Rare Earth Borohydrides—Crystal Structures and Thermal Properties
by
 ,
,
Christoph Frommen
1, 
Magnus H. Sørby
1,
Michael Heere
1,2,
Terry D. Humphries
3,
Jørn E. Olsen
1 and
Bjørn C. Hauback
1,* 1
Institute for Energy Technology, Physics Department, P.O. Box 40, NO-2027 Kjeller, Norway
2
Research Neutron Source Munich (FRM2) and Karlsruhe Institute of Technology (KIT), Institute for Applied Materials—Energy Storage Systems (IAM-ESS), 76344 Eggenstein, Germany
3
Department of Physics and Astronomy, Fuels and Energy Technology Institute, Curtin University, GPO Box U1987, Perth 6845, Australia
*
Author to whom correspondence should be addressed.
Energies 2017, 10(12), 2115; https://doi.org/10.3390/en10122115
Submission received: 3 November 2017
/
Revised: 1 December 2017
/
Accepted: 4 December 2017
/
Published: 12 December 2017
Abstract
:Rare earth (RE) borohydrides have received considerable attention during the past ten years as possible hydrogen storage materials due to their relatively high gravimetric hydrogen density. This review illustrates the rich chemistry, structural diversity and thermal properties of borohydrides containing RE elements. In addition, it highlights the decomposition and rehydrogenation properties of composites containing RE-borohydrides, light-weight metal borohydrides such as LiBH4 and additives such as LiH.
1. Introduction
Metal borohydrides are being intensively studied as potential hydrogen and thermal energy storage materials [1,2,3,4,5,6,7] as well as solid state electrolytes [8,9,10,11]. Alkali and alkaline earth borohydrides such as LiBH4 and Mg(BH4)2 are of great interest due to their high gravimetric hydrogen storage capacity. Unfortunately, their decomposition temperatures are too high and rehydrogenation only occurs under severe conditions as a result of the strong ionic interaction between the metal cation and the BH4− anion. In contrast, complexes containing transition metal cations are too unstable with decomposition temperatures below or slightly above room temperature. In recent years, a correlation between the thermodynamic stability (decomposition temperature) and the Pauling electronegativity of the metal cation has been established [12]. According to reference [12] the hydrogen desorption temperature decreases with increasing electronegativity of the metal. This result suggests that the charge transfer from the metal cation to the BH4− anion is responsible for the stability of the metal borohydride. It has become apparent that borohydrides with Pauling electronegativities larger than 1.4 are too unstable for use in hydrogen storage applications. Tuning the thermodynamic properties of borohydrides has been attempted by the combination of different metal cations to form double-cation metal borohydrides Mn+Mm+(BH4)(n+m) (Mn+ = Li+, Na+, K+; Mm+ = transition metal cation). The search for new possible candidates for hydrogen storage applications has stimulated interest in materials containing rare earth (RE) metal cations. The gravimetric hydrogen storage density of monometallic RE-borohydrides ranges from 9.1 wt % H2 for Y(BH4)3 to 5.6 wt % H2 for Yb(BH4)3, respectively, and their thermodynamic stability is expected to lie between those of the alkali and transition metals. Compared to alkali or alkaline-earth borohydrides such as LiBH4 (18.5 wt % H2) or Mg(BH4)2 (14.9 wt % H2), RE-borohydrides possess lower gravimetric hydrogen density. However, they are able to release a major amount of hydrogen between 200 and 300 °C, which is significantly lower than for LiBH4 or Mg(BH4)2. As a result, a large number of novel RE-borohydrides have been synthesized and more than 50 papers have been published since 2008. The majority of publications are focused on the synthesis and structural characterization of the compounds and several extensive reviews already exist [1,4,7]. In contrast, less information is available about the thermal properties and rehydrogenation behavior of pure RE-borohydrides or as part of composites.
The current review article presents the state of the art on the crystal structure and thermodynamic properties of pure RE-borohydrides. In addition, we elucidate the thermal decomposition and rehydrogenation behavior of RE-borohydrides in composite mixtures containing LiBH4 and additives such as LiH. The layout is as follows: Section 2 details commonly applied synthesis procedures for RE-borohydrides; Section 3 summarizes the crystal structures of mono-, di- and trimetallic borohydrides containing rare earth elements; Section 4 presents the thermal properties of RE-borohydrides starting from monometallic and halide substituted compounds, followed by composite mixtures containing RE-borohydrides, LiBH4 and LiH. This is the first time that the state of the art for the decomposition and rehydrogenation behavior of composites containing RE-borohydrides has been summarized in literature.
2. Synthesis of Metal Borohydrides
The synthesis of borohydrides is mostly carried out by either solvent-based methods or by mechanochemistry. Both approaches offer distinct advantages, but also suffer from certain drawbacks. The first solvent-based synthesis of a metal borohydride, Mg(BH4)2, was reported by Wiberg and Bauer in 1950 and was based on the reaction of diethylmagnesium, Mg(CH2CH3)2, and diborane, B2H6, in ether [13]. However, the authors were not aware of a stable solvent adduct that was formed under the experimental conditions described in their paper. A direct synthesis route for Mg(BH4)2 from the elements was described in a German patent from 1958 [14]. However, attempts to reproduce the procedure were not successful in obtaining sufficiently high yields and it was impossible to separate the product from the rest of the reaction mixture by solvent extraction [15]. In order to circumvent the high reactivity of diborane towards air and moisture and its inherent toxicity, Koester [16] developed a synthesis method in 1957 based on the reaction of the respective metal hydride with N-alkylborazanes instead of diborane. This method has since then been successfully adapted to substitute diborane by safer and easier to handle borane complexes such as triethylamine borane, (CH3CH2)3NBH3, or dimethylsulfide borane, (CH3)2SBH3. The underlying principal of each of the synthesis methods named above, is that a metal hydride or metal alkyl reacts with a borane donor anion. This leads to the formation of a solvated metal borohydride complex, which in turn requires further thermal treatment in order to obtain the solvent-free product [15,17]. A major risk of this synthesis method lies in the decomposition of the borohydride during thermal treatment and solvent removal. The same risk also exists for the metathesis reactions between an alkali borohydride (mostly LiBH4) and a metal chloride carried out in organic solvents such as ether or toluene. However, this strategy has been successful in obtaining the desired borohydride, e.g. in the case of Al(BH4)3 and other compounds according to Equation (1):
3LiBH4 (s) + AlCl3 (s) → Al(BH4)3 (l) + 3LiCl (s)
The use of a weakly coordinating solvent that selectively dissolves the borohydride then allows for the removal of the metal halide by solvent extraction. The separation of the borohydride from the metal halide by-product is the major advantage of the solvent-based approach and allows for structural and thermal studies on the pure and halide-free materials.
Mechanochemistry is the most commonly applied approach for the preparation of borohydrides. High-energy ball milling can incite a chemical metathesis reaction between an alkali borohydride (Li, Na, K) and the respective metal chloride in the solid state. LiBH4 has so far proven to be the most efficient precursor for these reactions, while attempts with other metal borohydrides, e.g., NaBH4, generally offers poor yields at most [9,18,19,20,21,22,23,24,25,26,27,28,29,30,31].
The high energy impaction between the milling balls and the powder causes a continuous fracturing and welding process that can induce grain refinement, produce fresh and reactive surfaces and provide nucleation centers for the chemical growth of reaction products. The major advantage of the ball milling method lies in the possibility to obtain metastable compounds at ambient conditions as the process proceeds far away from thermal equilibrium. In addition, ball milling provides access to more complicated bi-/trimetallic borohydrides in contrast to the solvent-based approach, which typically only yields the monometallic compounds. However, special care has to be taken in order to avoid decomposition of the product during ball milling or the formation of halide-substituted compounds. Such unwanted reactions can be suppressed by adding sufficient pauses to the milling protocol and by avoiding prolonged milling times. The metathesis reaction involving stoichiometric ratios of a trivalent RE metal chloride and lithium borohydride results in the formation of a mono- or bimetallic RE-borohydride, or a chloride substituted RE-borohydride, LiRE(BH4)3Cl (Table 1), according to Equations (2)–(4):
RECl3 + 3LiBH4 → RE(BH4)3 + 3LiCl
RECl3 + 4LiBH4 → LiRE(BH4)4 + 3LiCl
RECl3 + 3LiBH4 → LiRE(BH4)3Cl + 2LiCl
Unfortunately, the mechanochemical metathesis approach suffers from three major drawbacks. Firstly, the reaction products after ball milling are often not very crystalline and therefore require additional careful heat treatment in order to heal out structure defects and improve their crystallinity. Secondly, the product mixture always consists of the borohydride and a metal halide, the latter simply being “dead weight”, thereby effectively lowering the gravimetric hydrogen capacity. Finally, the metal halide may influence the chemical and physical properties of the borohydride and may affect its decomposition temperature or modify the decomposition pathway.
In order to avoid the drawbacks of the mechanochemical metathesis approach and to allow for halide-free products to be obtained, wet-chemical methods have been developed. They are based on the selective dissolution of the RE-borohydride in an organic solvent such as diethylether, toluene or dimethyl sulfide, followed by removal of the unwanted by-products (e.g., LiCl) through filtering, and careful drying of the solvated product in vacuo. The interested reader is referred to reference [1] for a more detailed description of the various synthesis procedures for metal borohydrides and their derivatives.
3. Crystal Structures
3.1. Monometallic RE-Borohydrides
All of the known monometallic RE-borohydrides are summarized in Table 2 and their crystal structures are discussed below.
3.1.1. Distorted ReO3-Type (α-RE(BH4)3)
The distorted ReO3-type structure (space group Pa) was suggested for Y(BH4)3, Gd(BH4)3 and Dy(BH4)3 from laboratory powder X-ray diffraction (PXD) data by Sato et al. [18]. The RE3+ ions form a pseudo-cube with BH4− approximately at the center of the pseudo-cube edges (Figure 1). Y(BH4)3 was further investigated as a hydride and deuteride with high-resolution synchrotron radiation powder X-ray diffraction (SR-PXD) and powder neutron diffraction (PND), respectively. However, the deuterated PND sample contained highly neutron-absorbing natural boron and they were thus unable to refine the atomic positions of boron or deuterium. Frommen et al. [21] performed combined SR-PXD and PND investigation on a double-isotope substituted sample, Y(11BD4)3. Structure refinements revealed a significant distortion compared to the undistorted ReO3-type structure (see Section 3.1.2). The B‒Y‒B angle is for instance 160.9° compared to 180° for an undistorted ReO3-structure and the D‒B‒D angles take values between 103 and 115° (ideal value 109.5°).
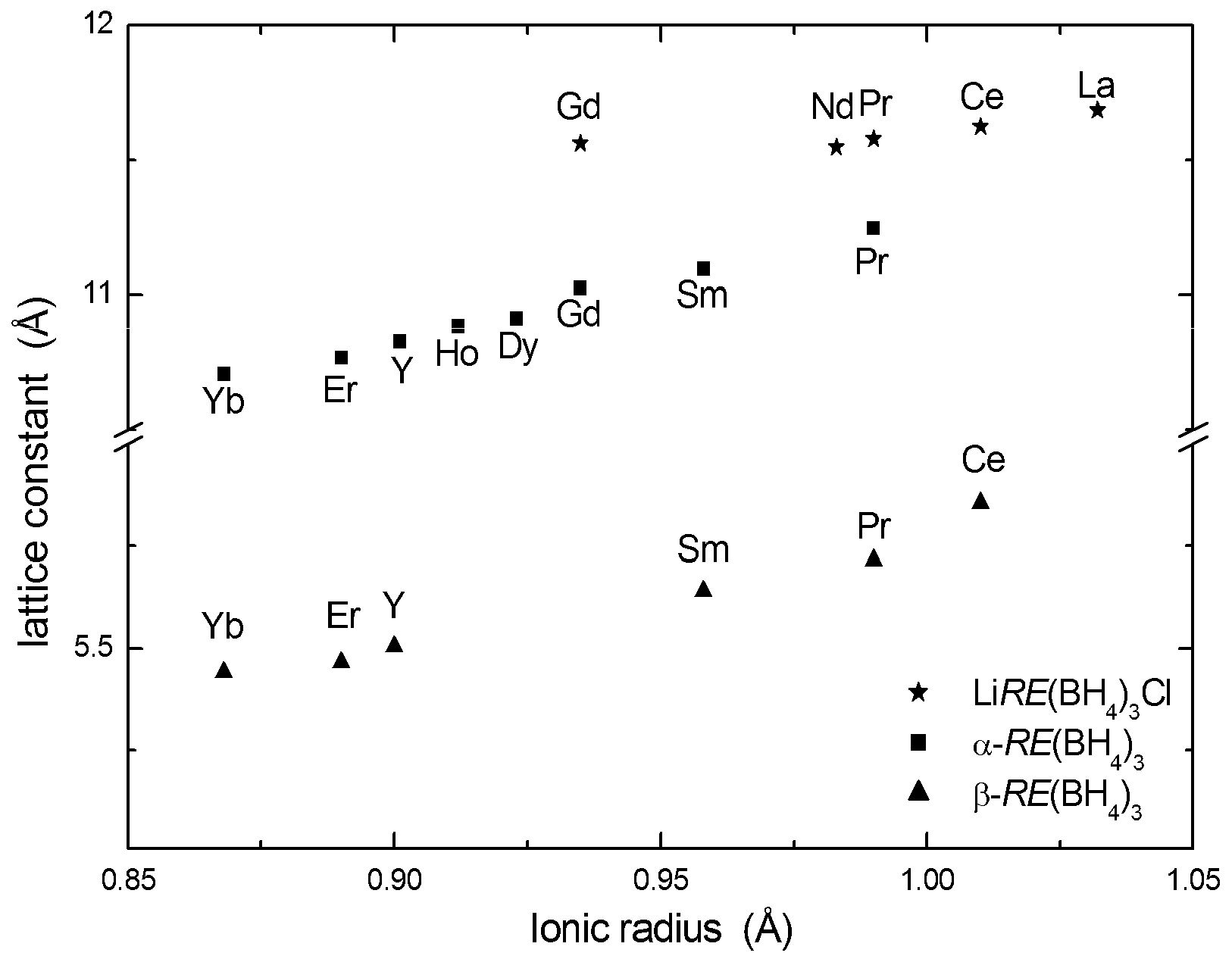
The same structure type has been reported from SR-PXD data for Sm(BH4)3, Gd(BH4)3, Tb(BH4)3, Dy(BH4)3, Er(BH4)3, Ho(BH4)3, Tm(BH4)3, Yb(BH4)3 [19,29,30,32,33]. Due to the challenges with extreme neutron absorption in Sm, Dy and Gd, in addition to the general problem with absorption in natural boron and incoherent scattering from H, these phases have not been investigated by PND and thus accurate positions for B and H have not been obtained. The unit cell parameters obtained from PXD show a near linear dependence on the radius of the RE3+ cation (Figure 2).
3.1.2. ReO3-Type (β-RE(BH4)3)
Differential scanning calorimetry (DSC) measurements of Y(BH4)3 indicated a structural phase transition between 199 and 488 °C [21]. The high-temperature phase was found to be metastable at ambient temperature in samples that were quenched after annealing above the phase transition temperature [21,22]. PXD data of the quenched material indicated a primitive cubic unit cell with a = 5.4547 Å and Rietveld refinements of the data showed excellent agreement with a regular ReO3-type structure (space group Pmm). Y is positioned in the unit cell corners, octahedrally coordinated by BH4− at the center of each unit cell edge (Figure 3). In the reported space group symmetry, the BH4− units are disordered with 8 half-occupied D sites around each boron [22]. Further investigation of a Y(11BD4)2 sample by PND measurement indicated a face-centered cubic unit cell with doubled unit cell axes (space group Fmc, a = 11.0086 Å). Rietveld refinement with this data yielded a model with identical Y and B distribution compared to the structure refined from PXD data alone, although the BH4− ions are now deemed to have an ordered orientation and are rotated by 90° relative to their closest neighbors in both the a, b and c-directions [21] (Figure 4). The strongest superstructure peak from the BH4− ordering in the PXD data has about 0.3% of the intensity of the strongest peak and is therefore virtually impossible to detect (Figure 5). In contrast, the superstructure peak with Miller index 531 is the strongest Bragg peak in the PND data (Figure 6).
SR-PXD measurements have revealed that β-RE(BH4)3 modifications also exist for Ce(BH4)3, Pr(BH4)3, Sm(BH4)3, Ho(BH4)3, Er(BH4)3 and Yb(BH4)3 [19,29,30,32,34]. Due to the chemical similarity of these RE ions and Y3+, it seems likely that these phases take structures with ordered BH4− orientations. However, due to lack of experimental verification, most of these structures were reported in the simplest model consistent with the PXD data, i.e., primitive cubic ReO3-type unit cells (space group Pmm) with disordered BH4− units (Table 2). The correlation between the unit cell parameters and the cation radii are shown in Figure 2.
3.1.3. Alkaline Earth Borohydride-Types
Sm, Eu, Tm and Yb can exist in the 2+ oxidation state and divalent borohydrides have been reported for all of them except Tm. Sm2+ and Eu2+ have ionic radii of 1.2 Å and 1.17 Å, respectively, which are similar to that of Sr2+ (1.18 Å). Sm(BH4)2 [19,27] and Eu(BH4)2 [27,35] have been reported to crystallize in the same structure type as the room temperature (RT) modification of Sr(BH4)2 [35] (space group Pbcn, α-PbO2 type structure). The RE2+ is octahedrally coordinated by 6BH4−, with the RE(BH4)6 octahedra sharing all six vertices and two edges with other RE(BH4)6 octahedra, creating zigzag chains along the c-axis. Each chain is again linked by vertices to four other chains. The borohydride tetrahedron is surrounded by three M2+ ions and forms nearly perfect trigonal planar coordination.
Eu(BH4)2 forms two additional polymorphs at higher temperatures; one tetragonal (space group P41212, superstructure of high temperature (HT)-ZrO2-type) and one cubic (space group Fmc, CaF2-related). Both are isostructural to HT polymorphs of Sr(BH4)2 [35]. Both HT polymorphs have 8-coordinated Eu2+. The tetragonal Eu(BH4)2 polymorph can be regarded as a distorted version of the CaF2-type polymorph. Thus, both consist of distorted (tetragonal polymorph) or regular (cubic polymorph) Eu(BH4)8 cubes that share all edges with other Eu(BH4)8 cubes.
Yb2+ has a similar ionic radius to Ca2+ (1.16 Å and 1.18 Å, respectively) and the three reported Yb(BH4)2 modifications are isostructural to known Ca(BH4)2 phases. An orthorhombic α-Yb(BH4)2 (space group F2dd), which is isostructural to α-Ca(BH4)2 is reported to be the stable polymorph at ambient temperature [1]. The two high-temperature polymorphs are isostructural to β-Ca(BH4)2 (space group P) and γ-Ca(BH4)2 (space group Pbca) [29]. Both of these HT phases have been shown to have partial substitution of BH4− by Cl‒ (37% and 15% substitution, respectively). However, it is not clear if Cl− substitution is a prerequisite for their stabilities. All of the Yb(BH4)2 polymorphs (with and without chlorine substitution) have Yb2+ octahedrally coordinated by six BH4− units.
