Lysyl Oxidase, A Critical Intra- and Extra-Cellular Target in the Lung for Cigarette Smoke Pathogenesis

Abstract
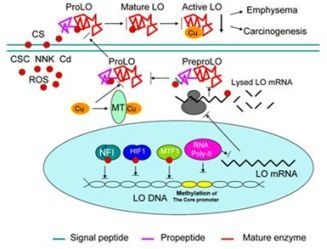
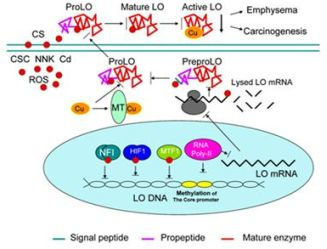
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
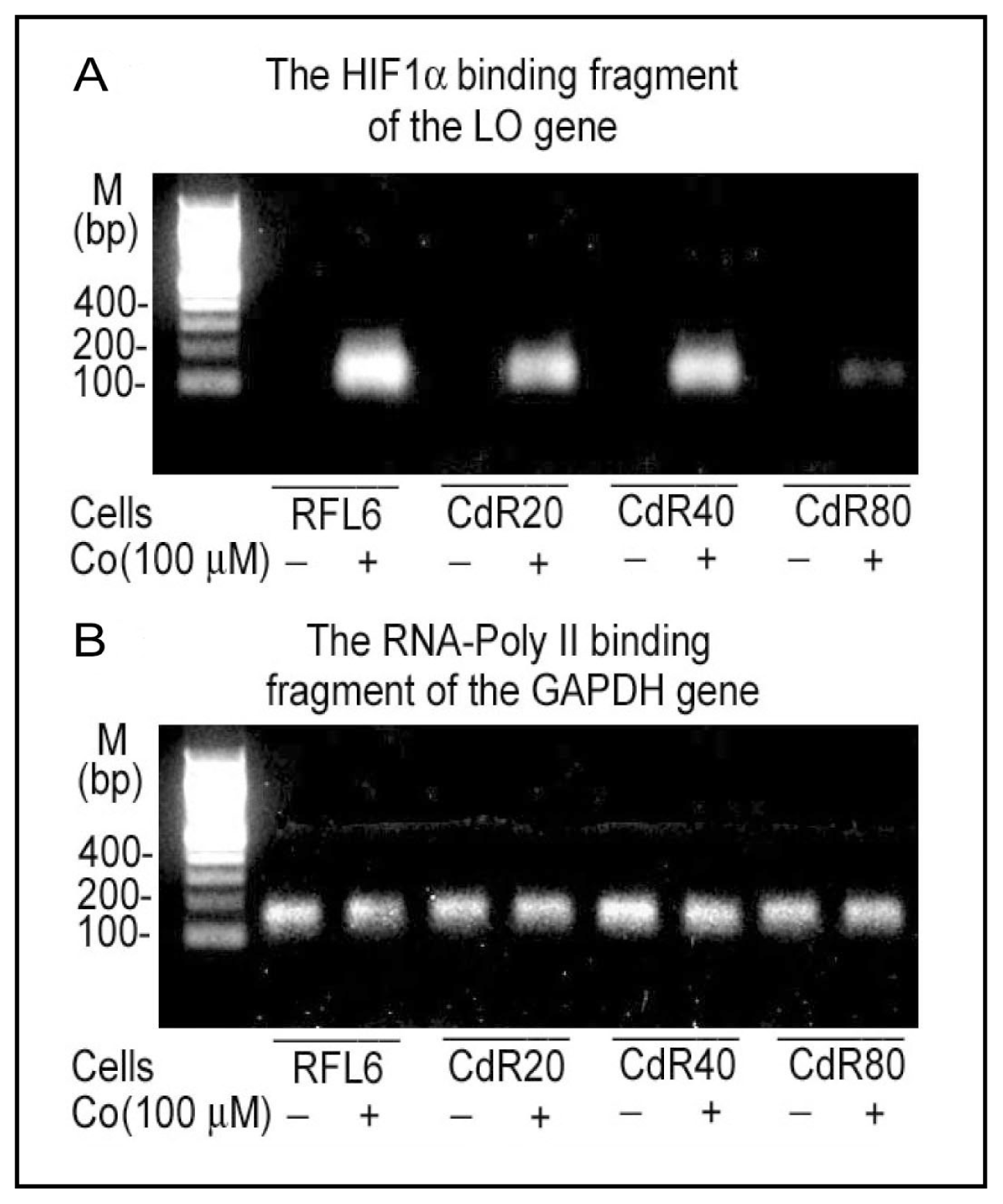{kind=link}
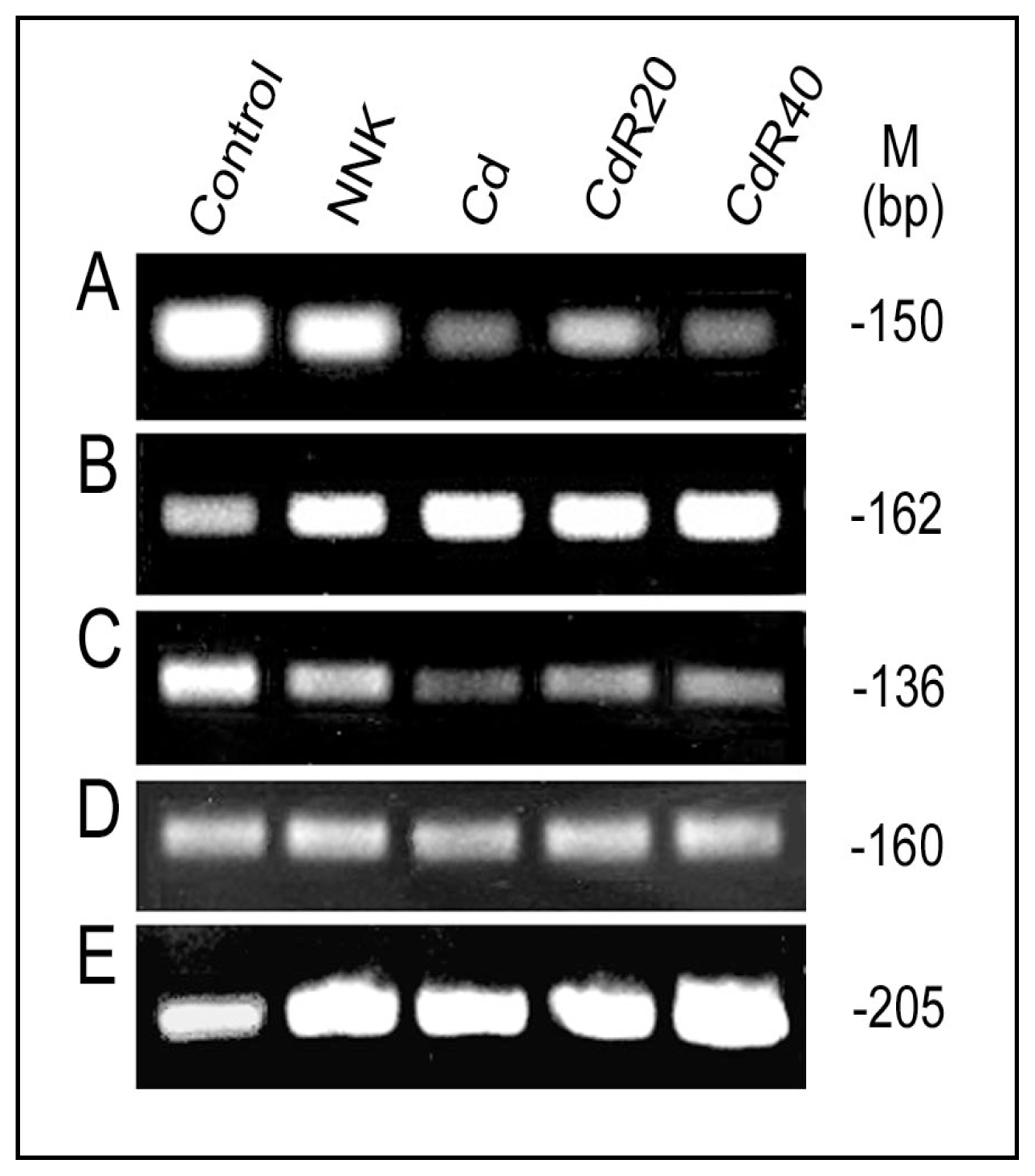{kind=link}
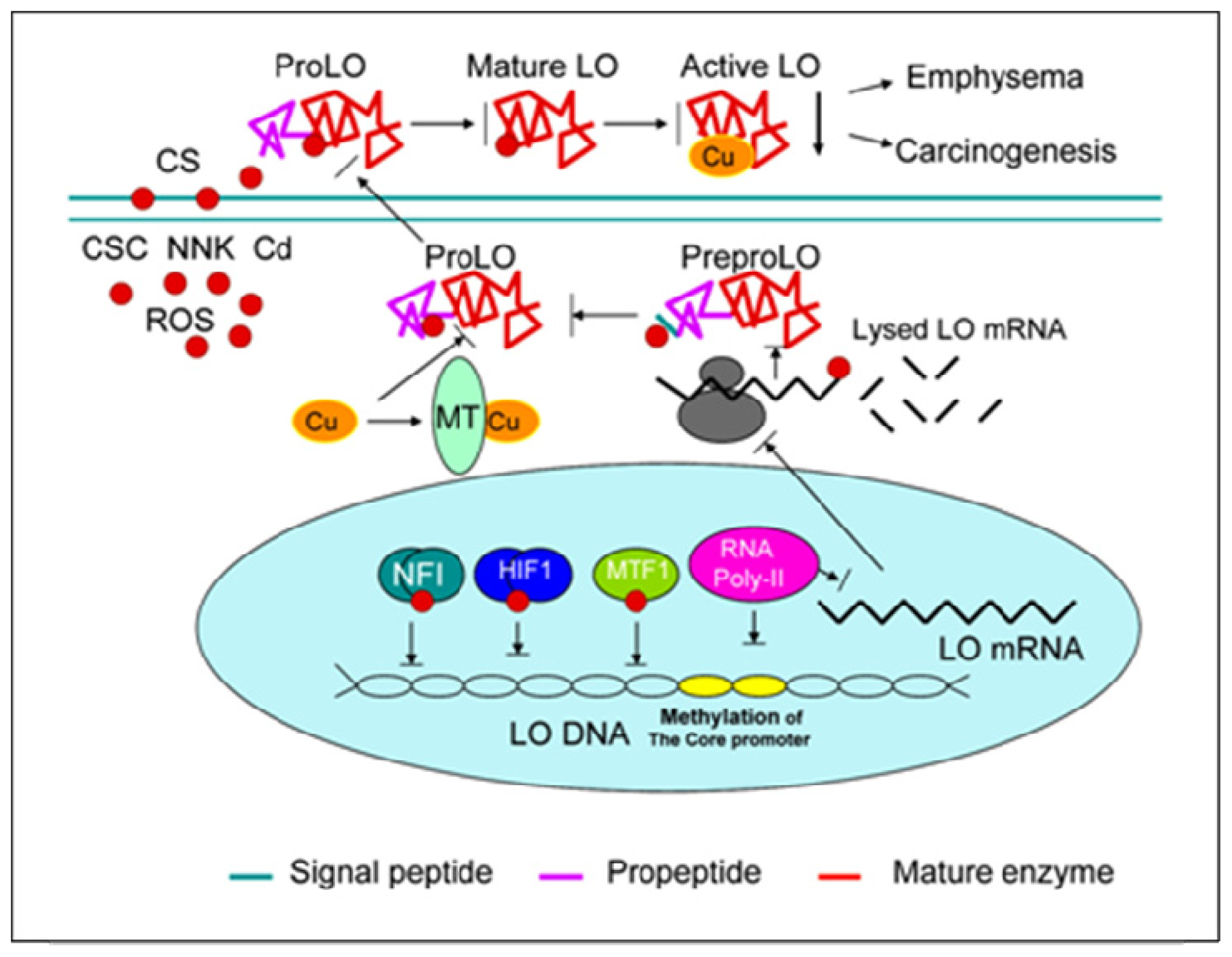
{kind=link}
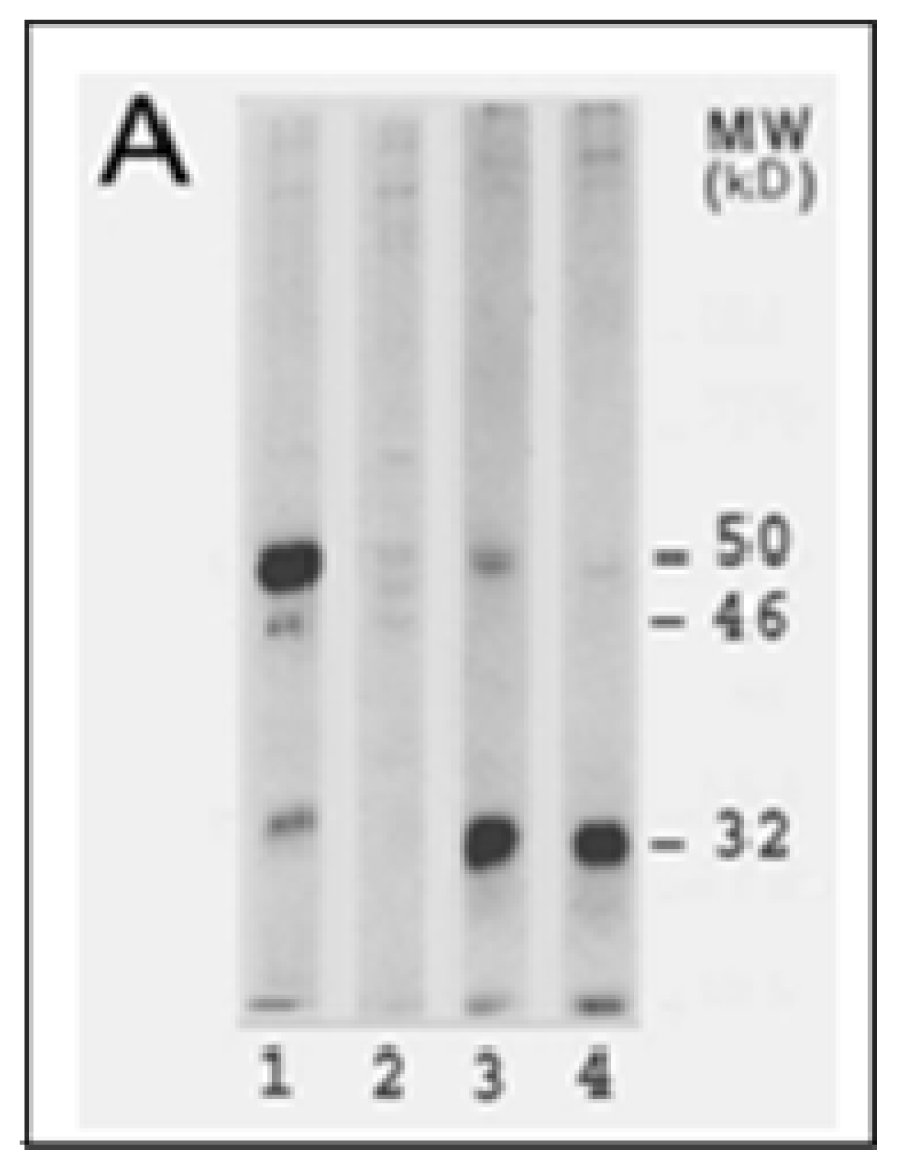
{kind=link}
1. Introduction
2. Major Pathogenic Agents of CS
2.1. CS, a Complex Chemical Mixture
2.2. NNK and Cd, Two of Major Pathogenic and Carcinogenic Components of CS
3. Lysyl Oxidase
3.1. Multiple Functions of LO in Biology
3.2. LO Molecular Structure
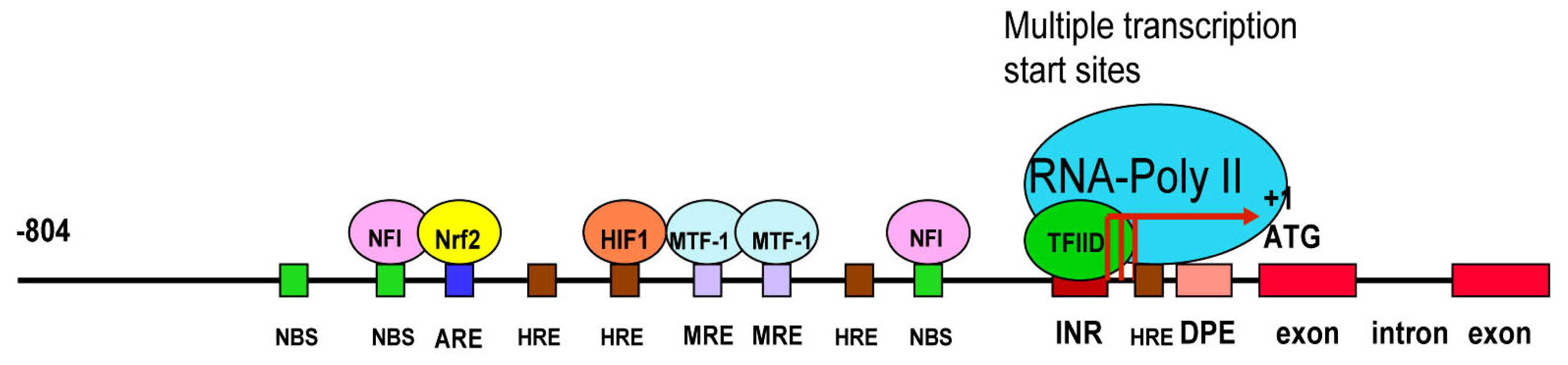
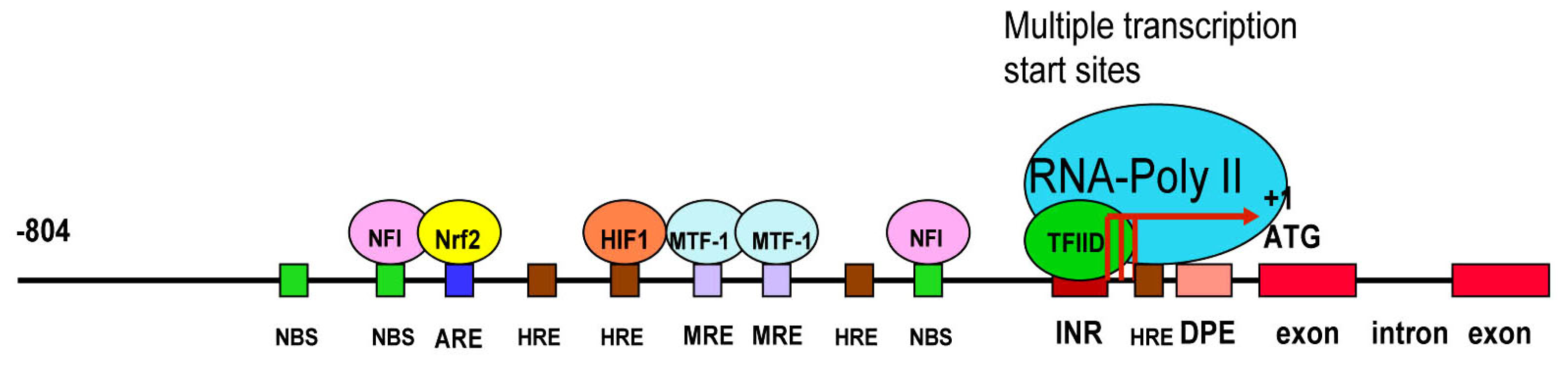
3.3. LO Gene Core Promoter
3.4. Redox-sensitive Cis-elements—the NFI-binding Site, HRE, MRE and ARE in the LO Promoter
4. LO as a Target of CS at Multiple Molecular Levels
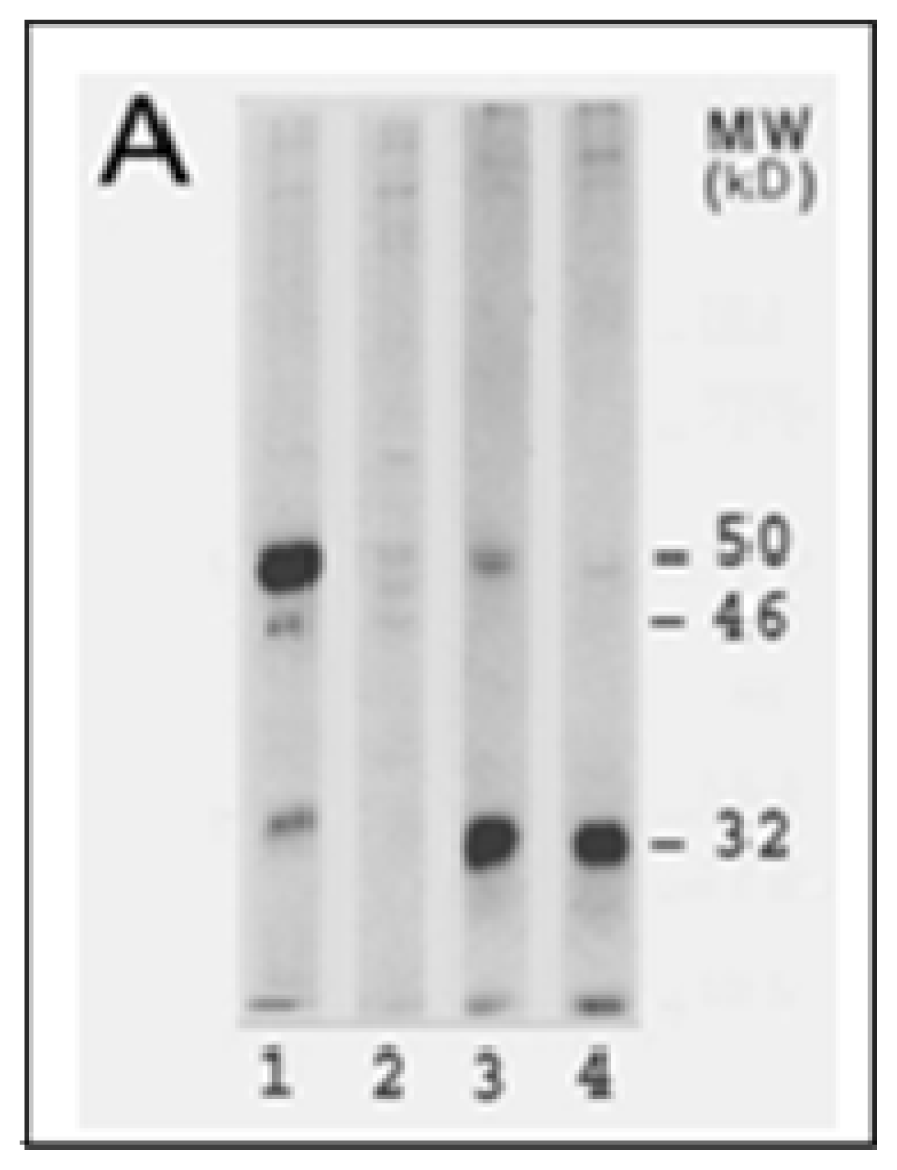
4.1. Inactivation of LO Catalytic Activity by CSC and Cd
4.2. Inhibition of LO Protein Synthesis and Processing by CSC and Cd
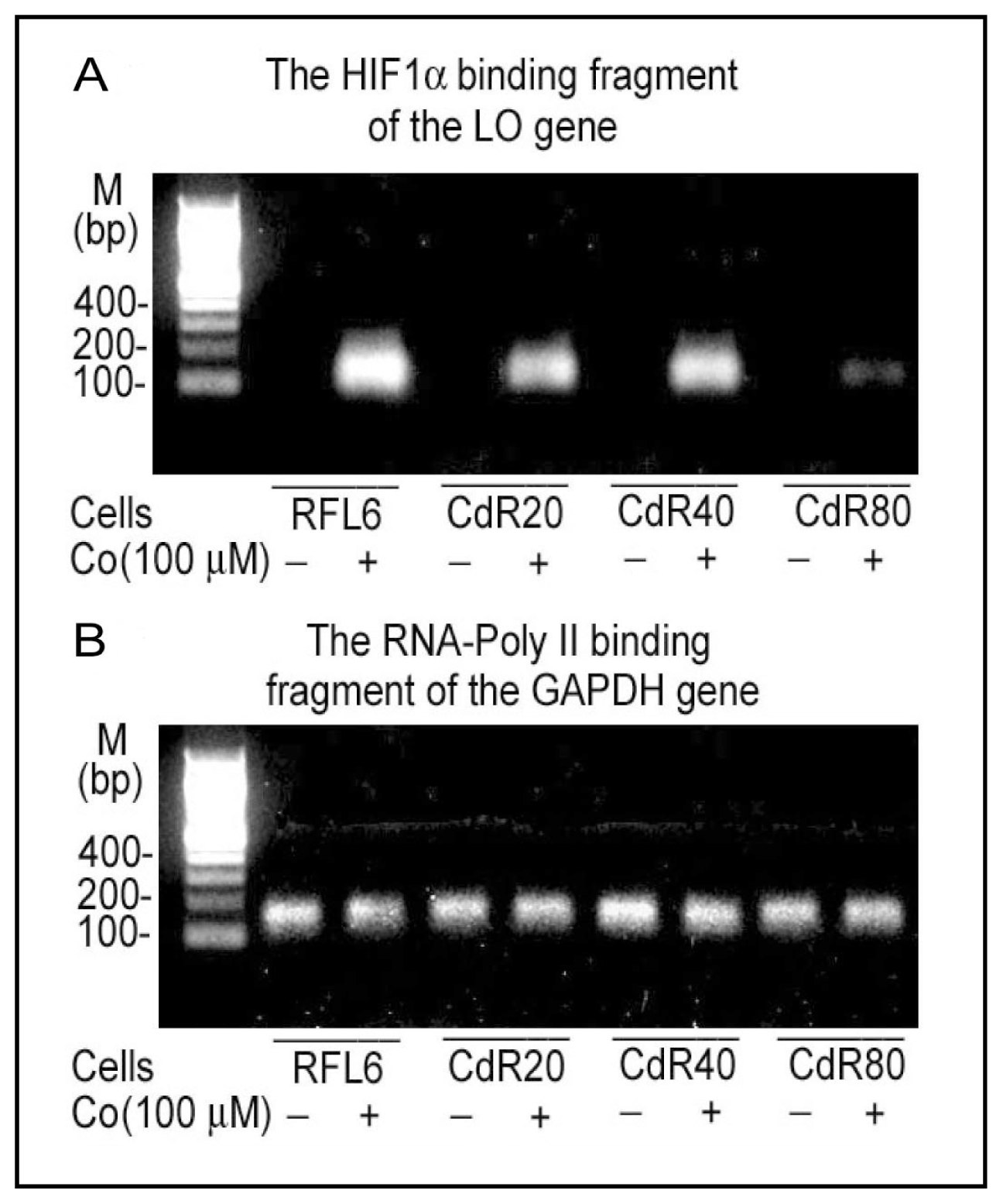
