A Comparison of Producer Gas, Biochar, and Activated Carbon from Two Distributed Scale Thermochemical Conversion Systems Used to Process Forest Biomass

Abstract
:1. Introduction
1.1. Background
1.2. Objectives
1.3. Overview of Pyrolysis and Gasification Products
2. Methods
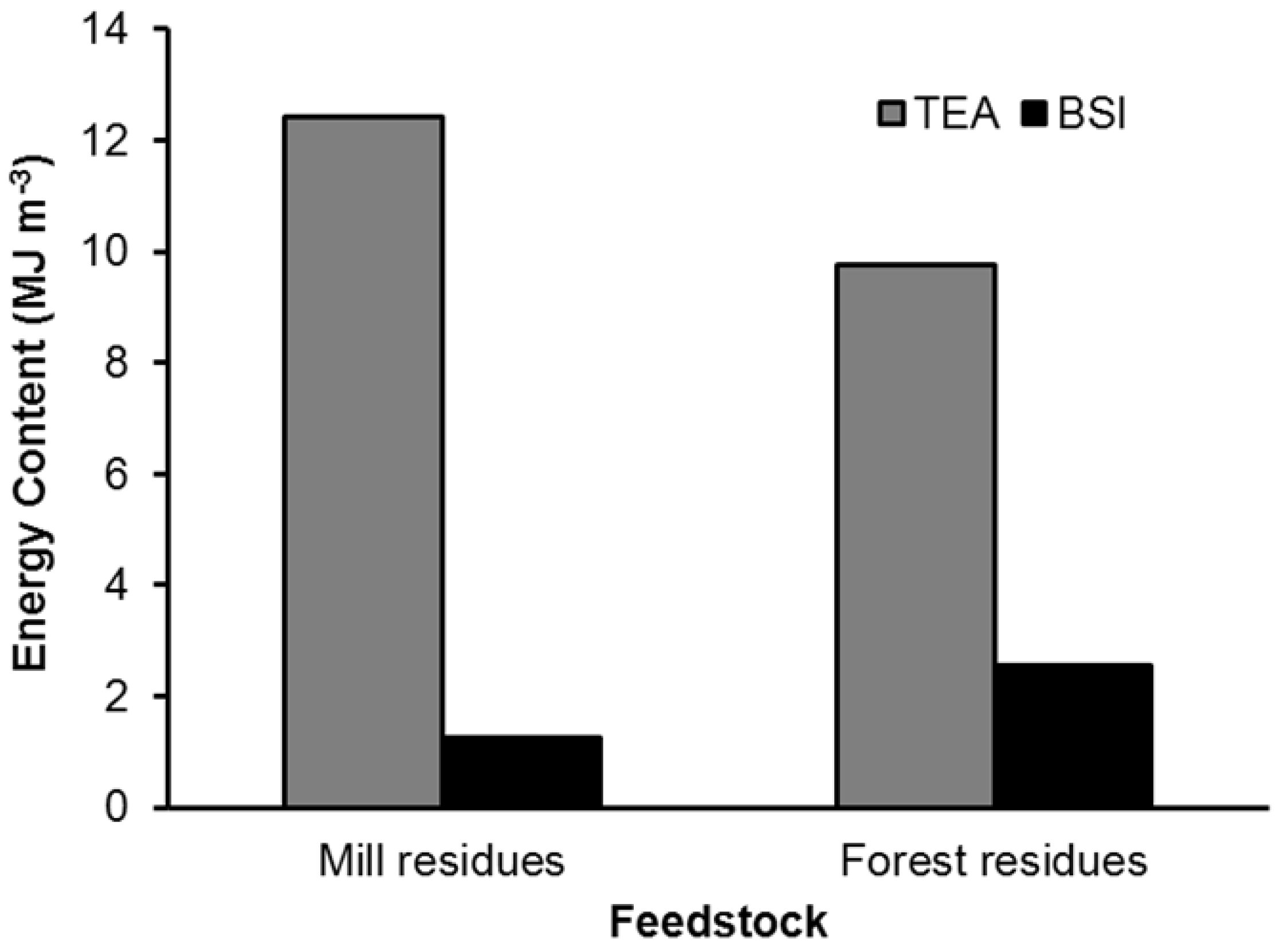
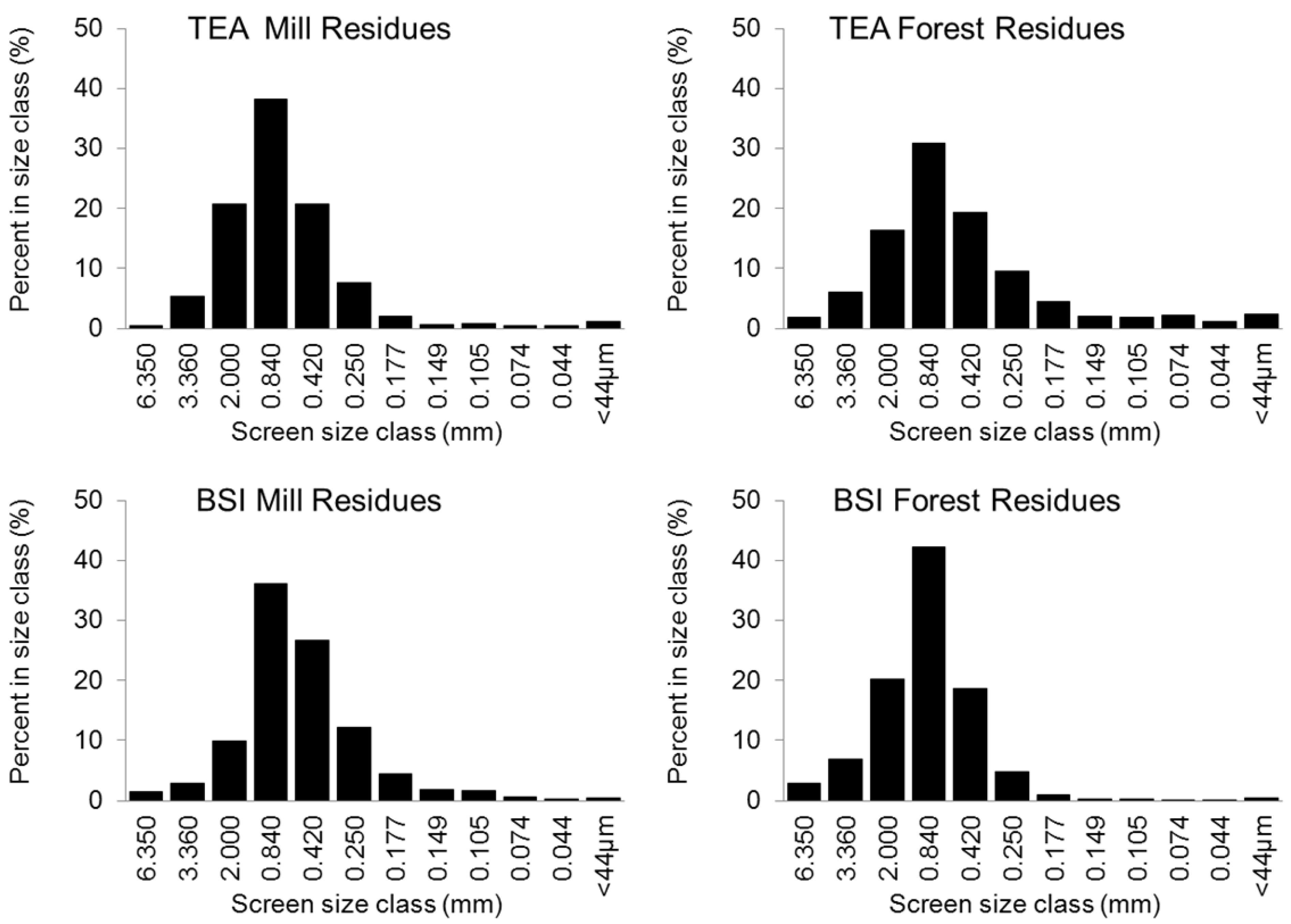
2.1. Biomass Feedstock

{kind=link}
{kind=link}
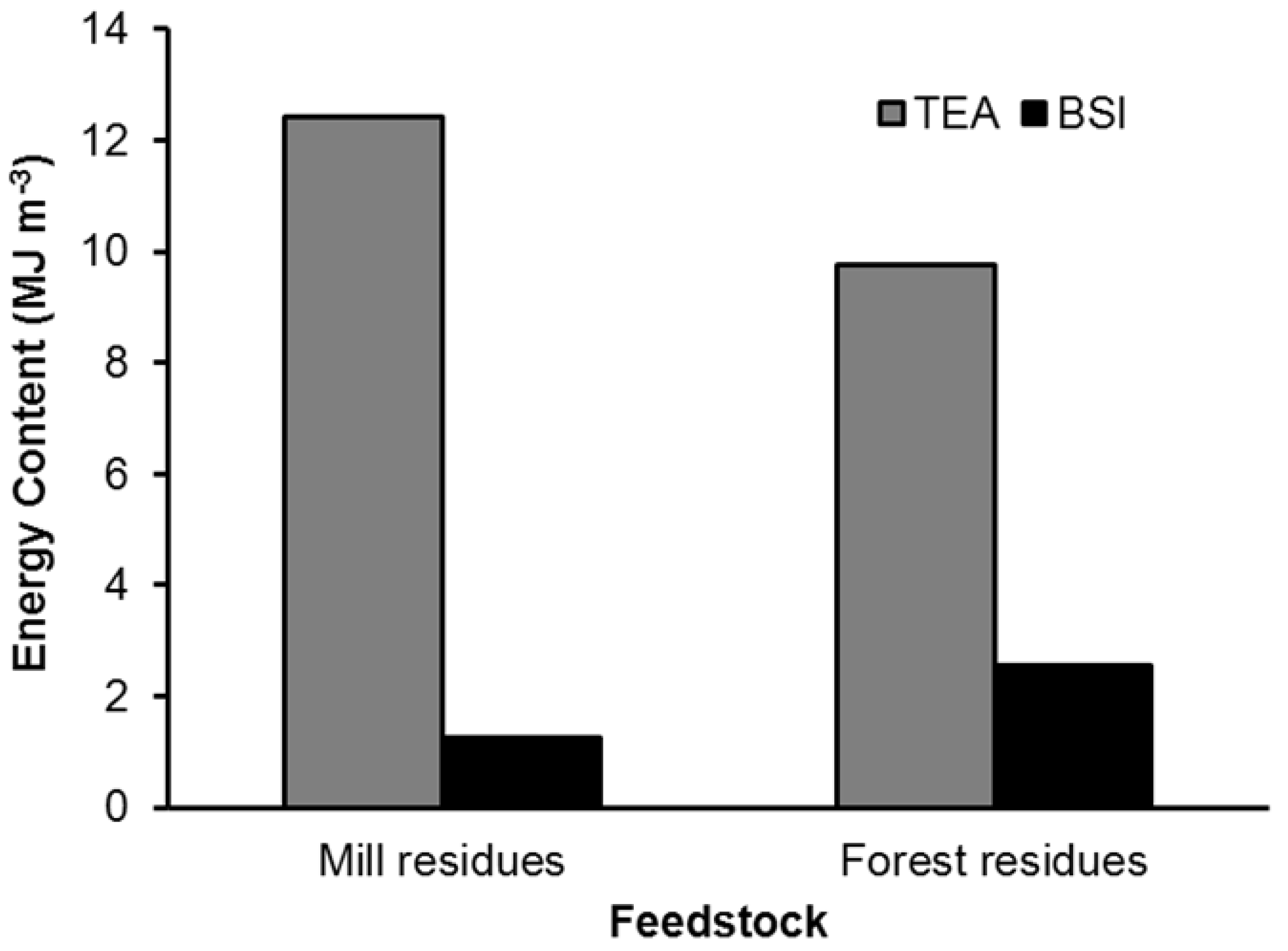{kind=link}
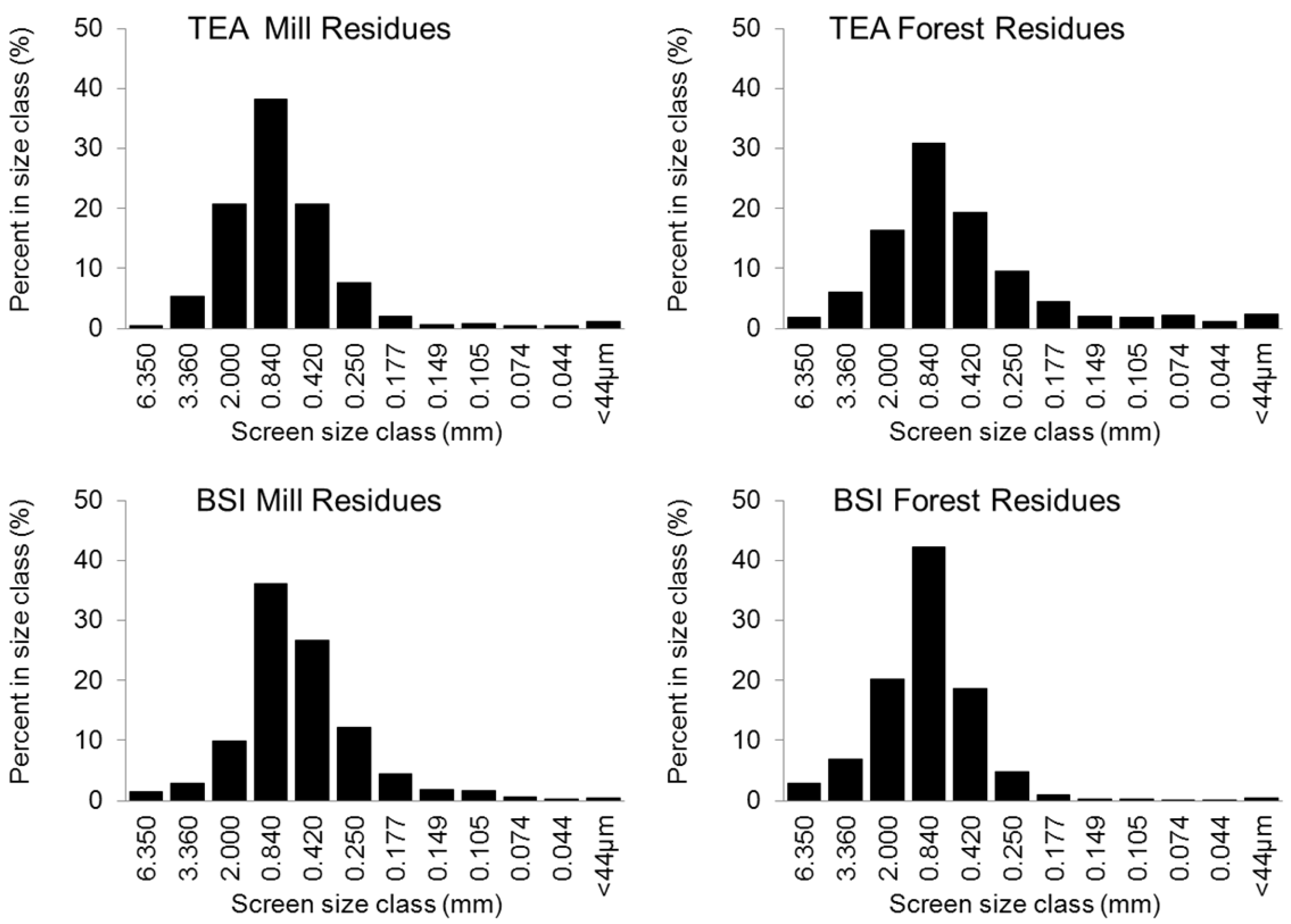{kind=link}
{kind=link}
| Feedstock | System | Moisture (%) b | Organic matter (%) | Mean C (%) | Mean N (%) | Total extractable cations (mg kg−1) a | ||
|---|---|---|---|---|---|---|---|---|