3.2. Halide-Containing RE-Borohydrides
The large RE elements La, Ce, Pr, Nd, Sm and Gd form cubic LiRE(BH4)3Cl compounds (space group I3m), usually by direct reaction between LiBH4 and RECl3. The first reported member of this series, LiCe(BH4)3Cl, was structurally characterized by combined SR-PXD and PND on a 11B and D substituted sample [36]. Ce3+ is octahedrally coordinated by three Cl− and three BH4− anions. The Ce3+ and Cl− form distorted, isolated Ce4Cl4 cubes, and with the three BH4− anions coordinating each Ce3+, these units form [Ce4(BH4)12Cl4]4− complex anions that are charge compensated by four Li+ cations (Figure 7). The Li+ was reported to fully occupy two crystallographic sites; one two-fold position (2a) in the center of the Ce4Cl4 cube and one six-fold position (6b) which is tetrahedrally coordinated four BH4− ions from two different [Ce4(BH4)12Cl4]4− units. Occupation of these two sites is the only way to place the required eight charge compensating Li+ in the unit cell in an ordered way. Disordered models were not considered during structure determination. Neither PXD nor PND are particularly sensitive to Li, but the proposed structure was found to be stable from density functional theory (DFT) calculations [36]. An alternative study of LiCe(11BD4)3Cl also employed SR-PXD, PND and DFT, but considered a variety of disordered distributions of Li+ within the structure. It was found that a 2/3 occupation of a 12-fold site (12d) both gave lower energy (DFT) and slightly better agreement with the diffraction data than the previously reported ordered Li-distribution. The fact that LiCe(BH4)3Cl exhibits a high Li-ion conductivity (0.1 mS/cm at RT), further supports the model with disordered Li+.
LiRE(BH4)3Cl with RE = La, Pr, Nd, Sm and Gd as well as LiLa(BH4)3X with X = Br and I have been investigated by lab- or SR-PXD confirming the same structure type as LiCe(BH4)3Cl for each of the heavy elements [9,19,37]. The Li-ion conductivities have been measured for RE = La, Gd and both compounds show higher conductivities (0.2 and 0.4 mS/cm, respectively) than LiCe(BH4)3Cl [9]. This indicates that the Li-ions also partially occupy the 12d site in these compounds.
Non-stoichiometric chloride-containing borohydrides, NaY(BH4)2−xCl2+x, LiYb(BH4)4−xClx and Yb(BH4)2−xClx, where chloride anions partly substitute BH4- have also been reported [29,31,38].
The halide containing compounds are summarized in Table 3.
3.3. Bi- and Trimetallic RE-Borohydrides
LiSc(BH4)4 (space group P2c) was the first reported bimetallic borohydride [39]. It consists of distorted tetrahedral [Sc(BH4)4]− complex anions with charge compensating Li+ (Figure 8). The Li+ atoms have a disorder distributed over half-occupied sites (4k) along the tetragonal c-axis. The same structure type has also been found for LiRE(BH4)4 where RE = Y [10,40], Yb [29] (with 25% of BH4− substituted by Cl−) and Lu [19]. The only difference from the LiSc(BH4)4 structure is that Li+ were reported in a fully occupied 2-fold site (2a), tetrahedrally coordinated by 4BH4− for RE = Y and Yb. This site is situated in the middle of the partly occupied sites in LiSc(BH4)4. No structure refinement has been performed for LiLu(BH4)4. Li-ion conductivity has not been measured for LiSc(BH4)3, but LiY(BH4)4 shows low conductivity (10−6 S/cm at RT).
NaSc(BH4)4 exists in an orthorhombic crystal structure (space group Cmcm) which contains tetrahedral [Sc(BH4)4]− complex anions that are less distorted than in LiSc(BH4)4 [41]. Na+ has a higher coordination number than Li+ due to its larger radius, and is octahedrally coordinated by six BH4− moieties. The Na(BH4)6 octahedra form edge-sharing chains along the c-axis, with the chains also being interconnected by egde-sharing Sc(BH4)4 tetrahedra. NaY(BH4)4 [10,40], NaEr(BH4)4 [42], KHo(BH4)4 [32], o-KY(BH4)4 [43], KEr(BH4)4 [44], and KYb(BH4)4 [45] are isostructural to NaSc(BH4)4. KSc(BH4)4 (space group Pnma) contains similar tetrahedral [Sc(BH4)4]− complexes while the coordination number of alkali metal increases further to eight [46].
The BH4− anion is isoelectronic with O2− and as such many borohydrides are structurally similar to oxides [33]. The various Ca(BH4)2 polymorphs are, for instance, similar to TiO2 polymorphs (Ti4+ being isoelectronic to Ca2+) [47]. A series of 20 RE-borohydrides with perovskite-like structures have been reported [38,48,49,50,51,52,53]. Seven of the compounds exhibit face-centered cubic double-perovskite-type structures (space group Fm). Despite the high symmetry, the compounds form ordered structures with large unit cell volumes ranging from 1424 Å3 (Rb2LiY(BH4)6) to 1710 Å3 (Cs3Gd(BH4)6). LT-KYb(BH4)3 (space group P3m) exhibits a primitive cubic structure with a much smaller unit cell volume of 176 Å3 and β-CsSm(BH4)3 takes the ideal perovskite structure (space group Pmm). The rest of the compounds are tetragonal, orthorhombic or monoclinic (Table 4). Using the same analogy to oxide structures, Li3K3La2(BH4)12 and Li3K3Ce2(BH4)12 (space group Iad) are reported to have a garnet-type structure [48].
All other reported bi- and tri-metallic borohydrides with structure types not discussed above, are included in Table 4.
4. Thermal Properties of Rare Earth Borohydrides
4.1. Monometallic and Halide Substituted Rare Earth Borohydrides
The metathesis reaction between stoichiometric quantities of lithium borohydride LiBH4 and a trivalent rare earth chloride RECl3 results in principle in the formation of either a monometallic trivalent RE-borohydride, RE(BH4)3, LiRE(BH4)4 or a chloride substituted compound, LiRE(BH4)3Cl, according to Equations (2)–(4) (see above). During the past seven years extensive efforts have been made to understand the thermal decomposition and rehydrogenation behavior of these compounds. The following sections summarize the thermal behavior of two compounds: Y(BH4)3 and LiCe(BH4)3Cl, which are representative of the monometallic and halide substituted rare earth borohydrides adopting the same structure type.
4.1.1. Yttrium borohydride
Y(BH4)3 has a high gravimetric hydrogen capacity of 9.1 wt %, and has therefore received considerable interest since 2008 as a potential hydrogen storage material [18]. The stability of crystalline Y(BH4)3 has been assessed by density functional theory (DFT), and calculations predict the decomposition into boron, yttrium hydride and hydrogen at elevated temperatures. Dai et al. [54] have recently employed first principles calculations to predict the influence of additional alloying elements such as Li, Na, K, Ti, Mn and Ni on the stability and dehydrogenation properties of Y(BH4)3. The authors concluded that all alloyed systems show smaller enthalpy of decomposition than pure Y(BH4)3 and that the alkali metals (especially K) may be suitable for improving the dehydrogenation properties of Y(BH4)3.
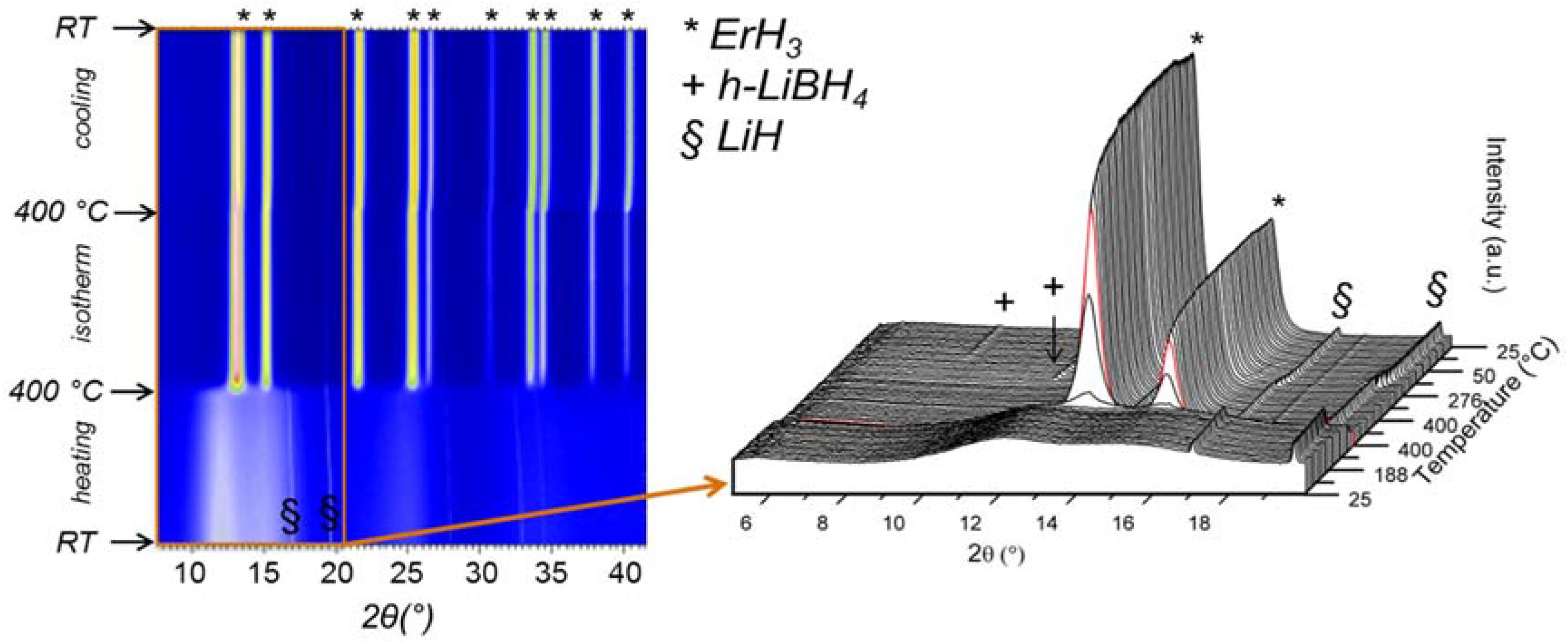
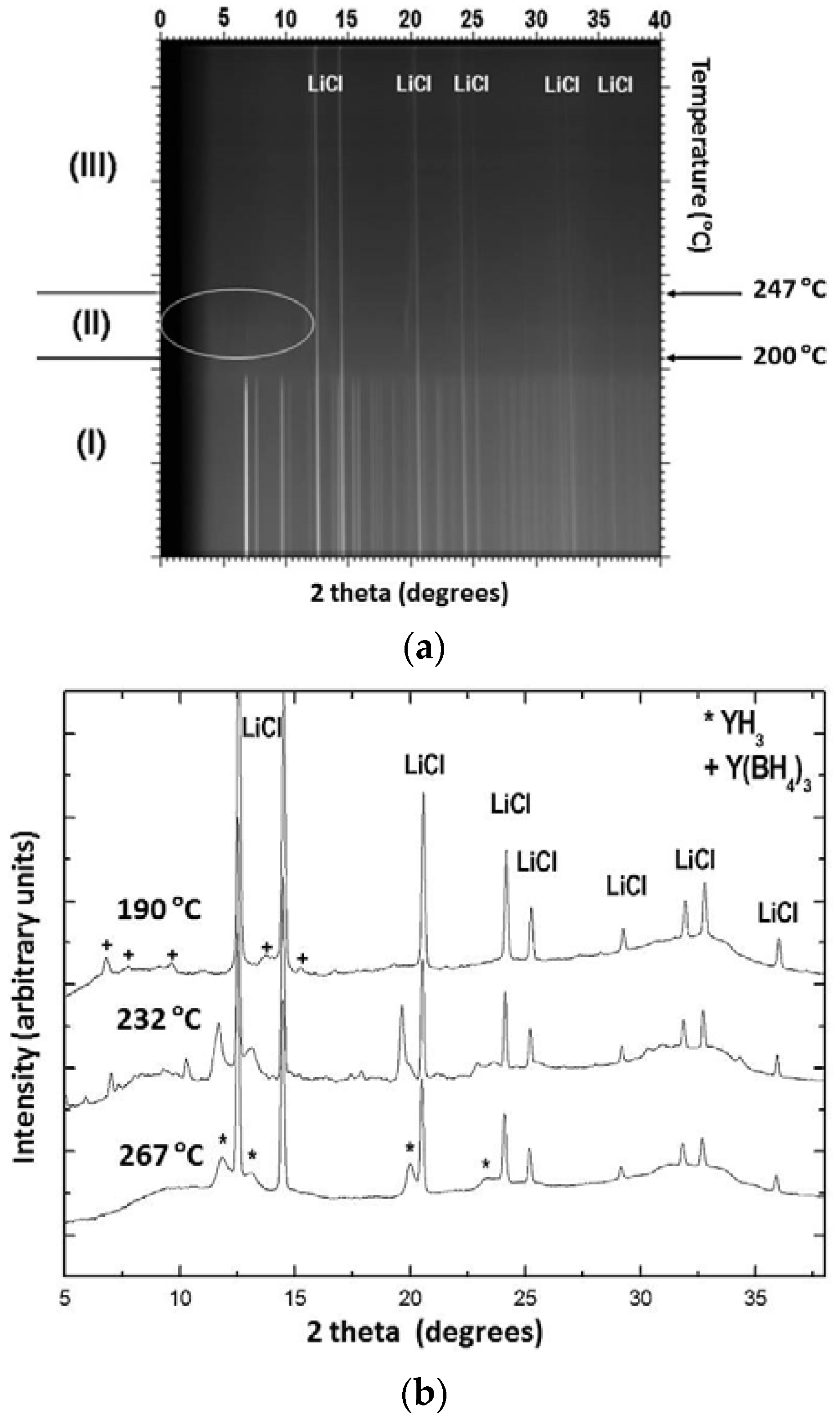
Experimental studies regarding the thermal properties of Y(BH4)3 prepared by liquid phase and mechanochemical synthesis have been described in references [21,22,23]. According to these reports, α-Y(BH4)3, which is the stable modification at RT, undergoes a structural transition from the distorted ReO3-type structure (cubic, Pa) into the ordered ReO3-type structure (cubic, Fmc) upon heating to 157–182 °C. In situ SR-PXD measurements on a LiBH4-Y(BH4)3 (3:1) sample heated under dynamic vacuum with a rate of 2 °C/min up to 450 °C shows the presence of three distinct temperature regions (Figure 9a,b) [21].
LiCl and α-Y(BH4)3 are the only crystalline phases present in region (I) which spans from RT to 200 °C. The peaks originating from Y(BH4)3 begin to decrease in intensity at 182 °C and completely disappear at 200 °C. The formation of β-Y(BH4)3 was not observed under the current experimental conditions, which suggests that the heating rate of 2 °C/min is probably too fast compared to the kinetics of the phase transformation. In region (III), above 262 °C, the pattern shows that LiCl and YH3 were the only crystalline materials present. In region (II), between 200 and 247 °C, the major crystalline phases are LiCl and YH3. Additionally, weaker peaks of a new phase emerge at around 200 °C gain in intensity up to 232 °C and diminish again until they finally disappear above 247 °C. Peak indexing and unit cell determination for this intermediate phase have indicated that it might be described in a primitive orthorhombic unit cell with lattice parameters a = 12.170(14) Å, b = 7.670(5) Å, and c = 7.478(6) Å. Similarly, Ravnsbæk et al. [22] have observed the formation of two unidentified compounds after the decomposition of Y(BH4)3 in the temperature range ~190 to 235 °C. The authors suggested that one of the phases has a composition similar to Y(BH4)Cl2 or Y(BH4)2Cl, while the other phase could be a decomposition product containing Y‒B‒Cl. Jaroń and Grochala [20] attempted to follow the thermal decomposition of Y(BH4)3 by heating samples in alumina crucibles inside a TG-DSC (Thermogravimetric-differential calorimeter) up to selected temperatures of 194, 210 and 229 °C followed by rapid cooling. The PXD patterns above 229 °C only show the presence of LiCl, with other decomposition products being amorphous. The partially decomposed samples possess a layered morphology and progressive color changes from white to light yellow, brown and finally black at high temperatures. This color change is attributed to the presence of amorphous decomposition products such as polymeric (BxHy)n compounds as well as H-poor Y‒B‒H phases. The same authors also studied the hydrogen desorption kinetics of α-Y(BH4)3 as well as that of the HT modification of β-Y(BH4)3. They observed significant differences below 260 °C for both phases, the decomposition being faster and more facile for the HT phase which they attributed to a three times lower activation energy for thermal decomposition as compared to the low-temperature polymorph [24]. Remhof et al. studied the thermal decomposition of pure Y(BH4)3 prepared by direct, solvent-free synthesis from YH3/YH2, B2H6 and H2 via reactive milling [55]. They observed the onset for hydrogen desorption at 187 °C and a maximum desorption rate of 0.12 wt % H2/min at 250 °C when the materials were heated with 1 °C/min under 1 bar H2. Time-resolved in situ PXD data with a low heating rate of 0.2 °C/min collected between RT and 450 °C under 1 bar of hydrogen showed only reflections from Y(BH4)3 and unreacted YH3 up to 207 °C, while at 247 °C the Bragg peaks originating from Y(BH4)3 disappeared and only YH3 remained visible. No structural transition from the α- to the β-phase was observed. Above 347 °C the decomposition was completed and no further change in the peak intensities was observed until the transformation of YH3 into YH2 proceeded at 447 °C. The authors also reported the appearance of at least one amorphous phase in addition to YH3. However, they did not provide any insight as to the nature of these additional phases. Finally, Park et al. have undertaken a comparative study between Y(BH4)3 synthesized from LiBH4-YCl3 (3:1 and 4:1 ratios) by mechanochemistry and via direct synthesis from YH3, B2H6 and H2 [56]. The authors arrived at the conclusion that both methods result in products that possess distinctly different polymorphic phase transformation, melting behavior and even altered decomposition pathways. In fact, it turned out that the side product LiCl in the metathesis reaction cannot be regarded as inert as it shifts the melting temperature of Y(BH4)3 and apparently promotes the formation of YB4 during decomposition which is otherwise absent in the case of Y(BH4)3 obtained from gas-solid reaction between YH3, B2H6 and H2.
4.1.2. Lithium Cerium Borohydride Chloride
LiCe(BH4)3Cl can be obtained via mechanochemical synthesis between three molar equivalents of LiBH4 and one molar equivalent of CeCl3 [8,36]. The thermal decomposition of LiCe(BH4)3Cl has been studied in reference [36] by a combination of TG-DSC and residual gas analysis (RGA) with a low heating rate of 2 °C/min under argon flow. According to this report, decomposition proceeds in a two-step process with minor gas release at about 215 °C followed by the major decomposition step at a peak temperature of 246 °C. RGA shows a more than 100 times higher partial pressure for hydrogen than for B2H6 and its fragments B2H5, B2H4 and BH3. This indicates that the decomposition proceeds almost exclusively by release of hydrogen and that boron is thus kept in the solid state, which is one of the prerequisites for reversible hydrogen storage in borohydrides. Ley et al. [8] have performed Sieverts measurements on a sample containing only LiCe(BH4)3Cl and LiCl which show a gas release of 3.75 wt % H2 during the first desorption and 1.25 wt % H2 during the second and third desorption. Samples containing one equivalent excess of LiBH4 show a different thermal behavior and exhibit a gas release of 0.5 wt % H2 up to 220 °C, while 3.25 wt % H2 are released between 220 and 270 °C. An additional amount of 0.8 wt % H2 is released between 270 and 500 °C, while during a subsequent isotherm (3 h) about 0.15 wt % H2 are released. Thus a total amount of 4.7 wt % H2 are desorbed during heating to 500 °C, while after rehydrogenation at 400 °C and 100 bar for 24 h, about 1.8 wt % of H2 are desorbed during the second dehydrogenation [8].
