
Developing a Dissociative Nanocontainer for Peptide Drug Delivery

Abstract
:
1. Introduction

2. Materials and Methods
2.1. Construction of P22-GFP Nanocontainers

2.2. Confocal Fluorescence Microscopy Characterization of P22-GFP Nanocontainers
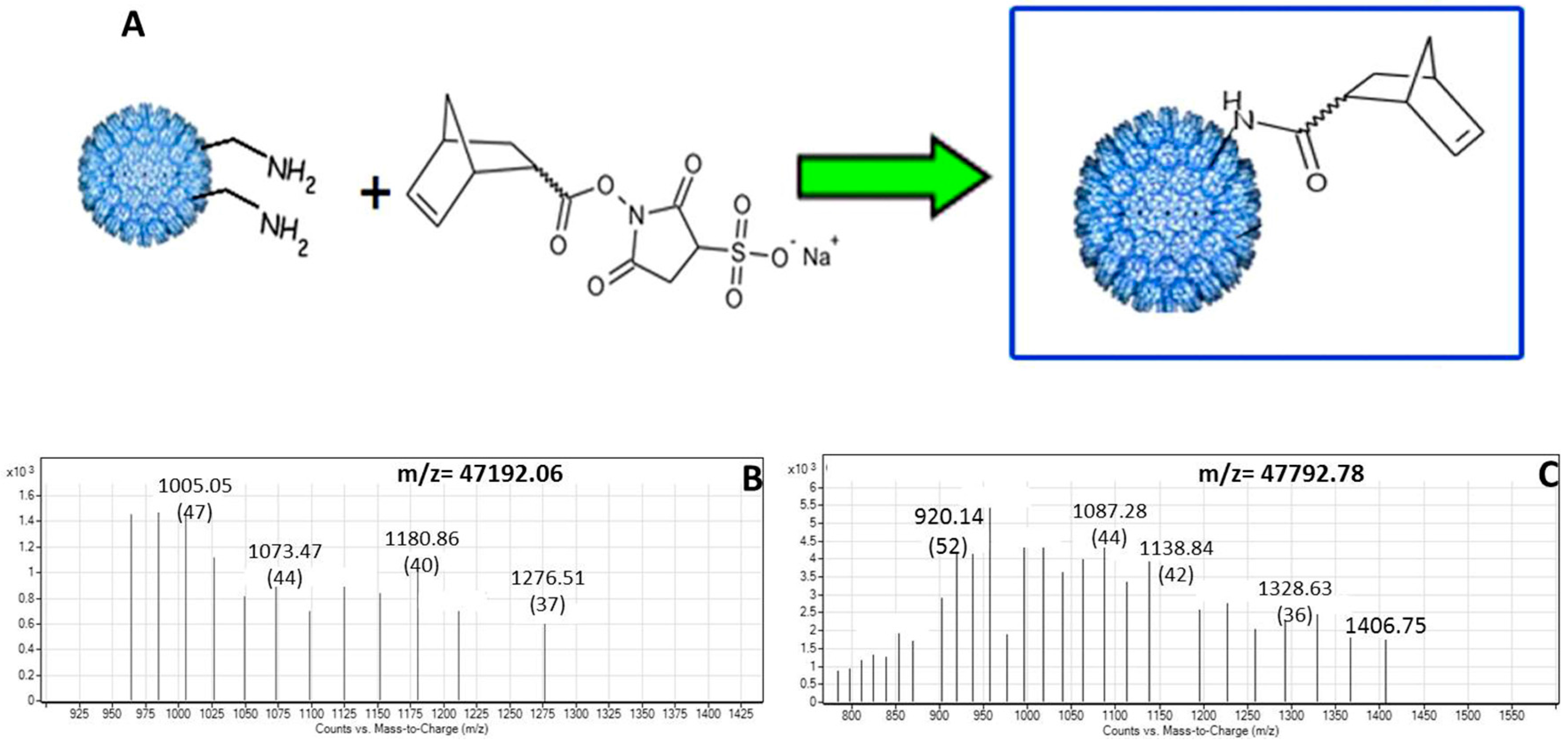
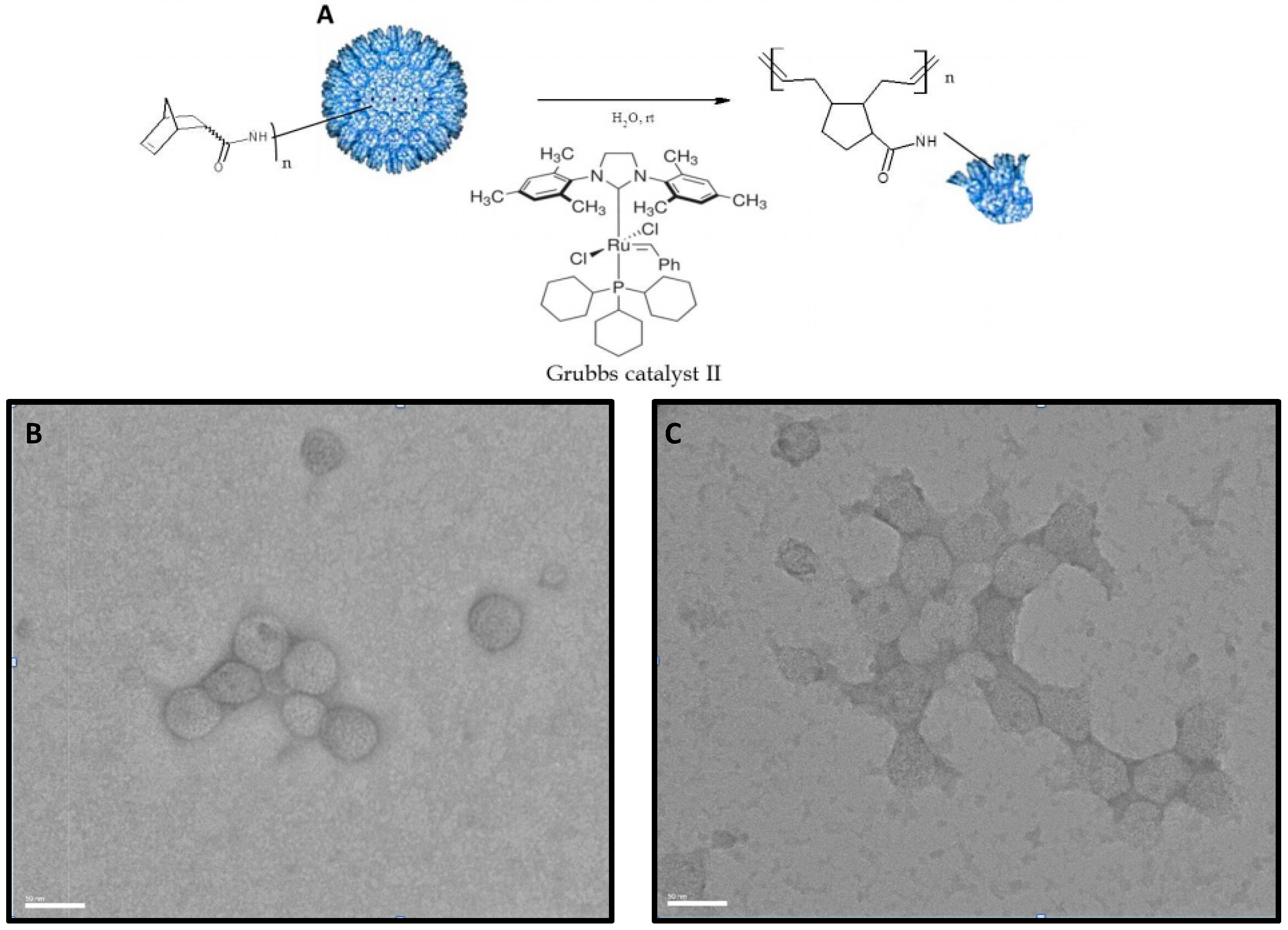
2.3. Conjugation of Norbornene to P22-GFP Nanocontainers
2.4. MS Characterization of P22-GFP-Norbornene Nanocontainers
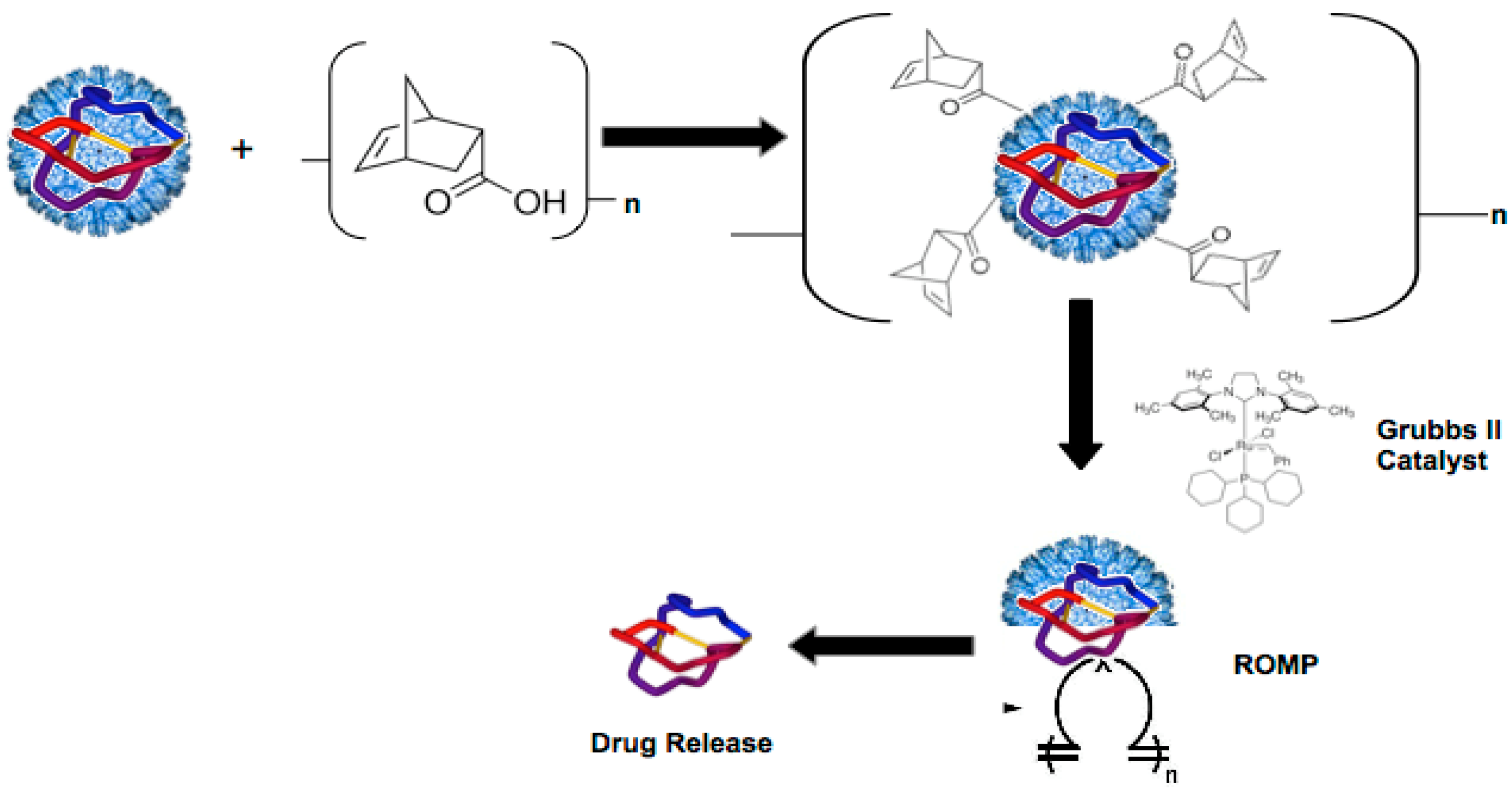
2.5. Ring Opening Metathesis Polymerization (ROMP) of P22-GFP-Norbornene Nanocontainers
2.6. Transmission Electron Microscopy (TEM) Characterization of P22-GFP Nanocontainers and Post-ROMP P22-GFP-Norbornene Nanocontainers
2.7. DLS Characterization of P22-GFP Nanocontainers
2.8. P22-GFP Heat-Activated Disassembly Monitored by Native Agarose Gels
3. Results
3.1. Characterization of P22-GFP Nanocontainers
3.2. Characterization of Norbornene-Conjugated P22-GFP Nanocontainers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mass | Vol % | No. of Norbornenes |
|---|---|---|---|
| 5 | 47,792.2998 | 26.8 | 5 |
| 4 | 47,672.4353 | 24.17 | 4 |
| 3 | 47,552.3638 | 20.9 | 3 |
| 2 | 47,672.0044 | 12.43 | 4 |
| 6 | 47,911.832 | 5.81 | 6 |
| 1 | 47,432.2401 | 5.24 | 2 |
| 7 | 47,911.4899 | 3.95 | 6 |

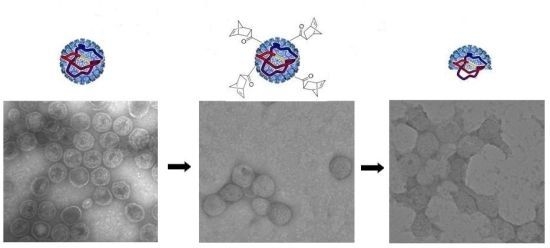
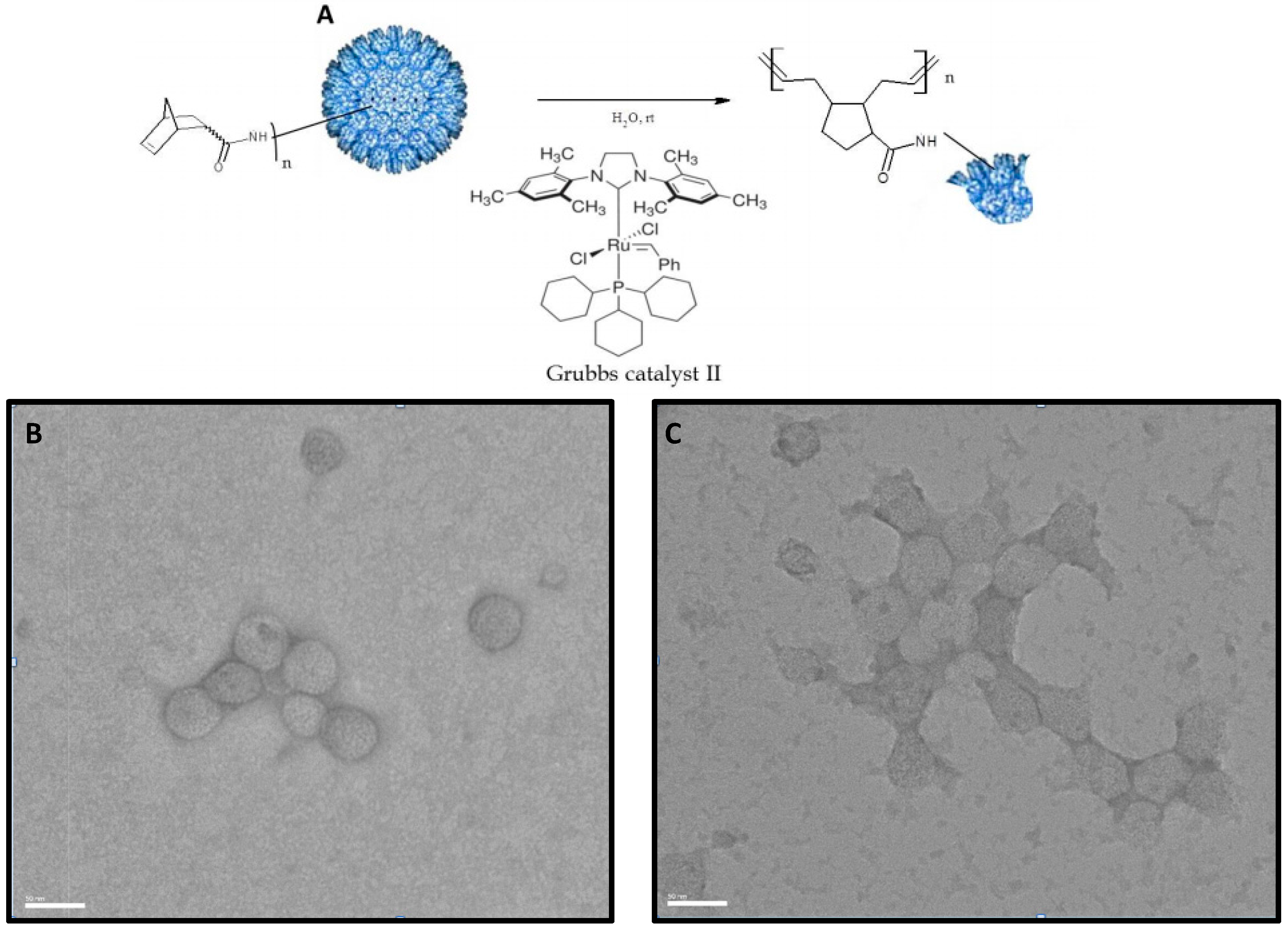
3.3. TEM Characterization of P22-GFP-Norbornene ROMP Reaction
