Mercury-Induced Externalization of Phosphatidylserine and Caspase 3 Activation in Human Liver Carcinoma (HepG2) Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Materials and Methods
Chemical and Growth Medium
Cell Culture
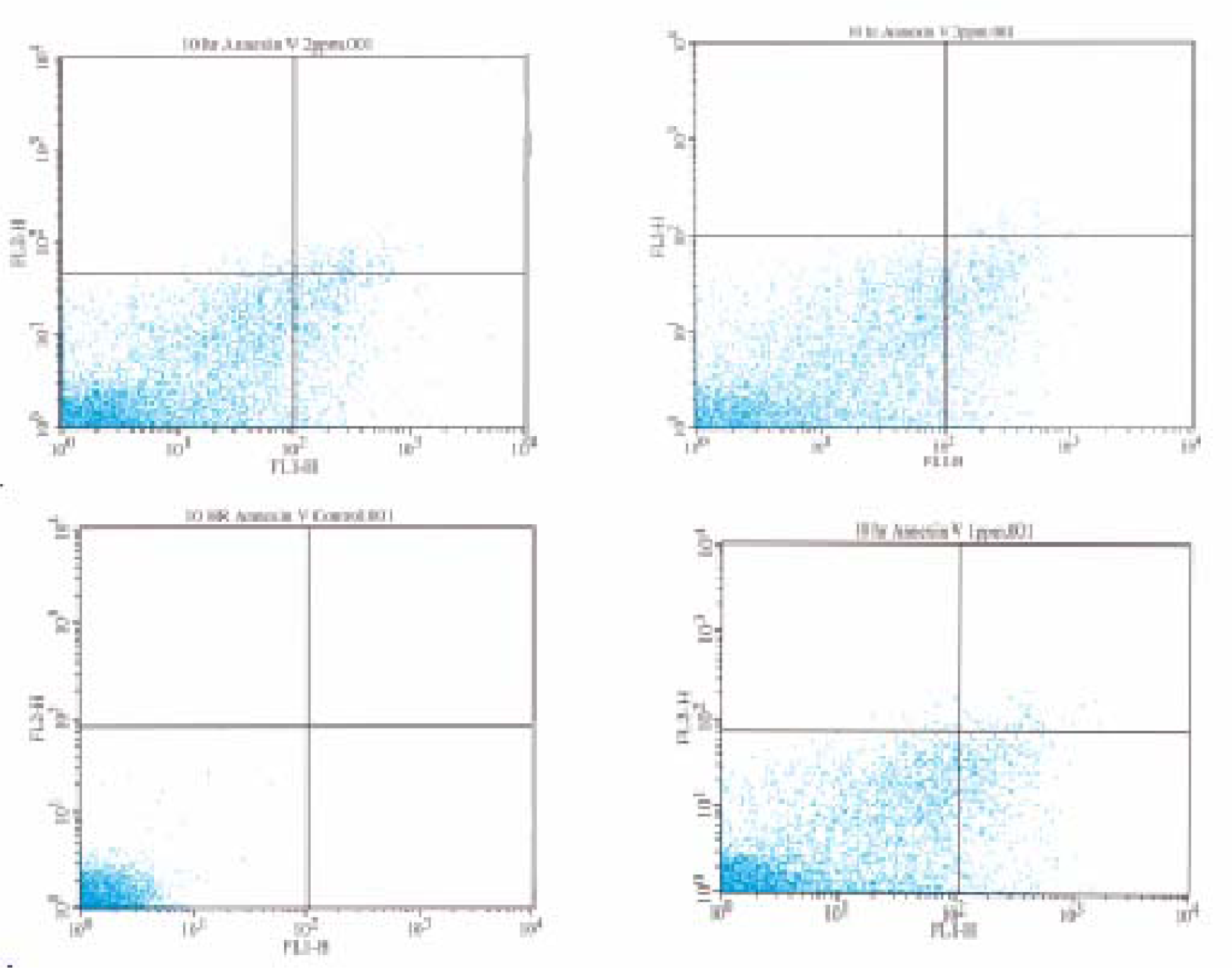
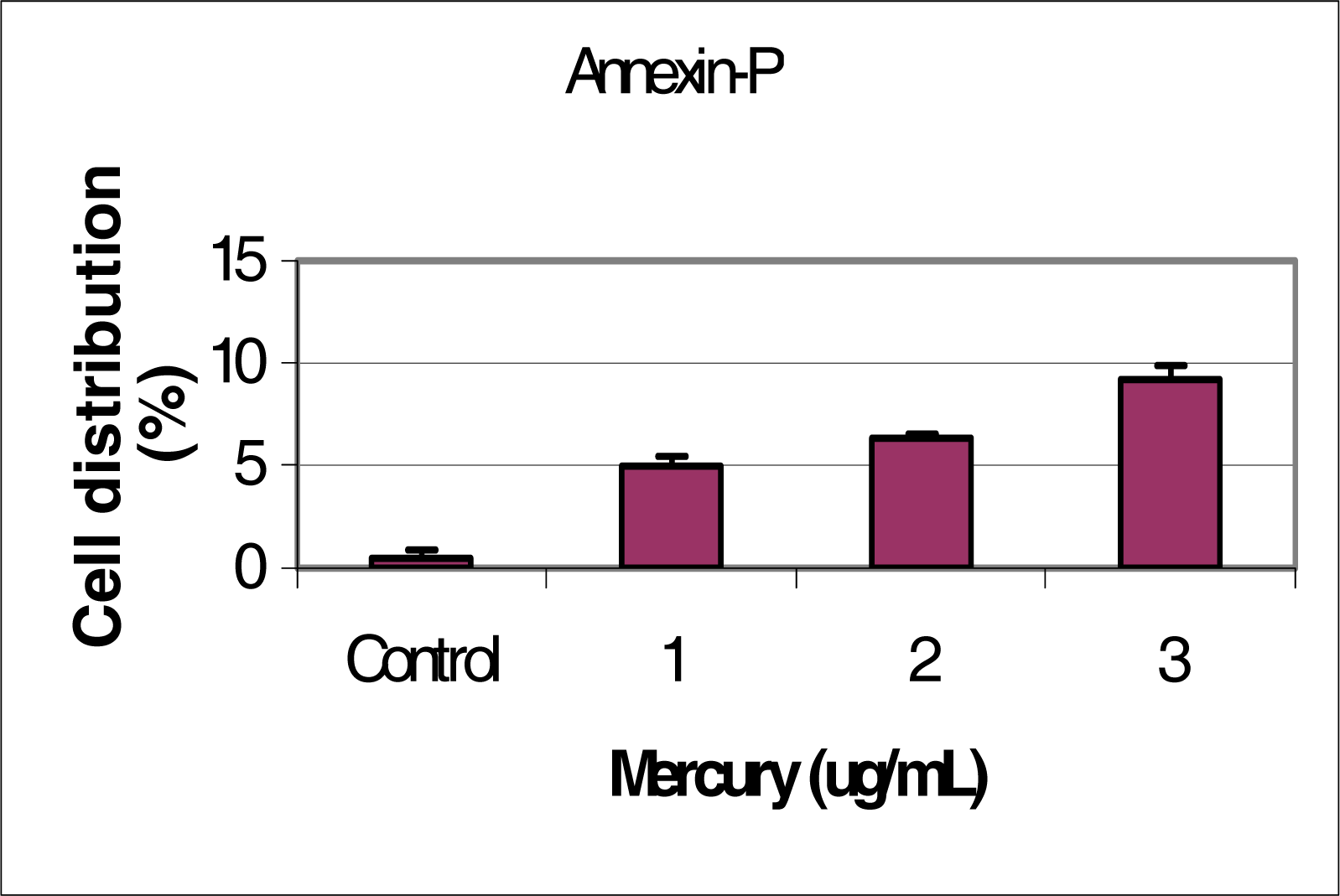
Flow Cytometric Analysis of Phosphatidylserine Externalization
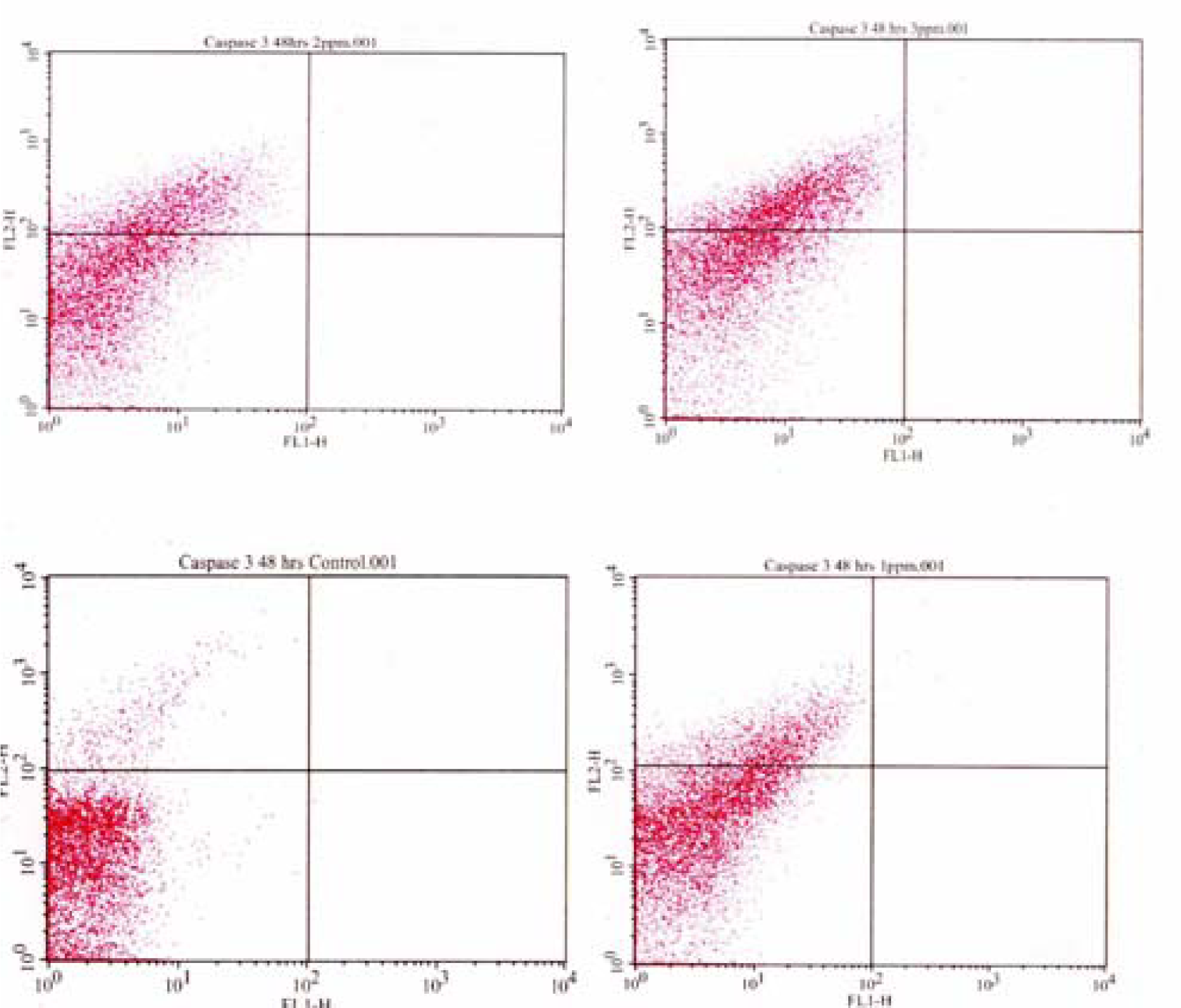
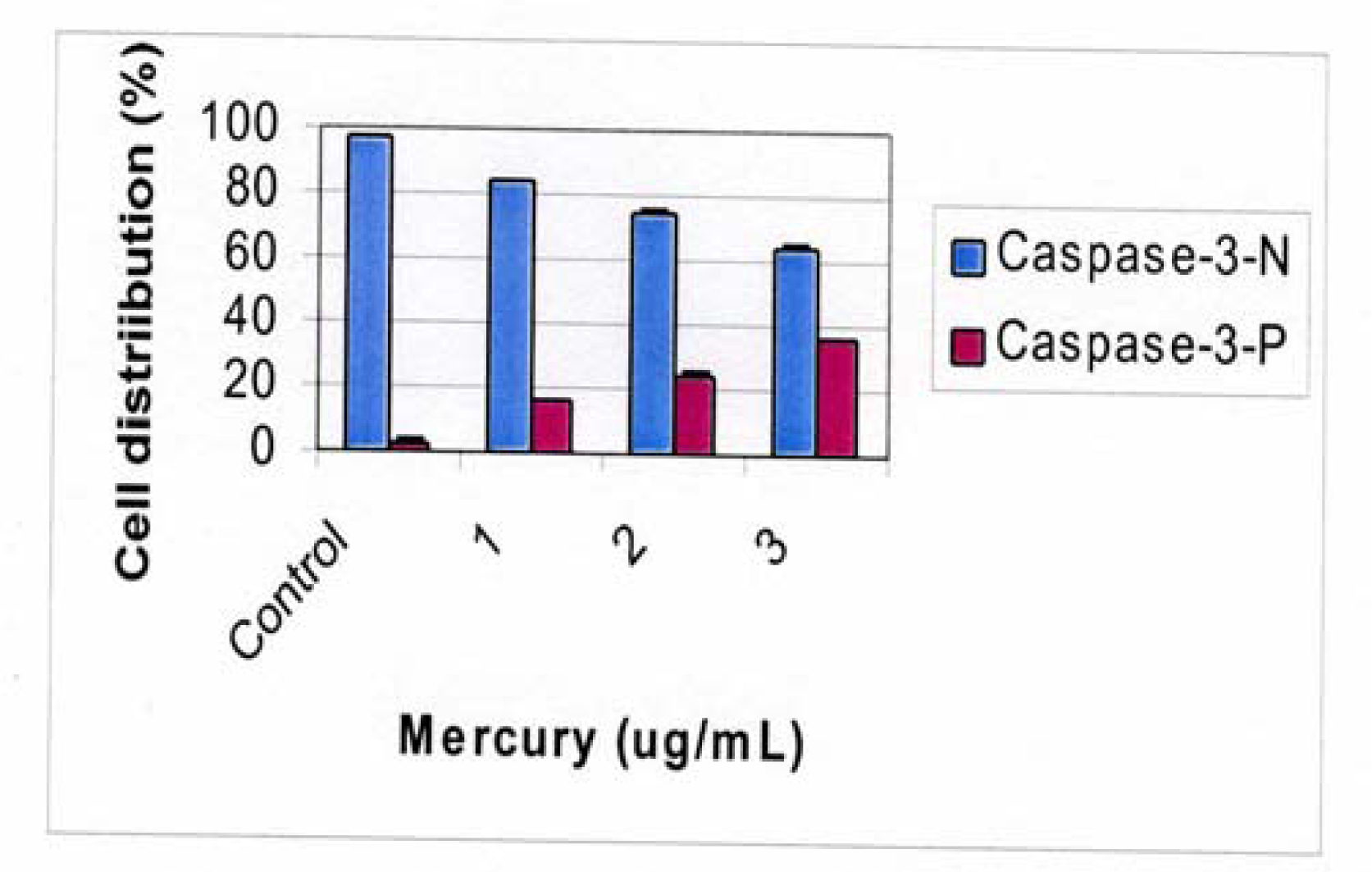
Flow Cytometric Analysis of Caspase 3 Activation
Results and Discussion
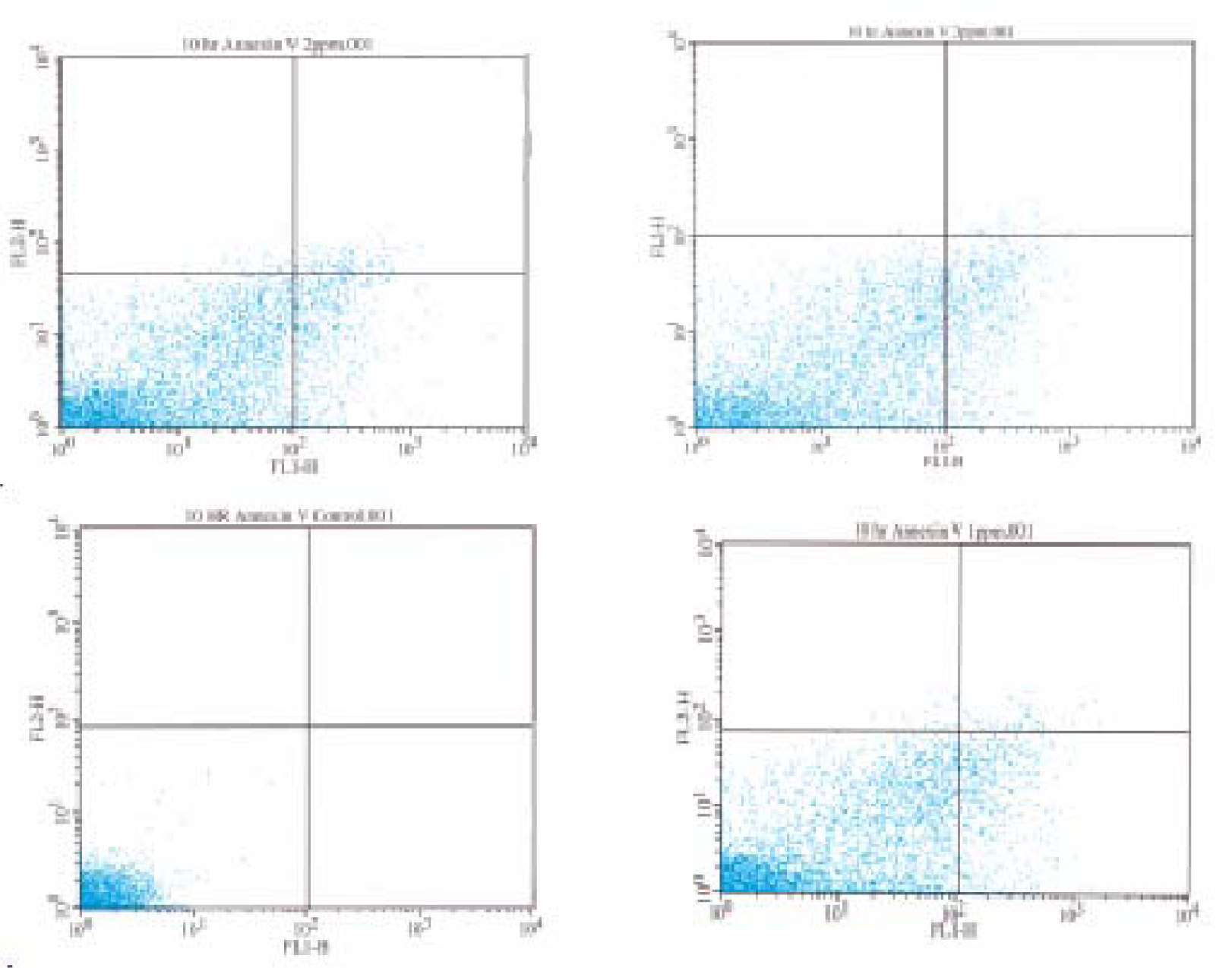
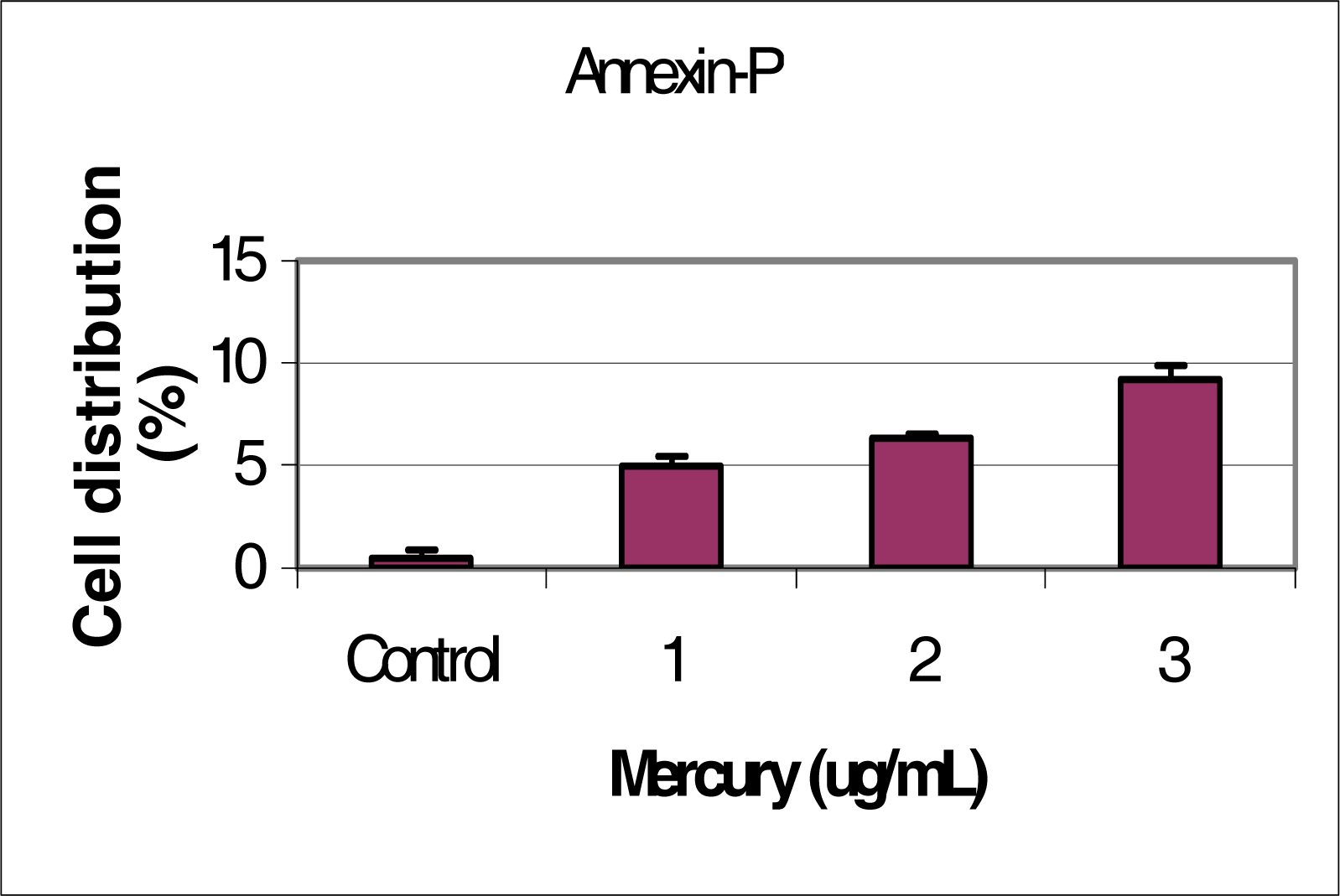
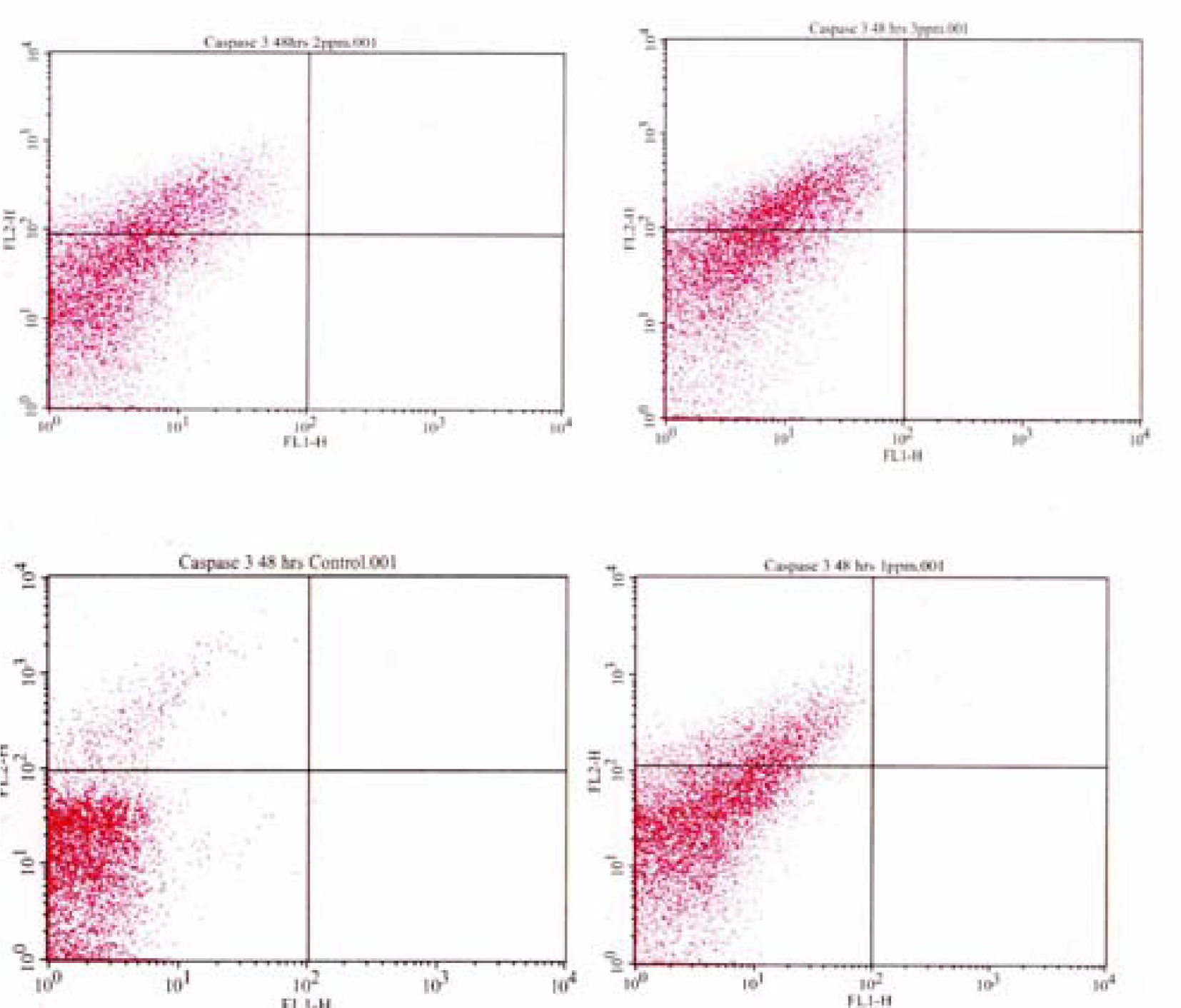
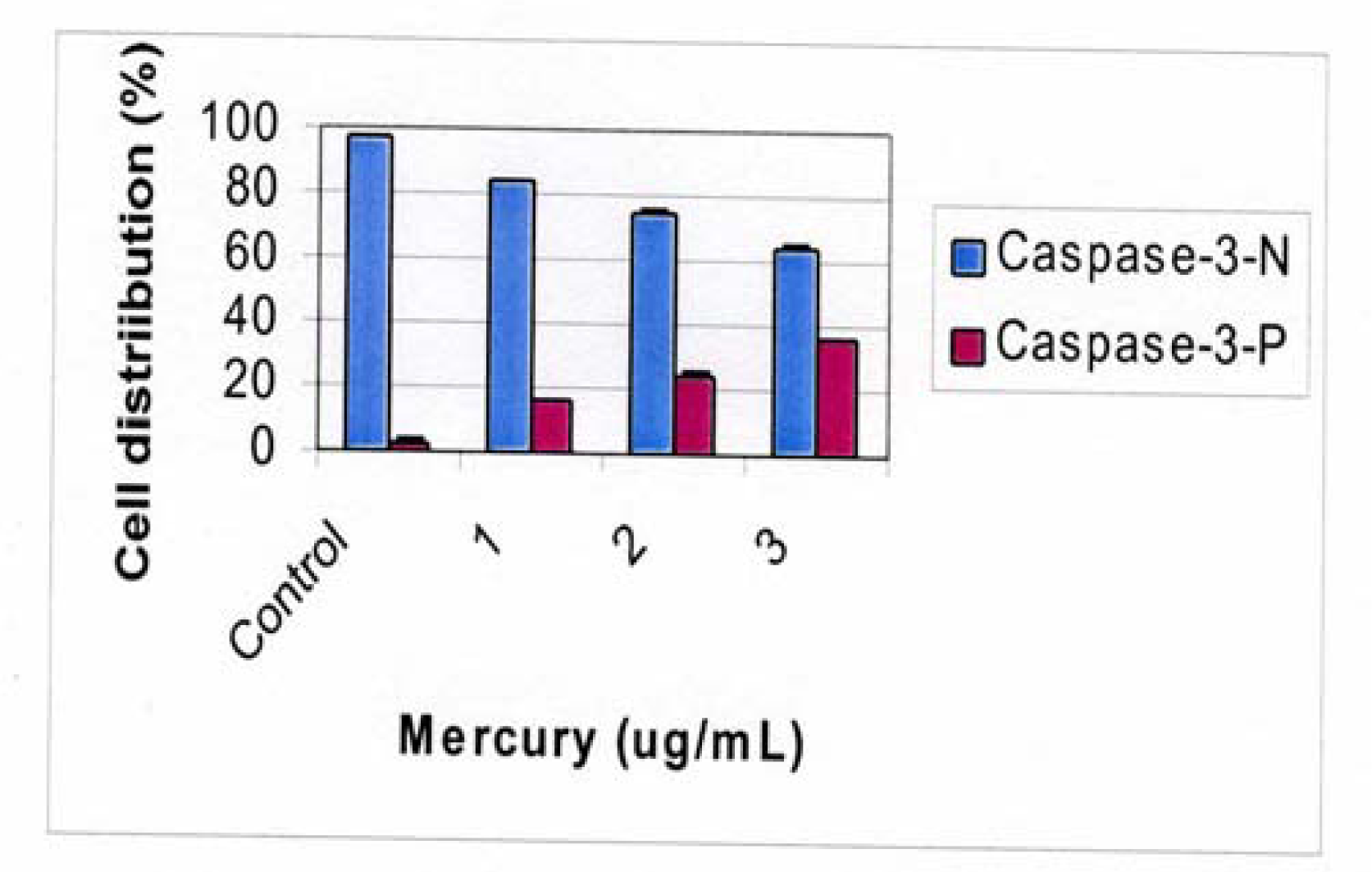
Acknowledgments
References
- Clarkson, TW; Magos, L; Myers, GJ. The toxicology of mercury – current exposures and clinical manifestations. N Engl J Med 2003, 349, 1731–1737. [Google Scholar]