In situ SR-PXD experiments have been conducted on the hydride and deuteride form of LiCe(BH4)3Cl in order to determine the decomposition products and formation of possible intermediates [8,36]. Figure 10a shows the evolution of powder patterns between 80 and 600 °C with a heating rate of 2 °C/min for a (3LiBD4-CeCl3) mixture obtained after ball milling. Figure 10b displays powder patterns at selected temperatures in more detail.
The only crystalline phases present after ball milling are LiCe(BD4)3Cl and LiCl. In the low-temperature region between 80 and 150 °C, the crystallinity of the material increases upon heating. As a result the X-ray profiles become narrower and the overall peak intensity increases. Peak intensities for LiCe(BD4)3Cl decrease rapidly above 230 °C up to its decomposition peak temperature of 240 °C. Between 270 and 450 °C the SR-PXD pattern exhibits peaks belonging to LiCl only, and a modulated background with a broad and featureless region around 2θ = 10–15°, which is characteristic of some amorphous boron-containing compounds such as Ce‒B‒D or B‒D species. Above 450 °C, new peaks emerge and gain intensity up to the maximum temperature of 600 °C. They can be assigned to CeD2, and have been highlighted in Figure 10b. The formation of cerium borides such as CeB4 or CeB6 could not be observed under the applied experimental conditions. Ley et al. performed in situ SR-PXD studies with a high heating rate of 8 °C/min up to 500 °C on several nLiBH4-CeCl3 mixtures (n = 3, 4) [8]. The authors observe the decomposition of LiCe(BH4)3Cl at 240 °C similar to [36], and the appearance of an unknown phase above 240 °C—possibly a ternary salt composed of lithium cerium and chloride. Major decomposition products at 500 °C are CeH2 and CeB6. Crystalline CeB6 has also been obtained by Liu et al. [26] while heating up a 3LiBH4-CeCl3 mixture to 600 °C for an extended period of time. None of the studies found any indication that favored the formation of boron over CeB6. Based on those findings the thermal decomposition of LiCe(BH4)3Cl is therefore believed to proceed via Equation (5) rather than via Equation (6):
LiCe(BH4)3Cl → ½CeH2 + ½CeB6 + 11/2H2 + LiCl
LiCe(BH4)3Cl → CeH2 + 3B + 5H2 + LiCl
The first reaction pathway is furthermore supported by TG-DSC experiments which show a weight loss of 3.5 wt % H2 for a 3LiBH4-CeCl3 hydride mixture after ball milling. This value is in excellent agreement with the expected weight loss of 3.6 wt % H2, assuming the formation of CeH2 and CeB6 in Equation (5).
Trends in the thermal decomposition of the monometallic and halide substituted RE-borohydrides are difficult to establish despite an increasing number of publications in the field. This complication is mainly due to the use of different preparation methods e.g., mechanochemistry vs. wet-chemistry. Ball milling of LiBH4-RECl3 mixtures always yields LiCl in addition to the RE-borohydride which cannot be simply regarded as “dead weight”. In fact, its presence can affect the melting point, phase transformation temperatures, induce halide substitution and even alter the decomposition pathway. Wet-chemical methods have therefore been developed which can separate the alkali metal chloride from the metal borohydride, e.g., through dissolution and/or filtering. However, they lead to the formation of solvated RE-borohydrides which have to be carefully dried at elevated temperatures and in vacuo. Here arises another risk in which the solvent cannot be fully removed or that the metal borohydride partially decomposes during the drying process. A second factor is based on the use of different characterization methods such as calorimetry and in situ powder diffraction in which vastly different experimental conditions are employed. In the case of calorimetric studies the purge gas (Ar/He/H2), heating rate, sample mass and choice of sample container (Al/Al2O3) have a profound effect on the number, intensity, shape and position of the registered heat flow signals. In the case of in situ diffraction the use of H2-backpresure vs. vacuum is known to alter the decomposition pathway. Furthermore, reactions between the borohydrides and the sample holder (mainly sapphire) at high temperature in connection with micro-leaks have led to the formation of oxide species on several occasions [57].
A correlation between the decomposition temperature of the metal borohydrides and their Pauling electronegativity was already suggested in 1955 [58,59] and has been thoroughly investigated by several research groups both experimentally and theoretically [12]. The more stable alkali metal borohydrides tend to show a linear correlation between the decomposition temperature and the electronegativity of the metal cation.
However, compounds formed by metals with higher electronegativity tend to deviate from this linear behavior. An exponential correlation between the decomposition temperature and the electronegativity of the metal has therefore been suggested instead. Table 5 and Table 6 summarize the thermal properties of selected monometallic rare earth borohydrides RE(BH4)n (n = 2, 3) and lithium rare earth borohydride chlorides LiRE(BH4)3Cl.
For the monometallic trivalent compounds the decomposition temperature decreases with increasing electronegativity of the metal. It reaches its highest value of 258 °C for La(BH4)3, 275 °C for Pr(BH4)3, and 260 °C for Gd(BH4)3 [30,34,60], and drops to 230 °C in the case of Er(BH4)3 [30]. Ultimately the decomposition reaction results in the formation of the corresponding rare earth hydrides and borides which is favored over the formation of elemental boron for technological applications. Crystalline decomposition products are not always formed, however, even when heating the materials to 400–500 °C, as is the case for Pr(BH4)3 and Ho(BH4)3 [30,32]. The thermal decomposition of the LiRE(BH4)3Cl-type compounds appears to be more complex and proceeds via a multi-step process involving at least two different steps over a temperature range of up to 50 °C. The highest decomposition temperature of 266 °C is observed for LiLa(BH4)3Cl while LiNd(BH4)3Cl possesses the lowest one with 241 °C. The variation in decomposition temperature is far less pronounced in the rare earth borohydrides as compared to alkali or alkaline earth borohydrides though. This is not unexpected since the range of electronegativity observed for the lanthanides is rather small and increases only moderately from 1.10 for Lanthanum to 1.23 for Erbium. In case of the LiRE(BH4)3Cl-type compounds the less electronegative alkali metal (Li) shows dominantly ionic interactions and the overall stability of compound is controlled by the more electronegative cation (RE) which is in fact part of a complex anion [RE4Cl4(BH4)12]4−. The difference of 25 °C in decomposition temperatures between LiLa(BH4)3Cl and LiNd(BH4)3Cl indicates that the decomposition temperatures are specific for these complex anions and directly reflects differences in their stability [1].
4.2. Composites Containing RE-Borohydrides and LiBH4
LiBH4 has the highest theoretical hydrogen storage capacity (18.5 wt % H2) of all borohydrides of which 13.9 wt % H2 are accessible through Equation (7):
LiBH4 → LiH + B + 3/2H2
However, harsh pressure and temperature conditions are necessary to reversibly release (>400 °C) and reabsorb (150 bar H2; 650 °C) hydrogen from LiBH4 due to its high thermal stability (ΔH = 74 kJ/mol H2) [61]. The RE-borohydrides on the other hand decompose between 200 and 300 °C, which is significantly lower than the decomposition temperature of LiBH4. There has been considerable interest in utilizing RE-borohydrides as part of a composite mixture with other light weight metal borohydrides such as LiBH4. The general idea behind the use of composite mixtures over pure compounds is to reduce the enthalpy and consequently the hydrogen desorption temperature of the more stable LiBH4. Indeed, additives such as MgH2 [62,63], CaH2 [64,65], CeH2/LaH2 [66] and Al [67,68] have been used successfully to lower the stability of LiBH4 by the formation of MgB2, CaB6 and AlB2, respectively. For a composite containing LiBH4-MgH2 (2:1), a two-step decomposition is observed at high temperatures (>400 °C) [62,63,69] while for LiBH4-CaH2 (6:1) decomposition occurs in a single step reaction, which is advantageous for reversible hydrogen storage applications [64]. In addition to hydrides, halides such as TiCl3, TiF3 and ZnF2 have been recently employed to reduce the decomposition temperature of LiBH4 through cation exchange and formation of an unstable transition metal borohydrides [70]. LiBH4 has also been destabilized by the addition of FeCl2, CoCl2 and NiCl2, however, in the case of CoCl2, diborane is released during dehydrogenation which results in capacity loss [71]. Gennari et al. showed that composites of 6LiBH4 + RECl3 (RE = Ce, Gd) form RE-hydrides during thermal decomposition which in turn destabilize the remaining LiBH4 [28]. A composite mixture of LiBH4-CeCl3 (6:1) showed a reversible capacity of 40% after rehydrogenation at 60 bar and 400 °C. In addition, a change in the decomposition pathway and increased formation of CeB6 and GdB4 has been reported for composites desorbed against 5 bar of hydrogen pressure as compared to vacuum [28]. In order to promote the reversibility of CeB6 formed during the decomposition of Ce(BH4)3, the same authors added LiH to as-milled LiBH4-CeCl3 (6:1) composites and then further milled for 1 h. PXD data showed the presence of LiCl and CeH2+x after the final milling step. Such a LiBH4-CeCl3-LiH (6:1:3) composite released 5.3 wt % H2 during the first dehydrogenation and 4.4 wt % H2 during the second cycle after rehydrogenation under relatively mild conditions (400 °C, 60 bar, 2 h) which corresponds to 80% reversible capacity.
Olsen et al. have intensively studied the structure, thermal behavior and hydrogen storage properties of a series of LiBH4-RECl3 (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb and Lu) composites obtained by ball milling LiBH4 and the respective RE-chlorides in a molar ratio of 6:1 [19]. The systematic study has shown that ball milling results in the formation of either LiRE(BH4)3Cl (RE = La, Ce, Pr, Nd, Sm, Gd), α,β-RE(BH4)3 (RE = Sm, Gd, Tb, Dy, Er Tm and Yb) or LiRE(BH4)4 (RE = Yb, Lu) together with excess LiBH4 [19]. TG-DSC combined with in/ex situ PXD show that the composites which contain LiRE(BH4)3Cl decompose at around 200 °C, and exhibit multiple decomposition steps with the formation of several intermediate phases. The remaining LiBH4 in these composites decomposes and releases hydrogen below 350 °C, which is much lower than the decomposition temperature of pure LiBH4 [61]. In contrast, the thermal decomposition of the composites which contain α,β-RE(BH4)3 and excess LiBH4, proceeds with only two major decomposition steps in the temperature range 250–350 °C. Common to all composites, is the formation of REH2 as the main decomposition product and REBn (n = 4, 6) species at temperatures in excess of 400 °C. It was concluded that the presence of REH2 has a destabilizing effect on the remaining LiBH4 in all the composites, as was already exemplary reported by Gennari et al. for RE = Ce and Gd [28]. Rehydrogenation of the La-system under 100 bar and 300 °C showed only very limited rehydrogenation capacity of 18%, whereas rehydrogenation of the Er-system (100 bar, 400 °C) showed a slightly higher capacity of 25% [19].
More recently, a selection of composites containing either La, Er or Pr have been synthesized to determine whether the composites obtained by mechanochemistry differ from those made by wet-chemical methods [30,72,73]; whether the composites which contain alkali chlorides as by-products show a different thermal behavior from those which are halide-free; and determine the role of additives such as LiH for the rehydrogenation behavior of the composites. Composite mixtures of RE-borohydrides of La, Er and LiBH4 and LiH were obtained by mechanochemical methods [72]. Ex situ PXD and TG-DSC measurements showed that heating to 400 °C yielded LaB6, ErB4 and REH2+δ (0 < δ < 1) as major decomposition products and that LiBH4 was destabilized by REH2+δ formed through decomposition of the parent borohydrides LiLa(BH4)3Cl and Er(BH4)3, respectively. Therefore, its decomposition temperature was reduced by as much as 100 °C compared to that of pure ball milled LiBH4. The La-containing composite showed only a limited reversible capacity of 20% (340 °C, 100 bar), probably caused by hydrogen uptake of some amorphous boron-containing phases; similar to the limited rehydrogenation capacity found in the 6LiBH4-LaCl3 composite after ball milling that did not contain additional LiH [19]. In contrast, the erbium-containing 6LiBH4-ErCl3-3LiH composite showed more promising hydrogen storage properties and could be rehydrogenated at 340 °C and 100 bar hydrogen to a large extent. Such a composite mixture showed a reversible capacity of 66% when desorbed in vacuo, which increased to 80% when desorbed against 5 bar of hydrogen backpressure.
The structure and thermal properties of halide-free RE-borohydrides (RE = Pr, Er) have also been obtained by solvent-based synthesis instead of mechanochemistry [30,73]. Both compounds showed a simple decomposition pathway into the corresponding hydrides and borides according to Equations (8) and (9):
Er(BH4)3 → 0.75ErB4 + 0.25ErH2 + 5.75H2
Pr(BH4)3 → 0.5PrB6 + 0.5PrH2 + 5.5H2
Rehydrogenation of both materials was monitored by ex situ and in situ SR-PXD measurements at 340 °C and hydrogen pressures of up to 100 bar. However, no new crystalline phases were observed. In addition, TG-DSC experiments of the samples exposed to 100 bar of hydrogen for 21 h showed the absence of any mass loss and heat flow signals, indicating that rehydrogenation was unsuccessful. These findings are in disagreement with Gennari et al. who reported that Er(BH4)3 synthesized by ball milling had a reversible hydrogen capacity of 20% (rehydrogenation at 400 °C, 60 bar H2) [74]. This “partial reversibility” for several RE-borohydride containing composites of the mechanomilled materials is most likely due to the rehydrogenation of LiBH4 and not due to the formation of the RE-borohydrides from the corresponding RE-hydrides or borides.
The decomposition and rehydrogenation behavior of composite mixtures containing the halide-free Er(BH4)3 ball milled for one hour in the presence of LiBH4 and/or LiH has been further investigated in reference [73]. Two composites were studied in detail: Er(BH4)3-LiH (1:6) and LiBH4-Er(BH4)3-LiH (3:1:3). Hydrogen sorption measurements in a Sieverts-type apparatus for the 3:1:3 composite showed a hydrogen release of 4.2, 3.7 and 3.5 wt % H2 during three desorption-absorption cycles (des: 400 °C, 5–10 bar H2; abs: 340 °C, 100 bar H2) and thus good rehydrogenation behavior with reversible hydrogenation capacity in the range of 80–85%. This halide-free composite showed similar behavior to a 6LiBH4-ErCl3-3LiH composite mixture [72] obtained by ball milling. In both systems, the major decomposition products were ErH2+δ (0 < δ < 1) and ErB4 while rehydrogenation leads to the formation of ErH3 and the rehydrogenation of LiBH4. Between the two studies, minor differences were observed in the number and variety of ErH3 polymorphs formed upon rehydrogenation. While cubic-ErH3 was the only polymorph reported in [72], a mixture of tetragonal-ErH3 and cubic-ErH3 polymorphs were reported after rehydrogenation (340 °C, 93 bar H2) in [73].
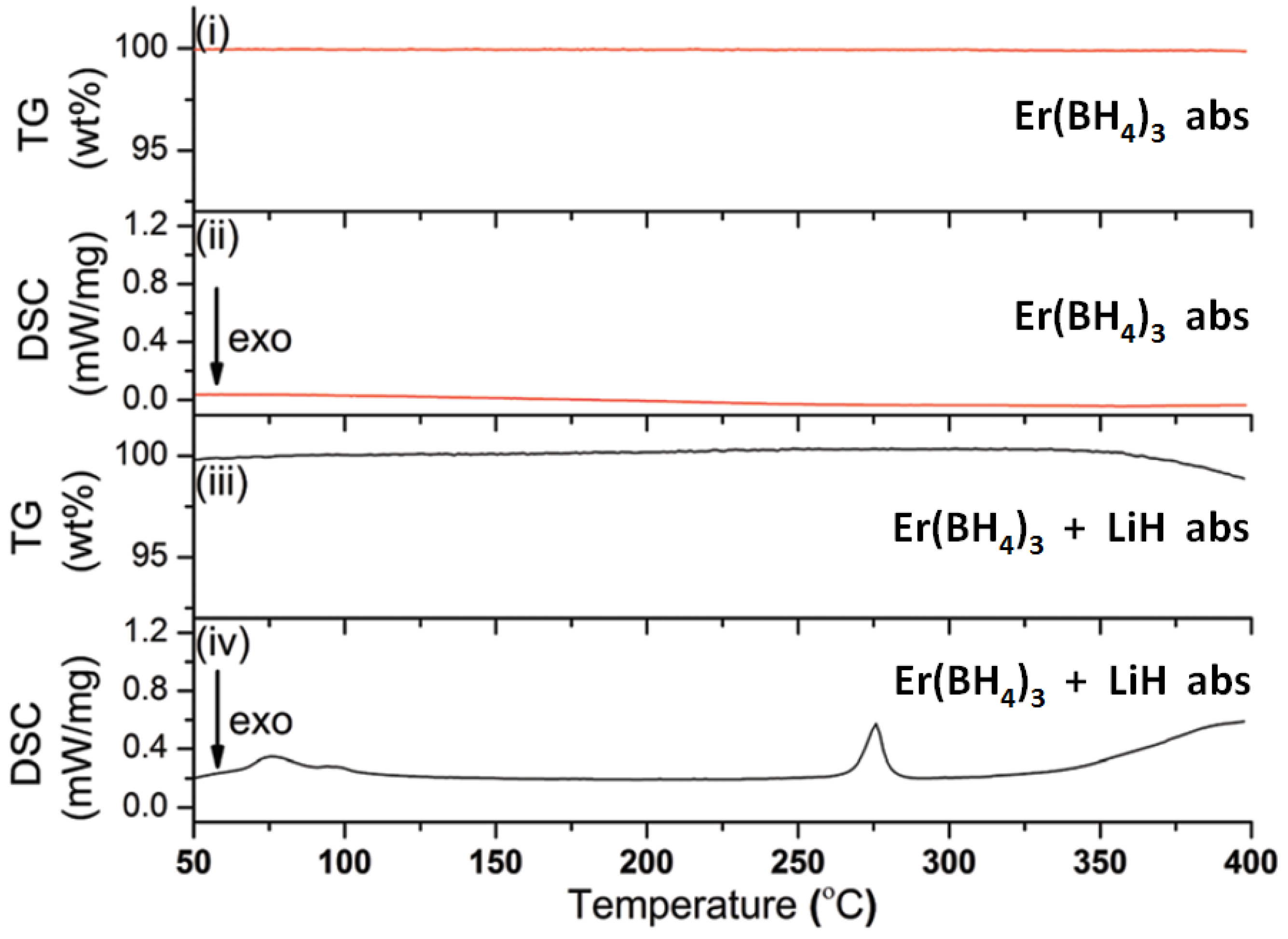
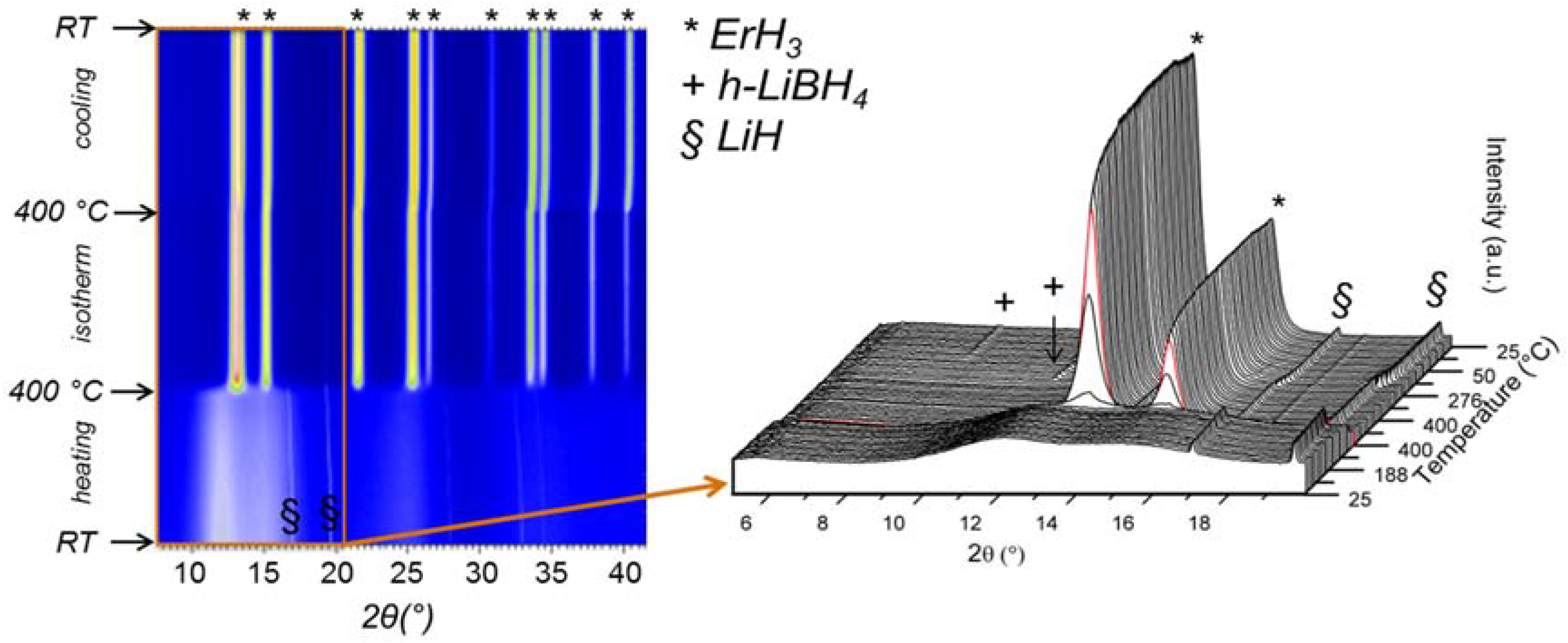
The influence of additives such as LiH on the rehydrogenation behavior of Er(BH4)3 was investigated in reference [30]. For this purpose, halide-free Er(BH4)3 was decomposed at 400 °C and rehydrogenated at 340 °C and 100 bar of hydrogen. PXD experiments on the desorbed and rehydrogenated samples did not show any crystalline phases while TG-DSC data measured for the rehydrogenated sample did not show any mass loss and no heat flow signal up to 400 °C (Figure 11). In two control experiments, either 100 wt % of LiCl or 50 wt % of LiH were added to the halide-free Er(BH4)3 samples, and decomposed and rehydrogenated as before.