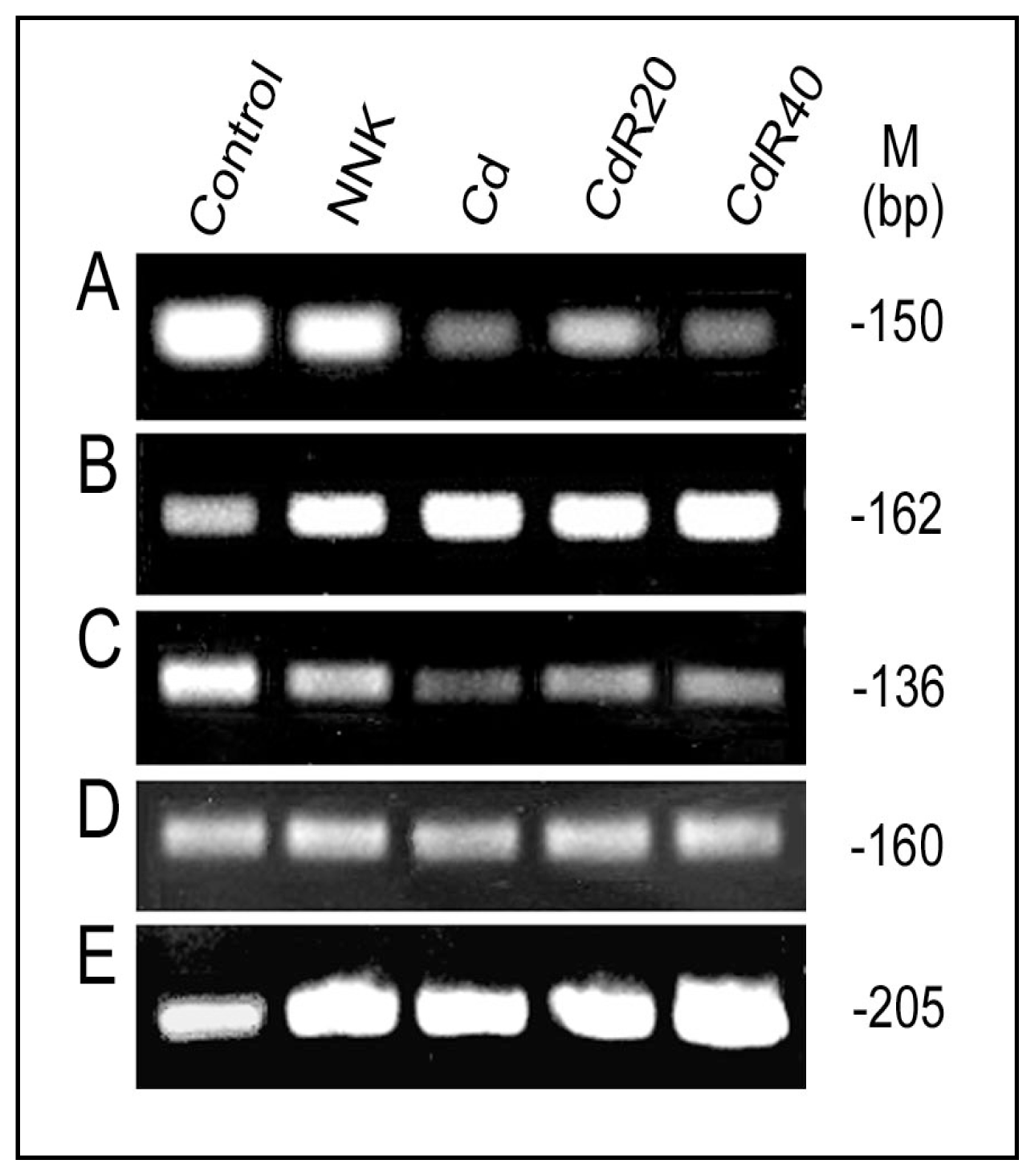
4.3. Downregulation of LO Transcription and Posttranscription by CSC/NNK and Cd
5. Downregulation of LO and Pathogenesis of the Lung
5.1. LO Deficiency and Emphysema
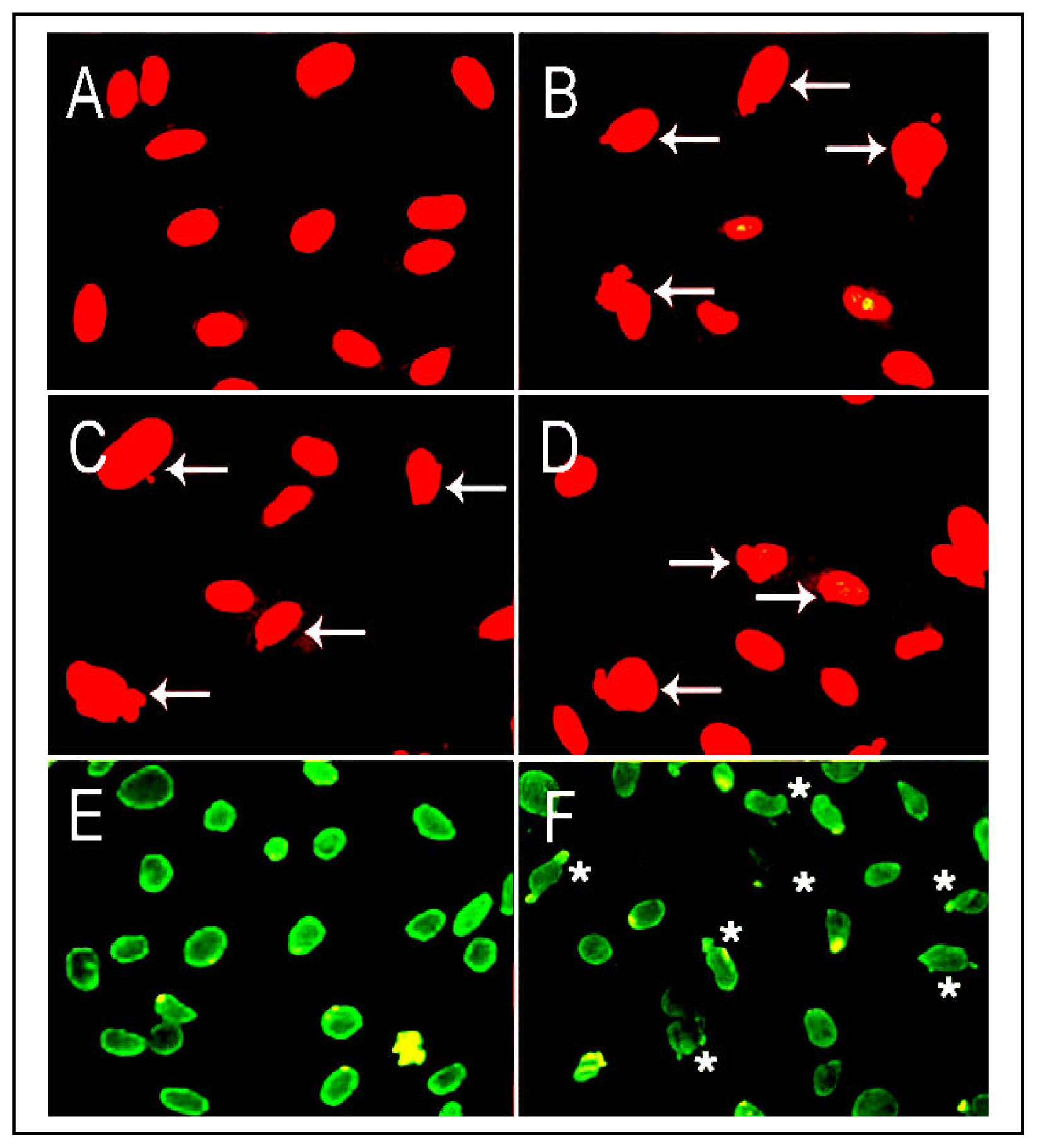
5.2. LO Deficiency and Carcinogenesis in the Lung and Other Organs
6. Conclusions
Acknowledgements
References
- American Lung Association in Washington. Lung Disease, 2011. Available online: http://www.alaw.org/lungdisease (accessed on 17 January 2011).
- E-MedicineHealth. Lung Cancer, 2011. Available online: http://www.emedicinehealth.com/lung_cancer/article_em.htm (accessed on 17 January 2011).
- Mackay, J; Eriksen, M. The Tobacco Atlas; WHO; Geneva, Switzerland, 2002. [Google Scholar]
- Hecht, SS. Cigarette smoke and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol 2002, 3, 461–469. [Google Scholar]
- Leischow, SJ; Djordjevic, MV. Smoking reduction and tobacco-related cancers: The more things change, the more they stay the same. J. Natl. Cancer Inst 2004, 96, 86–87. [Google Scholar]
- American Cancer Society. Tobacco-related Cancers Fact Sheet, 2011. Available online: http://www.cancer.org/Cancer/CancerCauses/TobaccoCancer/tobacco-related-cancer-fact-sheet (accessed on 17 January 2011).
