Neuregulin 1-Βeta Cytoprotective Role in AML 12 Mouse Hepatocytes Exposed to Pentachlorophenol


Abstract
:Introduction
Materials and Methods
Chemicals
Cell Culture
Cell Viability Experiments
Sample Collection and Protein Determination
Western Blot Analysis for Identification of Specific Cellular Proteins
Statistical Analysis
Results
Comparison of Untreated and NRG1-βTtreated Mouse Hepatocytes
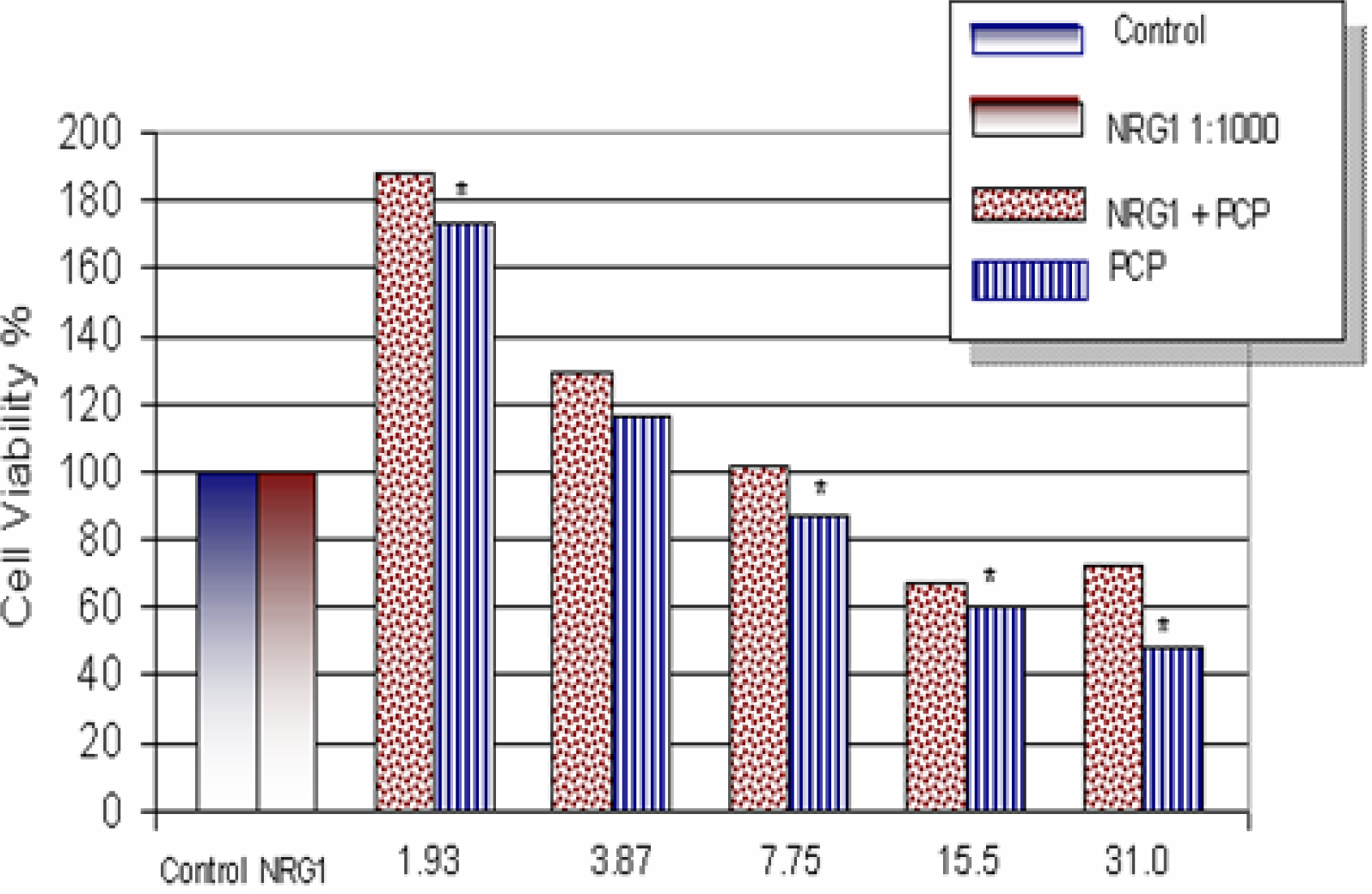
Effects of NRG1-β on PCP-Treated Mouse Hepatocytes
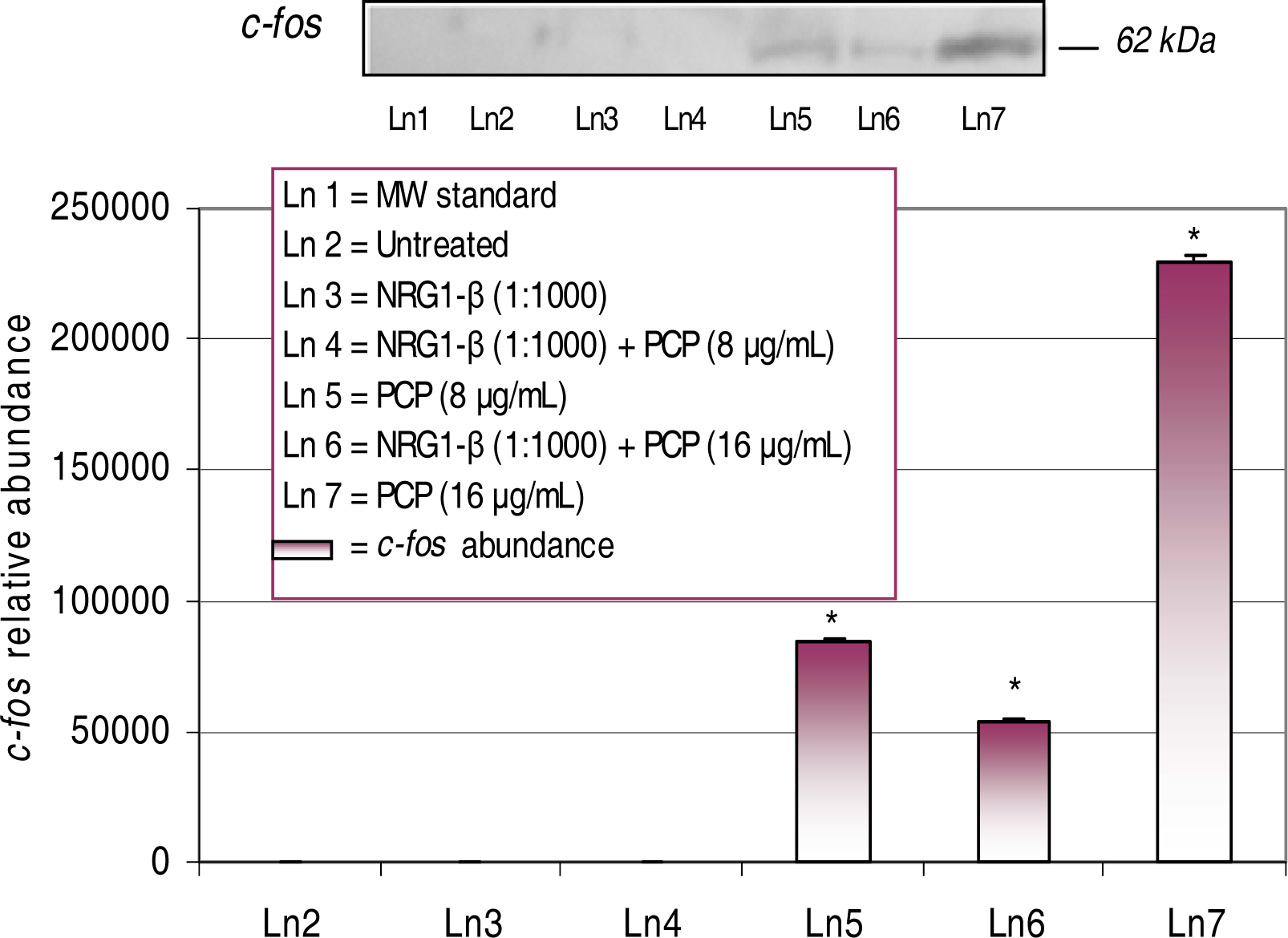
Western Blot and Densitometric Analyses for c-fos Expression
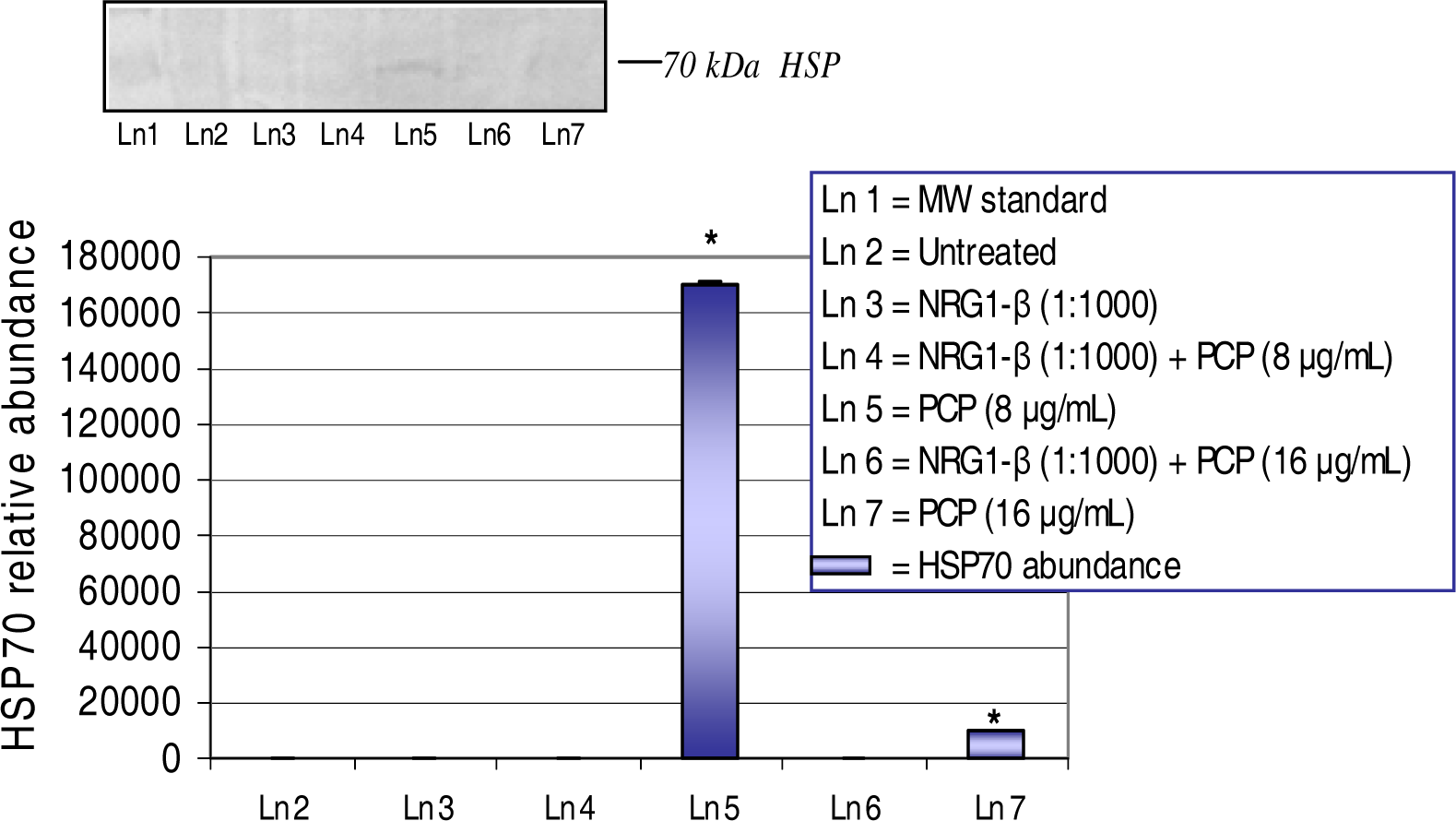
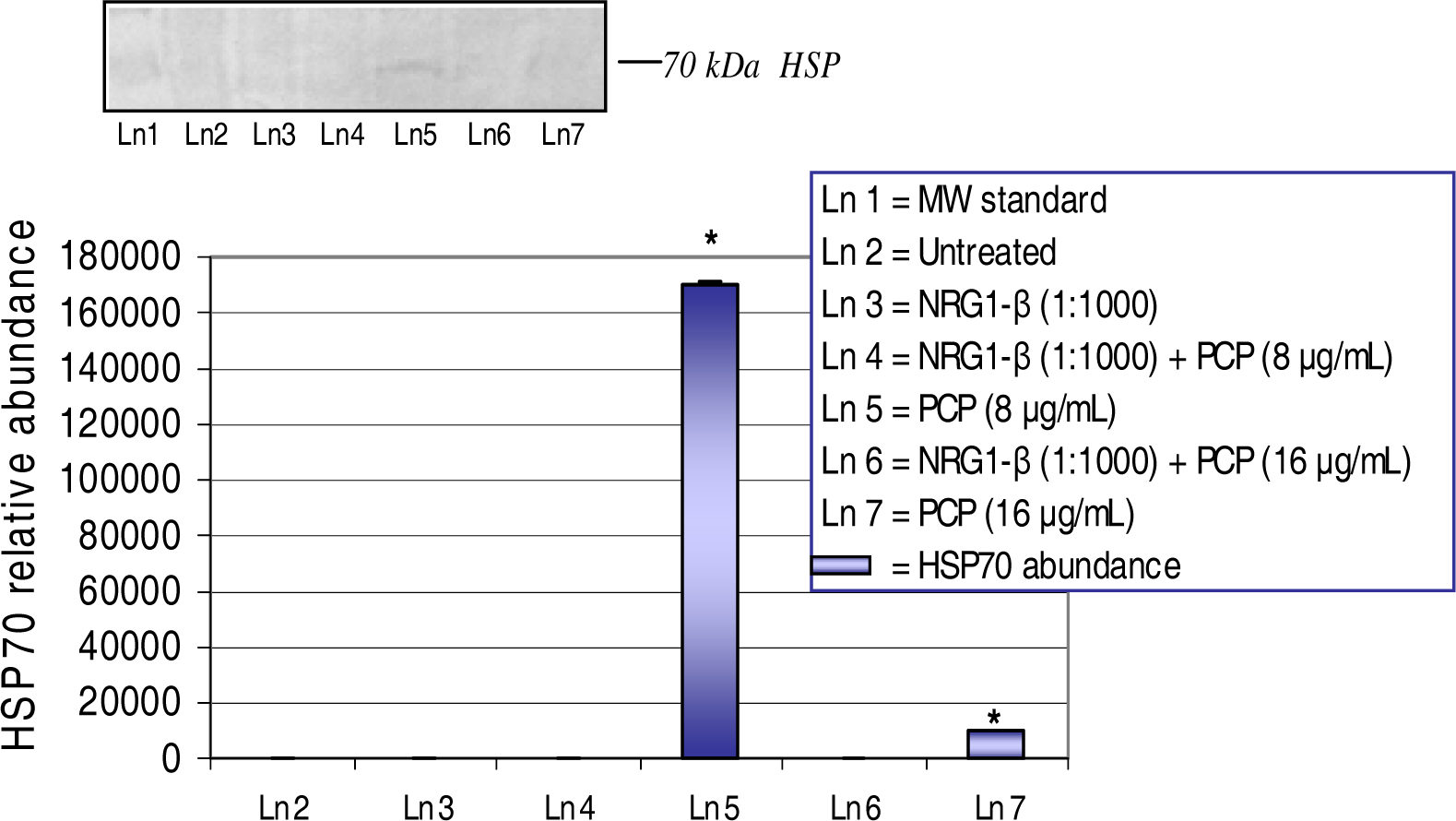
Western Blot and Densitometric Analyses for HSP70 Expression
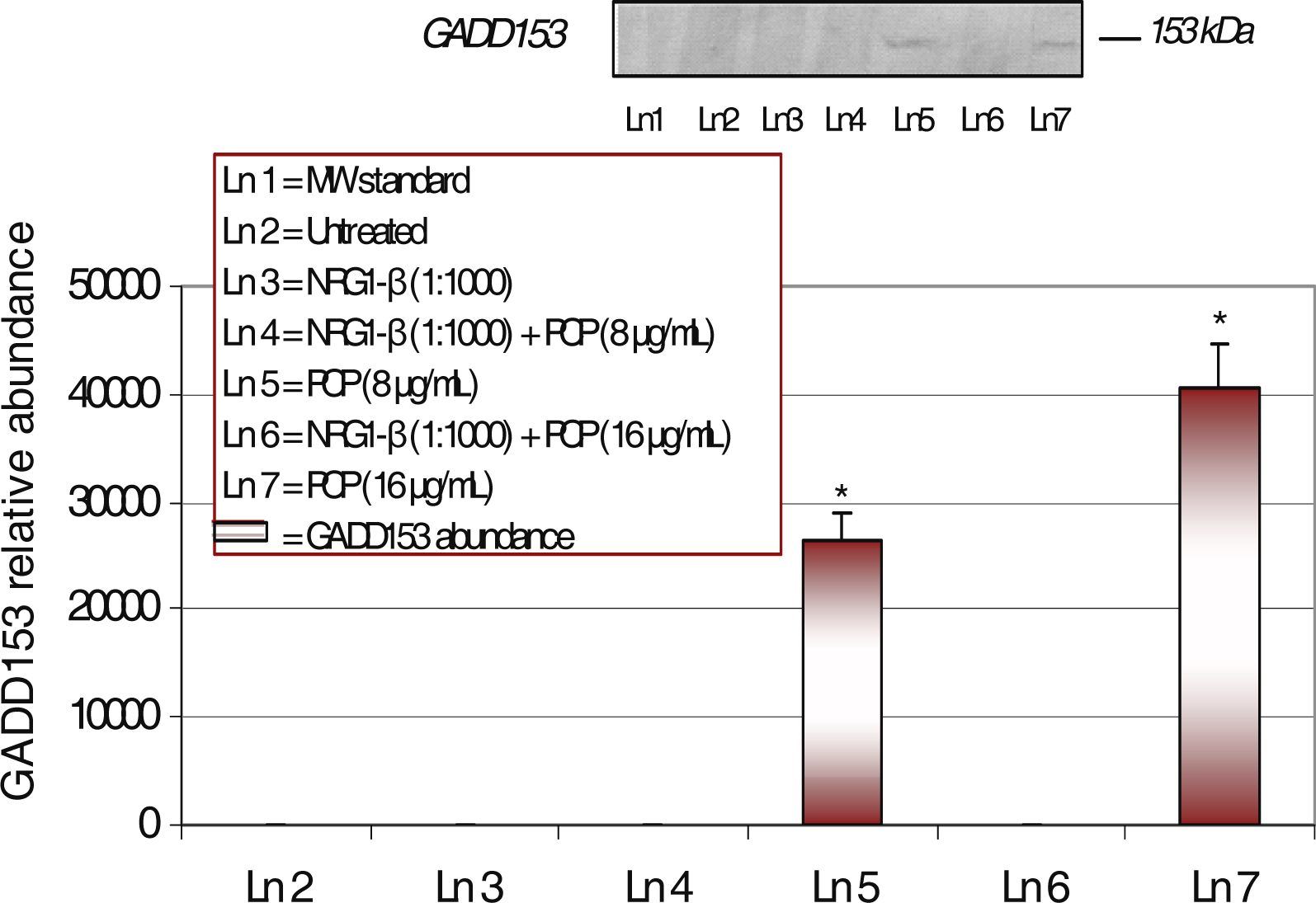
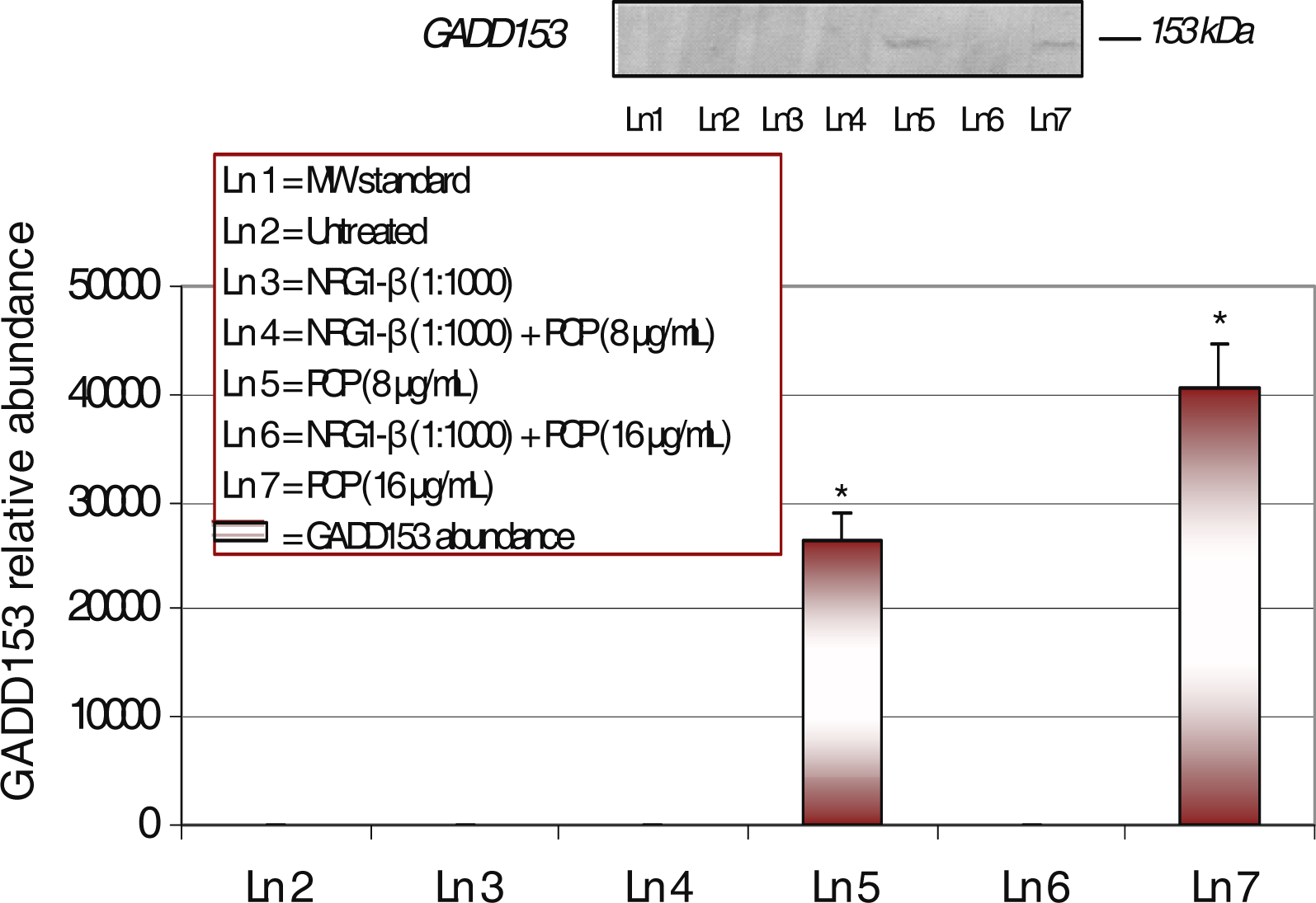
Western-Blot and Densitometric Analyses for GADD153 Expression
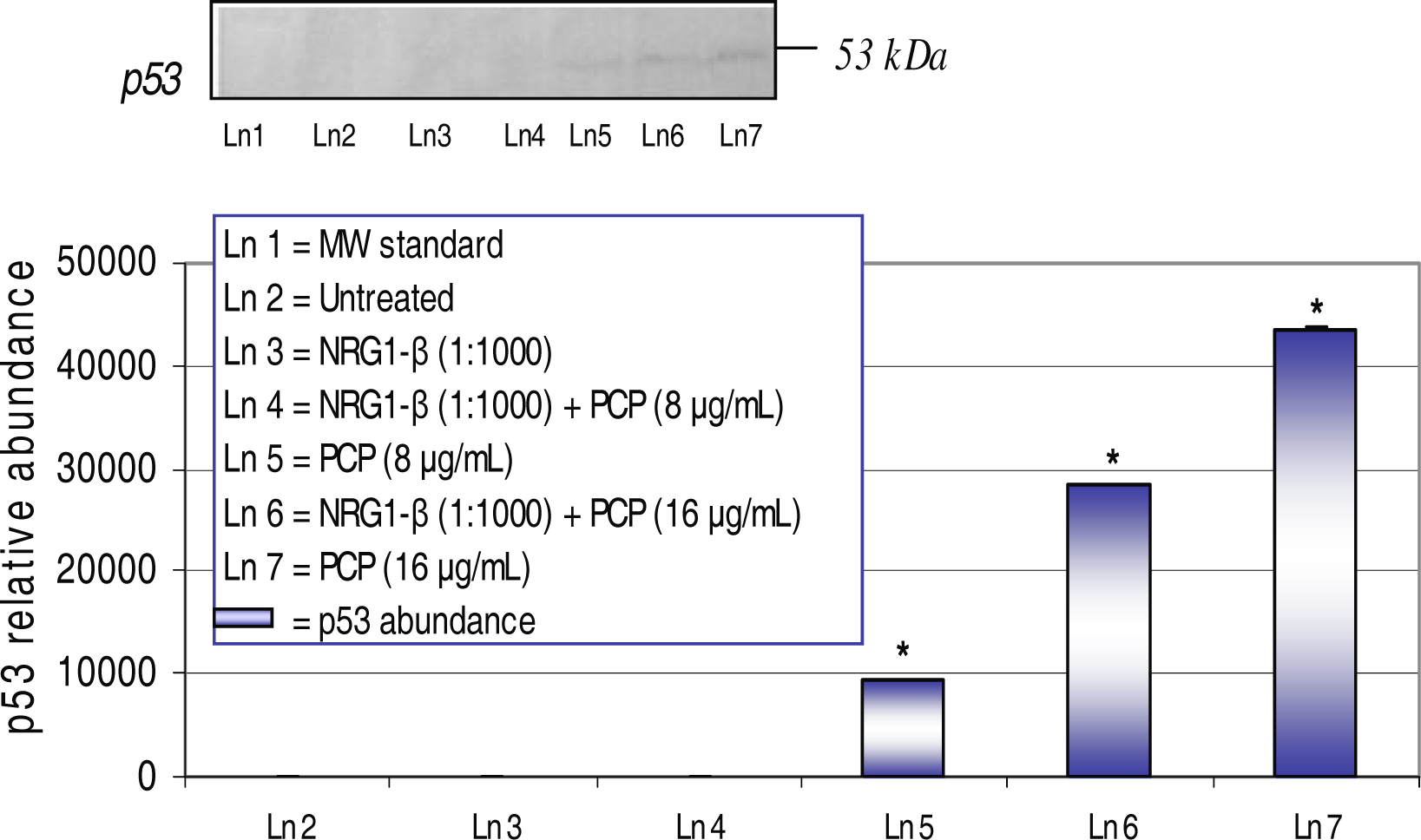