- Clarkson, TW. The three modern faces of mercury. Environ Health Perspect Suppl.Suppl.1 2002, 10, 11–23. [Google Scholar]
- Sutton, DJ; Tchounwou, PB; Ninashvili, N; Shen, E. Mercury induces cytotoxicity and transcriptionally activates stress genes in human liver carcinoma (HepG2) cells. Int. J. Mol. Sci 2002, 3, 965–984. [Google Scholar]
- Steuerwald, U; Weibe, P; Jorgensen, P; Bjerve, K; Brock, J; Heinzow, B; Budta-Jorgensen, E; Grandjean, P. Maternal seafood diet, Methylmercury exposure and neonatal neurologic function. J. Pediatr 2000, 5, 599–605. [Google Scholar]
- Olivieri, G; Brack, C; Muller-Spahn, F; Stahelin, HB; Herrmann, M; Renards, P; Brockhaus, M; Hock, C. Mercury induces cell cytotoxicity and oxidative stress and increases β-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem 2000, 74, 231–236. [Google Scholar]
- Baskin, DS; Nago, H; Didenko, V. Thimerol induces DNA breaks, caspase 3 activation, membrane damage, and cell death in cultured human neurons and fibroblasts. Toxicological Sciences 2003, 74, 361–368. [Google Scholar]
- Rossi, AD; Viviani, B; Zhivotovsky, B; Manzo, L; Orrenius, S; Vahter, M; Nicotera, P. Inorganic mercury modifies Ca2+ signals, triggers apoptosis and potentiates NMDA toxicity in cerebellar granule neurons. Cell Death and Differentation 1997, 4, 317–324. [Google Scholar]
- U.S. EPA. Mercury study report to congress volume I. Office of Air Quality Planning and Standards and Office of Research and Development. U. S. Environmental Protection Agency 1997. [Google Scholar]