- Snider, GL; Lucey, EC; Stone, PJ. Animal models of emphysema. Am. Rev. Respir. Dis 1986, 133, 149–169. [Google Scholar]
- Hautamaki, RD; Kobayashi, DK; Senior, RM; Shapiro, SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997, 277, 2002–2004. [Google Scholar]
- Shapiro, SD; Goldstein, NM; Houghton, AM; Kobayashi, DK; Kelley, D; Belaaouaj, A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol 2003, 163, 2329–2335. [Google Scholar]
- Wright, JL; Churg, A. Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. Am. J. Physiol 1995, 268, L17–20. [Google Scholar]
- D’Armiento, JL; Dalal, SS; Okada, Y; Berg, RA; Chada, K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 1992, 71, 955–961. [Google Scholar]
- Kuper, H; Boffetta, P; Adami, HO. Tobacco use and cancer causation: association by tumour type. J. Intern. Med 2002, 252, 206–224. [Google Scholar]
- Mulcahy, S. The toxicology of cigarette smoke and environmental tobacco smoke. 2011. Available online: http://www.skynet.ie/~stephen/reports/bc4927.html (accessed on 17 January 2011).
- Hoffmann, D; Wynder, EL. Marquardt, H, Schaefer, SG, McClellan, R, Welsch, F, Eds.; Active and passive smoking. In Toxicology; Academic Press: New York, NY, USA, 1999; pp. 879–898. [Google Scholar]
- Hoffmann, D; Hoffmann, I; El-Baypumy, K. The less harmful cigarette: a controversial issue. A tribute to Ernst L. Wynder. Chem. Res. Toxicol 2001, 14, 767–790. [Google Scholar]
- Church, T; Pryor, WA. Free-radical chemistry of cigarette smoke and its toxilogical implications. Environ. Health Perspect 1985, 64, 111–126. [Google Scholar]
- Schick, S; Glantz, S. Philip Morris toxicological experiments with fresh sidestream smoke: More toxic than mainstream smoke. Tob. Control 2005, 14, 396–404. [Google Scholar]
- Hecht, SS. Biochemistry, biology and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol 1998, 11, 560–603. [Google Scholar]
- Chepiga, T; Morton, M; Murphy, P; Avalos, J; Bombick, B; Doolittle, D; Borgerding, M; Swauger, J. A comparison of the mainstream smoke chemistry and mutagenicity of a representative sample of the US cigarette market with two Kentucky reference cigarettes (K1R4F and K1R5F). Food Chem. Toxicol 2000, 38, 949–962. [Google Scholar]