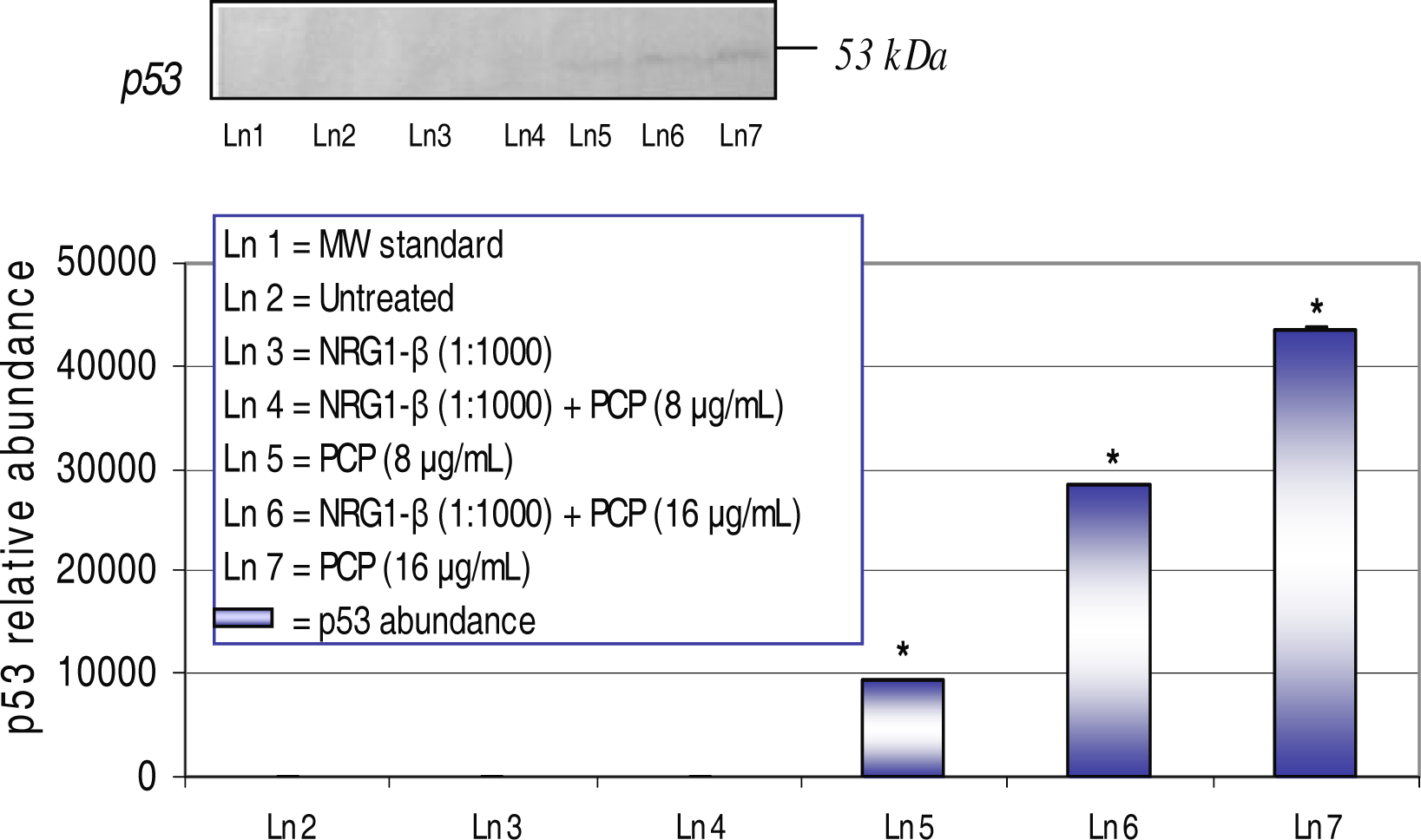
Western-Blot and Densitometric Analyses for p53 Expression
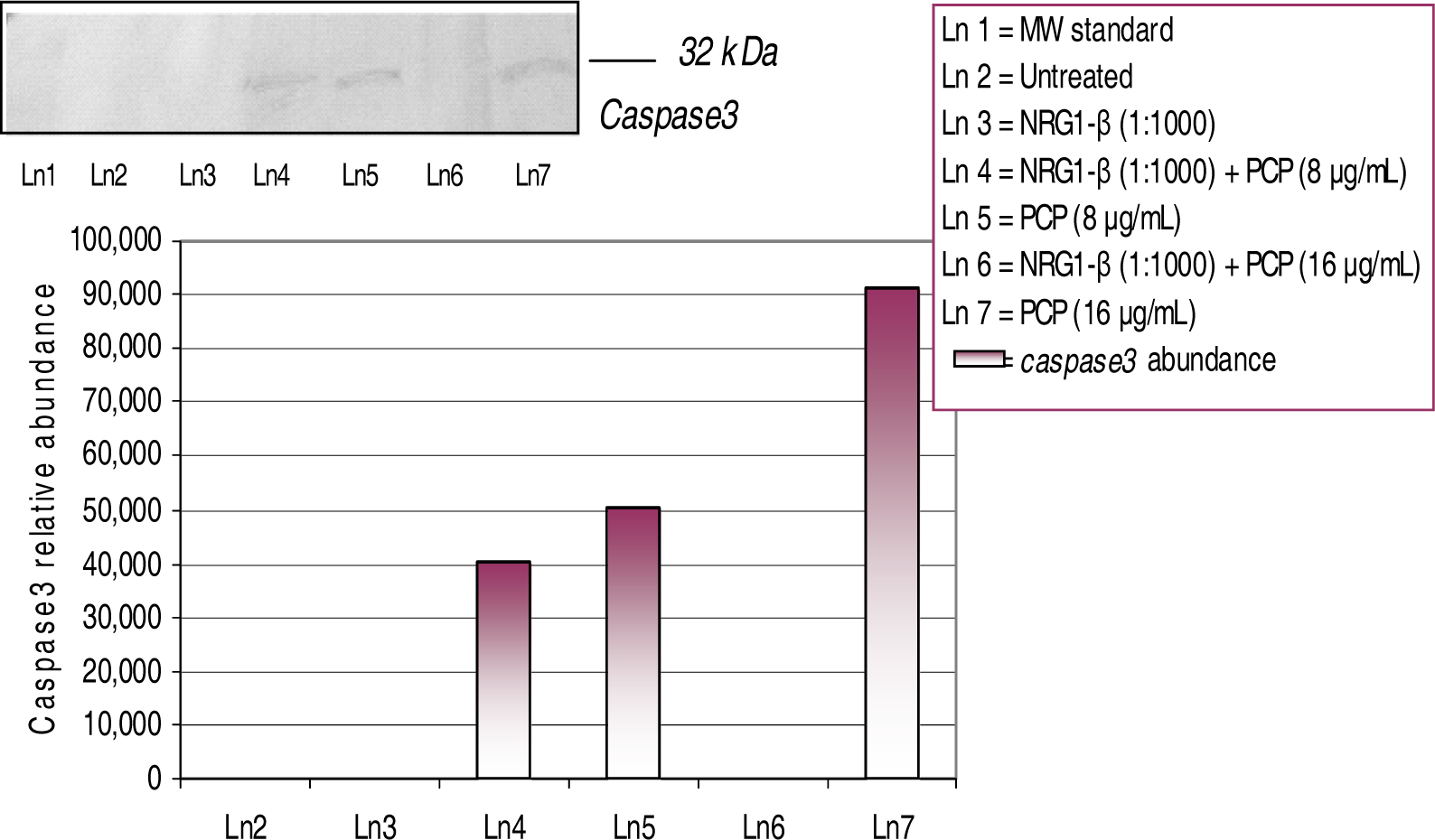
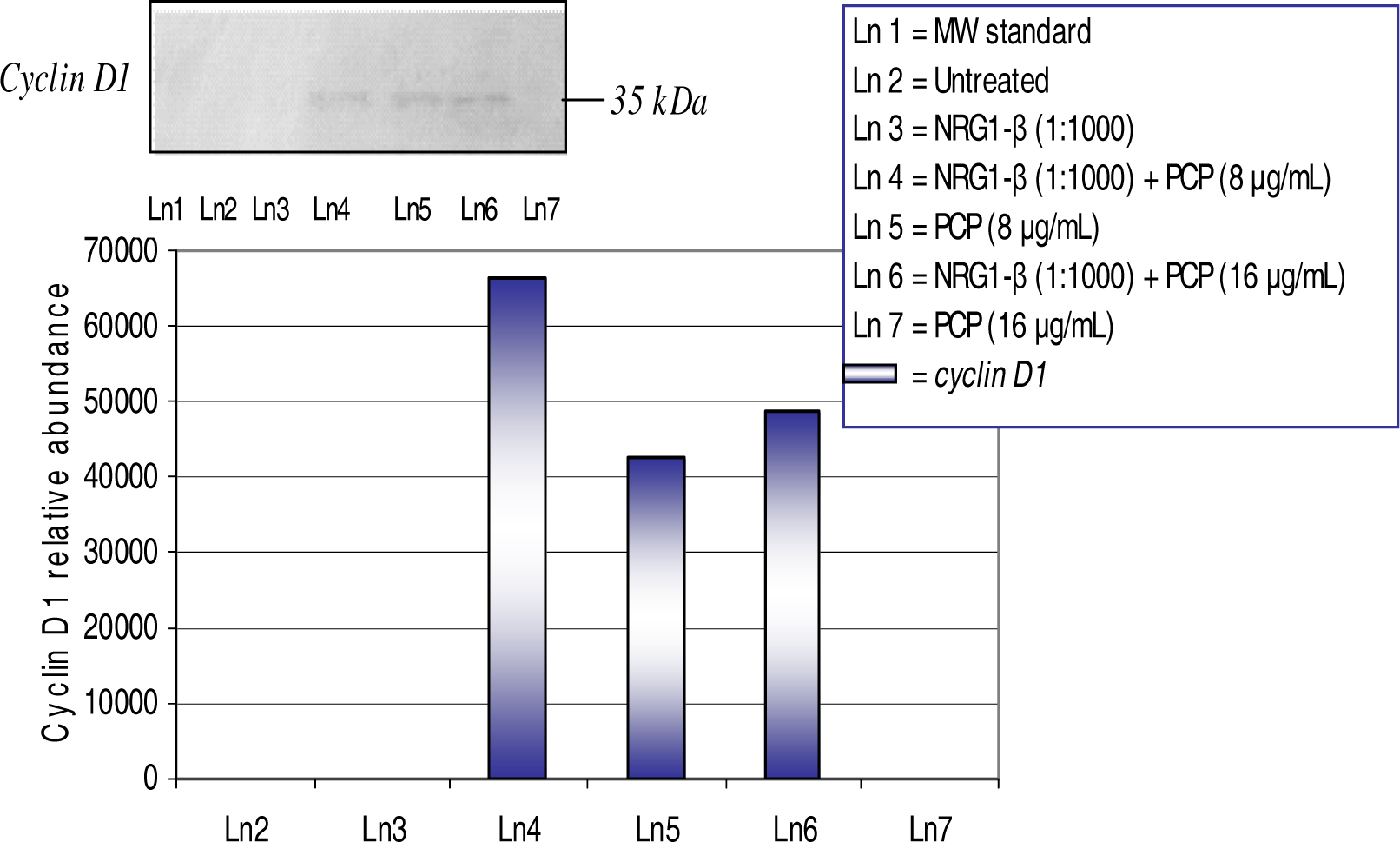
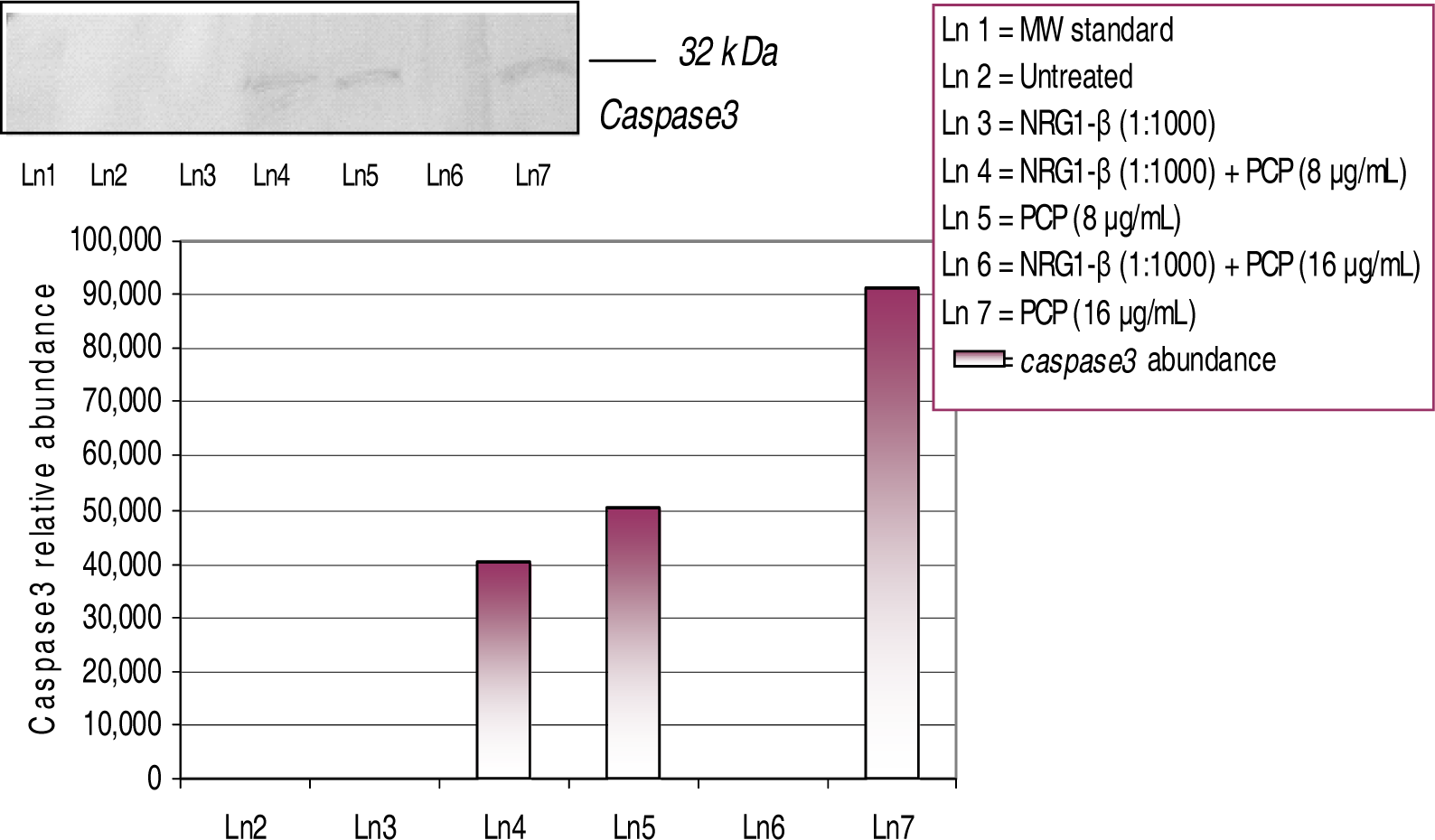
Western-Blot and Densitometric Analyses for Cyclin D1 and Caspase-3 Expression
Discussion
Neuregulin 1-βeta-Induced Protective Effects
NRG1-β Effects on PCP-Induced c-fos Expression
NRG1-β Effect on PCP-Induced HSP70 Expression
NRG1-β Effect on PCP-Induced GADD153 Expression
NRG1-β Effect on PCP-Induced on p53 Expression
NRG1-β Effect on PCP-Induced cyclin D1
NRG1-β Effects on PCP-induced Caspase-3 Expression
Conclusions
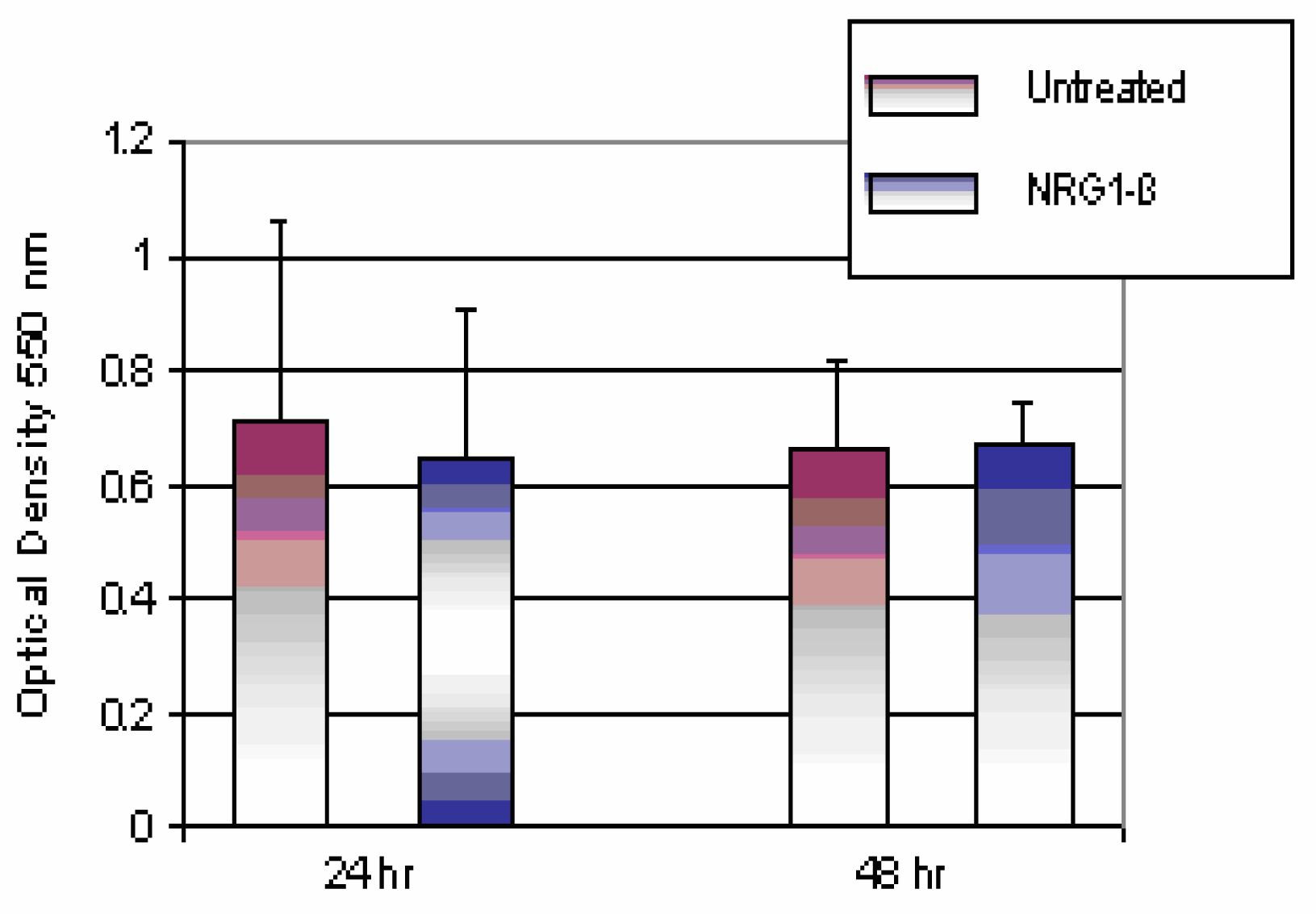




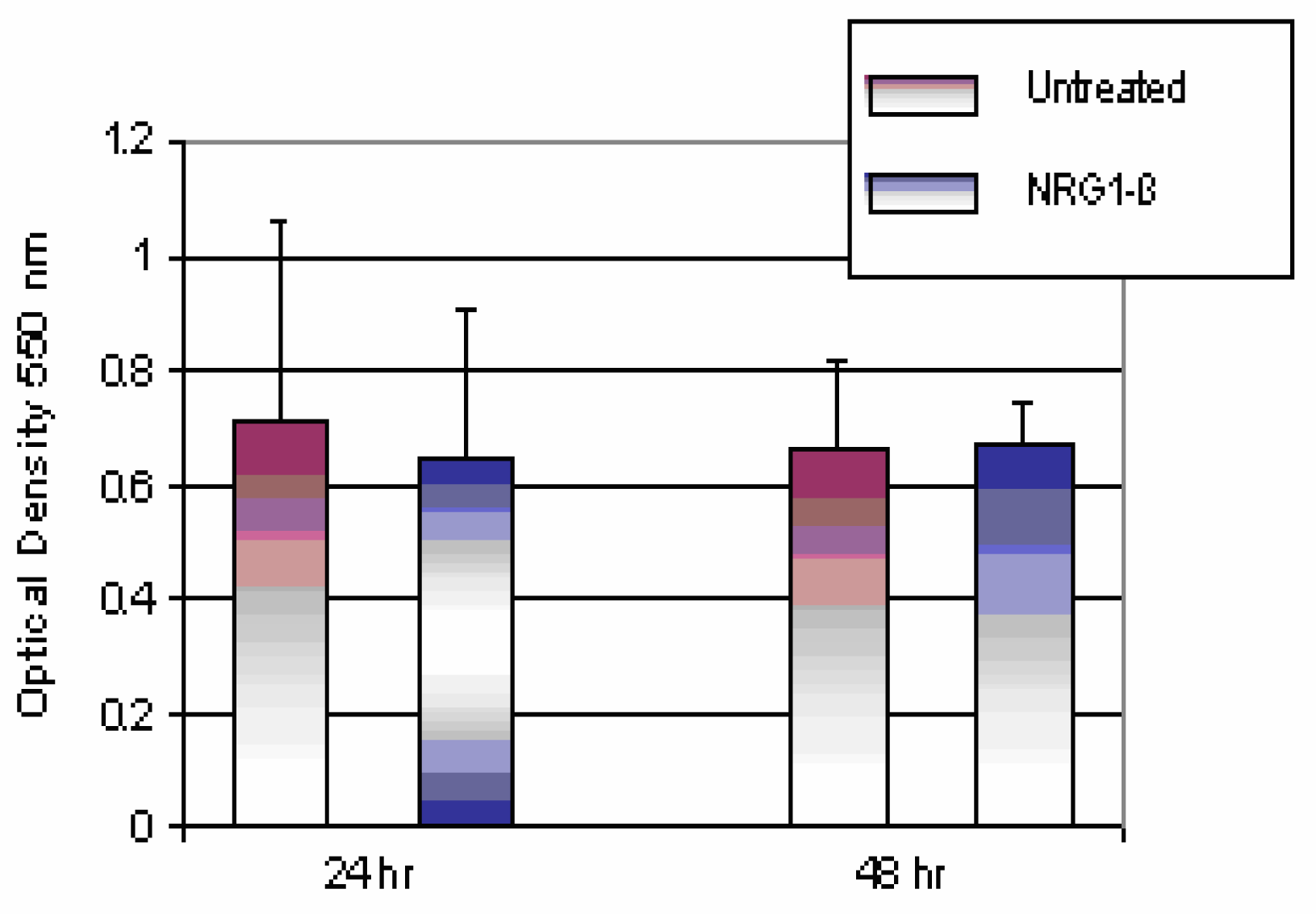{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Protein | Biological Function / Response | NRG1+PCP 8μg/mL | PCP 8μg/mL | NRG1+PCP 16μg/mL | PCP 16μg/mL |
|---|---|---|---|---|---|