| Ca | Mg | K | ||||||
| Mill residues | TEA | 8.17 | 98.19 | 47.4 | 0.1566 | 1250.0 | 146.5 | 577.5 |
| Mill residues | BSI | 7.04 | c | 47.4 | 0.2062 | 1919.6 | 296.4 | 781.0 |
| Forest residues | TEA | 5.71 | 91.51 | 45.8 | 0.2904 | 1299.6 | 396.3 | 898.4 |
| Forest residues | BSI | 7.25 | c | 48.4 | 0.2124 | 2405.8 | 290.5 | 1097.6 |
2.2. Conversion


2.3. Sampling and Laboratory Analysis
3. Results
| Feedstock | Mill residues | Forest residues | ||
|---|---|---|---|---|
| TEA | BSI | TEA | BSI | |
| H2 (%) | 8.46 | 1.84 | 7.39 | 2.63 |
| CO (%) | 39.03 | 4.04 | 34.05 | 7.83 |
| CO2 (%) | 7.66 | 0.89 | 5.97 | 1.90 |
| CH4 (%) | 12.68 | 7.35 | 10.38 | 12.08 |
| C2 (%) | 3.43 | 0.24 | 1.63 | 0.64 |
| C2H6 (%) | 0.14 | 0.08 | 0.03 | 0.15 |
| C2H4 (%) | 2.77 | 0.15 | 1.18 | 0.44 |
| C2H2 (%) | 0.51 | 0.01 | 0.42 | 0.05 |
| C3 (%) | 0.06 | 0.07 | 0.01 | 0.17 |
| C4–C5 (%) | 0.00 | 0.02 | 0.00 | 0.05 |
| Other (%) | 28.68 | 85.55 | 40.57 | 74.69 |
| Feedstock | System | H2O (%) | Bulk density, dry (Mg m−3) | pH | C (%) | N (%) | C:N | BET surface area (m2 g−1) | Energy (MJ kg−1) |
|---|---|---|---|---|---|---|---|---|---|
| Mill residues | TEA | 2.94 | 0.165 | 10.2 | 91.5 | 0.89 | 102.8 | 15.0 | 33.98 |
| Mill residues | BSI | 1.31 | 0.150 | 9.0 | 82.1 | 0.83 | 98.9 | 203.0 | 35.71 |
| Forest residues | TEA | 1.68 | 0.183 | 8.9 | 70.5 | 0.81 | 87.0 | 11.8 | 33.40 |