- Belinsky, SA; Foley, JF; White, CM; Anderson, MW; Maronpot, RR. Dose-response relationship between O6-methylguanine formation in Clara cells and induction of pulmonary neoplasia in rat by 4-(methyl nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res 1990, 50, 3772–3780. [Google Scholar]
- Jalas, JR; Ding, X; Murphy, SE. Comparative metabolism of the tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol by rat cytochrome P450 2A3 and human cytochrome P450 2A13. Drug Metab. Dispos 2003, 31, 1199–1202. [Google Scholar]
- Lin, RK; Hsieh, YS; Lin, P; Hsu, HS; Chen, CY; Tang, YA; Lee, CF; Wang, YC. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Invest 2010, 120, 521–532. [Google Scholar]
- Castonguay, A; Foiles, PG; Trushin, N; Hecht, SS. Study of DNA methylation by tobacco-specific N-nitrosamines.
- Pfeifer, GP; Denissenko, MF; Olivier, M; Tretyakova, N; Hecht, SS; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar]
- Beryllin, Cadmium. Mercury and Exposures in the Glass Manufacturing Industry. Monographs on the Evaluation of Carcinogenic Risk to Humans; IARC: Lyon, France, 1993; 58, pp. 119–236.
- Tobacco Smoke and Involuntary Smoking. In Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans; IARC: Lyon, France, 2004; Volume 83.