| c-fos | AP-1 component Signaling-immediate early gene | - | 83,714 | 54,115 | 229,374 |
| DNA-damage Molecular chaperone | |||||
| HSP70 | Oxidative stress Protein alterations | - | 170,584 | - | 10,000 |
| Cell cycle regulator | |||||
| GADD153 | DNA repair | - | 26,441 | - | 40,626 |
| DNA-damage | |||||
| p53 | Cell cycle- regulator Apoptosis | - | 9,502 | 28,389 | 43,458 |
| DNA-damage |
References
- Crovello, CS; Lai, C; Cantley, LC; Carraway, KL, III. Differential signaling by the epidermal growth factor-like growth factors neuregulin-1 and neuregulin -2. J Biol Chem 1998, 273, 26954–26961. [Google Scholar]
- Burden, S; Yarden, Y. Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron 1997, 18, 847–855. [Google Scholar]
- Nakano, N; Higashiyama, S; Ohmoto, H; Ishiguro, H; Taniguchi, N; Wada, Y. The N-terminal region of NTAK/neuregulin-2 isoforms has an inhibitory activity on angiogenesis. J Biol Chem 2004, 279(12), 11465–11470. [Google Scholar]
- Parkinson, DB; Langner, K; Namini, SS; Jessen, KR; Mirksy, R. β-Neuregulin and autocrine mediated survival of Schwann cells requires activity of Ets family transcription factors. Molecular and Cellular Neuroscience 2002, 20, 154–167. [Google Scholar]
- Murphy, S; Krainock, R; Tham, M. Neuregulin signaling via ErbB receptor assemblies in the nervous system. Molecular Neurobiology 2002, 25, 67–78. [Google Scholar]
- Adlkofer, K; Lai, C. Role of neuregulins in glial cell development. Glia 2000, 15, 104–111. [Google Scholar]
- Marchionni, MA; Cannella, B; Hoban, C; Gao, YL; Garcia-Arnas, R; Lawson, D; Happel, E; Noel, F; Tofilon, P; Gwynne, D; Raine, CS. Neuregulin in neuron/glial interactions in the central nervous system: GGF2 diminishes auto-immune demyelination, promotes oligodendrocytes progenitor expansion, and enhances remyelination. Adv. Exp. Med. Biol 1999, 468, 283–295. [Google Scholar]
