Pectenotoxin-2 from Marine Sponges: A Potential Anti-Cancer Agent—A Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
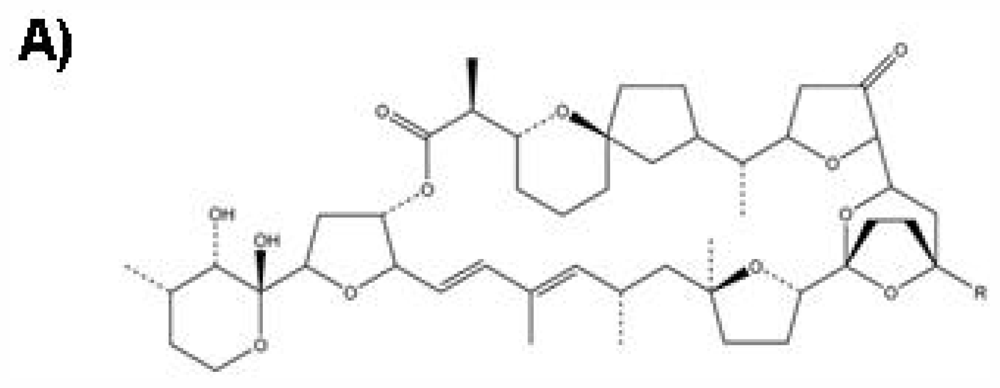
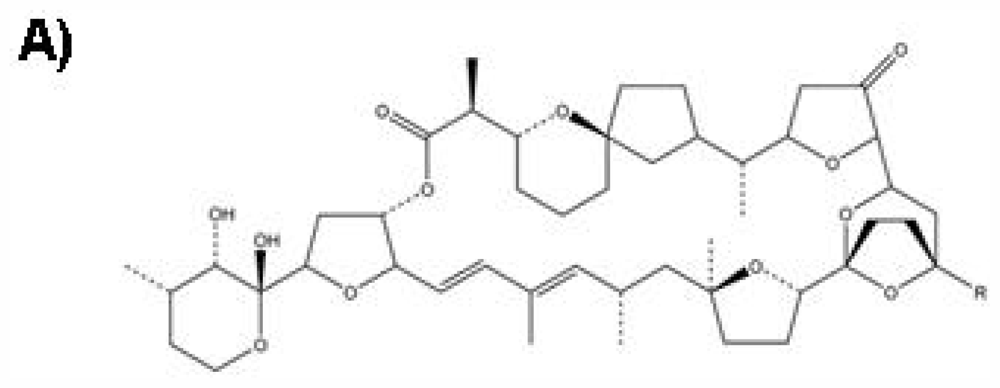
:1. Introduction
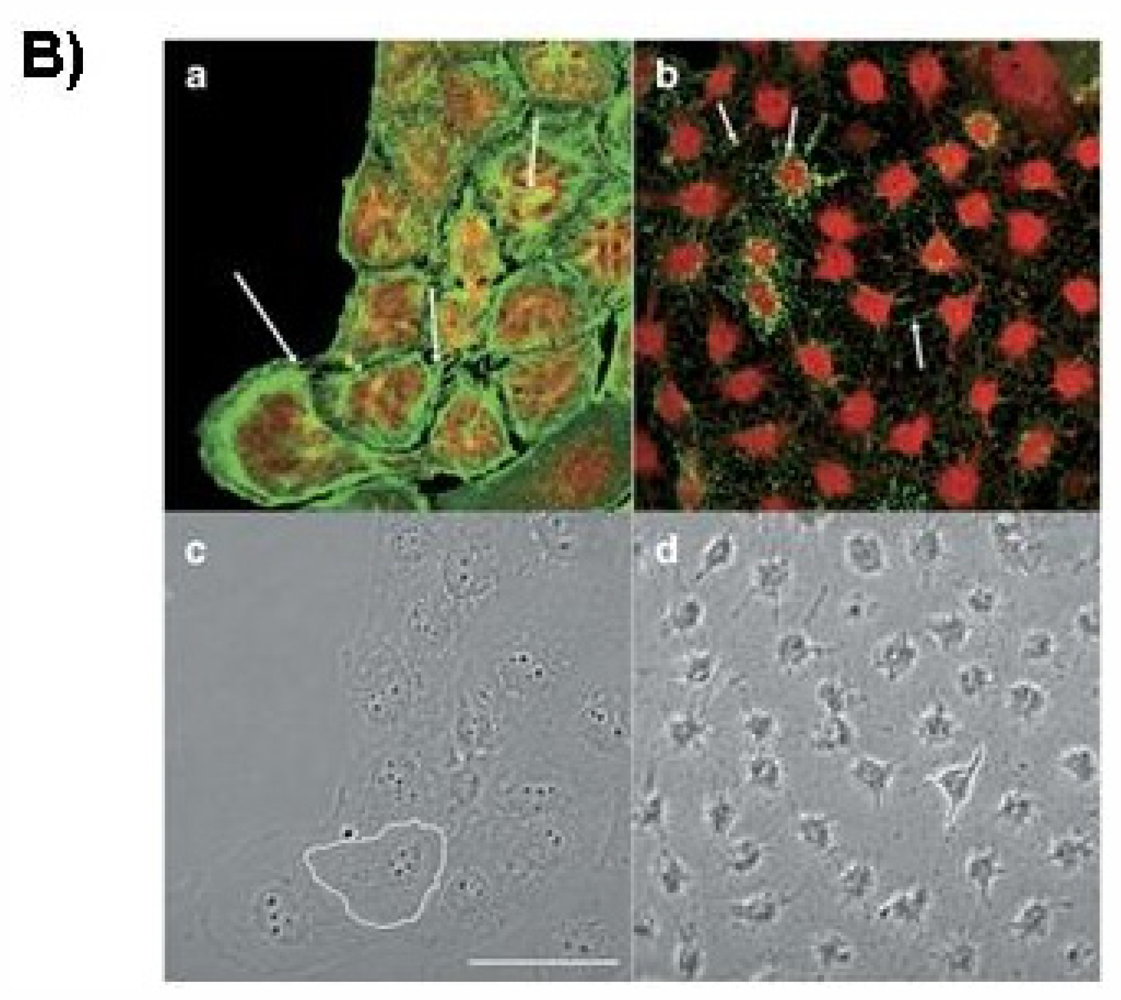
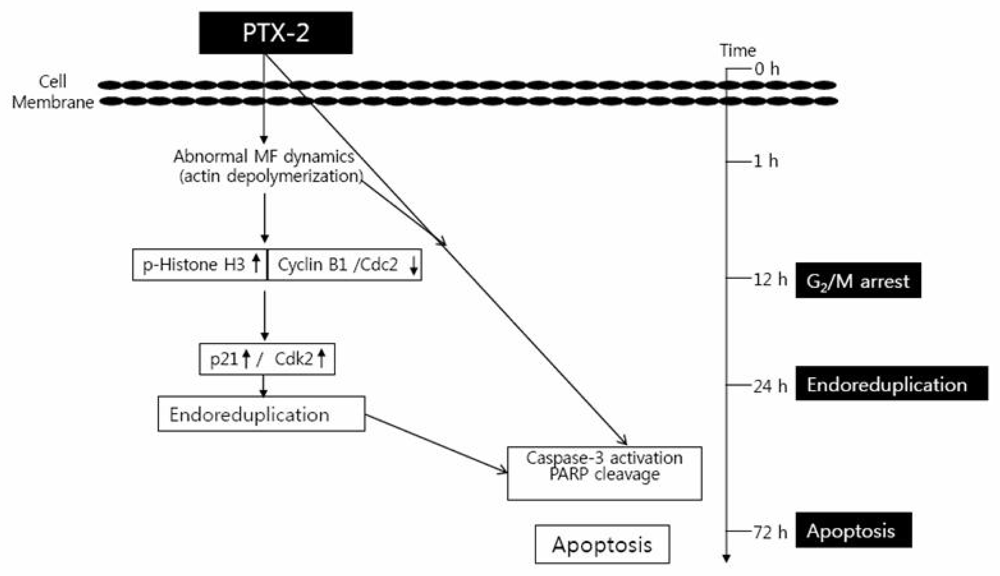
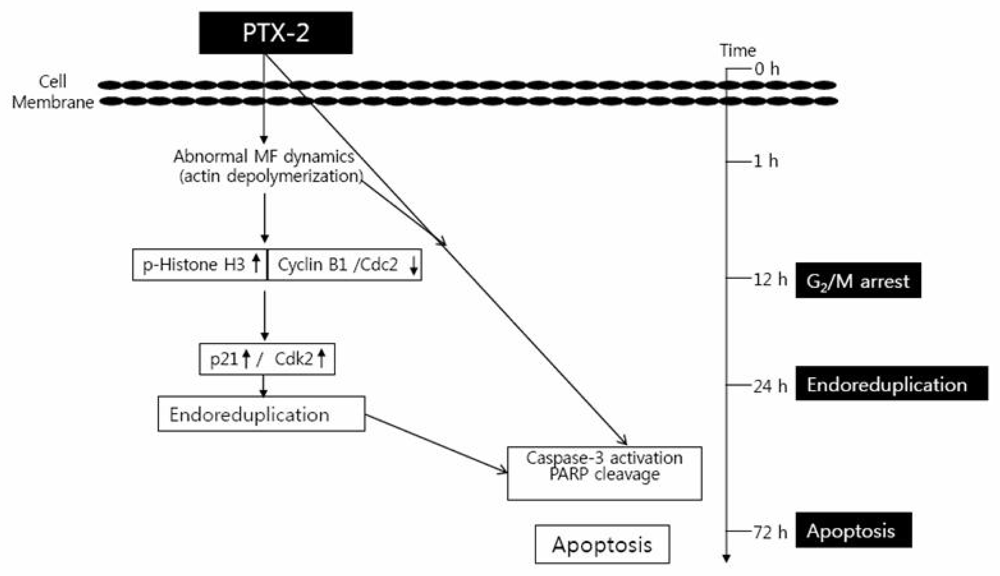
2. Effect of PTX-2 on Cell Cycle Arrest and Endoreduplication
2.1. G2/M Phase Cell Cycle Arrest
2.2. Endoreduplication
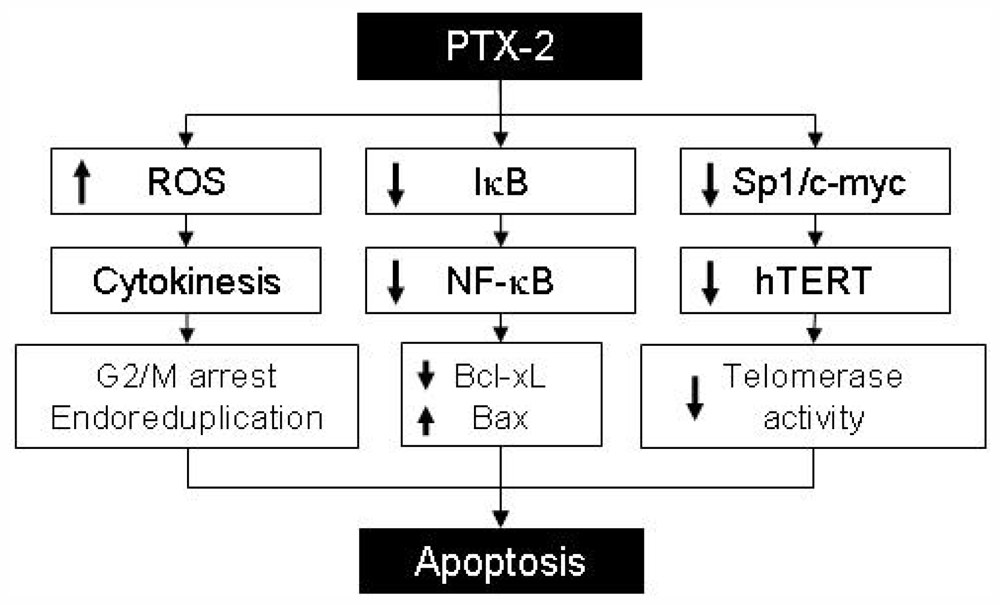
2.3. Effect of PTX-2 on NF-κB and Its Related-Gene Products
3. Effect of PTX-2 on Apoptosis
4. Effect of PTX-2 on Telomerase Activity
5. Conclusion
Acknowledgments
References
- Spector, I.; Braet, F.; Shochet, N.R.; Bubb, M.R. New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Microsc. Res. Tech 1999, 47, 18–37. [Google Scholar]
- Leira, F.; Cabado, A.G.; Vieytes, M.R.; Roman, Y.; Alfonso, A.; Botana, L.M.; Yasumoto, T.; Malaguti, C.; Rossini, G.P. Characterization of F-actin depolymerization as a major toxic event induced by pectenotoxin-6 in neuroblastoma cells. Biochem. Pharmacol 2002, 63, 1979–1988. [Google Scholar]
- Allingham, J.S.; Miles, C.O.; Rayment, I. A structural basis for regulation of actin polymerization by pectenotoxins. J. Mol. Biol 2007, 371, 959–970. [Google Scholar]