| Forest residues | BSI | 2.23 | 0.131 | 8.7 | 75.9 | 0.45 | 168.7 | 129.0 | 33.46 |
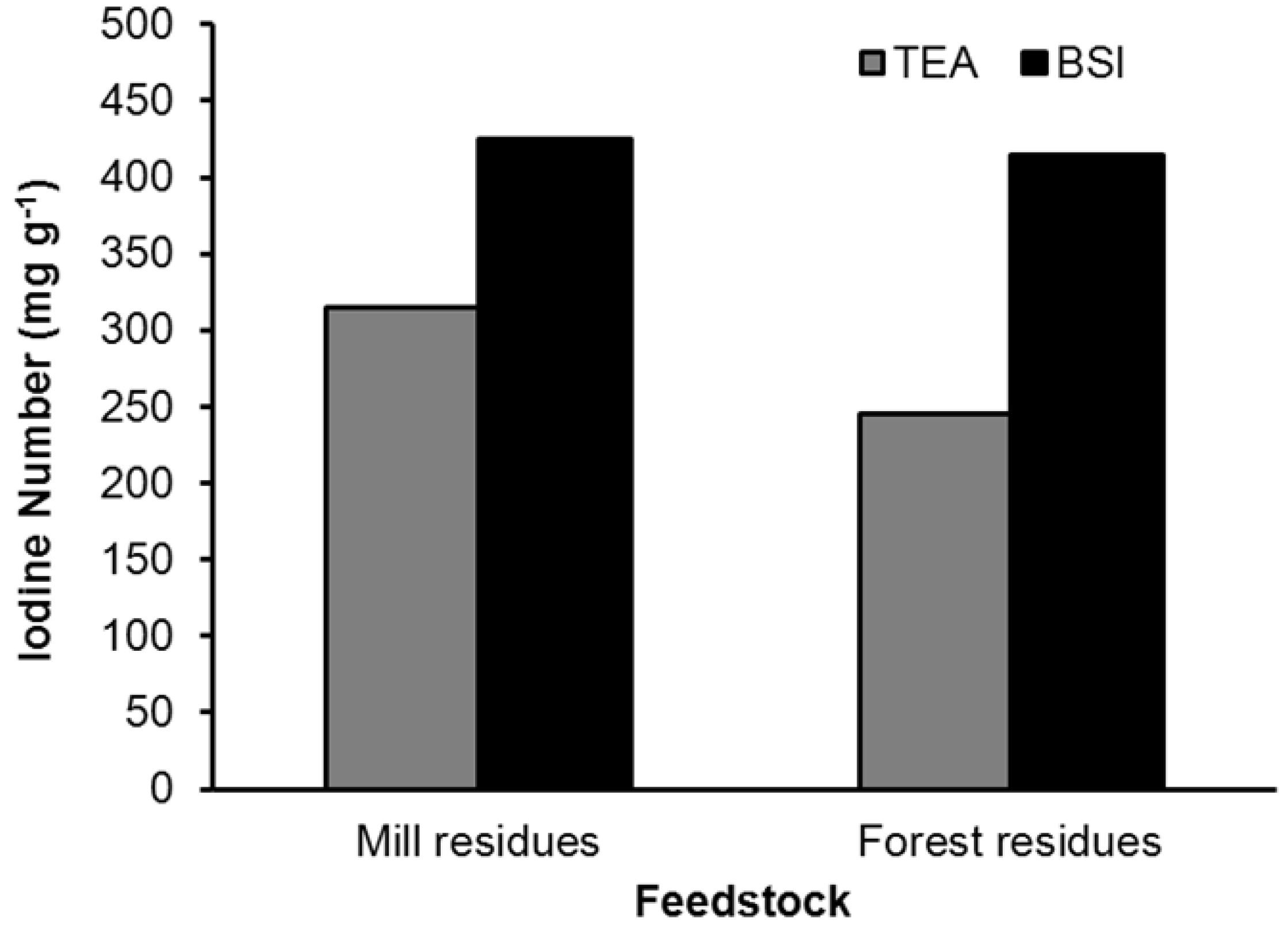
| Feedstock | Product | BET surface (m2 g−1) | Total pore volume (cc g−1) | Porosity (per g of sample) | Ave. pore diameter (Å) | Med. PV pore dia. (Å) | Med. SA pore dia. (Å) |
|---|---|---|---|---|---|---|---|
| Mill residues | Biochar | 15.0 | 0.0161 | 0.0158 | 42.79 | 45.90 | 35.97 |
| Forest residues | Biochar | 11.8 | 0.0359 | 0.0347 | 122.09 | 175.98 | 60.92 |
| Mill residues | AC | 1283.0 | 0.9591 | 0.4895 | 29.90 | 48.15 | 26.97 |
| Forest residues | AC | 575.9 | 0.4441 | 0.3075 | 30.85 | 51.32 | 27.28 |
4. Discussion
4.1. Producer Gas
4.2. Biochar
| Feedstock | BET surface (m2 g−1) | Pyrolysis temp. (°C) | Activat. type | Activat. temp. (°C) | Pore volume (cm3 g−1) | Iodine # (mg g−1) | Source |
|---|---|---|---|---|---|---|---|
| Subbituminous coal | 988 | 700–950 | CO2 | 750 | 0.482 | a | [47] |
| Bituminous coal | 536 | 500 | H3PO4 | 50 | 0.030 | a | [48] |
| Apricot stones | 566 | 200 | H2SO4 | 200 | a | 548 | [49] |
| Wood | 1780 | 440 | H3PO4 + Steam | 440 | 0.130 | a | [50] |
5. Conclusions
Acknowledgments
References
- U.S. Department of Energy. U.S. Billion-Ton Update: Biomass Supply for a Bioenergy and Bioproducts Industry; Perlack, R.D., Stokes, B.J., Eds.; ORNL/TM-2011/224; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 2011; pp. 7–15.