- Fischbach, GD; Rosen, KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci 1997, 20, 429–458. [Google Scholar]
- Carraway, KL, III; Burden, SJ. Neuregulins and their receptors. Curr Opin Neurobiol 1995, 5, 606–612. [Google Scholar]
- Carraway, KL, III; Cantley, LC. A neu acquaintance for erbB3 and erbB4: a role for receptor heterodimerization in growth signaling. Cell 1994, 78, 5–8. [Google Scholar]
- Fallon, KB; Havlioglu, N; Hamilton, LH; Cheng, TP; Carroll, SL. Constitutive activation of the neuregulin-1/erbB signaling path way promotes proliferation of a human peripheral neuroepithelioma cell line. J Neurooncol 2004, 66, 273–284. [Google Scholar]
- Xu, Z; Jiang, J; Ford, G; Ford, BD. Neuregulin-1 is neuroprotective and attenuates inflammatory responses induced by ischemic stroke. Biochem Biophys Res Commun 2004, 322, 440–446. [Google Scholar]
- Xu, Z; Ford, GD; Croslan, DR; Jiang, J; Gates, A; Allen, R; Ford, BD. Neuroprotection by neuregulin-1 following focal stroke is associated with the Attenuation of ischemia-induced pro-inflammatory and stress gene expression. Neurobiology of Disease 2005, 19, 461–470. [Google Scholar]
- Fukazawa, R; Miller, TA; Kuramochi, Y; Frantz, S; Kim, YD; Marchionni, MA; Kelly, RA; Sawyer, DB. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J Mol Cell Cardio 2003, 35(12), 1473–1479. [Google Scholar]
- Negro, A; Brar, K; Lee, KF. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog Horm Res 2004, 59, 1–12. [Google Scholar]
- Sawyer, DB; Zuppinger, C; Miller, TA; Eppenberger, HM; Suter, TM. Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1 beta and anti-erbB2: potential mechanism fortrastuzumab-induced cardiotoxicity. Circulation 2002, 105(13), 1551–1554. [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological Profile for Pentachlorophenol; U.S. Public Health Service, U.S. Department of Health and Human Services: Atlanta, GA, 1992.
