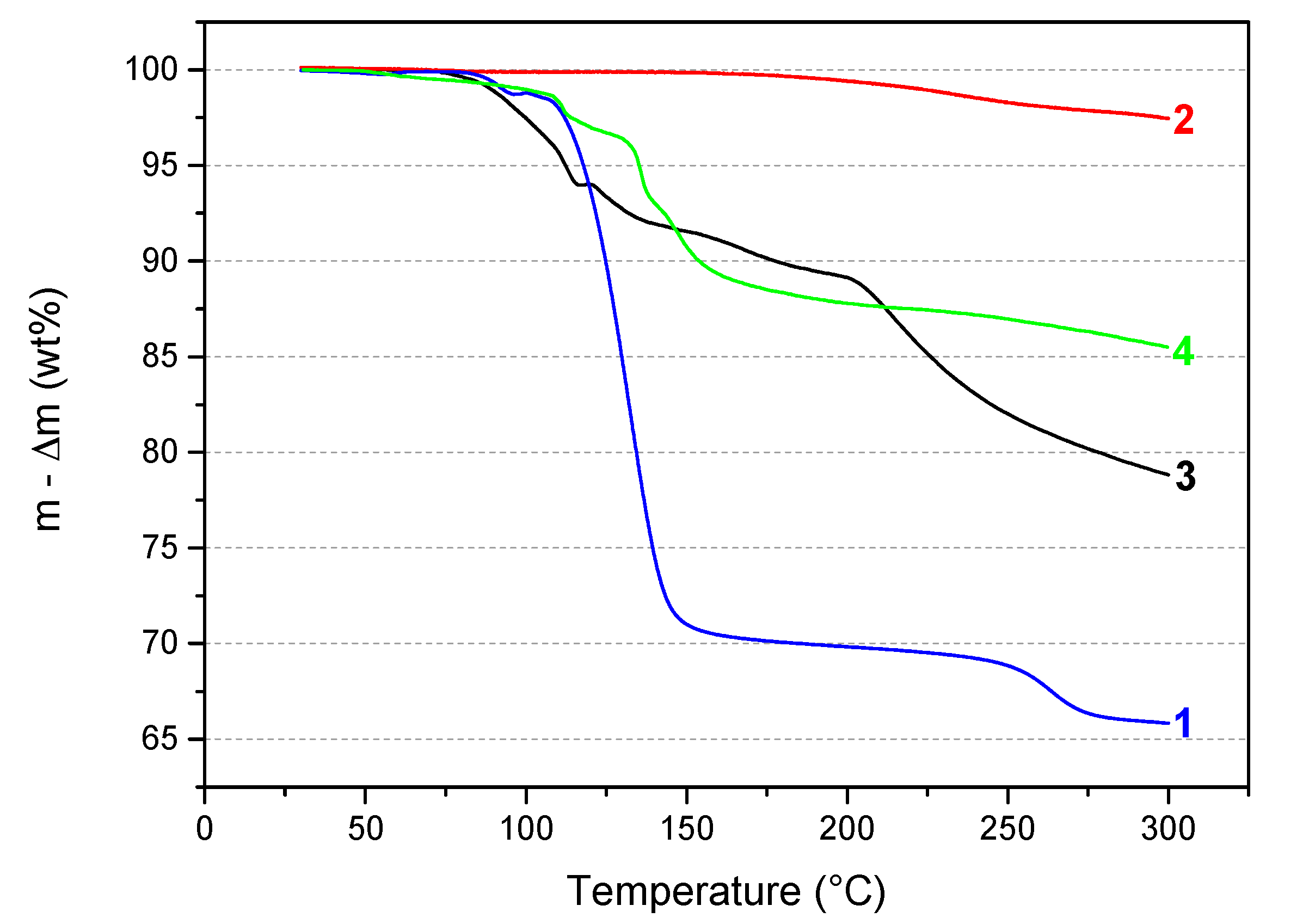
Figure 6.