- Peterson, D.; Haase, S. Market Assessment of Biomass Gasification and Combustion Technology for Small- and Medium-Scale Applications; Technical Report NREL/TP-7A2-46190; National Renewable Energy Laboratory: Golden, CO, USA, 2009; pp. 1–32. [Google Scholar]
- Turnbull, J. Use of biomass in electric power generation: The California experience. Biomass Bioenergy 1993, 4, 75–84. [Google Scholar] [CrossRef]
- Morgan, T. An Assessment of Forest-Based Woody Biomass Supply and Use in Montana; Forestry Assistance Bureau, Forestry Division, Montana Department of Natural Resources and Conservation: Missoula, MT, USA, 2009.
- Jones, J.G.; Loeffler, D.; Calkin, D.; Chung, W. Forest residues for thermal energy compared with disposal by onsite burning: Emissions and energy return. Biomass Bioenergy 2010, 34, 737–746. [Google Scholar] [CrossRef]
- Korb, J.; Johnson, N.; Covington, W. Pile burning effects on soil biotic and chemical properties and plant establishment: Recommendations for amelioration. Restor. Ecol. 2004, 12, 52–62. [Google Scholar] [CrossRef]
- Fransham, P.; Badger, P. Use of mobile fast pyrolysis plants to densify biomass and reduce biomass handling cost—A preliminary assessment. Biomass Bioenergy 2006, 30, 321–325. [Google Scholar] [CrossRef]
- Garcia-Perez, M.; Lewis, T.; Kruger, C. Methods for Producing Biochar and Advanced Biofuels in Washington State, Part 1: Literature Review of Pyrolysis Reactors; Department of Biological Systems Engineering and the Center for Sustaining Agriculture and National Resources, Washington State University: Pullman, WA, USA, 2010; pp. 1–137. [Google Scholar]
- El-Khattam, W.; Salama, M. Distributed generation technologies, definitions and benefits. Electric. Power Syst. Res. 2004, 71, 119–128. [Google Scholar] [CrossRef]
- Biochar Solutions, Incorporated (BSI). BSI Biochar Base Unit: Technical Specifications, Version 3.0.; Biochar Solutions, Incorporated: Carbondale, CO, USA, 2011. [Google Scholar]
- PHG Energy. Industrial Grade Downdraft Gasification; PHG Energy: LaVergne, TN, USA, 2011; Available online: http://www.phgenergy.com (accessed on 7 January 2013).
- Mohan, D.; Pittman, C., Jr.; Steele, P. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Kumar, A.; Jones, D.; Hanna, M. Thermochemical biomass gasification: A review of the current status of the technology. Energies 2009, 2, 556–581. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Gerhauser, H.; Kiel, J.; Ptasinski, K. Biomass upgrading by torrefaction for the production of biofuels: A review. Biomass Bioenergy 2011, 35, 3748–3762. [Google Scholar]
- Uslu, A.; Faaij, A.; Bergman, P. Pre-treatment technologies, and their effect on international bioenergy supply chain logistics. Techno-economic evaluation of torrefaction, fast pyrolysis and pelletisation. Energy 2008, 33, 1206–1223. [Google Scholar] [CrossRef]
- Prins, M.; Ptasinski, K.; Janssen, F. Torrefaction of wood Part 2. Analysis of products. J. Anal. Appl. Pyrolysis 2006, 77, 35–40. [Google Scholar] [CrossRef]
- Rath, J.; Staudinger, G. Cracking reactions of tar from pyrolysis of spruce wood. Fuel 2001, 80, 1379–1389. [Google Scholar] [CrossRef]
- Hamelinck, C.; Faaij, A. Production of Methanol from Biomass. In Alcoholic Fuels; Miller, S., Ed.; Taylor and Francis Group: New York, NY, USA, 2006; pp. 7–50. [Google Scholar]
- Tijmensen, M.; Faaij, A.; Hamelinck, C.; van Hardeveld, M. Exploration of the possibilities for production of Fischer Tropsch liquids and power via biomass gasification. Biomass Bioenergy 2002, 23, 129–152. [Google Scholar] [CrossRef]
- Jenkins, B.; Baxter, L.; Miles, T., Jr.; Miles, T. Combustion properties of biomass. Fuel Proc. Technol. 1998, 54, 17–46. [Google Scholar] [CrossRef]
- Biochar Solutions, Incorporated (BSI). Biochar Solutions: Overview; Biochar Solutions, Incorporated: Carbondale, CO, USA, 2011; Available online: http://www.biocharsolutions.com/overview.html (accessed on 7 January 2013).