- Dorsey, WC; Tchounwou, PB; Ishaque, AB; Shen, E. Transcriptional activation of stress genes and cytotoxicity in human liver carcinoma (HepG2) exposed to pentachlorophenol. International J Molecular Sci 2002, 3, 989–1004. [Google Scholar]
- Dorsey, WC; Tchounwou, PB. Pentachlorophenol-induced cytotoxic, mitogenic, and endocrine-disrupting activities in channel catfish, Ictalurus punctatus. International J Environ Research and Public Health 2004, 1(2), 74–83. [Google Scholar]
- Dorsey, WC; Tchounwou, PB; Sutton, D. Mitogenic and cytotoxic effects of pentachlorophenol to AML 12 mouse hepatocytes. International J Environ Research and Public Health 2004, 1(2), 100–105. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: applications to proliferation and cytotoxicity assays. J Immunol Methods 1983, 65, 55–63. [Google Scholar]
- Holbrook, NJ; Fornace, AJ. Response to adversity: molecular control of gene activation following genotoxic stress. New Biol 1991, 3, 825–833. [Google Scholar]
- Dorsey, WC; Tchounwou, PB. CYP1A1, HSP70, p53, and c-Fos expression in human liver carcinoma cells (HepG2) exposed to pentachlorophenol. ISA-Biomedical Sciences Instrumentation 2003, 39, 389–396. [Google Scholar]
- Jolly, C; Morimoto, RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. Journal of the National Cancer Institute 2000, 92(19), 1564–1572. [Google Scholar]
- Zinszer, H; Kuroda, M; Wang, X; Batchvarova, N; Lightfoot, RT; Remotti, H; Stevens, JL; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & Development 1998, 12(7), 982–995. [Google Scholar]
- Quelle, FW; Wang, J; Feng, J; Wang, D; Cleveland, JL; Ihle, JN; Zambetti, GP. Cytokine rescue of p53-dependent apoptosis and cell cycle arrest is mediated by distinct Jakkinase signaling pathways. Genes & Development 1998, 12(8), 1099–1107. [Google Scholar]
- Albrecht, JH; Hansen, LK. Cyclin D1 promotes mitogenic-independent cell cycle progression in hepatocytes. Cell Growth & Differentiation 1999, 10, 397–404. [Google Scholar]
- Tormanen-Napankangas, U; Soini, Y; Kahlos, K; Kinnula, V; Paakko, P. Expression of Caspases-3, -6 and -8 and their relation to apoptosis in non-small cell lung carcinoma. Int J Cancer 2001, 93(2), 192–98. [Google Scholar]
- Adlkofer, K; Lai, C. Role of neuregulins in glial cell development. Glia 2000, 29, 104–111. [Google Scholar]
- Fukazawa, R; Miller, TA; Kuramochi, Y; Frantz, S; Kim, YD; Marchionni, MA; Kelly, RA; Sawyer, DB. Neuregulin-1 protects ventricular myocytes from anthracyline-induced apoptosis via erbB4-dependent activation of PI3-Akt. J Mol Cell Cardiol 2003, 35(12), 1473–1479. [Google Scholar]
- Kuramochi, Y; Lim, CC; Guo, X; Colucci, WS; Liao, R; Sawyer, DB. Myocyte contractile activity modulates norepinephrine cytotoxicity and survival effects of neuregulin-1 beta. Am J Physiol Cell Physiol 2004, 286(2), C222–229. [Google Scholar]
- Carpenter, CL; Cantley, LC. Phosphoinositide 3-kinase and the regulation of cell growth. Biochem Biophys Acta 1996, 1288, M11–M16. [Google Scholar]
- Di Segni, A; Shaharabani, E; Stein, R; Pinkas-Kramarski, R. Neuregulins rescue PC12-ErbB-4 cells from cell death induced by ß-amyloid 34.
- Parkinson, DB; Langner, K; Namini, SS; Jessen, KR; Mirsky, R. beta- Neuregulin and autocrine mediated survival of Schwann cells requires activity of Ets family transcription factors. Mol Cell Neurosci 2002, 20(1), 154–167. [Google Scholar]
- Xie, F; Raetzman, LT; Siegal, RE. Neuregulin induces GABAA receptor β2 subunit expression in cultured rat cerebellar granule neurons by activating multiple signal pathways. Journal of Neurochemistry 2004, 90, 1521–1529. [Google Scholar]
- Carver, RS; Sliwkowski, MX; Sitaric, S; Russell, WE. Insulin regulates heregulin binding and ErbB3 expression in rat hepatocytes. J Biol Chem 1996, 271(23), 13491–13496. [Google Scholar]
- Carver, RS; Stevenson, MC; Scheving, LA; Russell, WE. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology 2002, 123(6), 2017–2027. [Google Scholar]
- Schreiber, M; Kolbus, A; Piu, F; Szabowski, A; Mohle-Steinlein, U; Tian, J; Karin, M; Angel, P; Wagner, EF. Control of cell cycle progression by c-jun is p53 dependent. Genes & Development 1999, 13, 617–619. [Google Scholar]
- Deschamps, J; Meijlink, F; Verma, IM. Identification of a transcriptional enhancer element upstream from the proto-oncogene fos. Science 1985, 230, 1174–1177. [Google Scholar]
- Kovary, K; Bravo, R. The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol Cell Biol 1991, 11, 4466–4472. [Google Scholar]
- Takeuchi, K; Shibamoto, S; Nagamine, K; Shigemori, I; Omura, S; Kitamura, N; Ito, F. Signaling pathways leading to transcription and translation cooperatively regulate the transient increase in expression of c-fos protein. J Biol Chem 2001, 276(280), 26077–26083. [Google Scholar]
- Schenk, H. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-*B and AP-1. Proc Natl Acad Sci USA 1994, 91, 1672–1676. [Google Scholar]
- Jackson, SP; Jeggo, PA. DNA double-strand break repair and V(D)J recombination: Involvement of DNA-PK. Trends Biochem Sci 1995, 20, 412–415. [Google Scholar]
- Brown, JR; Nigh, E; Lee, RJ; Ye, H; Thompson, MA; Saudou, F; Pestell, RG; Greenburg, ME. fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol 1998, 18, 5609–5619. [Google Scholar]
- Miao, GG; Curran, T. Cell transformation by c-fos requires an extended period of expression and is independent of the cell cycle. Mol Cell Biol 1994, 14, 4295–4310. [Google Scholar]
- Yamauchi, K; Holt, K; Pessin, JE. Phosphatidylinositol 3-kinase functions upstream of Ras and Raf in mediating insulin stimulation of c-fos transcription. J Biol Chem 1993, 268, 14597–14600. [Google Scholar]
- Jhun, BH; Rose, DW; Seely, BL; Rameh, L; Cantley, L; Saltiel, AR; Olefsky, JM. Microinjection of the SH2 domain of the 85-kilodalton subunit phosphatidylinositol 3-kinase inhibits insulin induced DNA synthesis and c-fos expression. Mol Cell Biol 1994, 14, 7466–7475. [Google Scholar]
- Wang, Y; Falasca, M; Schlessinger, J; Malstrom, S; Tsichlis, P; Settlemen, J; Hu, W; Lim, B; Prywes, R. Activation of the c-fos serum response element by phosphatidyl inositol 3-kinase and rho pathways in HeLa cells. Cell Growth Differ 1998, 9, 513–522. [Google Scholar]
- Jolly, C; Morimoto, RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Nation. Cancer Inst 2000, 92, 1564–1572. [Google Scholar]
- Kozutsumi, Y; Segal, M; Normington, K; Gething, MJ; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar]
- Kaufman, RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes & Development 1999, 13, 1211–1233. [Google Scholar]
- Jaattela, M; Wissing, D; Kokholm, K; Kallunki, T; Egeblad, M. Hsp70 exerts its anti-apoptotic function downstream of Caspase-3-like proteases. EMBO J 1998, 17(21), 6124–6134. [Google Scholar]
- Ramage, L; Guy, K. Expression of C-reactive protein and heat-shock protein-70 in the lung epithelial cell line A549, in response to PM10 exposure. Inhal Toxicol 2004, 16(6–7), 447–452. [Google Scholar]