- Hori, M.; Matsuura, Y.; Yoshimoto, R.; Ozaki, H.; Yasumoto, T.; Karaki, H. Actin depolymerizing action by marine toxin, pectenotoxin-2. Nippon Yakurigaku Zasshi 1999, 114, 225–229. [Google Scholar]
- Allingham, J.S.; Klenchin, V.A.; Rayment, I. Actin-targeting natural products: structures, properties and mechanisms of action. Cell. Mol. Life Sci 2006, 63, 2119–2134. [Google Scholar]
- Espiña, B.; Louzao, M.C.; Ares, I.R.; Cagide, E.; Vieytes, M.R.; Vega, F.V.; Rubiolo, J.A.; Miles, C.O.; Suzuki, T.; Yasumoto, T.; et al. Cytoskeletal toxicity of pectenotoxins in hepatic cells. Br. J. Pharmacol 2008, 155, 934–944. [Google Scholar]
- Moon, D.O.; Kim, M.O.; Kang, S.H.; Lee, K.J.; Heo, M.S.; Choi, K.S.; Choi, Y.H.; Kim, G.Y. Induction of G2/M arrest, endoreduplication, and apoptosis by actin depolymerization agent pextenotoxin-2 in human leukemia cells, involving activation of ERK and JNK. Biochem. Pharmacol 2008, 76, 312–321. [Google Scholar]
- Shin, I.J.; Ahn, Y.T.; Kim, Y.; Kim, J.M.; An, W.G. Actin disruption agents induce phosphorylation of histone H2AX in human breast adenocarcinoma MCF-7 cells. Oncol. Rep 2011, 25, 1313–1319. [Google Scholar]
- Draetta, G.; Eckstein, J. Cdc25 protein phosphatases in cell proliferation. Biochim. Biophys. Acta 1997, 1332, 53–63. [Google Scholar]
- Myer, D.L.; Bahassi, E.M.; Stambrook, P.J. The Plk3-Cdc25 circuit. Oncogene 2005, 24, 299–305. [Google Scholar]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar]