- Bergman, R. Drying and Control of Moisture Content and Dimensional Changes. In Wood Handbook—Wood as an Engineering Material; General Technical Report FPL-GTR-113; USDA Forest Service, Forest Products Laboratory: Madison, WI, USA, 2010. [Google Scholar]
- Nelson, D.; Sommers, L. Total Carbon, Organic Carbon, and Organic Matter. In Methods of Soil Analysis, Part 3—Chemical Methods; Soil Science Society of America, American Society of Agronomy: Madison, WI, USA, 1996; pp. 961–1069. [Google Scholar]
- American Society for Testing and Materials (ASTM). ASTM D4607-94 (2011) Standard Test Method for Determination of Iodine Number of Activated Carbon; ASTM International: West Conshohocken, PA, USA, 2011. [Google Scholar]
- Downie, A.; Crosky, A.; Munroe, P. Physical Properties of Biochar. In Biochar for Environmental Management Science and Technology; Lehmann, J., Joseph, S., Eds.; Earthscan: London, UK, 2006; pp. 13–32. [Google Scholar]
- Elder, T.; Groom, W. Pilot-scale gasification of woody biomass. Biomass Bioenergy 2011, 35, 3522–3528. [Google Scholar] [CrossRef]
- Son, Y.; Yoon, S.; Kim, Y.; Lee, J. Gasification and power generation characteristics of woody biomass utilizing a downdraft gasifier. Biomass Bioenergy 2011, 35, 4215–4220. [Google Scholar] [CrossRef]
- Jenkins, B. Chapter 3.2.2: Pyrolysis Gas. In Handbook of Agricultural Engineering Volume V: Energy and Biomass Engineering; Kitani, O., Ed.; American Society of Agricultural Engineers: St. Joseph, MI, USA, 1999. [Google Scholar]
- KiOR. KiOR Production Facilities; KiOR, Inc.: Columbus, MS, USA, 2011; Available online: http://www.kior.com/content/?s=6&s2=56&p=56&t=Production-Facilities/ (accessed on 7 January 2013).
- Bergman, P.; Boersma, A.; Kiel, J.; Zwart, R. Development of Torrefaction for Biomass Co-Firing in Existing Coal-Fired Power Stations; BIOCOAL Concept Version, ECN Report; Energy Research Center of the Netherlands: Petten, The Netherlands, 2005. [Google Scholar]
- Abdullah, H.; Wu, H. Biochar as a fuel: 1. Properties and grindability of biochars produced from the pyrolysis of mallee wood under slow-heating conditions. Energy Fuels 2009, 23, 4174–4181. [Google Scholar] [CrossRef]
- Antal, M., Jr.; Croiset, E.; Dai, X.; DeAlmeida, C.; Shu-Lai Mok, W.; Norberg, N. High-yield biomass charcoal. Energy Fuels 1996, 10, 652–658. [Google Scholar] [CrossRef]
- Ertas, M.; Alma, M. Pyrolysis of laurel (Laurusnobilis L.) extraction residues in a fixed-bed reactor: Characterization of bio-oil and bio-char. J. Anal. Appl. Pyrolysis 2010, 88, 22–29. [Google Scholar] [CrossRef]
- Sukiran, M.; Kheang, L.; Bakar, N.; May, C. Production and characterization of bio-char from the pyrolysis of empty fruit bunches. Am. J. Appl. Sci. 2011, 8, 984–988. [Google Scholar] [CrossRef]
- Federal Energy Management Program (FEMP). Biomass Cofiring in Coal-Fired Boilers; Federal Technology Alert DOE/EE-0288; U.S. Department of Energy, Energy Efficiency and Renewable Energy: Washington, DC, USA, 2004.