3.4. Characterization of Heat-Induced Dissociation of P22 Nanocontainer

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marx, V. Watching peptide drugs grow up. Chem. Eng. News 2005, 83, 17–24. [Google Scholar]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kini, R.M. From snake venom toxins to therapeutics—Cardiovascular examples. Toxicon 2012, 59, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Holford, M. The terebridae and teretoxins: Combining phylogeny and anatomy for concerted discovery of bioactive compounds. BMC Chem. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.; Campbell, L.; Jenner, R. Quo vadis venomics? A roadmap to neglected venomous invertebrates. Toxins 2014, 6, 3488–3551. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.G. ω-Conotoxin MVIIA: From marine snail venom to analgesic drug. In Drugs from the Sea; Fusetani, N., Ed.; Karger: Basel, Switzerland, 2000; pp. 75–85. [Google Scholar]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; O’Neil, A.; Lin, E.; Douglas, T.; Holford, M. Tailored delivery of analgesic ziconotide across a blood brain barrier model using viral nanocontainers. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhu, J.; Chen, T.; Lin, S.; Cai, C.; Zhang, L.; Zhuang, Y.; Wang, X.S. Drug releasing behavior of hybrid micelles containing polypeptide triblock copolymer. Biomaterials 2009, 30, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Lian, T.; Ho, R.J. Trends and developments in liposome drug delivery systems. J. Pharm. Sci. 2001, 90, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A logic-gated nanorobot for targeted transport of molecular payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Virus-based nanocarriers for drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, L.; van Hest, J.C.M. Functionalization of protein-based nanocages for drug delivery applications. Nanoscale 2014, 6, 7124–7141. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sek, M.W.; Lim, L.Y. Folic acid-conjugated protein cages of a plant virus: A novel delivery platform for doxorubicin. Bioconjug. Chem. 2007, 18, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.J.; Hsiao, S.C.; Carrico, Z.M.; Francis, M.B. Viral capsid DNA aptamer conjugates as multivalent cell targeting vehicles. J. Am. Chem.Soc. 2010, 131, 11174–11178. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Uchida, M.; Oneil, A.; Li, R.; Prevelige, P.E.; Douglas, T. Implementation of P22 viral capsids as nanoplatforms. Biomacromolecules 2010, 11, 2804–2809. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.H.; Casjens, S.; Prevelige, P.E. Functional domains of bacteriophage P22 scaffolding protein. J. Mol. Biol. 1998, 281, 69–79. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, A.; Reichhardt, C.; Johnson, B.; Prevelige, P.E.; Douglas, T. Genetically programmed in vivo packaging of protein cargo and its controlled release from bacteriophage P22. Angew. Chem. Int. Ed. Engl. 2011, 50, 7425–7428. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Prevelige, P.E.; Douglas, T. Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano 2012, 6, 5000–5009. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.B.; Raines, R.T. Olefin metathesis for chemical biology. Curr. Opin. Chem. Biol. 2008, 12, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Brendgen, T.; Fahlbusch, T.; Frank, M.; Schühle, D.T.; Seßler, M.; Schatz, J. Metathesis in pure water mediated by supramolecular additives. Adv. Synth. Catal. 2009, 351, 303–307. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Teschke, C.M.; McGough, A.; Thuman-Commike, P.A. Penton release from P22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophys. J. 2003, 84, 2585–2592. [Google Scholar] [CrossRef]
- Kaiser, C.R.; Flenniken, M.L.; Gillitzer, E.; Harmsen, A.L.; Harmsen, A.G.; Jutila, M.A.; Douglas, T.; Young, M.J. Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo. Int. J. Nanomedicine 2007, 2, 715–733. [Google Scholar] [PubMed]
- Patel, P.R.; Kiser, R.C.; Lu, Y.Y.; Fong, E.; Ho, W.C.; Tirrell, D.A.; Grubbs, R.H. Synthesis and cell adhesive properties of linear and cyclic RGD functionalized polynorbornene thin films. Biomacromolecules 2012, 13, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Trnka, T.M.; Grubbs, R.H. The Development of L2X2Ru=CHR Olefin metathesis catalysts: An organometallic success story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Nováková, O. DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist. Updat. 2006, 9, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platin. Met. Rev. 2001, 45, 62–69. [Google Scholar]
- Lipshutz, B.H.; Aguinaldo, G.T.; Ghorai, S.; Voigtritter, K. Olefin cross-metathesis reactions at room temperature using the nonionic amphiphile “PTS”: Just add water. Org. Lett. 2008, 10, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Recent advances in olefin metathesis by molybdenum and tungsten imido alkylidene complexes. J. Mol. Catal. A Chem. 2004, 213, 21–30. [Google Scholar] [CrossRef]
- Schrock, R.R.; Hoveyda, A.H. Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts. Angew. Chem. Int. Ed. Engl. 2003, 42, 4592–4633. [Google Scholar] [CrossRef] [PubMed]
- Ben-Asuly, A.; Aharoni, A.; Diesendruck, C.E.; Vidavsky, Y.; Goldberg, I.; Straub, B.F.; Lemcoff, N.G. Photoactivation of ruthenium olefin metathesis initiators. Organometallics 2009, 28, 4652–4655. [Google Scholar] [CrossRef]