- IRIS, Methyl Mercury. Integrated Rick Information Service; Washington, D.C; U. S. Environmental Protection Agency, 2001. [Google Scholar]
- Diamond, GL; Zalups, RK. Understanding renal toxicity of heavy metals. Toxicol.Pathol 1998, 26, 92–103. [Google Scholar]
- Nielsen, JB; Hultman, P. Mercury-induced autoimmunity in mice. Environ.Health Perspect. Suppl. 5 2002, 110, 877–881. [Google Scholar]
- Markovich, D; James, KM. Heavy metals mercury, cadmium, and chromium inhibit the activity of the mammalian liver and kidney sulfate transporter sat-1. Toxicology and Applied Pharmacology 1999, 154, 181–187. [Google Scholar]
- Sweet, LI; Zelikoff, JF. Toxicology and immunotoxicology of mercury: a review in fish and humans. J. Toxicol Environ. Health B Crit. Rev 2001, 2, 161–205. [Google Scholar]
- Kirkland, RA; Franklin, JL. Evidence for redox regulation of cytochrome c release during programmed neuronal death: Antioxidant effects of protein synthesis and caspase inhibition. Journal of Neuroscience 2001, 6, 1949–1963. [Google Scholar]
- Liu, L; Hammar, P; Smith, JS; Inoue, S; Keefe, DL. Mitochondrial modulation of calcium signaling at the initiation of development. Cell Calcium 2001, 6, 423–433. [Google Scholar]
- Kono, DH; Balomenos, D; Pearson, DL; Park, MS; Hildebrand, B; Hultman, P; Pollard, KM. The prototypic Th2 autoimmunity induced by mercury is dependent on IFN-γ and not Th1/Th2 imbalance. Journal of Immunology 1998, 161, 234–240. [Google Scholar]
- Shenker, BJ; Guo, TL; Shapiro, IM. Mercury induced-apoptosis in human lymphoid cells: evidence that the apoptotic pathway is mercurial species dependent. Environ Res 2000, 2, 89–99. [Google Scholar]