- Eymin, B.; Claverie, P.; Salon, C.; Brambilla, C.; Brambilla, E.; Gazzeri, S. p14ARF triggers G2 arrest through ERK-mediated Cdc25C phosphorylation, ubiquitination and proteasomal degradation. Cell Cycle 2006, 5, 759–765. [Google Scholar]
- Moon, D.O.; Kim, M.O.; Nam, T.J.; Kim, S.K.; Choi, Y.H.; Kim, G.Y. Pectenotoxin-2 induces G2/M phase cell cycle arrest in human breast cancer cells via ATM and Chk1/2-mediated phosphorylation of cdc25C. Oncol. Rep 2010, 24, 271–276. [Google Scholar]
- Singh, S.V.; Herman-Antosiewicz, A.; Singh, A.V.; Lew, K.L.; Srivastava, S.K.; Kamath, R.; Brown, K.D.; Zhang, L.; Baskaran, R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J. Biol. Chem 2004, 279, 25813–25822. [Google Scholar]
- Niculescu, A.B.; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell Biol 1998, 18, 629–643. [Google Scholar]
- Landry, J.; Huot, J. Modulation of actin dynamics during stress and physiological stimulation by a signaling pathway involving p38 MAP kinase and heat-shock protein 27. Biochem. Cell. Biol 1995, 73, 703–707. [Google Scholar]
- Espina, B.; Louzao, M.C.; Ares, I.R.; Cagide, E.; Vievtes, M.R.; Vega, F.V.; Rubiolo, J.A.; Miles, C.O.; Suzuki, T.; Yasumoto, T.; Botana, L.M. Cytoskeletal toxicity of pectenotoxins in hepatic cells. Br. J. Pharmacol 2008, 155, 934–944. [Google Scholar]