- Post, C; Johansson, B; Allenmark, S. Organ distribution and protein binding of cadmium in autopsy material from heavy smokers. Environ. Res 1984, 34, 29–37. [Google Scholar]
- Paakko, P; Kokkonen, P; Anttila, S; Kalliomaki, P-L. Cadmium and chromium as markers of smoking in human lung tissue. Environ. Res 1989, 49, 197–207. [Google Scholar]
- Coultas, DB; Samet, JM. Hensley, MJ, Saunders, NA, Eds.; Cigarette smoking. In Clinical Epidemiology of Chronic Obstractive Pulmonary Disease; Marcel Dekker: New York, NY, USA, 1989; pp. 109–138. [Google Scholar]
- Davision, AG; Newman-Taylor, AJ; Darbyshire, J; Chettle, DR; Guthrie, CJG; O’Malley, D; Mason, HJ; Fayers, PM; Venables, KM; Pickering, CAC; Franklin, D; Scott, MC; Holden, H; Wright, AL. Cadmium fume inhalation and emphysema. Lancet 1988, 1, 663–667. [Google Scholar]
- Kazantzis, G; Blacks, RG. Cook, ME, Hiscock, SA, Morrow, H, Volpe, RA, Eds.; A mortality study of cadmium exposed workers. Proceedings of the Seventh International Cadmium Conference, New Orleans, LA, USA, 6–8 April 1992, Cadmium Association/Cadmium Council: New Orleans, LA, USA/Reston, VA, USA, 1992; pp. 150–157. [Google Scholar]
- Lane, RE; Campbell, ACP. Fatal emphysema in two men making a copper cadmium alloy. Brit. J. Indust. Med 1954, 11, 118–122. [Google Scholar]
- Kimbel, P. Petty, TL, Ed.; Proteolytic damage and emphysema pathogenesis. In Chronic Obstructive Pulmonary Disease; Marcel Dekker: New York, NY, USA, 1985; pp. 105–127. [Google Scholar]
- Snider, GL; Hayes, JA; Korthy, AL; Lewis, GP. Centrilobular emphysema experimentally induced by cadmium chloride aerosol. Am. Rev. Respir. Dis 1973, 108, 40–47. [Google Scholar]
- Waisberg, M; Joseph, P; Hale, B; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar]
- Ninth Report on Carcinogens. In National Toxicology Program; Research Triangle Park: NC, USA, 2000.
- Glaser, U; Hochrainer, D; Otto, FJ; Oldiges, H. Carcinogenicity and toxicity of four cadmium compounds inhaled by rats. Toxicol. Environ. Chem 1990, 27, 153–162. [Google Scholar]
- Takenaka, S; Oldiges, H; Konig, H; Hochrainer, D; Oberdorster, G. Carcinogenicity of cadmium chloride aerosols in W. rats. J. Natl. Cancer Inst 1983, 70, 367–373. [Google Scholar]
- Gao, S; Chen, K; Zhao, Y; Rich, CB; Chen, L; Li, SJ; Toselli, P; Stone, P; Li, W. Transcriptional and posttranscriptional inhibition of lysyl oxidase expression by cigarette smoke condensate in cultured rat fetal lung fibroblasts. Toxicol. Sci 2005, 87, 197–203. [Google Scholar]
- Chen, L-J; Zhao, Y; Gao, S; Chou, I-N; Toselli, P; Stone, P; Li, W. Downregulation of lysyl oxidase and upregulation of cellular thiols in rat fetal lung fibroblasts treated with cigarette smoke condensate. Toxicol. Sci 2005, 83, 372–379. [Google Scholar]
- Kagan, HM; Li, W. Lysyl oxidase: Properties, specificity and biological roles inside of the cell. J. Cell. Biochem 2003, 88, 660–672. [Google Scholar]
- Giampuzzi, M; Oleggini, R; Di Donato, A. Demonstration of in vitro interaction between tumor suppressor lysyl oxidase and histone H1 and H2: Definition of the regions involved. Biochim. Biophys. Acta 2003, 1647, 245–251. [Google Scholar]
- Kagan, HM; Williams, MA; Calaman, SD; Berkowitz, EM. Histone H1 is a substrate for lysyl oxidase and contains endogenous sodium borotritide-reducible residues. Biochem. Biophys. Res. Commun 1983, 115, 186–192. [Google Scholar]
- Li, W; Nugent, MA; Zhao, Y; Chau, AN; Li, SJ; Chou, IN; Liu, G; Kagan, HM. Lysyl oxidase oxidizes basic fibroblast growth factor and inactivates its mitogenic potential. J. Cell Biochem 2003, 88, 152–164. [Google Scholar]
- Li, W; Nellaiappan, K; Strassmaier, T; Graham, L; Thomas, KM; Kagan, HM. Localization and activity of lysyl oxidase within nuclei of fibrogenic cells. Proc. Natl. Acad. Sci. USA 1997, 94, 12817–12822. [Google Scholar]
- Mello, MLS; Contente, S; Vidal, BC; Planding, W; Schenck, U. Modulation of ras transformation affecting chromatin supraorganization as assessed by image analysis. Exp. Cell Res 1995, 220, 374–382. [Google Scholar]
- Nellaiappan, K; Risitano, A; Liu, G; Nicklas, G; Kagan, HM. Fully processed lysyl oxidase catalyst translocates from the extracellular space into nuclei of aortic smooth muscle cells. J. Cell. Biochem 2000, 79, 576–582. [Google Scholar]
- Lazarus, HM; Cruikshank, WW; Narasimhan, N; Kagan, HM; Center, DM. Induction of human monocyte motility by lysyl oxidase. Matrix Biol 1995, 14, 727–731. [Google Scholar]
- Lucero, HA; Ravid, K; Grimsby, JL; Rich, CB; DiCamillo, SJ; Mäki, JM; Myllyharju, J; Kagan, HM. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascular smooth muscle cells. J. Biol. Chem 2008, 283, 24103–24117. [Google Scholar]
- Li, W; Liu, G; Chou, I-N; Kagan, HM. Hydrogen peroxide-mediated, lysyl oxidase-dependent chemotaxis of vascular smooth muscle cells. J. Cell. Biochem 2000, 78, 550–557. [Google Scholar]
- Erler, JT; Bennewith, KL; Nicolau, M; Dornhofer, N; Kong, C; Le, QT; Chi, JT; Jeffrey, SS; Giaccia, AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar]
- Kenyon, K; Contente, S; Trackman, PC; Tang, J; Kagan, HM; Friedman, RM. Lysyl oxidase and rrg messenger RNA. Science 1991, 253, 802. [Google Scholar]
- Kosonen, T; Uriu-Hare, JY; Clegg, MS; Keen, CL; Rucker, RB. Incorporation of copper into lysyl oxidase. Biochem. J 1997, 327, 283–289. [Google Scholar]
- Wan, SX; Mure, M; Medzihradszky, KF; Burlingame, AL; Brown, DE; Dooley, DM; Smith, AJ; Kagan, HM; Klinman, JP. A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science 1996, 273, 1078–1084. [Google Scholar]
- Gacheru, SN; Trackman, PC; Shah, MA; O’Gara, CY; Spacciapoli, P; Greenaway, FT; Kagan, HM. Structural and catalytic properties of copper in lysyl oxidase. J. Biol. Chem 1990, 265, 19022–19027. [Google Scholar]
- Csiszar, K. Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol 2001, 70, 1–32. [Google Scholar]
- Bazan, JF. A novel family of growth factor receptors: A common binding domain in the growth hormone, prolactin, erythropoietin and IL-6 receptors and the p75 IL-2 receptor β-chain. Biochem. Biophys. Res. Commun 1989, 164, 788–795. [Google Scholar]
- Bazan, JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc. Natl. Acad. Sci. USA 1990, 87, 6934–6938. [Google Scholar]
- Gao, S; Zhao, Y; Kong, L; Toselli, P; Chou, I-N; Stone, P; Li, W. Cloning and characterization of the rat lysyl oxidase gene promoter: Identification of core promoter elements and functional nuclear factor I binding sites. J. Biol. Chem 2007, 282, 25322–25337. [Google Scholar]
- Corden, J; Wasylyk, B; Buchwalder, A; Sassone-Corsi, P; Kedinger, C; Chambon, P. Promoter sequences of eukaryotic protein-coding genes. Science 1980, 209, 1406–1414. [Google Scholar]
- Smale, ST; Kadonaga, JT. The RNA polymerase II core promoter. Annu. Rev. Biochem 2003, 72, 449–479. [Google Scholar]