TGA results-based proposition of a decomposition mechanism of LiN2H3BH3·0.25NH3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Space group (s.g.), unit cell parameters, goodness of fit, and R-values for the refined structures for 3 (LiN2H3BH3·xNH3) and 2 (LiNH2) at room temperature.
Table 1.
Space group (s.g.), unit cell parameters, goodness of fit, and R-values for the refined structures for 3 (LiN2H3BH3·xNH3) and 2 (LiNH2) at room temperature.
| LiN2H3BH3·xNH3 | LiNH2 | |
|---|---|---|
| s.g. | P21/n (N° 14) | (N° 82) |
| Z | 4 | 8 |
| a (Å) | 7.6498(18) | 5.1158(11) |
| b (Å) | 7.482(3) | 5.1158(11) |
| c (Å) | 5.968(17) | 10.103(3) |
| β (°) | 97.803(12) | - |
| V (Å3) | 338.91(17) | 264.41(12) |
| R.P.A. (wt%) 1 | 95.4(5)% | 4.6(6)% |
| GoF | 2.94 | 2.94 |
| Rp | 3.66 | 3.66 |
| wRp | 486 | 4.86 |
| R(obs)/R(all) | 13.85/15.47 | 11.17/12.49 |
| wR(obs)/wR(all) | 11.66/11.77 | 11.92/12.00 |
1 Relative phase amounts in weight.
Table 2.
Experimental structural parameters of 3 (LiN2H3BH3·xNH3) and 2 (LiNH2) at room temperature. The atomic positions for LiNH2 were kept fixed during the refinement [19].
Table 2.
Experimental structural parameters of 3 (LiN2H3BH3·xNH3) and 2 (LiNH2) at room temperature. The atomic positions for LiNH2 were kept fixed during the refinement [19].
| Sample | Atom | Site | Occupancy | x | y | z | Uiso (Å2) |
|---|---|---|---|---|---|---|---|
| 3 | Li1_1 | 4e | 1 | 0.4025(11) | 0.4401(9) | 0.767(2) | 0.0229(1) |
| B2_1 | 4e | 1 | 0.7210(11) | 0.3147(17) | 0.5489(13) | 0.0213(1) | |
| N3_1 | 4e | 1 | 0.6451(11) | 0.2980(11) | 0.7827(11) | 0.0202(1) | |
| N4_1 | 4e | 1 | 0.6373(18) | 0.1096(14) | 0.867(3) | 0.0202(1) | |
| 2 | Li1_2 | 2a | 1 | 0.00000 | 0.500000 | 0.25000 | 0.0177(1) |
| Li2_2 | 2d | 1 | 0.00000 | 0.00000 | 0.00000 | 0.0177(1) | |
| Li3_2 | 4e | 1 | 0,00000 | 0.00000 | 0.25300 | 0.0177(1) | |
| N4_2 | 8g | 1 | 0.23400 | 0.25400 | 0.13700 | 0.0065(1) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ould-Amara, S.; Granier, D.; Chiriac, R.; Toche, F.; Yot, P.G.; Demirci, U.B. Lithium Hydrazinidoborane Ammoniate LiN2H3BH3·0.25NH3, a Derivative of Hydrazine Borane. Materials 2017, 10, 750. https://doi.org/10.3390/ma10070750
AMA Style
Ould-Amara S, Granier D, Chiriac R, Toche F, Yot PG, Demirci UB. Lithium Hydrazinidoborane Ammoniate LiN2H3BH3·0.25NH3, a Derivative of Hydrazine Borane. Materials. 2017; 10(7):750. https://doi.org/10.3390/ma10070750
Chicago/Turabian StyleOuld-Amara, Salem, Dominique Granier, Rodica Chiriac, François Toche, Pascal G. Yot, and Umit B. Demirci. 2017. "Lithium Hydrazinidoborane Ammoniate LiN2H3BH3·0.25NH3, a Derivative of Hydrazine Borane" Materials 10, no. 7: 750. https://doi.org/10.3390/ma10070750
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.