- Dybdahl, B; Slordahl, SA; Waage, A; Kierulf, P; Espevik, T; Sundan, A. Myocardial ischaemia and the inflammatory response: release of heat shock protein 70 after myocardial infarction. Heart 2005, 91, 299–304. [Google Scholar]
- Bartlett, JD; Luethy, JD; Carlson, SG; Sollott, SJ; Holbrook, NJ. Calcium ionophore A23187 induces expression of the growth arrest and DNA damage inducible CCAAT/enhancer-binding protein (C/EBP)-related gene, gadd153. Ca2+ increases transcriptional activity and mRNA stability. J Biol Chem 1992, 267(28), 20465–20470. [Google Scholar]
- Los, G; Benbatoul, K; Gately, DP; Barton, R; Christen, R; Robbins, KT; Vicario, D; Kirmani, S; Orloff, LA; Weisman, R; Howell, SB. Quantitation of the change in GADD153 messenger RNA Level as a molecular marker of tumor response in head and neck cancer. Clinical Cancer Research 1999, 5, 1610–1618. [Google Scholar]
- Gately, DP; Jones, JA; Christen, R; Barton, R; Los, G; Howell, SB. Induction of growth arrest and DNA damage inducible gene gadd153 by cisplatin in vitro and in vivo. Br J Cancer 1994, 70, 1102–1106. [Google Scholar]
- Eymim, B; Dubrez, L; Allouche, M; Solary, E. Increased gadd153 messenger RNA level is associated with apoptosis in human leukemic cells treated with etoposide. Cancer Res 1997, 57, 686–695. [Google Scholar]
- Friedman, AD. GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells. Cancer Res 1996, 56, 3250–3256. [Google Scholar]
- Jackman, J; Alamo, I, Jr; Fornace, AJ, Jr. Genotoxic stress confers preferential and coordinate messenger RNA stability on the five gadd genes. Cancer Res 1994, 54, 5656–5662. [Google Scholar]
- Levine, AJ. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar]
- Bargonetti, J; Friedman, PN; Kern, SE; Vogelstein, B; Prives, C. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 1991, 65(6), 1083–91. [Google Scholar]
- Kern, SE; Kinzler, KW; Bruskin, A; Jarosz, D; Friedman, P; Prives, C; Vogelstein, B. Identification of p53 as a sequence-specific DNA-binding protein. Science 1991, 252(5013), 1708–1711. [Google Scholar]
- Midgley, CA; Lane, D. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene 1997, 15, 1179–1189. [Google Scholar]
- Kastan, MB; Onyekwere, O; Sidransky, D; Vogelstein, B; Craig, RW. Participation of p53 protein in cellular response to DNA damage. Cancer Res 1991, 51, 6304–6311. [Google Scholar]
- Lane, DP. p53, guardian of the genome. Nature 1992, 258, 15–16. [Google Scholar]
- Muraoka, RS; Lenferink, AEG; Law, B; Hamilton, E; Brantley, DM; Roebuck, LR; Arteaga, CL. ErbB2/neu-induced, cyclin D1-dependent transformation is accelerated in p27-haploinsufficient mammary epithelial cells but impaired in p27-null cells. Molecular and Cellular Biology 2002, 22(7), 2204–2219. [Google Scholar]
- Gillett, C; Smith, P; Gregory, W; Richards, M; Millis, R; Peters, G; Barnes, D. Cyclin D1 and prognosis in human breast cancer. Int J Cancer 1996, 69, 92–99. [Google Scholar]
- Hall, M; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and cdk inhibitors in human cancer. Adv Cancer Res 1996, 68, 67–108. [Google Scholar]
- Uto, H; Ido, A; Moriuchi, A; Onaga, Y; Nagata, K; Onaga, M; Tahara, Y; Hori, T; Hirono, S; Hayashi, K; Tsubouchi, H. Transduction of antisense cyclin D1 using two-step gene transfer inhibits the growth of rat hepatoma cells. Cancer Res 2001, 61, 4779–4783. [Google Scholar]
- Jiang, W; Kahn, SM; Zhou, P; Zhang, YJ; Cacace, AM; Infante, AS; Doi, S; Santella, RM; Weinstein, IB. Overexpression of cyclin D1 in rat fibroblasts causes abnormalities in growth control, cell cycle progression and gene expression. Oncogene 1993, 8, 3447–3457. [Google Scholar]
- Lovec, H; Sewing, A; Lucibello, FC; Müller, R; Möröy, T. Oncogenic activity of cyclin D1 revealed through cooperation with Ha-ras: link between cell cycle con trol and malignant transformation. Oncogene 1994, 9, 323–326. [Google Scholar]
- Kranenburg, O; van der Eb, AJ; Zentema, A. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J 1996, 15, 46–54. [Google Scholar]
- Sofer-Levi, Y; Resnitzky, D. Apoptosis induced by ectopic expression of cyclin D1 but not cyclin E. Oncogene 1996, 13, 2431–2437. [Google Scholar]
- Hiyama, H; Reeves, SA. Role for cyclin D1 in UVC-induced and p53-mediated apoptosis. Cell Death and Differentiation 1999, 6, 565–569. [Google Scholar]
- Lenferink, AEG; Busse, D; Flanagan, WM; Yakes, FM; Arteaga, CL. ErbB2/ neu kinase modulates cellular p27Kip1 and cyclin D1 through multiple pathways. Cancer Res 2001, 61, 6583–6581. [Google Scholar]
- Michalopoulos, GK; De Frances, MC. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar]
- Albrecht, JH; Hu, MY; Cerra, FB. Distinct patterns of cyclin D1 regulation in models of liver regeneration and human liver. Biochem. Biophys. Res. Commun 1995, 209, 648–655. [Google Scholar]
- Ehrenfried, JA; Ko, TC; Thompson, EA; Evers, BM. Cell cycle-mediated regulation of hepatic regeneration. Surgery 1997, 122, 927–935. [Google Scholar]
- Utz, PJ; Anderson, P. Life and death decisions: regulation of apoptosis by proteolysis of signaling molecules. Cell Death Differ 2000, 7(7), 589–602. [Google Scholar]
- Ohshima, K; Sugihara, M; Haraoka, S; Suzumiya, J; Kanda, M; Kawasaki, C; Shimazaki, K; Kikuchi, M. Possible immortalization of Hodgkin and Reed-Sternberg cells: telomerase expression, lengthening of telomere, and inhibition of apoptosis by NF-kappa B expression. Leuk Lymphoma 2001, 41, 367–376. [Google Scholar]
- Hussein, MR; Haemel, AK; Wood, GS. Apoptosis and melanoma: molecular mechanisms. J Pathol 2003, 199, 275–288. [Google Scholar]
- Shirin, H; Pinto, JT; Kawabata, Y; Soh, J; Delohery, T; Moss, SF; Murty, V; Rivlin, RS; Holt, PR; Weinstein, IB. Antiproliferative effects of s-allylmer- captocysteine on colon cancer cells when tested alone or in combination with sulindac sulfide. Cancer Res 2001, 61, 725–731. [Google Scholar]
- Hirota, J; Furuichi, T; Mikoshiba, K. Inositol 1,4,5–triphosphate receptor type 1 is a for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J Biol Chem 1999, 274, 34433–34437. [Google Scholar]
- Voss, OH; Kim, S; Wewers, MD; Doseff, AI. Regulation of monocyte apoptosis by the protein kinase Cδ-dependent phosphorylation of caspase-3. J Biol Chem 2005, 17371–17379. [Google Scholar]
© 2006 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dorsey, W.C.; Tchounwou, P.B.; Ford, B.D. Neuregulin 1-Βeta Cytoprotective Role in AML 12 Mouse Hepatocytes Exposed to Pentachlorophenol. Int. J. Environ. Res. Public Health 2006, 3, 11-22. https://doi.org/10.3390/ijerph2006030002
Dorsey WC, Tchounwou PB, Ford BD. Neuregulin 1-Βeta Cytoprotective Role in AML 12 Mouse Hepatocytes Exposed to Pentachlorophenol. International Journal of Environmental Research and Public Health. 2006; 3(1):11-22. https://doi.org/10.3390/ijerph2006030002
Chicago/Turabian StyleDorsey, Waneene C., Paul B. Tchounwou, and Byron D. Ford. 2006. "Neuregulin 1-Βeta Cytoprotective Role in AML 12 Mouse Hepatocytes Exposed to Pentachlorophenol" International Journal of Environmental Research and Public Health 3, no. 1: 11-22. https://doi.org/10.3390/ijerph2006030002