- Gronostajski, RM. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar]
- Bandyopadhyay, S; Gronostajski, RM. Identification of a conserved oxidation-sensitive cysteine residue in the NFI family of DNA binding proteins. J. Biol. Chem 1994, 269, 29949–29955. [Google Scholar]
- Bandyopadhyay, S; Starke, DW; Mieyal, JJ; Gronostajski, RM. Thioltranferase (Glutaredoxin) reactivates the DNA-binding activity of oxidation-inactivated Nuclear Factor I. J. Biol. Chem 1998, 273, 392–397. [Google Scholar]
- Semenza, GL. Expression of hypoxia-inducible factor 1: Mechanism and consequences. Biochem. Pharmacol 2000, 59, 47–53. [Google Scholar]
- Déry, MA; Michaud, MD; Richard, DE. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol 2005, 37, 535–540. [Google Scholar]
- Giedroc, DP; Chen, X; Apuy, JL. Metal response element (MRE) binding transcription factor-1 (MTF-1): Structure, function, and regulation. Antioxid. Redox Signal 2001, 3, 577–596. [Google Scholar]
- Stuart, GW; Searle, PF; Chen, HY; Brinster, R; Palmiter, RD. A 12-base pair DNA motif that is repeated several times in metallothione gene promoters confers metal regulation to a heterologous. Proc. Natl. Acad. Sci. USA 1984, 81, 7318–7322. [Google Scholar]
- Lichtlen, P; Wang, Y; Belser, T; Georgiev, O; Certa, U; Sack, R; Schaffner, W. Target gene search for the metal-responsive transcription factor MTF-1. Nuc. Aci Res 2001, 29, 1514–1523. [Google Scholar]
- Copple, IM; Goldring, CE; Kitteringham, NR; Park, BK. The Nrf2-Keap1 defence pathway: Role in protection against drug-induced toxicity. Toxicology 2008, 246, 24–33. [Google Scholar]
- Wang, GL; Jiang, B-H; Rue, EA; Semenza, GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar]
- Weissmann, N. Hypoxia-driven mechanisms in lung biology and disease: a new review series of the ERS Lung Science Conference. Eur. Respir. J 2008, 3, 697–698. [Google Scholar]
- Yasuo, M; Mizuno, S; Kraskauskas, D; Bogaard, HJ; Natarajan, R; Cool, CD; Zamora, M; Voelkel, NF. Hypoxia inducible factor-1 {alpha} in human emphysema lung tissue. Eur. Respir. J 2010. [Google Scholar] [CrossRef]
- Wang, GL; Semenza, GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar]
- Epstein, AC; Gleadle, JM; McNeill, LA; Hewitson, KS; O’Rourke, J; Mole, DR; Mukherji, M; Metzen, E; Wilson, MI; Dhanda, A; Tian, YM; Masson, N; Hamilton, DL; Jaakkola, P; Barstead, R; Hodgkin, J; Maxwell, PH; Pugh, CW; Schofield, CJ; Ratcliffe, PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar]
- Zhao, Y; Gao, S; Chou, I-N; Toselli, P; Stone, P; Li, W. Inhibition of the expression of lysyl oxidase and its substrates in cadmium-resistant rat fetal lung fibroblasts. Toxicol. Sci 2006, 90, 478–489. [Google Scholar]
- Chou, DK; Zhao, Y; Gao, S; Chou, I-N; Toselli, P; Stone, P; Li, W. Perturbation of copper (Cu) homeostasis and expression of Cu-binding proteins in cadmium-resistant lung fibroblasts. Toxicol Sci 2007, 99, 267–276. [Google Scholar]
- Dubic, MA; Keen, CL; Rucker, RB. Elastin metabolism during prenatal lung development in the copper-deficient rat. Exp. Lung Res 1985, 8, 227–241. [Google Scholar]
- Harris, ED. Biochemical defect in chick lung resulting from copper deficiency. J. Nutr 1986, 116, 252–258. [Google Scholar]
- Soskel, NT; Watanabe, S; Sandberg, LB. Mechanisms of lung injury in the copper-deficient hamster model of emphysema. Chest 1984, 85, 70s–72s. [Google Scholar]
- Richards, MP. Recent developments in trace element metabolism and function: Role of metallothionein in copper and zinc metabolism. J. Nutr 1989, 119, 1062–1070. [Google Scholar]
- Brouwer, M; Hoexum-Brouwer, T; Cashon, RE. A putative glutathione-binding site in CdZn-metallothionein identified by epuilibrium binding and molecular modelling studies. Biochem. J 1993, 294, 219–225. [Google Scholar]
- Briggs, RW; Armitage, IM. Evidence for site-selective metal binding in calf liver metallothionein. J. Biol. Chem 1982, 257, 1259–1262. [Google Scholar]
- Morris, PE; Bernard, GR. Significance of glutathione in lung disease and implications for therapy. Am. J. Med. Sci 1994, 307, 119–127. [Google Scholar]
- Li, XY; Donaldson, K; Rahman, I; MacNee, W. An investigation of the role of glutathione in the increased permeability induced by cigarette smoke in vivo and in vitro. Am. Rev. Respir. Crit. Care Med 1994, 149, 1518–1525. [Google Scholar]
- Rahman, I; Li, XY; Donaldson, K; Harrison, HJ; MacNee, W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am. J. Physiol 1995, 269, L285–L292. [Google Scholar]
- Rahman, I. Regulation of glutathione in inflammation and chronic lung diseases. Mutat. Res 2005, 579, 58–80 . [Google Scholar]Int. J. Environ. Res. Public Health 2011, 8, 182.
- Zhao, Y; Chen, L; Gao, S; Toselli, P; Stone, P; Li, W. The critical role of the cellular thiol homeostasis in cadmium perturbation of the lung extracellular matrix. Toxicology 2010, 267, 60–69. [Google Scholar]
- Li, W; Chou, I-N; Boak, A; Kagan, HM. Down-regulation of lysyl oxidase in cadmium-resistant fibroblasts. Am. J. Respir. Cell Mol. Biol 1995, 13, 418–425. [Google Scholar]
- Li, W; Zhao, Y; Toselli, P; Stone, P. Cigarette smoke condensate downregulation of lysyl oxidase transcription mediated by inactivation of nuclear factor I. Toxicologist 2008, 102, 227, (abstract). [Google Scholar]
- Li, W; Zhao, Y; Gao, S; Toselli, P. Transcriptional regulation of the lysyl oxidase gene by the hypoxia-inducible factor 1. FASEB J 2010, 24. Meeting Abstract Supplement, No. 833.16, (abstract). [Google Scholar]
- Galan, A; Garcia-Bermejo, L; Troyano, A; Vilaboa, NE; Fernandez, C; de Blas, E; Aller, P. The role of intracellular oxidation in death induction (apoptosis and necrosis) in human promonocytic cells treated with stress inducers (cadmium, heat, X-ray). Eur. J. Cell Biol 2001, 80, 312–320. [Google Scholar]
- Zhao, Y; Gao, S; Zhou, J; Toselli, P; Li, W. NNK, a tobacco-specific carcinogen, inhibits the expression of lysyl oxidase, a tumor suppressor gene. Toxicologist 2011, (abstract in press). [Google Scholar]
- Bestor, TH. The DNA methyltransferase of mammals. Hum. Mol. Genet 2000, 9, 2395–2402. [Google Scholar]
- Ng, HH; Bird, A. DNA methylation and chromatin modification. Curr. Opin. Genet. Dev 1999, 9, 158–163. [Google Scholar]
- Takiguchi, M; Achanzar, WE; Qu, W; Li, G; Waalkes, MP. Effects of cadmium on DNA-(cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp. Cell Res 2003, 286, 355–365. [Google Scholar]
- Downs, JA. Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA Repair 2008, 7, 2020–2024. [Google Scholar]
- Xu, G-L; Bestor, TH; Bourchis, D; Hsieh, CL; Tommerrup, N; Bugge, M; Hulten, M; Qu, X; Russo, JJ; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar]
- Gisselsson, D; Bjork, J; Hoglund, M; Mertens, F; Cin, PD; Akerman, M; Mandahl, N. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol 2001, 158, 199–206. [Google Scholar]