- Fortin, D.L.; Banghart, M.R.; Dunn, T.W.; Borges, K.; Wagenaar, D.A.; Gaudry, Q.; Karakossian, M.H.; Otis, T.S.; Kristan, W.B.; Trauner, D.; Kramer, R.H. Photochemical control of endogenous ion channels and cellular excitability. Nat. Methods 2008, 5, 331–338. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, P.; Anand, P.; Uvaydov, A.; Chakravartula, S.; Sherpa, C.; Pires, E.; O’Neil, A.; Douglas, T.; Holford, M. Developing a Dissociative Nanocontainer for Peptide Drug Delivery. Int. J. Environ. Res. Public Health 2015, 12, 12543-12555. https://doi.org/10.3390/ijerph121012543
Kelly P, Anand P, Uvaydov A, Chakravartula S, Sherpa C, Pires E, O’Neil A, Douglas T, Holford M. Developing a Dissociative Nanocontainer for Peptide Drug Delivery. International Journal of Environmental Research and Public Health. 2015; 12(10):12543-12555. https://doi.org/10.3390/ijerph121012543
Chicago/Turabian StyleKelly, Patrick, Prachi Anand, Alexander Uvaydov, Srinivas Chakravartula, Chhime Sherpa, Elena Pires, Alison O’Neil, Trevor Douglas, and Mandë Holford. 2015. "Developing a Dissociative Nanocontainer for Peptide Drug Delivery" International Journal of Environmental Research and Public Health 12, no. 10: 12543-12555. https://doi.org/10.3390/ijerph121012543
APA StyleKelly, P., Anand, P., Uvaydov, A., Chakravartula, S., Sherpa, C., Pires, E., O’Neil, A., Douglas, T., & Holford, M. (2015). Developing a Dissociative Nanocontainer for Peptide Drug Delivery. International Journal of Environmental Research and Public Health, 12(10), 12543-12555. https://doi.org/10.3390/ijerph121012543