The composite mixture containing additional LiCl did not show any crystalline phases after rehydrogenation. Neither heat flow signals nor weight loss could be detected after rehydrogenation which clearly demonstrated that the presence of LiCl had no beneficial effect on the rehydrogenation behavior. The composite mixture containing 50 wt % LiH was studied by in situ SR-PXD (Figure 12). While the ball milled sample mainly showed the presences of LiH and a modulation of the background indicative of some amorphous boron-containing phases such as ErB4 when heated from RT to 400 °C, the rehydrogenated material (100 bar H2) showed the evolution of peaks belonging to ErH3 at 400 °C and upon cooling to RT the appearance of peaks originating from h-LiBH4.
The unsuccessful rehydrogenation of the composites containing LiCl vs. LiH clearly demonstrates that the influence of LiH as an additive goes beyond simply being a source of Li-ions. The successful formation of LiBH4 for the LiH-containing composite is an analogous to that of similar reactive hydride composites with LiH, such as LiH + MgB2. In the latter case, however, much harsher conditions or the use of additional catalysts is necessary [62,75].
5. Conclusions
The past 10 years have led to the discovery of a multitude of new borohydrides containing RE elements which show rich chemistry, structural diversity and a wide range of compositions. Their application ranges from hydrogen and thermal heat storage to solid state electrolytes. Lately, exciting new fluorescent and magnetocaloric properties have been discovered in several RE-borohydrides such as CsEu(BH4)3 and K2Gd(BH4)5 [52,53]. In the future, it will hopefully be possible to design new advanced functional materials based on RE-borohydrides with desired compositions and new exciting properties that make their entrance into many more applications and research areas. A promising new field could be the use of RE-borohydrides as advanced fast neutron shields [76] for compact and portable neutron sources [77]. Such mobile units are in high demand for materials testing and neutron capture therapy, which is a promising approach to cancer therapy where conventional radiation therapies fail.
Acknowledgments
The research leading to these results has received funding from the People Program (Marie Curie Actions) of the European Union’s Seventh Framework Program FP7/2007–2013/ under REA grant agreement n_ 607040 (Marie Curie ITN ECOSTORE) and is thankfully acknowledged. The Research Council of Norway is acknowledged for financial support through the FRIENERGI project and the SYNKNØYT program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Cerny, R.; Ravnsbæk, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacherf, F.; Jensen, T.R. Metal borohydrides and derivatives—Synthesis, structure and properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef] [PubMed]
- Mohtadi, R.; Orimo, S.I. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2017, 2, 16091. [Google Scholar] [CrossRef]
- Callini, E.; Atakli, Z.O.K.; Hauback, B.C.; Orimo, S.; Jensen, C.; Dornheim, M.; Grant, D.; Cho, Y.W.; Chen, P.; Hjörvarsson, B.; et al. Complex and liquid hydrides for energy storage. Appl. Phys. A Mater. Sci. Process. 2016, 122, 353. [Google Scholar]
- Li, H.W.; Yan, Y.G.; Orimo, S.; Züttel, A.; Jensen, C.M. Recent Progress in Metal Borohydrides for Hydrogen Storage. Energies 2011, 4, 185–214. [Google Scholar] [CrossRef]
- Orimo, S.I.; Nakamori, Y.; Eliseo, J.R.; Züttel, A.; Jensen, C.M. Complex hydrides for hydrogen storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.A.; Paskevicius, M.; Humphries, T.D.; Felderhoff, M.; Capurso, G.; von Colbe, J.B.; Dornheim, M.; Klassen, T.; Ward, P.A.; Teprovich, J.A.; et al. Metal hydrides for concentrating solar thermal power energy storage. Appl. Phys. A Mater. Sci. Process. 2016, 122, 395. [Google Scholar] [CrossRef]
- Møller, K.; Sheppard, D.; Ravnsbæk, D.; Buckley, C.; Akiba, E.; Li, H.-W.; Jensen, T. Complex Metal Hydrides for Hydrogen, Thermal and Electrochemical Energy Storage. Energies 2017, 10, 1645. [Google Scholar] [CrossRef]
- Ley, M.B.; Ravnsbæk, D.B.; Filinchuk, Y.; Lee, Y.S.; Janot, R.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. LiCe(BH4)3Cl, a New Lithium-Ion Conductor and Hydrogen Storage Material with Isolated Tetranuclear Anionic Clusters. Chem. Mater. 2012, 24, 1654–1663. [Google Scholar] [CrossRef]
- Ley, M.B.; Boulineau, S.; Janot, R.; Filinchuk, Y.; Jensen, T.R. New Li Ion Conductors and Solid State Hydrogen Storage Materials: LiM(BH4)3Cl, M = La, Gd. J. Phys. Chem. C 2012, 116, 21267–21276. [Google Scholar] [CrossRef]
- Roedern, E.; Lee, Y.S.; Ley, M.B.; Park, K.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. Solid state synthesis, structural characterization and ionic conductivity of bimetallic alkali-metal yttrium borohydrides MY(BH4)4 (M = Li and Na). J. Mater. Chem. A 2016, 4, 8793–8802. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Ley, M.B.; Jensen, T.R.; Filinchuk, Y. Nuclear Magnetic Resonance Studies of BH4 Reorientations and Li Diffusion in LiLa(BH4)3Cl. J. Phys. Chem. C 2013, 117, 14965–14972. [Google Scholar] [CrossRef]
- Nakamori, Y.; Miwa, K.; Ninomiya, A.; Li, H.W.; Ohba, N.; Towata, S.I.; Züttel, A.; Orimo, S.I. Correlation between thermodynamical stabilities of metal borohydrides and cation electronegativites: First-principles calculations and experiments. Phys. Rev. B 2006, 74, 045126. [Google Scholar] [CrossRef]
- Wiberg, E.; Bauer, R. Zur Kenntnis eines Magnesium-Bor-Wasserstoffs Mg(BH4)2. Z. Fur Naturforschung Sect. B J. Chem. Sci. 1950, 5, 397–398. [Google Scholar] [CrossRef]
- Goerrig, D. Verfahren zur Herstellung von Boranaten. German Patent DBP 1,077,644, 27 December 1958. [Google Scholar]
- Chlopek, K.; Frommen, C.; Leon, A.; Zabara, O.; Fichtner, M. Synthesis and properties of magnesium tetrahydroborate, Mg(BH4)2. J. Mater. Chem. 2007, 17, 3496–3503. [Google Scholar] [CrossRef]
- Koester, R. Neue herstellungsmethoden für metallborhydride. Angew. Chem. 1957, 3, 94. [Google Scholar] [CrossRef]
- Zanella, P.; Crociani, L.; Masciocchi, N.; Giunchi, G. Facile high-yield synthesis of pure, crystalline Mg(BH4)2. Inorg. Chem. 2007, 46, 9039–9041. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Miwa, K.; Nakamori, Y.; Ohoyama, K.; Li, H.W.; Noritake, T.; Aoki, M.; Towata, S.I.; Orimo, S.I. Experimental and computational studies on solvent-free rare-earth metal borohydrides R(BH4)3 (R = Y, Dy, and Gd). Phys. Rev. B 2008, 77, 104114. [Google Scholar] [CrossRef]
- Olsen, J.E.; Frommen, C.; Jensen, T.R.; Riktor, M.D.; Sørby, M.H.; Hauback, B.C. Structure and thermal properties of composites with RE-borohydrides (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb or Lu) and LiBH4. RSC Adv. 2014, 4, 1570–1582. [Google Scholar] [CrossRef]
- Jaroń, T.; Grochala, W. Y(BH4)3-an old-new ternary hydrogen store aka learning from a multitude of failures. Dalton Trans. 2010, 39, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Frommen, C.; Aliouane, N.; Deledda, S.; Fonneløp, J.E.; Grove, H.; Lieutenant, K.; Llamas-Jansa, I.; Sartori, S.; Sørby, M.H.; Hauback, B.C. Crystal structure, polymorphism, and thermal properties of yttrium borohydride Y(BH4)3. J. Alloys Compd. 2010, 496, 710–716. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Filinchuk, Y.; Cerny, R.; Ley, M.B.; Haase, D.; Jakobsen, H.J.; Skibsted, J.; Jensen, T.R. Thermal Polymorphism and Decomposition of Y(BH4)3. Inorg. Chem. 2010, 49, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.G.; Li, H.W.; Sato, T.; Umeda, N.; Miwa, K.; Towata, S.; Orimo, S. Dehydriding and rehydriding properties of yttrium borohydride Y(BH4)3 prepared by liquid-phase synthesis. Int. J. Hydrogen Energy 2009, 34, 5732–5736. [Google Scholar] [CrossRef]
- Jaroń, T.; Kozminskia, W.; Grochala, W. Phase transition induced improvement in H2 desorption kinetics: The case of the high-temperature form of Y(BH4)3. Phys. Chem. Chem. Phys. 2011, 13, 8847–8851. [Google Scholar] [CrossRef] [PubMed]
- Gennari, F.C.; Esquivel, M.R. Synthesis and dehydriding process of crystalline Ce(BH4)3. J. Alloys Compd. 2009, 485, L47–L51. [Google Scholar] [CrossRef]
- Zhang, B.J.; Liu, B.H.; Li, Z.P. Destabilization of LiBH4 by (Ce, La)(Cl, F)3 for hydrogen storage. J. Alloys Compd. 2011, 509, 751–757. [Google Scholar] [CrossRef]
- Humphries, T.D.; Ley, M.B.; Frommen, C.; Munroe, K.T.; Jensen, T.R.; Hauback, B.C. Crystal structure and in situ decomposition of Eu(BH4)2 and Sm(BH4)2. J. Mater. Chem. A 2015, 3, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Gennari, F.C.; Albanesi, L.F.; Puszkiel, J.A.; Larochette, P.A. Reversible hydrogen storage from 6LiBH4-MCl3 (M = Ce, Gd) composites by in-situ formation of MH2. Int. J. Hydrogen Energy 2011, 36, 563–570. [Google Scholar] [CrossRef]
- Olsen, J.E.; Frommen, C.; Jensen, T.R.; Riktor, M.D.; Sørby, M.H.; Hauback, B.C. Crystal structures and properties of solvent-free LiYb(BH4)4−xClx, Yb(BH4)3 and Yb(BH4)2−xClx. RSC Adv. 2013, 3, 10764–10774. [Google Scholar] [CrossRef]
- Heere, M.; GharibDoust, S.H.P.; Frommen, C.; Humphries, T.D.; Ley, M.B.; Sørby, M.H.; Jensen, T.R.; Hauback, B.C. The influence of LiH on the rehydrogenation behavior of halide free rare earth (RE) borohydrides (RE = Pr, Er). Phys. Chem. Chem. Phys. 2016, 18, 24387–24395. [Google Scholar] [CrossRef] [PubMed]
- Ravnsbæk, D.B.; Ley, M.B.; Lee, Y.S.; Hagemann, H.; D’Anna, V.; Cho, Y.W.; Filinchuk, Y.; Jensen, T.R. A mixed-cation mixed-anion borohydride NaY(BH4)2Cl2. Int. J. Hydrogen Energy 2012, 37, 8428–8438. [Google Scholar] [CrossRef]
- Wegner, W.; Jaroń, T.; Grochala, W. Polymorphism and hydrogen discharge from holmium borohydride, Ho(BH4)3, and KHo(BH4)4. Int. J. Hydrogen Energy 2014, 39, 20024–20030. [Google Scholar] [CrossRef]
- Cerny, R.; Schouwink, P. The crystal chemistry of inorganic metal borohydrides and their relation to metal oxides. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 619–640. [Google Scholar] [CrossRef] [PubMed]
- Ley, M.B.; Jørgensen, M.; Cerny, R.; Filinchuk, Y.; Jensen, T.R. From M(BH4)3 (M = La, Ce) Borohydride Frameworks to Controllable Synthesis of Porous Hydrides and Ion Conductors. Inorg. Chem. 2016, 55, 9748–9756. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Didelot, E.; Spyratou, A.; Daku, L.M.L.; Cerny, R.; Hagemann, H. Halide Free M(BH4)2 (M = Sr, Ba, and Eu) Synthesis, Structure, and Decomposition. Inorg. Chem. 2016, 55, 7090–7097. [Google Scholar] [CrossRef] [PubMed]
- Frommen, C.; Sørby, M.H.; Ravindran, P.; Vajeeston, P.; Fjellvåg, H.; Hauback, B.C. Synthesis, Crystal Structure, and Thermal Properties of the First Mixed-Metal and Anion-Substituted Rare Earth Borohydride LiCe(BH4)3Cl. J. Phys. Chem. C 2011, 115, 23591–23602. [Google Scholar] [CrossRef] [Green Version]
- GharibDoust, S.P.; Brighi, M.; Sadikin, Y.; Ravnsbæk, D.B.; Cerny, R.; Skibsted, J.; Jensen, T.R. Synthesis, Structure, and Li-Ion Conductivity of LiLa(BH4)3X, X = Cl, Br, I. J. Phys. Chem. C 2017, 121, 19010–19021. [Google Scholar] [CrossRef]