- Davidson, JM. Biochemistry and turnover of lung interstitium. Eur. Respir. J 1990, 3, 1048–1063. [Google Scholar]
- Campa, JS; Harrison, NK; Laurent, G. Jolles, G, Karlsson, J-A, Taylor, J, Eds.; Regulation of matrix production in the airways. In T-Lymphocyte and Inflammatory Cell Research in Asthma; Academic Press: London, UK, 1993; pp. 221–235. [Google Scholar]
- Snider, GL. Emphysema: the first two centuries-and beyond. Am. Rev. Respir. Dis 1992, 146, 1615–1622. [Google Scholar]
- Soskel, NT; Watanabe, S; Hammond, E; Sandberg, LB; Renzetti, AD, Jr; Crapo, JD. A copper-deficient, zinc-supplemented diet produces emphysema in pig. Am. Rev. Respir. Dis 1982, 126, 316–325. [Google Scholar]
- Chvapil, M; Misiorowski, R. In vivo inhibition of lysyl oxidase by high dose of zinc. Proc. Soc. Exp. Biol. Med 1980, 164, 137–141. [Google Scholar]
- Grange, DK; Kaler, SG; Albers, GM; Petterchak, JA; Thorpe, CM; DeMello, DE. Severe bilateral panlobular emphysema and pulmonary arterial hypoplasia: unusual manifestations of Menkes disease. Am. J. Med. Genet 2005, 139A, 151–155. [Google Scholar]
- Kuhn, C; Starcher, BC. The effects of lathyrogens on the evolution of elastase-induced emphysema. Am. Rev. Respir. Dis 1980, 122, 453–460. [Google Scholar]
- Niewoehner, DE; Hoidal, JR. Lung fibrosis and emphysema: Divergent responses to a common injury. Science 1982, 217, 359–360. [Google Scholar]
- Fisk, DE; Kuhn, C. Emphysema-like changes in the lungs of blotchy mouse. Am. Rev. Respir. Dis 1976, 113, 787–797. [Google Scholar]
- Warburton, D; Shi, W. Lo, and the niche is knit: Lysyl oxidase activity and maintenance of lung, aorta, and skin integrity. Am. J. Pathol 2005, 167, 921–922. [Google Scholar]
- Liu, X; Zhao, Y; Gao, J; Pawlyk, B; Starcher, B; Spencer, JA; Yanagisawa, H; Zuo, J; Li, T. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat. Genet 2004, 36, 178–182. [Google Scholar]
- Maki, JM; Sormunen, R; Lippo, S; Kaarteenaho-Wiik, R; Soininen, R; Myllyharju, J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol 2005, 167, 927–936. [Google Scholar]
- Laurent, P; Janoff, A; Kagan, HM. Cigarette smoke blocks cross-linking of elastin. in vitro. Am. Rev. Respir. Dis 1983, 127, 189–192. [Google Scholar]
- Jackson, LE; Faris, B; Martin, BM; Jones, HV; Rich, CB; Foster, JA; Franzblau, C. The effect of β-aminopropionitrile on elastin gene expression in smooth muscle cell cultures. Biochem. Biophys. Res. Commun 1991, 179, 939–944. [Google Scholar]
- Giampuzzi, M; Botti, G; Duca, MD; Arata, L; Ghiggeri, G; Gusmano, R; Ravazzolo, R; Donato, AD. Lysyl oxidase activates the transcription activity of human collagene III promoter. J. Biol. Chem 2000, 275, 36341–36349. [Google Scholar]
- Giampuzzi, M; Botti, G; Gilli, M; Gusmano, R; Borel, A; Sommer, P; Di Donato, A. Down-regulation of lysyl oxidase-induced tumorigenic transformation in NRK-49F cells characterized by constitutive activation of ras-proto-oncogene. J. Biol. Chem 1995, 276, 29226–29232. [Google Scholar]
- Hamalainen, ER; Kemppainen, R; Kuivaniemi, H; Tromp, G; Vaheri, A; Pihlajaniemi, T; Kivirikko, KI. Quantitative polymerase chain reaction of lysyl oxidase mRNA in malignantly transformed human cell lines demonstrates that their low lysyl oxidase activity is due to low quantities of its mRNA and low levels of transcription of the respective gene. J. Biol. Chem 1995, 270, 21590–21593. [Google Scholar]
- Woznick, AR; Braddock, AL; Dulai, M; Seymour, ML; Callahan, RE; Welsh, RJ; Chmielewski, GW; Zelenock, GB; Hanley, SCJ. Lysyl oxidase expression in bronchogenic carcinoma. Am. J. Surg 2005, 189, 297–301. [Google Scholar]
- Kaneda, A; Wakazono, K; Tsukamoto, T; Watanabe, N; Yagi, Y; Tatematsu, M; Kaminishi, M; Sugimura, T; Ushijima, T. Lysyl oxidase is a tumor suppressor gene inactivated by methylation and loss of heterozygosity in human gastric cancers. Cancer Res 2004, 64, 6410–6415. [Google Scholar]
- Rost, T; Pyritz, V; Rathcke, IO; Gorogh, T; Dunne, AA; Werner, JA. Reduction of LOX- and LOXL2-mRNA expression in head and neck squamous cell carcinomas. Anticancer Res 2003, 23, 1565–1573. [Google Scholar]
- Wu, M; Min, C; Wang, X; Yu, Z; Kirsch, KH; Trackman, PC; Sonenshein, GE. Repression of BCL2 by the tumor suppressor activity of the lysyl oxidase propeptide inhibits transformed phenotype of lung and pancreatic cancer cells. Cancer Res 2007, 67, 6278–6285. [Google Scholar]
- Ren, C; Yang, G; Timme, TL; Wheeler, TM; Thompson, TC. Reduced lysyl oxidase messenger RNA levels in experimental and human prostate cancer. Cancer Res 1998, 58, 1285–1290. [Google Scholar]
- Decitre, M; Gleyzal, C; Raccurt, M; Peyrol, S; Aubert-Foucher, E; Csiszar, K; Sommer, P. Lysyl oxidase-like protein localizes to sites of de novo fibrinogenesis and in the early stromal reaction of ductal breast carcinomas. Lab. Invest 1998, 78, 143–151. [Google Scholar]
- Peyrol, S; Raccurt, M; Gerard, F; Gleyzal, C; Grimaud, JA; Sommer, P. Lysyl oxidase gene expression in the stromal reaction to in situ and invasive ductal breast carcinoma. Am. J. Pathol 1997, 150, 497–507. [Google Scholar]
- Bouez, C; Reynaud, C; Noblesse, E; Thépot, A; Gleyzal, C; Kanitakis, J; Perrier, E; Damour, O; Sommer, P. The lysyl oxidase (LOX) is absent in basal and squamous cell carcinomas and its knockdown induces an invading phenotype in a skin equivalent model. Clin. Cancer Res 2006, 12, 1463–1469. [Google Scholar]
- Csiszar, K; Fong, FST; Ujfalusi, A; Krawetz, SA; Salvati, EP; Mackenzie, JW; Boyd, CD. Somatic mutations of the lysyl oxidase gene on chromosome 5q23.1 in colorectal tumors. Int. J. Cancer 2002, 97, 636–642. [Google Scholar]
- Li, W; Li, SJ; Zhao, Y; Kagan, HM. Oxidation of histone H1 within cell nuclei by lysyl oxidase: Implications for changes in chromosomal stability. The First Meeting Bulletin of the American Society for Matrix Biology 2002, 1, 119, (abstract). [Google Scholar]

© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, W.; Zhou, J.; Chen, L.; Luo, Z.; Zhao, Y. Lysyl Oxidase, A Critical Intra- and Extra-Cellular Target in the Lung for Cigarette Smoke Pathogenesis. Int. J. Environ. Res. Public Health 2011, 8, 161-184. https://doi.org/10.3390/ijerph8010161
Li W, Zhou J, Chen L, Luo Z, Zhao Y. Lysyl Oxidase, A Critical Intra- and Extra-Cellular Target in the Lung for Cigarette Smoke Pathogenesis. International Journal of Environmental Research and Public Health. 2011; 8(1):161-184. https://doi.org/10.3390/ijerph8010161
Chicago/Turabian StyleLi, Wande, Jing Zhou, Lijun Chen, Zhijun Luo, and Yinzhi Zhao. 2011. "Lysyl Oxidase, A Critical Intra- and Extra-Cellular Target in the Lung for Cigarette Smoke Pathogenesis" International Journal of Environmental Research and Public Health 8, no. 1: 161-184. https://doi.org/10.3390/ijerph8010161