- Takemoto, D.; Hardham, A.R. The cytoskeleton as a regulator and target of biotic interactions in plants. Plant Physiol 2004, 136, 3864–3876. [Google Scholar]
- Chang, B.D.; Broude, E.V.; Fang, J.; Kalinichenko, T.V.; Abdryashitov, R.; Poole, J.C.; Roninson, I.B. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosiscontrol proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene 2000, 19, 2165–2170. [Google Scholar]
- Reiterer, G.; Yen, A. Inhibition of the janus kinase family increases extracellular signal-regulated kinase 1/2 phosphorylation and causes endoreduplication. Cancer Res 2006, 66, 9083–9089. [Google Scholar]
- Lee, K.; Song, K. Actin dysfunction activates ERK1/2 and delays entry into mitosis in mammalian cells. Cell Cycle 2007, 6, 1487–1495. [Google Scholar]
- Hombach-Klonisch, S.; Paranjothy, T.; Wiechec, E.; Pocar, P.; Mustafa, T.; Seifert, A.; Zahl, C.; Gerlach, K.L.; Biermann, K.; Steger, K.; et al. Cancer stem cells as targets for cancer therapy: Selected cancers as examples. Arch. Immunol. Ther. Exp. (Warsz) 2008, 56, 165–180. [Google Scholar]
- Donat, S.; Streker, K.; Schirmeister, T.; Rakette, S.; Stehle, T.; Liebeke, M.; Lalk, M.; Ohlsen, K. Transcriptome and functional analysis of the eukaryotic-type serine/threonine kinase PknB in Staphylococcus aureus. J. Bacteriol 2009, 191, 4056–4069. [Google Scholar]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar]