- Sadikin, Y.; Stare, K.; Schouwink, P.; Ley, M.B.; Jensen, T.R.; Meden, A.; Cerny, R. Alkali metal—Yttrium borohydrides: The link between coordination of small and large rare-earth. J. Solid State Chem. 2015, 225, 231–239. [Google Scholar] [CrossRef]
- Hagemann, H.; Longhini, M.; Kaminski, J.W.; Wesolowski, T.A.; Cerny, R.; Penin, N.; Sørby, M.H.; Hauback, B.C.; Severa, G.; Jensen, C.M. LiSc(BH4)4: A novel salt of Li+ and discrete Sc(BH4)4− complex anions. J. Phys. Chem. A 2008, 112, 7551–7555. [Google Scholar] [CrossRef] [PubMed]
- Jaroń, T.; Wegner, W.; Fijałkowski, K.J.; Leszczyński, P.J.; Grochala, W. Facile Formation of Thermodynamically Unstable Novel Borohydride Materials by a Wet Chemistry Route. Chem. A Eur. J. 2015, 21, 5689–5692. [Google Scholar] [CrossRef] [PubMed]
- Cerny, R.; Severa, G.; Ravnsbæk, D.B.; Filinchuk, Y.; D’Anna, V.; Hagemann, H.; Haase, D.; Jensen, C.M.; Jensen, T.R. NaSc(BH4)4: A Novel Scandium-Based Borohydride. J. Phys. Chem. C 2010, 114, 1357–1364. [Google Scholar] [CrossRef]
- GharibDoust, S.H.P.; Ravnsbæk, D.B.; Cerny, R.; Jensen, T.R. Synthesis, structure and properties of bimetallic sodium rare-earth (RE) borohydrides, NaRE(BH4)4, RE = Ce, Pr, Er or Gd. Dalton Trans. 2017, 46, 13421–13431. [Google Scholar] [CrossRef] [PubMed]
- Jaroń, T.; Grochala, W. Probing Lewis acidity of Y(BH4)(3) via its reactions with MBH4 (M = Li, Na, K, NMe4). Dalton Trans. 2011, 40, 12808–12817. [Google Scholar] [CrossRef] [PubMed]
- Heere, M.; GharibDoust, S.H.P.; Sørby, M.H.; Frommen, C.; Jensen, T.R.; Hauback, B.C. In situ investigations of bimetallic potassium erbium borohydride. Int. J. Hydrogen Energy 2017, 42, 22468–22474. [Google Scholar] [CrossRef]
- Wegner, W.; Jaroń, T.; Grochala, W. MYb(BH4)4 (M = K, Na) from laboratory X-ray powder data. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2013, 69, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Cerny, R.; Ravnsbæk, D.B.; Severa, G.; Filinchuk, Y.; D’ Anna, V.; Hagemann, H.; Haase, D.; Skibsted, J.; Jensen, C.M.; Jensen, T.R. Structure and Characterization of KSc(BH4)4. J. Phys. Chem. C 2010, 114, 19540–19549. [Google Scholar] [CrossRef]
- Aeberhard, P.C.; Refson, K.; Edwards, P.P.; David, W.I.F. High-pressure crystal structure prediction of calcium borohydride using density functional theory. Phys. Rev. B 2011, 83, 174102. [Google Scholar] [CrossRef]
- Brighi, M.; Schouwink, P.; Sadikin, Y.; Cerny, R. Fast ion conduction in garnet-type metal borohydrides Li3K3Ce2(BH4)12 and Li3K3La2(BH4)12. J. Alloys Compd. 2016, 662, 388–395. [Google Scholar] [CrossRef]
- GharibDoust, S.H.P.; Heere, M.; Sørby, M.H.; Ley, M.B.; Ravnsbæk, D.B.; Hauback, B.C.; Cerny, R.; Jensen, T.R. Synthesis, structure and properties of new bimetallic sodium and potassium lanthanum borohydrides. Dalton Trans. 2016, 45, 19002–19011. [Google Scholar] [CrossRef] [PubMed]
- Jaroń, T.; Wegner, W.; Grochala, W. M Y(BH4)4 and M2LiY(BH4)6−xClx (M = Rb, Cs): New borohydride derivatives of yttrium and their hydrogen storage properties. Dalton Trans. 2013, 42, 6886–6893. [Google Scholar] [CrossRef] [PubMed]
- Møller, K.T.; Jørgensen, M.; Fogh, A.S.; Jensen, T.R. Perovskite alkali metal samarium borohydrides: Crystal structures and thermal decomposition. Dalton Trans. 2017, 46, 11905–11912. [Google Scholar] [CrossRef] [PubMed]
- Schouwink, P.; Didelot, E.; Lee, Y.S.; Mazet, T.; Cerny, R. Structural and magnetocaloric properties of novel gadolinium borohydrides. J. Alloys Compd. 2016, 664, 378–384. [Google Scholar] [CrossRef]
- Schouwink, P.; Ley, M.B.; Tissot, A.; Hagemann, H.; Jensen, T.R.; Smrcok, L.; Cerny, R. Structure and properties of complex hydride perovskite materials. Nat. Commun. 2014, 5, 5706. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.H.; Chen, Y.Y.; Xie, R.W.; Hu, Z.; Song, Y. Influence of alloying elements on the stability and dehydrogenation properties on Y(BH4)3 by first principles calculations. Int. J. Hydrogen Energy 2016, 41, 1662–1671. [Google Scholar] [CrossRef]
- Remhof, A.; Borgschulte, A.; Friedrichs, O.; Mauron, P.; Yan, Y.; Züttel, A. Solvent-free synthesis and decomposition of Y(BH4)3. Scr. Mater. 2012, 66, 280–283. [Google Scholar] [CrossRef]
- Park, K.; Lee, H.S.; Remhof, A.; Lee, Y.S.; Yan, Y.G.; Kim, M.Y.; Kim, S.J.; Züttel, A.; Cho, Y.W. Thermal properties of Y(BH4)3 synthesized via two different methods. Int. J. Hydrogen Energy 2013, 38, 9263–9270. [Google Scholar] [CrossRef]
- Riktor, M.D.; Filinchuk, Y.; Vajeeston, P.; Bardaji, E.G.; Fichtner, M.; Fjellvåg, H.; Sørby, M.H.; Hauback, B.C. The crystal structure of the first borohydride borate, Ca3(BD4)3(BO3). J. Mater. Chem. 2011, 21, 7188–7193. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Ermittlung des Ionencharakters mit Hilfe der Stabilitatsquotiententheoriet. Naturwissenschaften 1955, 42, 43. [Google Scholar]
- Schrauzer, G.N. Die chemische Bestimmung des Ionencharakters im Lithiumborhydridt. Naturwissenschaften 1955, 42, 43–44. [Google Scholar]
- Ley, M.B.; Paskevicius, M.; Schouwink, P.; Richter, B.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Novel solvates M(BH4)3S(CH3)2 and properties of halide-free M(BH4)3 (M = Y or Gd). Dalton Trans. 2014, 43, 13333–13342. [Google Scholar] [CrossRef] [PubMed]
- Mauron, P.; Buchter, F.; Friedrichs, O.; Remhof, A.; Bielmann, M.; Zwicky, C.N.; Züttel, A. Stability and reversibility of LiBH4. J. Phys. Chem. B 2008, 112, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Vajo, J.J.; Skeith, S.L.; Mertens, F. Reversible storage of hydrogen in destabilized LiBH4. J. Phys. Chem. B 2005, 109, 3719–3722. [Google Scholar] [CrossRef] [PubMed]
- Bosenberg, U.; Doppiu, S.; Mosegaard, L.; Barkhordarian, G.; Eigen, N.; Borgschulte, A.; Jensen, T.R.; Cerenius, Y.; Gutfleisch, O.; Klassen, T.; et al. Hydrogen sorption properties of MgH2-LiBH4 composites. Acta Mater. 2007, 55, 3951–3958. [Google Scholar] [CrossRef]
- Lim, J.H.; Shim, J.H.; Lee, Y.S.; Suh, J.Y.; Cho, Y.W.; Lee, J. Rehydrogenation and cycle studies of LiBH4-CaH2 composite. Int. J. Hydrogen Energy 2010, 35, 6578–6582. [Google Scholar] [CrossRef]
- Jin, S.A.; Lee, Y.S.; Shim, J.H.; Cho, Y.W. Reversible hydrogen storage in LiBH4-MH2 (M = Ce, Ca) composites. J. Phys. Chem. C 2008, 112, 9520–9524. [Google Scholar] [CrossRef]
- Gennari, F.C. Destabilization of LiBH4 by MH2 (M = Ce, La) for hydrogen storage: Nanostructural effects on the hydrogen sorption kinetics. Int. J. Hydrogen Energy 2011, 36, 15231–15238. [Google Scholar] [CrossRef]
- Jin, S.A.; Shim, J.H.; Cho, Y.W.; Yi, K.W.; Zabara, O.; Fichtner, M. Reversible hydrogen storage in LiBH4-Al-LiH composite powder. Scr. Mater. 2008, 58, 963–965. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, Q.F.; Zhang, J.; Liu, S.S.; Sun, L.X. The Dehydrogenation Reactions and Kinetics of 2LiBH4-Al Composite. J. Phys. Chem. C 2009, 113, 18424–18430. [Google Scholar] [CrossRef]
- Vajo, J.J.; Salguero, T.T.; Gross, A.E.; Skeith, S.L.; Olson, G.L. Thermodynamic destabilization and reaction kinetics in light metal hydride systems. J. Alloys Compd. 2007, 446, 409–414. [Google Scholar] [CrossRef]
- Au, M.; Jurgensen, A.R.; Spencer, W.A.; Anton, D.L.; Pinkerton, F.E.; Hwang, S.J.; Kim, C.; Bowman, R.C. Stability and Reversibility of Lithium Borohydrides Doped by Metal Halides and Hydrides. J. Phys. Chem. C 2008, 112, 18661–18671. [Google Scholar] [CrossRef]
- Zhang, B.J.; Liu, B.H. Hydrogen desorption from LiBH4 destabilized by chlorides of transition metal Fe, Co, and Ni. Int. J. Hydrogen Energy 2010, 35, 7288–7294. [Google Scholar] [CrossRef]
- Frommen, C.; Heere, M.; Riktor, M.D.; Sørby, M.H.; Hauback, B.C. Hydrogen storage properties of rare earth (RE) borohydrides (RE = La, Er) in composite mixtures with LiBH4 and LiH. J. Alloys Compd. 2015, 645, S155–S159. [Google Scholar] [CrossRef] [Green Version]
- Heere, M.; GharibDoust, S.H.P.; Brighi, M.; Frommen, C.; Sørby, M.H.; Cerny, R.; Jensen, T.R.; Hauback, B.C. Hydrogen Sorption in Erbium Borohydride Composite Mixtures with LiBH4 and/or LiH. Inorganics 2017, 5, 31. [Google Scholar] [CrossRef]
- Gennari, F.C. Mechanochemical synthesis of erbium borohydride: Polymorphism, thermal decomposition and hydrogen storage. J. Alloys Compd. 2013, 581, 192–195. [Google Scholar] [CrossRef]
- Barkhordarian, G.; Klassen, T.; Dornheim, M.; Bormann, R. Unexpected kinetic effect of MgB2 in reactive hydride composites containing complex borohydrides. J. Alloys Compd. 2007, 440, L18–L21. [Google Scholar] [CrossRef]
- Dhliwayo, T. Development of advanced shield systems for fast neutrons. Int. Nuclear Saf. J. 2014, 3, 49–53. [Google Scholar]
- Fantidis, J.G. The comparison between simple and advanced shielding materials for the shield of portable neutron sources. Int. J. Radiat. Res. 2015, 13, 287–295. [Google Scholar]
Figure 1.
The crystal structure of α-RE(BH4)3, space group Pa. The pseudo-cubic arrangement of RE3+ (grey spheres) and their octahedral coordination of 6 BH4− (blue tetrahedra) are outlined.
Figure 1.
The crystal structure of α-RE(BH4)3, space group Pa. The pseudo-cubic arrangement of RE3+ (grey spheres) and their octahedral coordination of 6 BH4− (blue tetrahedra) are outlined.

Figure 2.
Reported unit cell parameters for α-RE(BH4)3, β-RE(BH4)3, LiRE(BH4)3Cl as a function of ionic radii of the RE3+ ions. The unit cell parameter for the β-Y(BH4)3 superstructure is divided by 2 to be comparable with the other β-RE(BH4)3 phases.
Figure 2.
Reported unit cell parameters for α-RE(BH4)3, β-RE(BH4)3, LiRE(BH4)3Cl as a function of ionic radii of the RE3+ ions. The unit cell parameter for the β-Y(BH4)3 superstructure is divided by 2 to be comparable with the other β-RE(BH4)3 phases.
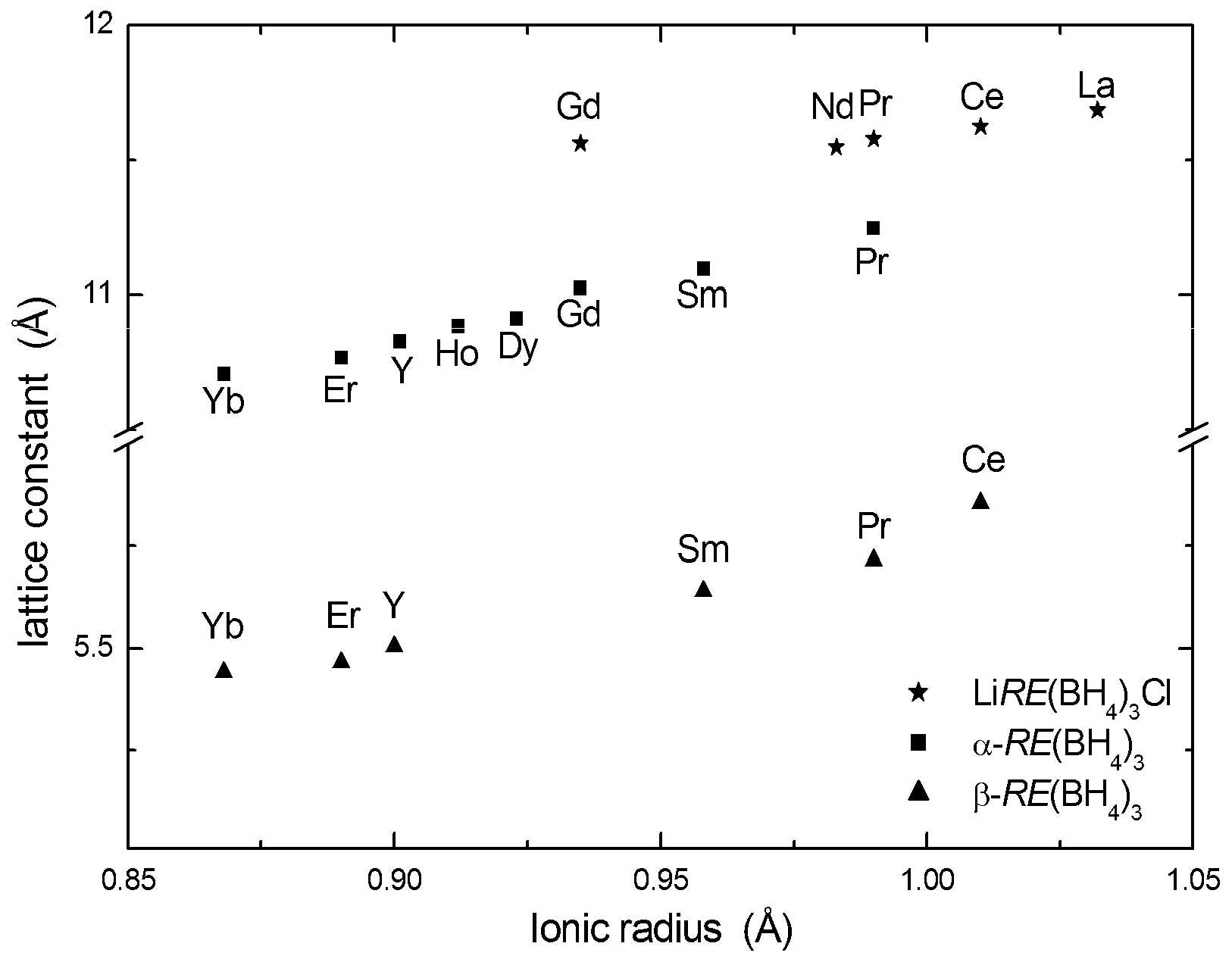
Figure 3.
Crystal structure of β-RE(BH4)3, space group Pmm. The boron atoms have a cubic coordination (blue) of half-occupied hydrogen sites (yellow half-spheres). This represent two different orientations of the BH4-tetrahdra as outlined for one boron where green and orange bonds represent two different BH4− orientations.
Figure 3.
Crystal structure of β-RE(BH4)3, space group Pmm. The boron atoms have a cubic coordination (blue) of half-occupied hydrogen sites (yellow half-spheres). This represent two different orientations of the BH4-tetrahdra as outlined for one boron where green and orange bonds represent two different BH4− orientations.
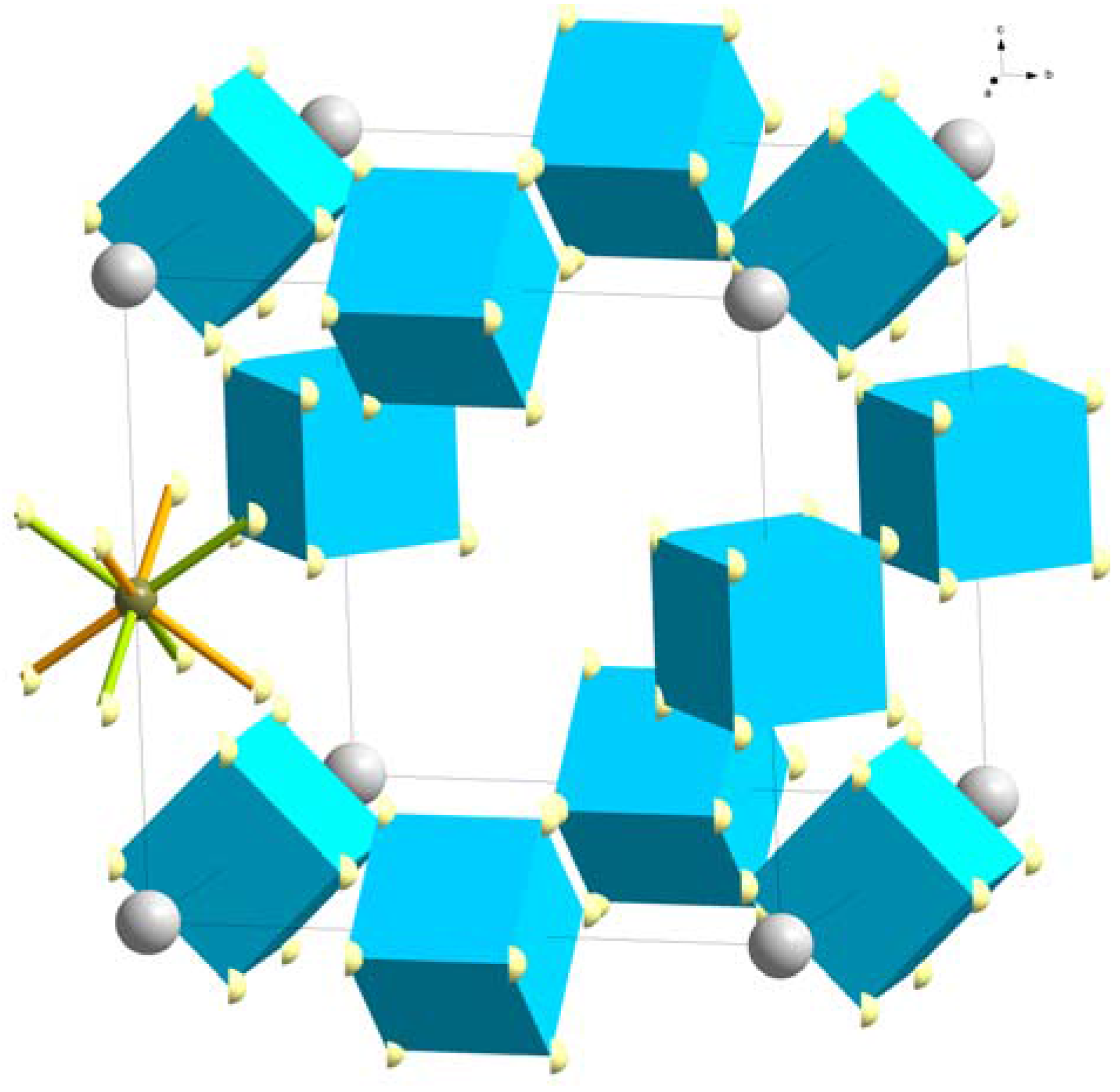
Figure 4.
Crystal structure of β-Y(BH4)3, space group Fmc. The octahedral coordination of one Y3+ (white) is outlined. Note the alternating orientations of the BH4− ions (light blue) along the three axes of the unit cell.
Figure 4.
Crystal structure of β-Y(BH4)3, space group Fmc. The octahedral coordination of one Y3+ (white) is outlined. Note the alternating orientations of the BH4− ions (light blue) along the three axes of the unit cell.

Figure 5.