- Johnson, EM; Desjmukh, L. Staurosporine-induced neuronal death: multiple mechanisms and methodological implications. Cell Death and Differentation 2000, 7, 250–261. [Google Scholar]
- Parran, DK; Mundy, WR; Barone, S, Jr. Effects of Methylmercury and mercuric chloride on differentation and cell viability in PC12 cells. Toxicological Sciences 2001, 59, 278–290. [Google Scholar]
- Gulbins, E; Jekle, A; Ferlinz, K; Grassme, H; Lang, F. Physiology of apoptosis. Am. J. Physiol. Renal Physiol 2000, 279, F605–F615. [Google Scholar]
- Hajela, RK; Peng, SQ; Athison, WD. Comparative effects of methylmercury and Hg2+ on human neuronal N and R type high voltage activated calcium channels transiently expressed in human embryonic kidney 293 cells. Journal of Pharmacology and Experimental Therapeutics 2003, 306, 1129–1136. [Google Scholar]
- Waalkes, MP; Fox, DA; States, JC; Patierno, SR; McCabe, MJ, Jr. Metals and disorders of cell accumulation: Modulation of apoptosis and cell proliferation. Toxicological Sciences 2000, 56, 255–261. [Google Scholar]
- Weed, R; Eber, J; Rothstein, A. Interaction of mercury with human erythrocytes. Journal of General Physiology 1962, 45, 395–410. [Google Scholar]
- Comparison of neurobehavioral changes I three inbred strain of mice prenatally exposed to methylmercury. Neurotoxicology and Teratology 2000, 22, 397–403.
- Graff, RD; Falconer, MM; Brown, DL; Reuhl, KR. Altered sensitivity of posttranslationally modified microtubules to Methylmercury in differentiating embryonal carcinoma-derived neurons. Toxicol Appl. Pharmacol 1997, 2, 215–224. [Google Scholar]
© 2006 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sutton, D.J.; Tchounwou, P.B. Mercury-Induced Externalization of Phosphatidylserine and Caspase 3 Activation in Human Liver Carcinoma (HepG2) Cells. Int. J. Environ. Res. Public Health 2006, 3, 38-42. https://doi.org/10.3390/ijerph2006030005
Sutton DJ, Tchounwou PB. Mercury-Induced Externalization of Phosphatidylserine and Caspase 3 Activation in Human Liver Carcinoma (HepG2) Cells. International Journal of Environmental Research and Public Health. 2006; 3(1):38-42. https://doi.org/10.3390/ijerph2006030005
Chicago/Turabian StyleSutton, Dwayne J., and Paul B. Tchounwou. 2006. "Mercury-Induced Externalization of Phosphatidylserine and Caspase 3 Activation in Human Liver Carcinoma (HepG2) Cells" International Journal of Environmental Research and Public Health 3, no. 1: 38-42. https://doi.org/10.3390/ijerph2006030005