- Tillman, D. Biomass cofiring: The technology, the experience, the combustion consequences. Biomass Bioenergy 2000, 19, 365–384. [Google Scholar] [CrossRef]
- Demirbas, A. Sustainable cofiring of biomass with coal. Energy Convers. Manag. 2003, 44, 1465–1479. [Google Scholar] [CrossRef]
- Lehmann, J. Bio-energy in the black. Front. Ecol. Environ. 2007, 5, 381–387. [Google Scholar] [CrossRef]
- Laird, D.A. The charcoal vision: A win-win-win scenario for simultaneously producing bioenergy, permanently sequestering carbon, while improving soil and water quality. Agron. J. 2008, 100, 178–181. [Google Scholar] [CrossRef]
- Mbagwu, J.; Piccolo, A. Effects of Humic Substances from Oxidized Coal on Soil Chemical Properties and Maize Yield. In The Role of Humic Substances in Ecosystems and in Environmental Protection; Drozd, J., Gonet, S.S., Senesi, N., Weber, J., Eds.; Polish Society of Humic Substances: Wroclaw, Poland, 1997; pp. 921–925. [Google Scholar]
- Brodowski, S.; John, B.; Flessa, H.; Amelung, W. Aggregate-occluded black carbon in soil. Eur. J. Soil Sci. 2006, 57, 539–546. [Google Scholar] [CrossRef]
- Lehmann, J. Biochar for Environmental Management; Earthscan: London, UK, 2009. [Google Scholar]
- McElligott, K. Biochar Amendments to Forest Soils: Effects on Soil Properties and Tree Growth. MSc Thesis, University of Idaho, Moscow, ID, USA, 2011. [Google Scholar]
- Meurisse, R.T. Propoerties of Anidsols Important for Forestry. 1985. In Western Hemlock Management Conference; Atkinson, W.A., Zozoski, R.T., Eds.; University of Washington: Seattle, WA, USA, 1976. [Google Scholar]
- McElligott, K.; Page-Dumroese, D.; Coleman, M. Bioenergy Production Systems and Biochar Application in Forests: Potential for Renewable Energy, Soil Enhancement, and Carbon Sequestration; Research Note RMRS-RN-26; Rocky Mountain Research Station: Moscow, ID, USA, 2011. [Google Scholar]
- Pollard, S.; Fowler, G.; Sollars, C. Low-cost adsorbents for waste and wastewater treatment: A review. Sci. Total Environ. 1992, 116, 31–52. [Google Scholar] [CrossRef]
- Teng, H.; Lin, H. Activated carbon production from low ash subbituminous coal with CO2 activation. Mater. Interfaces Electrochem. Phenom. 1998, 44, 1170–1177. [Google Scholar]
- Teng, H.; Yeh, T.; Hsu, L. Preparation of activated carbon from bituminous coal with phosphoric acid activation. Carbon 1998, 36, 1387–1395. [Google Scholar] [CrossRef]
- Kobya, M.; Demirbas, E.; Senturk, E.; Ince, M. Adsorption of heavy metal ions from aqueous solutions by activated carbon prepared from apricot stone. Bioresour. Technol. 2005, 96, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Benaddi, H.; Bandosz, T.; Jagiello, J.; Schwarz, J.; Rouzaud, J.; Legras, D.; Beguin, F. Surface functionality and porosity of activated carbons obtained from chemical activation of wood. Carbon 2000, 38, 669–674. [Google Scholar] [CrossRef]
- Azargohar, R.; Dalai, A. Biochar as a precursor of activated carbon. Appl. Biochem. Biotechnol. 2006, 131, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Halliop, E.; MacDonald, J. The production of chemically-activated carbon. Carbon 1999, 37, 269–274. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Anderson, N.; Jones, J.G.; Page-Dumroese, D.; McCollum, D.; Baker, S.; Loeffler, D.; Chung, W. A Comparison of Producer Gas, Biochar, and Activated Carbon from Two Distributed Scale Thermochemical Conversion Systems Used to Process Forest Biomass. Energies 2013, 6, 164-183. https://doi.org/10.3390/en6010164
Anderson N, Jones JG, Page-Dumroese D, McCollum D, Baker S, Loeffler D, Chung W. A Comparison of Producer Gas, Biochar, and Activated Carbon from Two Distributed Scale Thermochemical Conversion Systems Used to Process Forest Biomass. Energies. 2013; 6(1):164-183. https://doi.org/10.3390/en6010164
Chicago/Turabian StyleAnderson, Nathaniel, J. Greg Jones, Deborah Page-Dumroese, Daniel McCollum, Stephen Baker, Daniel Loeffler, and Woodam Chung. 2013. "A Comparison of Producer Gas, Biochar, and Activated Carbon from Two Distributed Scale Thermochemical Conversion Systems Used to Process Forest Biomass" Energies 6, no. 1: 164-183. https://doi.org/10.3390/en6010164