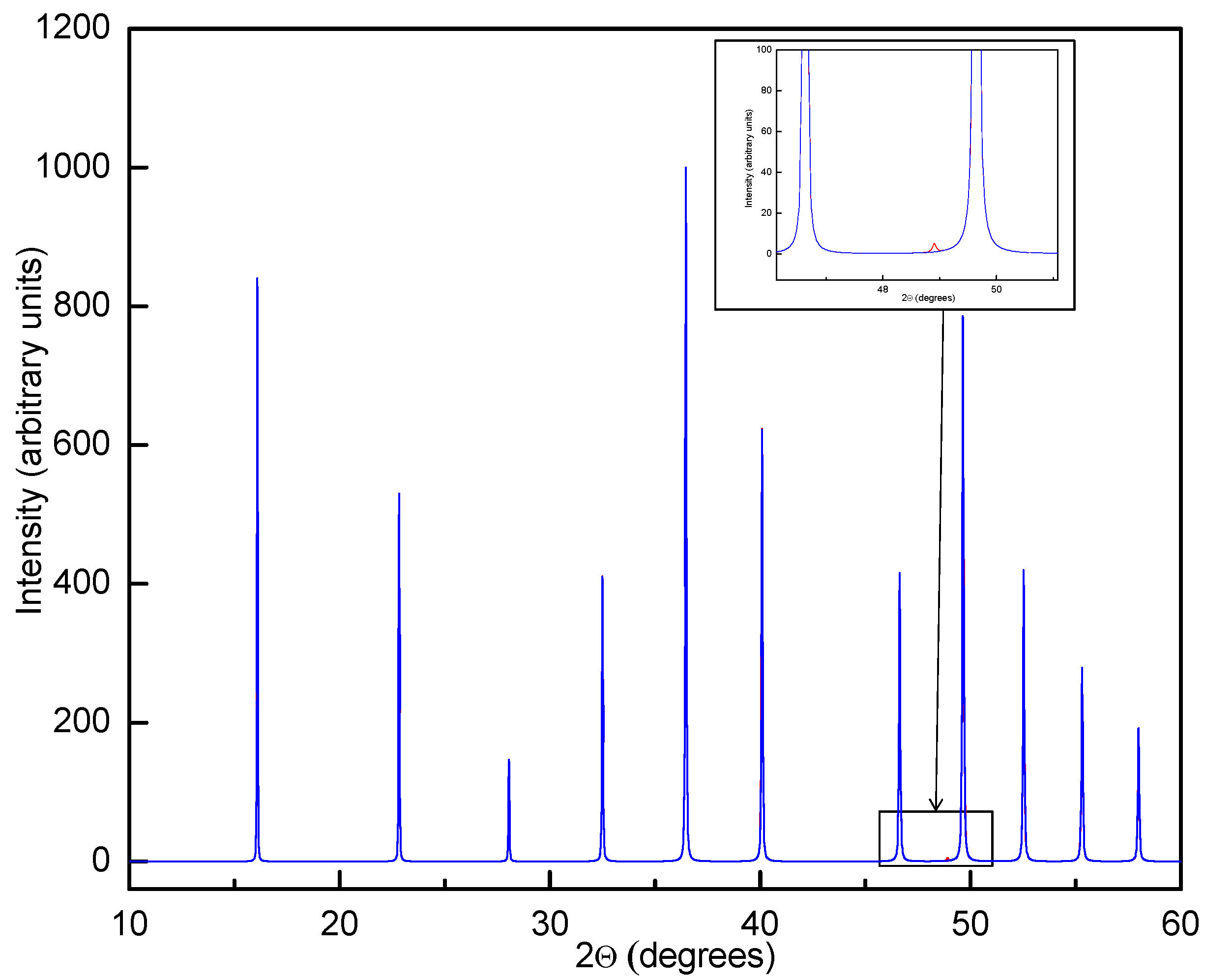
Simulated powder X-ray diffraction (PXD) data (CuKα1) for β-Y(BH4)3 with disordered structure and space group Pmm (blue curve) and a ordered structure with doubled unit cell edges and space group Fmc (red curve). The red curve is mostly not visible due almost perfect overlap with the blue curve. The strongest superstructure peak 531 is shown inset and barely visible even in the noise-free simulated data.
Figure 5.
Simulated powder X-ray diffraction (PXD) data (CuKα1) for β-Y(BH4)3 with disordered structure and space group Pmm (blue curve) and a ordered structure with doubled unit cell edges and space group Fmc (red curve). The red curve is mostly not visible due almost perfect overlap with the blue curve. The strongest superstructure peak 531 is shown inset and barely visible even in the noise-free simulated data.
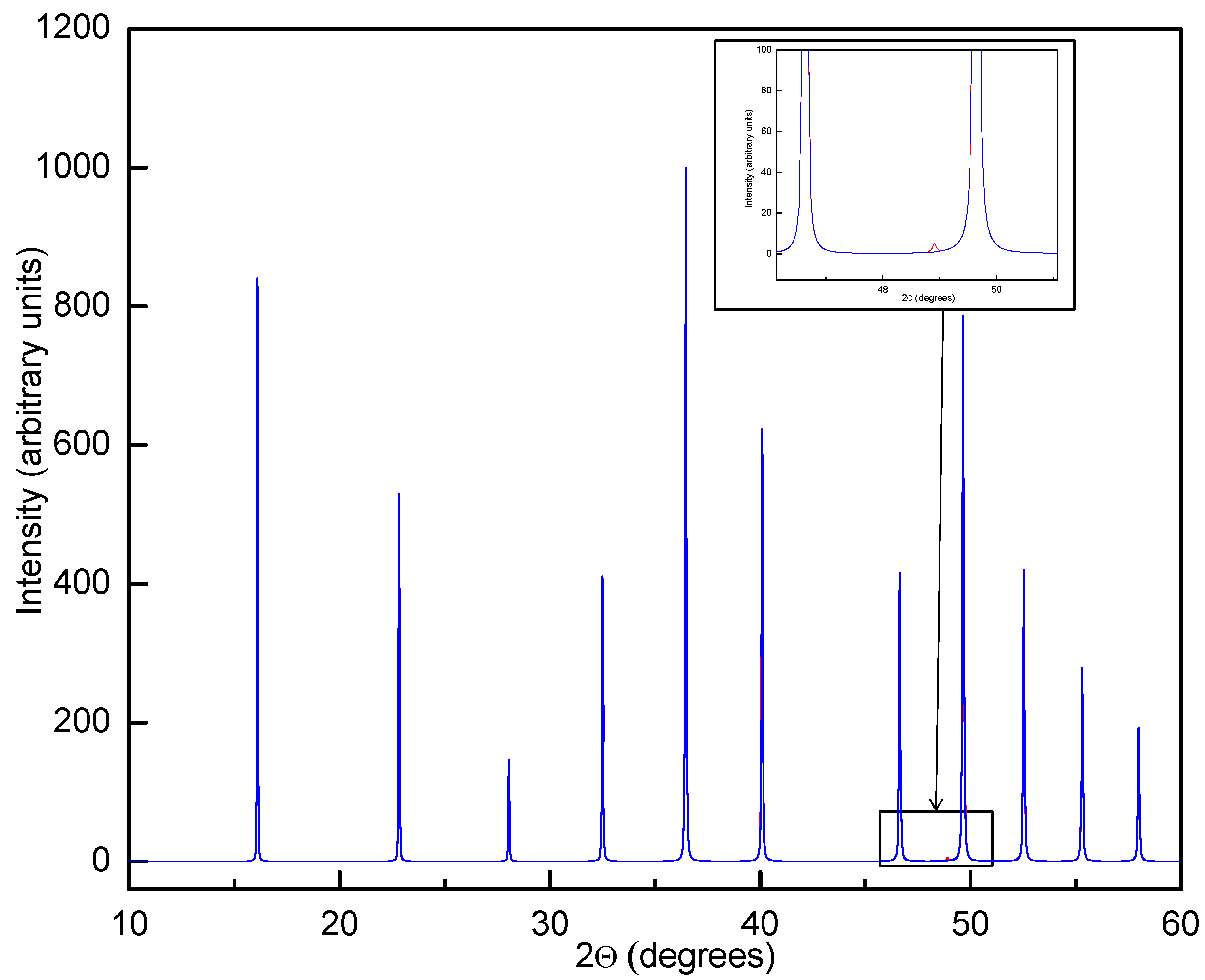
Figure 6.
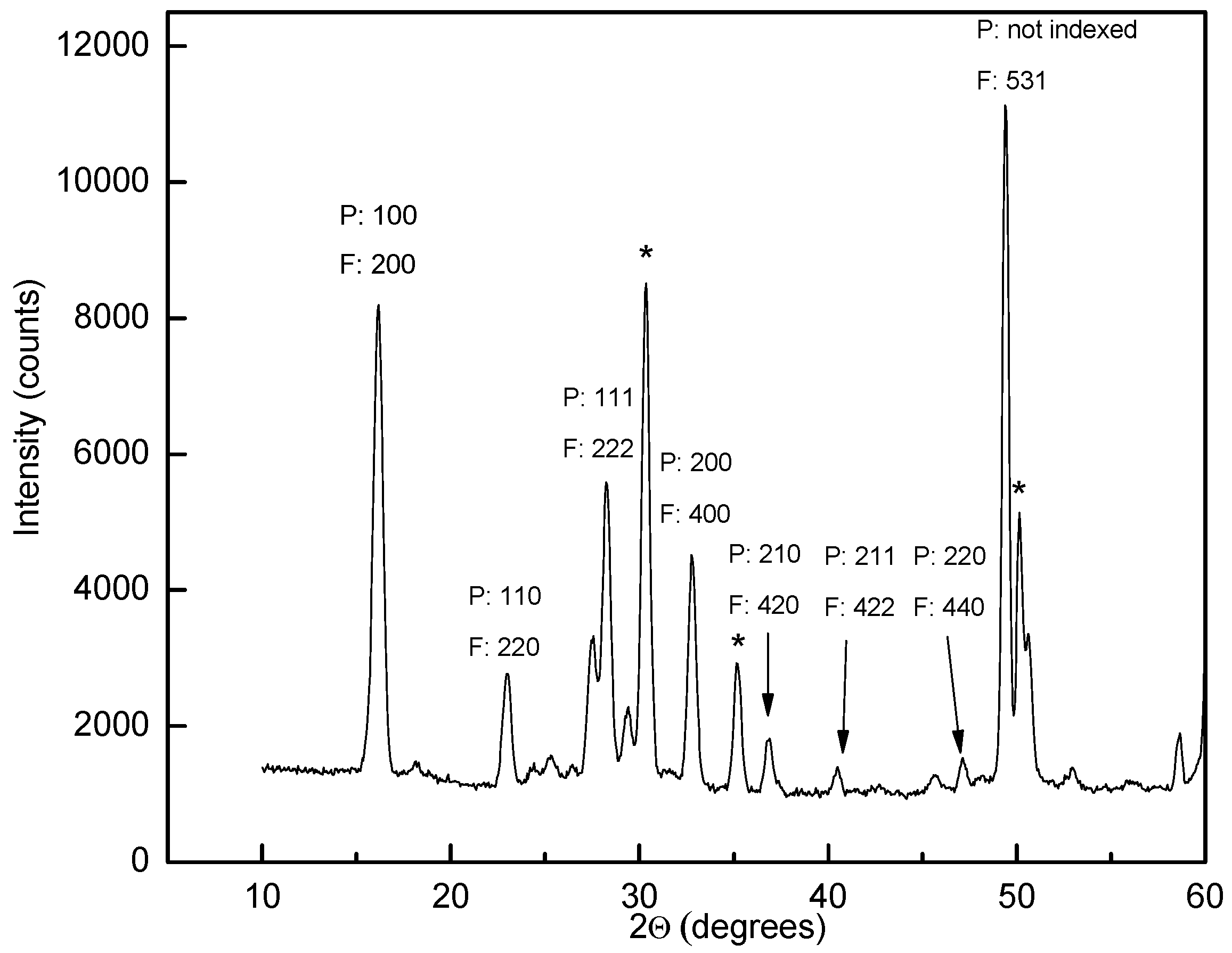
Powder neutron diffraction (PND) data for β-Y(11BD4)3. Miller indices for the small disordered unit cell (P) and the large ordered unit cell (F) are indicated. LiCl peaks are marked with * and unindexed peaks are from Li11BD4. Note the high intensity of the superstructure peak (“F: 531”). λ = 1.5558 Å.
Figure 6.
Powder neutron diffraction (PND) data for β-Y(11BD4)3. Miller indices for the small disordered unit cell (P) and the large ordered unit cell (F) are indicated. LiCl peaks are marked with * and unindexed peaks are from Li11BD4. Note the high intensity of the superstructure peak (“F: 531”). λ = 1.5558 Å.
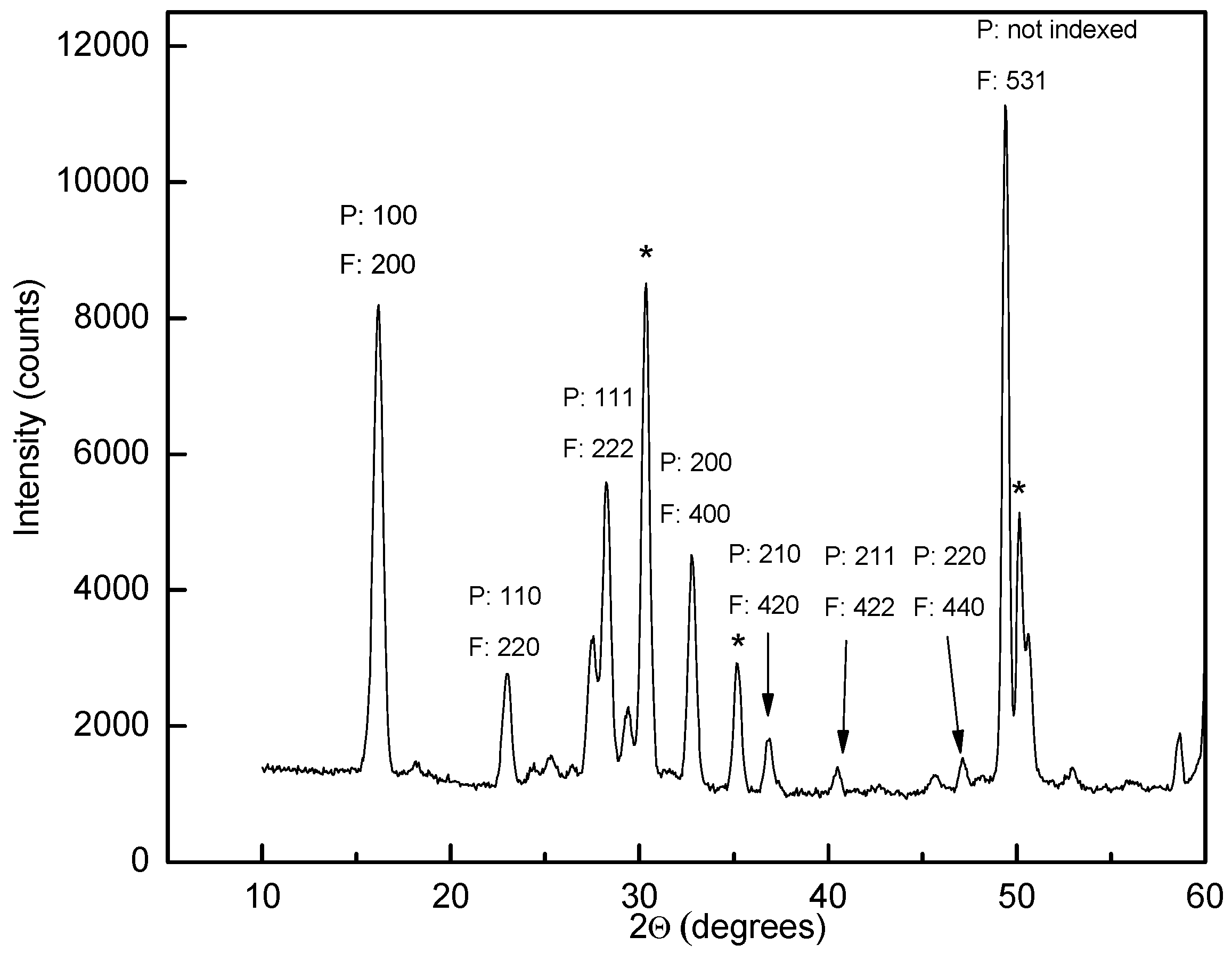
Figure 7.
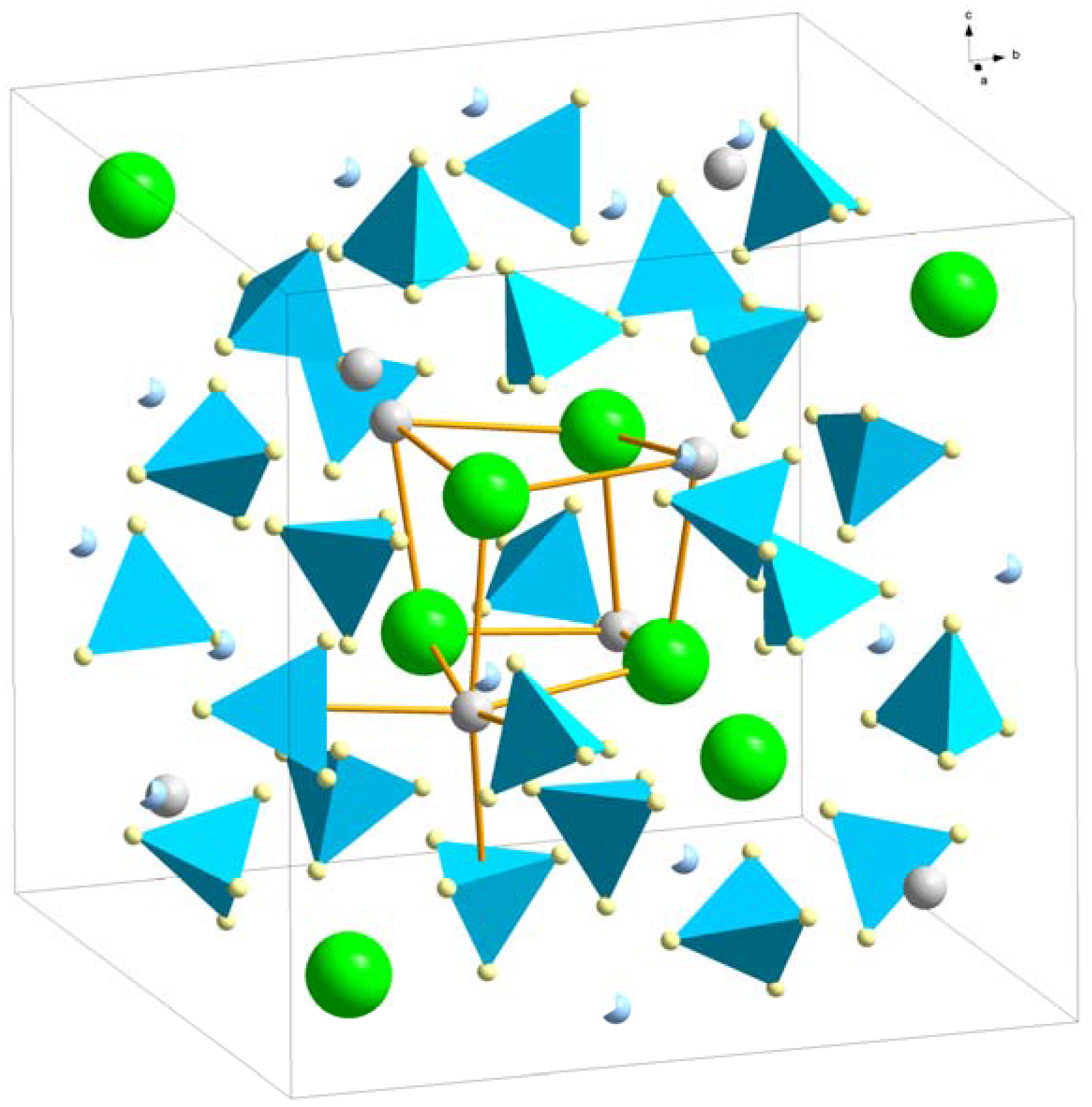
Crystal structure of LiRE(BH4)3Cl, space group I3m. The pseudocube of three Cl− (green) and three RE (white) is outlined as well as the octahedral coordination of RE3+ by three Cl− and three BH4− (blue tetrahedra). Li+ (blue) in partly occupied 12d sites are indicated.
Figure 7.
Crystal structure of LiRE(BH4)3Cl, space group I3m. The pseudocube of three Cl− (green) and three RE (white) is outlined as well as the octahedral coordination of RE3+ by three Cl− and three BH4− (blue tetrahedra). Li+ (blue) in partly occupied 12d sites are indicated.
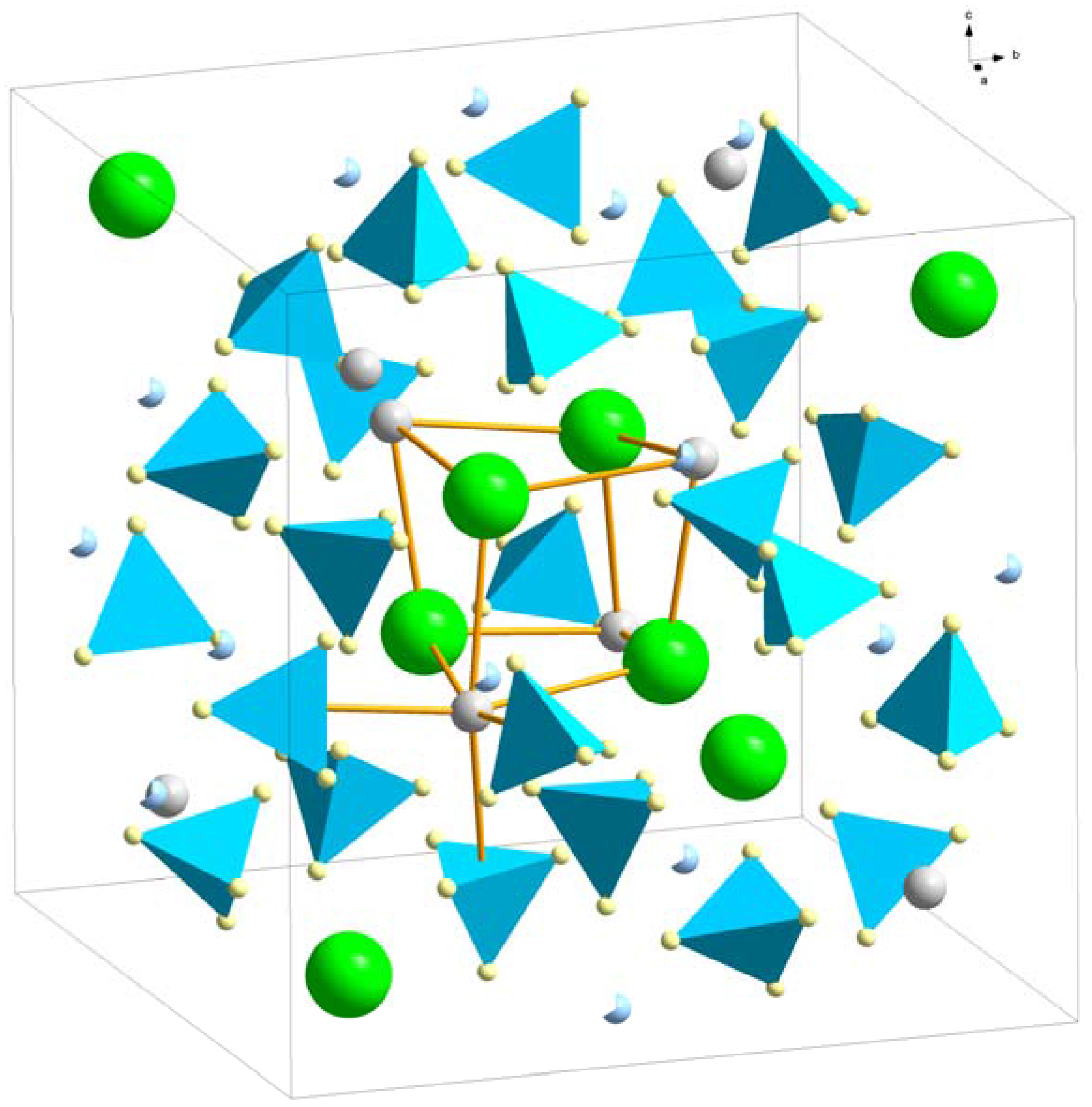
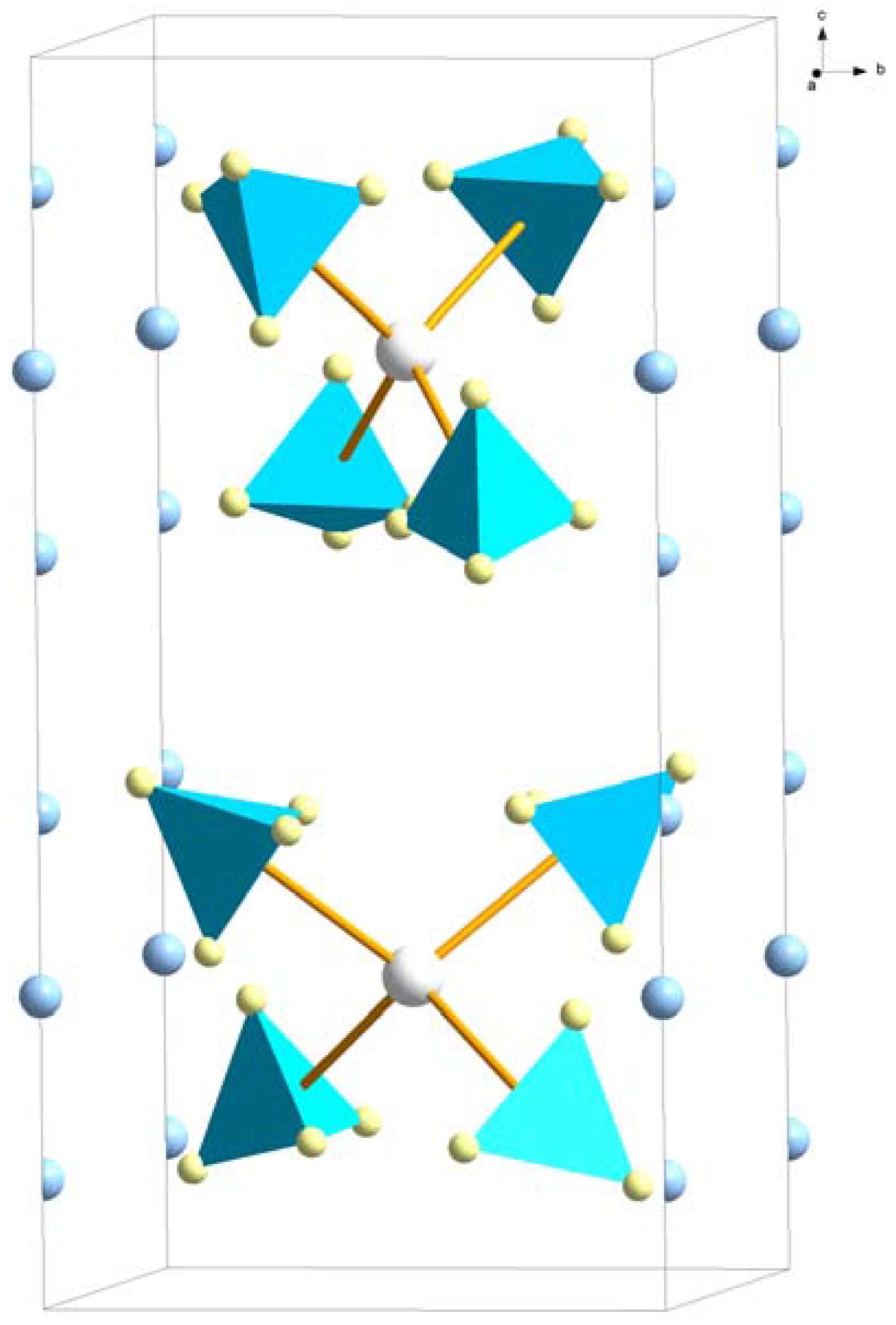
Figure 8.
Crystal structure of LiRE(BH4)4, space group P2c. The Sc(BH4)4− complex ions are outlined (Sc3+—white, BH4−—blue tetrahedra). Both the half-occupied 4k site (RE = Sc) and the fully occupied 2a site (RE = Y, Yb, Lu) for Li+ (blue) are indicated.
Figure 8.
Crystal structure of LiRE(BH4)4, space group P2c. The Sc(BH4)4− complex ions are outlined (Sc3+—white, BH4−—blue tetrahedra). Both the half-occupied 4k site (RE = Sc) and the fully occupied 2a site (RE = Y, Yb, Lu) for Li+ (blue) are indicated.
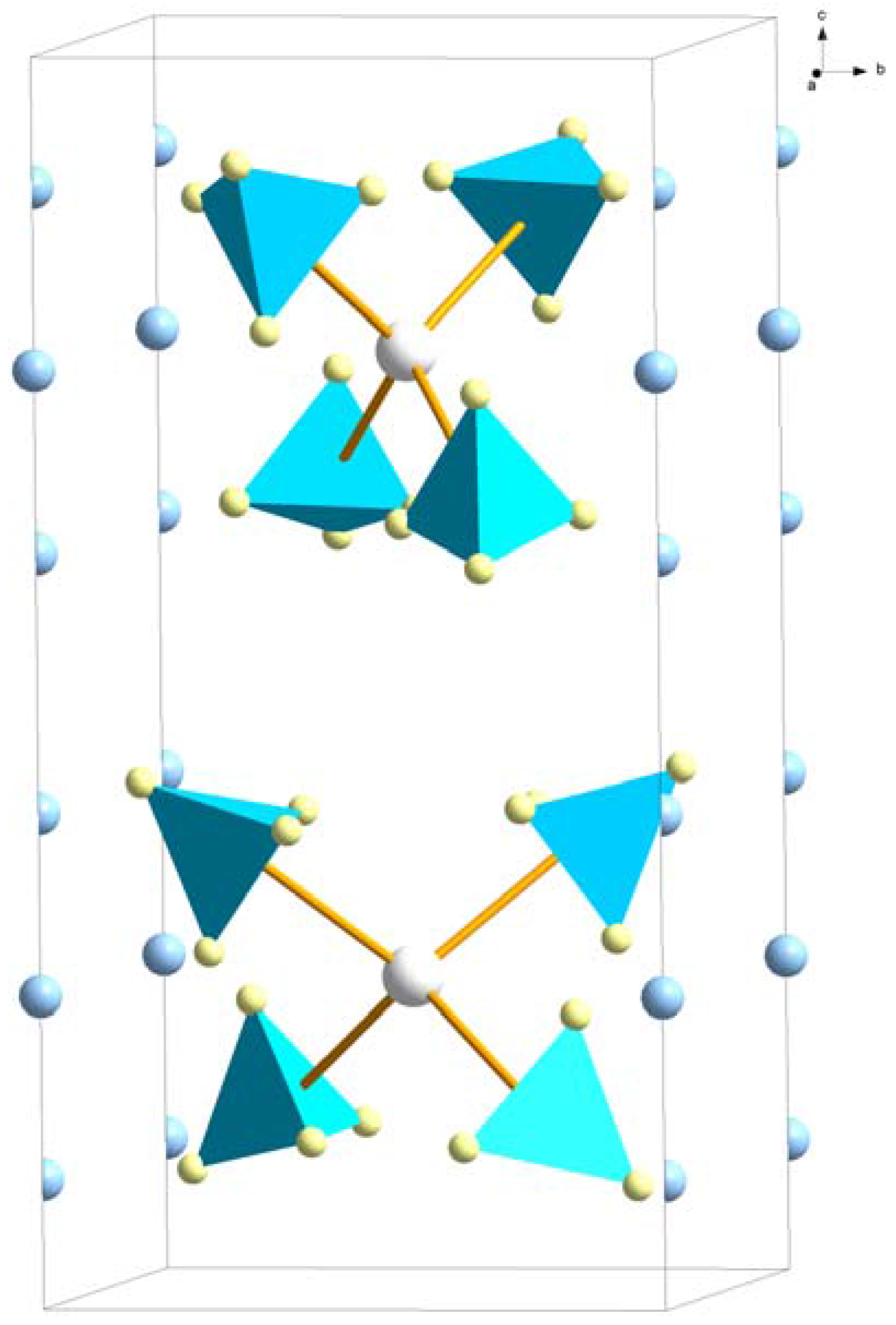
Figure 9.
(a) SR-PXD pattern of Y(BH4)3 in the temperature range of 22–450 °C. The sample was heated under dynamic vacuum with a heating rate of 2 °C/min. Three distinct regions have been highlighted and the temperature region for the appearance of an intermediate phase has been encircled; (b) Representative SR-PXD patterns at selected temperatures: the top pattern at 190 °C shows remanence of the low-temperature LT phase of Y(BH4)3, the bottom pattern at 267 °C shows YH3, and the middle pattern (232 °C) exhibits YH3 and an additional intermediate phase.
Figure 9.
(a) SR-PXD pattern of Y(BH4)3 in the temperature range of 22–450 °C. The sample was heated under dynamic vacuum with a heating rate of 2 °C/min. Three distinct regions have been highlighted and the temperature region for the appearance of an intermediate phase has been encircled; (b) Representative SR-PXD patterns at selected temperatures: the top pattern at 190 °C shows remanence of the low-temperature LT phase of Y(BH4)3, the bottom pattern at 267 °C shows YH3, and the middle pattern (232 °C) exhibits YH3 and an additional intermediate phase.
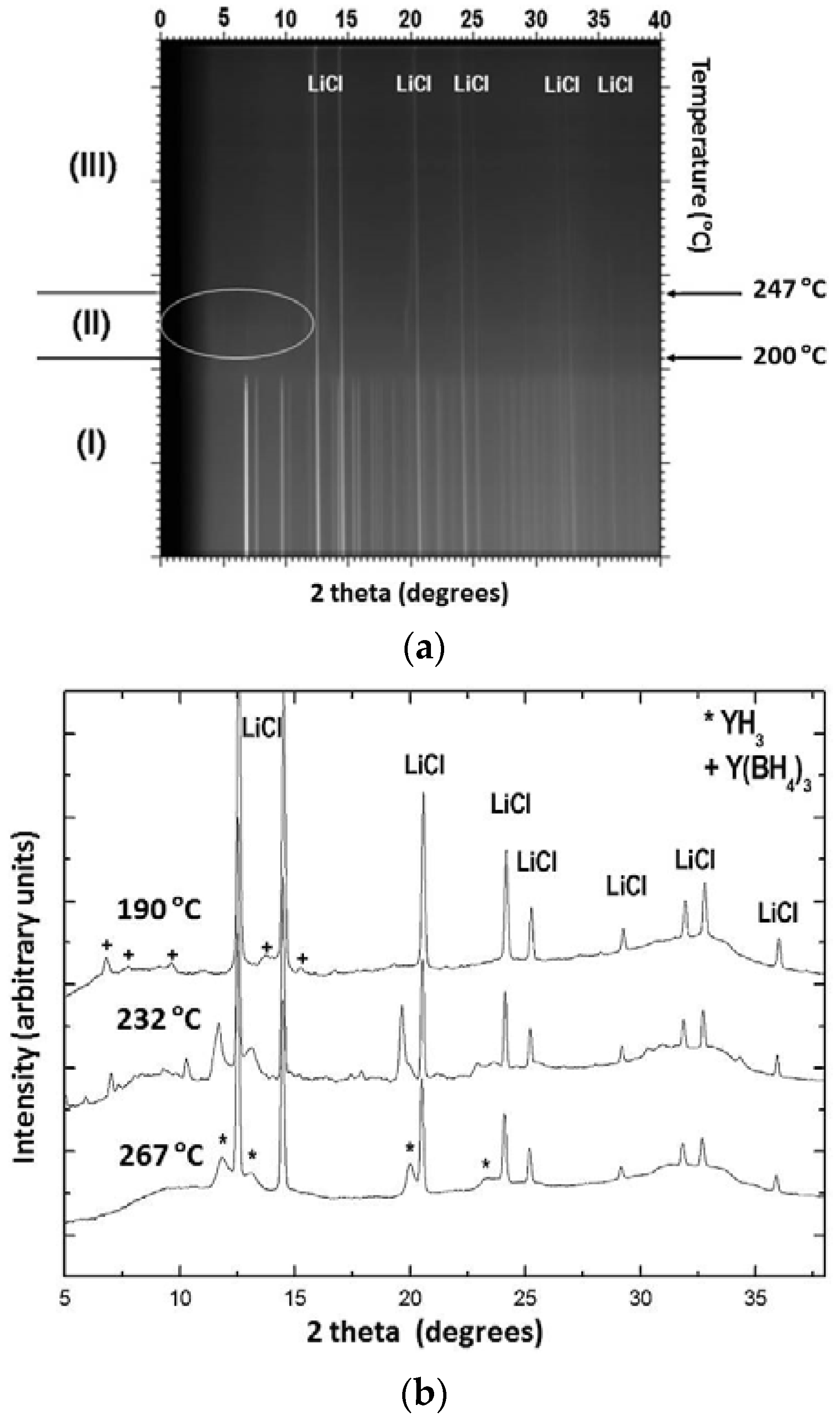
Figure 10.
(a) In situ SR-PXD data between 80 and 600 °C for a (3LiBD4-CeCl3) mixture after ball milling (6 h); (b) Selected powder patterns between 130 and 590 °C. The heating rate was 2 °C/min. Peak positions of major phases are indicated in the plots.
Figure 10.
(a) In situ SR-PXD data between 80 and 600 °C for a (3LiBD4-CeCl3) mixture after ball milling (6 h); (b) Selected powder patterns between 130 and 590 °C. The heating rate was 2 °C/min. Peak positions of major phases are indicated in the plots.
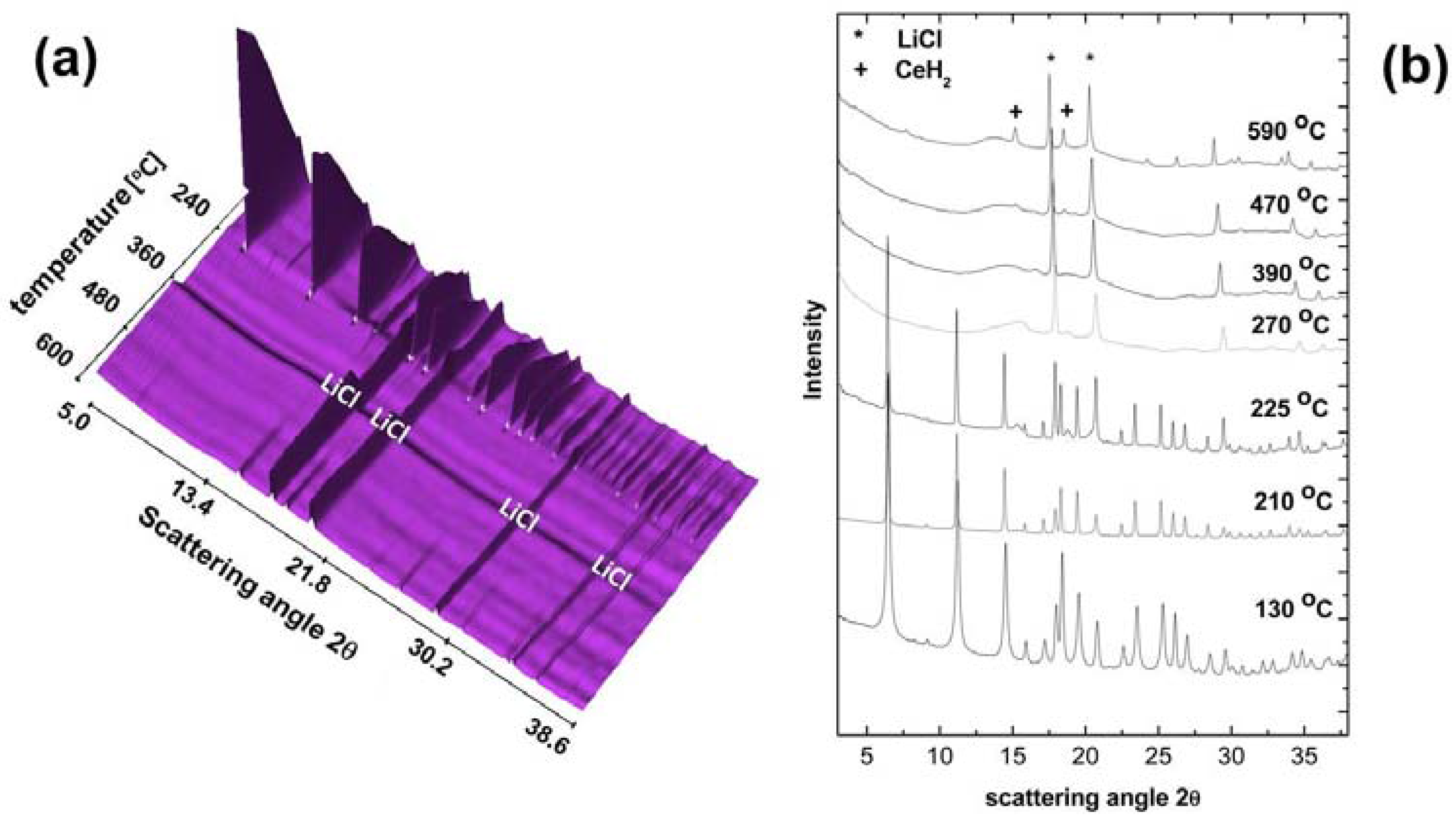
Figure 11.
TG data ((i) and DSC heat flow signal (ii) of halide-free Er(BH4)3 after one desorption (des)-absorption (abs) cycle; TG-data (iii) and DSC heat flow signal (iv) of a mixture containing Er(BH4)3 and 50 wt % LiH after one des-abs cycle (des: 400 °C, 5 bar H2; abs: 340 °C, 100 bar H2 for both cases) [30].
Figure 11.
TG data ((i) and DSC heat flow signal (ii) of halide-free Er(BH4)3 after one desorption (des)-absorption (abs) cycle; TG-data (iii) and DSC heat flow signal (iv) of a mixture containing Er(BH4)3 and 50 wt % LiH after one des-abs cycle (des: 400 °C, 5 bar H2; abs: 340 °C, 100 bar H2 for both cases) [30].

Figure 12.
In situ SR-PXD data of a powder mixture containing desorbed Er(BH4)3 + 50 wt % LiH. The composite was heated from RT to 400 °C followed by a 1 h isotherm and cooled to RT in p(H2) = 100 bar (temperature ramp of 5 °C/min). The inset illustrates a magnification of the 2θ range from 5 to 20° to enhance the visibility of Bragg peaks for h-LiBH4 (+). The red curve in the magnified section is the diffraction pattern measured at 400 °C. λ = 0.69733 Å.
Figure 12.
In situ SR-PXD data of a powder mixture containing desorbed Er(BH4)3 + 50 wt % LiH. The composite was heated from RT to 400 °C followed by a 1 h isotherm and cooled to RT in p(H2) = 100 bar (temperature ramp of 5 °C/min). The inset illustrates a magnification of the 2θ range from 5 to 20° to enhance the visibility of Bragg peaks for h-LiBH4 (+). The red curve in the magnified section is the diffraction pattern measured at 400 °C. λ = 0.69733 Å.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Structure types for binary and ternary lithium rare earth borohydrides formed by metathesis reactions between LiBH4 and RECl3.
Table 1.
Structure types for binary and ternary lithium rare earth borohydrides formed by metathesis reactions between LiBH4 and RECl3.
| RE Ion | Ion Radius [Å] | LiRE(BH4)4 (CuAuCl4) | α-RE(BH4)3 (ReO3) | β-RE(BH4)3 (ReO3) | LiRE(BH4)3Cl (Spinel) |
|---|---|---|---|---|---|
| Sc3+ | 0.745 | X | |||
| Y3+ | 0.90 | X | X | ||
| La3+ | 1.172 | X | |||
| Ce3+ | 1.15 | X | X | ||
| Pr3+ | 1.13 | X | X | X | |
| Nd3+ | 1.123 | X | |||
| Sm3+ | 1.098 | X | X | X | |
| Gd3+ | 1.078 | X | X | ||
| Tb3+ | 1.063 | X | |||
| Dy3+ | 1.052 | X | |||
| Ho3+ | 1.041 | X | X | ||
| Er3+ | 1.03 | X | X | ||
| Yb3+ | 1.008 | X | X | X | |
| Lu3+ | 1.001 | X |
Table 2.
Monometallic rare earth borohydrides. RT = room temperature; HT = high temperature polymorph; meta = metastable.
Table 2.
Monometallic rare earth borohydrides. RT = room temperature; HT = high temperature polymorph; meta = metastable.
| Cation | Polymorph | Hydrogen Capacity (wt %) | Stability | Crystal System | Space Group | Structure Type | Ref. |
|---|---|---|---|---|---|---|---|
| Y3+ | α-Y(BH4)3 β-Y(BH4)3 | 9.1 | RT RT, HT | Cubic Cubic | Pa Fmc | Distorted ReO3 ReO3 | [18] [21] |
| La3+ | La(BH4)3 | 6.6 | RT | Trigonal | Rc | Distorted ReO3 | [34] |
| Ce3+ | Ce(BH4)3 | 6.6 | RT | Cubic Trigonal | Fmc Rc | ReO3 Distored ReO3 | [34] [34] |
| Pr3+ | Pr(BH4)3 | 6.5 | RT | Cubic | Pa Pmm | Distorted ReO3 ReO3 | [30] |
| Sm2+ | o-Sm(BH4)2 | 4.5 | ~500 K | Orthorhombic | Pbcn | ort-Sr(BH4)2 | [19,27] |
| Sm3+ | α-Sm(BH4)3 β-Sm(BH4)3 | 6.2 | RT RT | Cubic Cubic | Pa Pmm | Distorted ReO3 ReO3 | [19] [19] |
| Eu2+ | o-Eu(BH4)2 t-Eu(BH4)2 c-Eu(BH4)2 | 4.4 | ~430 K >668 K >668 K | Orthorhombic Tetragonal Cubic | Pbcn P41212 Pmm | orthorombic Sr(BH4)2 tetragonal Sr(BH4)2 cubic Sr(BH4)2 | [27] [35] [35] |
| Gd3+ | Gd(BH4)3 | 6.0 | RT | Cubic | Pa | Distorted ReO3 | [18,19] |
| Tb3+ | Tb(BH4)3 | 5.9 | RT | Cubic | Pa | Distorted ReO3 | [19] |
| Dy3+ | Dy(BH4)3 | 5.8 | RT | Cubic | Pa | Distorted ReO3 | [18] |
| Ho3+ | α-Ho(BH4)3 β-Ho(BH4)3 | 5.8 | RT RT | Cubic Cubic | Pa Fmc | Distorted ReO3 ReO3 | [32] [32] |
| Er3+ | α-Er(BH4)3 β-Er(BH4)3 | 5.7 | RT RT | Cubic Cubic | Pa Pmm | Distorted ReO3 ReO3 | [19] [19] |
| Tm3+ | α-Tm(BH4)3 | 5.7 | Cubic | Pa | Distorted ReO3 | [33] | |
| Yb2+ | α-Yb(BH4)2 β-Yb(BH4)2 γ-Yb(BH4)2 | 4.0 | RT >523 K 473–573 K | Orthorhombic Tetragonal Orthorhombic | F2dd P Pbca | α-Ca(BH4)2 β-Ca(BH4)2 γ-Ca(BH4)2 | [1] [29] [29] |
| Yb3+ | α-Yb(BH4)3 β-Yb(BH4)3 | 5.6 | RT RT, meta | Cubic Cubic | Pa Pmm | Distorted ReO3 ReO3 | [29] [29] |
Table 3.
Halide containing rare earth borohydrides.
| Cation | Composition | Crystal System | Space Group | Ref. | |
|---|---|---|---|---|---|
| Li+ | La3+ | LiLa(BH4)3Cl | Cubic | I3m | [9,19,37] |
| Li+ | La3+ | LiLa(BH4)3Br | Cubic | I3m | [37] |
| Li+ | La3+ | LiLa(BH4)3I | Cubic | I3m | [37] |
| Li+ | Ce3+ | LiCe(BH4)3Cl | Cubic | I3m | [8,19,36] |
| Li+ | Pr3+ | LiPr(BH4)3Cl | Cubic | I3m | [19] |
| Li+ | Nd3+ | LiNd(BH4)3Cl | Cubic | I3m | [19] |
| Li+ | Sm3+ | LiSm(BH4)3Cl | Cubic | I3m | [19] |
| Li+ | Gd3+ | LiGd(BH4)3Cl | Cubic | I3m | [9] |
| Na+ | Y3+ | NaY(BH4)2−xCl2+x | Monoclinic | P2/c | [31,38] |
| Li+ | Yb3+ | LiYb(BH4)4−xClx | Tetragonal (x ~ 1.0) | P2c | [29] |
| Yb2+ | Yb(BH4)2−xClx | Orthorhombic (x = 0.3) | Pbca | [29] | |
| Tetragonal (x = 0.76) | P | [29] | |||
Table 4.
Bi- and tri-metallic rare earth borohydrides.
| Cations | Polymorph | wt % H | Crystal System | Space Group | Structure Type | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Li+ | Sc3+ | LiSc(BH4)4 | 14.5 | Tetragonal | P2c | New | [39] | |
| Li+ | Y3+ | LiY(BH4)4 | 10.4 | Tetragonal | P2c | LiSc(BH4)4 | [10,40] | |
| Li+ | Yb3+ | LiYb(BH4)4 | 6.7 | Tetragonal | P2c | LiSc(BH4)4 | [29] | |
| Li+ | Lu3+ | LiLu(BH4)4 | 6.7 | Tetragonal | P2c | LiSc(BH4)4 | [19] | |
| Na+ | Sc3+ | NaSc(BH4)4 | 12.7 | Orthorhombic | Cmcm | HT-CrVO4 | [41] | |
| Na+ | Y3+ | NaY(BH4)4 | 9.4 | Orthorhombic | Cmcm | NaSc(BH4)4 | [10,40] | |
| Na+ | La3+ | NaLa(BH4)4 | 7.3 | Orthorhombic | Pbcn | New | [49] | |
| Na+ | Ce3+ | NaCe(BH4)4 | 7.3 | Orthorhombic | Pbcn | NaLa(BH4)4 | [42] | |
| Na+ | Pr3+ | NaPr(BH4)4 | 7.2 | Orthorhombic | Pbcn | NaLa(BH4)4 | [42] | |
| Na+ | Er3+ | NaEr(BH4)4 | 6.5 | Orthorhombic | Cmcm | NaSc(BH4)4 | [42] | |
| Na+ | Yb3+ | NaYb(BH4)4 | 6.3 | Orthorhombic | Cmcm | NaSc(BH4)4 | [45] | |
| K+ | Sc3+ | KSc(BH4)4 | 11.3 | Orthorhombic | Pnma | BaSO4 | [46] | |
| K+ | Y3+ | o-KY(BH4)4 m-KY(BH4)4 | 8.6 | Orthorhombic Monoclinic | Cmcm C2/c | NaSc(BH4)4 rt-LaNbO4 | [38,43] [38] | |
| K+ | La3+ | K3La(BH4)6 | 7.0 | Monoclinic | P21/n | Double-perovskite | [49] | |
| K+ | Ce3+ | K3Ce(BH4)6 | 7.0 | Monoclinic | P21/c | Double-perovskite | [48] | |
| K+ | Sm2+ | KSm(BH4)3 | 5.2 | Monoclinic | P21cn | Perovskite-related | [51] | |
| K+ | Gd3+ | KGd(BH4)4 K2Gd(BH4)5 K3Gd(BH4)6 | 6.3 6.5 6.7 | Monoclinic Monoclinic Monoclinic | P21/c P21/m P21/c | LiMnF4 New Double-perovskite | [52] [52] [52] | |
| K+ | Ho3+ | KHo(BH4)4 | 6.1 | Orthorhombic | Cmcm | NaSc(BH4)4 | [32] | |
| K+ | Er3+ | KEr(BH4)4 | 6.1 | Orthorhombic | Cmcm | NaSc(BH4)4 | [44] | |
| K+ | Yb2+ | LT-KYb(BH4)3 HT-KYb(BH4)3 | 4.7 | Cubic Orthorhombic | P3m Pmc21 | Perovskite-related Perovskite-related | [53] [53] | |
| K+ | Yb3+ | KYb(BH4)4 | 5.9 | Orthorhombic | Cmcm | NaSc(BH4)4 | [45] | |
| Rb+ | Y3+ | o-RbY(BH4)4 m-RbY(BH4)4 Rb3Y(BH4)6 | 6.9 6.9 5.6 | Orthorhombic Monoclinic Cubic | Pnma P21/c Fm | KSc(BH4)4 Distorted KSc(BH4)4 Double-perovskite | [38] [50] [38,53] | |
| Rb+ | Ce3+ | Rb3Ce(BH4)6 | 5.0 | Monoclinic | P21/c | Double-perovskite | [53] | |
| Rb+ | Sm2+ | RbSm(BH4)3 | 4.3 | Orthorhombic | Pbn21 | Perovskite-related | [51] | |
| Rb+ | Eu2+ | RbEu(BH4)3 | 4.3 | Orthorhombic | Pna21 | Perovskite-related | [53] | |
| Cs+ | Y3+ | CsY(BH4)4 Cs2Y(BH4)5 Cs3Y(BH4)6 | 5.7 4.2 | Tetragonal Hexagonal Cubic | I41/a P63cm Fm | CaWO4 new Double-perovskite | [50] [38] [38,53] | |
| Cs+ | Sm2+ | α -CsSm(BH4)3 α’-CsSm(BH4)3 β-CsSm(BH4)3 | 3.7 | Orthorhomic Tetragonal Cubic | P21212 Pm2 Pmm | Perovskite-related Perovskite-related Ideal perovskite | [51] [51] [51] | |
| Cs+ | Eu2+ | CsEu(BH4)3 | 3.7 | Tetragonal | P4/mbm | Perovskite-related | [53] | |
| Cs+ | Gd3+ | Cs3Gd(BH4)6 | 3.8 | Cubic | Fm | Double-perovskite | [53] | |
| Li+ | K+ | La3+ | Li3K3La2(BH4)12 | 8.1 | Cubic | Iad | Garnet | [48] |
| Li+ | K+ | Ce3+ | Li3K3Ce2(BH4)12 | 8.1 | Cubic | Iad | Garnet | [48] |
| Li+ | Rb+ | Y3+ | Rb2LiY(BH4)6 | 6.8 | Cubic | Fm | Double-perovskite | [38,50] |
| Li+ | Cs+ | Y3+ | Cs2LiY(BH4)6 | 5.4 | Cubic | Fm | Double-perovskite | [38,50] |
| Li+ | Cs+ | Ce3+ | Cs2LiCe(BH4)6 | 4.8 | Cubic | Fm | Double-perovskite | [53] |
| Li+ | Cs+ | Gd3+ | Cs2LiGd(BH4)6 | 4.7 | Cubic | Fm | Double-perovskite | [53] |
Table 5.
Thermal properties of selected monometallic RE-borohydrides with trivalent cation.
| Cation | Compound | Tdec. (°C) | Decompositon Products | Comments | Ref. |
|---|---|---|---|---|---|
| La3+ | La(BH4)3 | 258 | Not specified | Material obtained from thermolysis of La(BH4)3 nS(CH3)2; 8% mass loss due to simultaneous release of B2H6 and H2 | [34] |
| Ce3+ | Ce(BH4)3 | 251 | Not specified | Material obtained from thermolysis of Ce(BH4)3 nS(CH3)2; 8.5% mass loss due to simultaneous release of B2H6 and H2 | [34] |
| Pr3+ | Pr(BH4)3 | 275 | No crystalline products | Sample synthesized in toluene and DMS. In situ SR-PXD showed no crystalline decomposition product until 500 °C. | [30] |
| Gd3+ | Gd(BH4)3 | 260 | Not specified | Sample obtained from thermal decomposition of Gd(BH4)3*nS(CH3)2; DSC shows endothermic event at 260 °C related to hydrogen desorption. MS shows minor contributions of B2H6 between 225 and 275 °C. | [60] |
| Tb3+ | Tb(BH4)3 | 213 and 253 | Not specified | 3LiBH4-TbCl3 mixture after ball milling (2 h). DSC trace shows two endothermic events with peak temperatures of 213 and 253 °C | [19] |
| Dy3+ | Dy(BH4)3 | No data | No data | Only structural information available; decomposition is believed to occur similar to Y(BH4)3 and Gd(BH4)3 | [18] |
| Ho3+ | Ho(BH4)3 | 170 225 255 | No crystalline products | Three distinct endothermic events visible in the DSC up to 400 °C; onset of hydrogen release observed at 170 °C with the fastest rate at 255 °C | [32] |
| Er3+ | Er(BH4)3 | 230 | ErH2 and ErB4 | Hydrogen desorption starts at 230 °C; multiple endothermic events visible in DSC; 3.2% mass loss observed up to 400 °C | [30] |
Table 6.
Thermal properties of lithium rare earth borohydride chlorides.
| Cation1 | Cation 2 | Compound | Tdec. (°C) | Decompositon Products | Comments | Ref |
|---|---|---|---|---|---|---|
| La3+ | Li+ | LiLa(BH4)3Cl | 226 and 266 | LaH2 and LaB6 | TG/DSC shows two endothermic events between 200 and 300 °C and total mass loss of 2.87% | [9] |
| Ce3+ | Li+ | LiCe(BH4)3Cl | a 215, a 246, b 240, b 266 | CeH2 and CeB6 | a 3LiBD4-CeCl3 mixture after ball milling; two endothermic events at 215 and 246 °C observed by DSC b 3LiBH4-CeCl3 mixture after ball milling: major DSC events at 213 (melting), 240, and 266 °C (both decomposition) | [8,36] |
| Pr3+ | Li+ | LiPr(BH4)3Cl | a 176 b 212 and 247 | PrH3 and PrB6 | a Synthesised in (C2H5)2O. Sample is a mixture between Pr(BH4)3 and LiPr(BH4)3Cl; Tdec determined by TG/DSC b 3LiBH4-PrCl3 mixture after ball milling.TG/DSC shows two endothermic events at 212 °C and 247 °C with 0.7% and 2.6% mass loss, respectively | [30] |
| Nd3+ | Li+ | LiNd(BH4)3Cl | 223 and 241 | Not specified | 3LiBH4-NdCl3 mixture after ball milling. TG/DSC shows two endothermic events at 223 °C and 241 °C with 0.4% and 2.4% mass loss respectively | [19] |
| Gd3+ | Li+ | LiGd(BH4)3Cl | 261 | GdB4 | Value determined from thermogravimetric experiment | [9] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Frommen, C.; Sørby, M.H.; Heere, M.; Humphries, T.D.; Olsen, J.E.; Hauback, B.C. Rare Earth Borohydrides—Crystal Structures and Thermal Properties. Energies 2017, 10, 2115. https://doi.org/10.3390/en10122115
AMA Style
Frommen C, Sørby MH, Heere M, Humphries TD, Olsen JE, Hauback BC. Rare Earth Borohydrides—Crystal Structures and Thermal Properties. Energies. 2017; 10(12):2115. https://doi.org/10.3390/en10122115
Chicago/Turabian StyleFrommen, Christoph, Magnus H. Sørby, Michael Heere, Terry D. Humphries, Jørn E. Olsen, and Bjørn C. Hauback. 2017. "Rare Earth Borohydrides—Crystal Structures and Thermal Properties" Energies 10, no. 12: 2115. https://doi.org/10.3390/en10122115
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.