- Jeong, W.S.; Kim, I.W.; Hu, R.; Kong, A.N. Modulatory properties of various natural chemopreventive agents on the activation of NF-κB signaling pathway. Pharm. Res 2004, 21, 661–670. [Google Scholar]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-κB controls cell growth differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol 1999, 19, 5785–5799. [Google Scholar]
- Kim, M.O.; Moon, D.O.; Heo, M.S.; Lee, J.D.; Jung, J.H.; Kim, S.K.; Choi, Y.H.; Kim, G.Y. Pectenotoxin-2 abolishes constitutively activated NF-κB, leading to suppression of NF-κB related gene products and potentiation of apoptosis. Cancer Lett 2008, 271, 25–33. [Google Scholar]
- Debatin, K.M. Apoptosis pathways in cancer and cancer therapy. Cancer Immunol. Immunother 2004, 53, 153–159. [Google Scholar]
- Jin, Z.; el-Deiry, W.S. Over view of cell death signaling pathways. Cancer Biol. Ther 2005, 4, 139–163. [Google Scholar]
- Chowdhury, I.; Tharakan, B.; Bhat, G.K. Current concepts in apoptosis: the physiological suicide program revisited. Cell. Mol. Biol. Lett 2006, 11, 506–525. [Google Scholar]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar]
- Halaby, M.J.; Yang, D.Q. p53 translational control: a new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 2007, 395, 1–7. [Google Scholar]
- Prasad, S.; Madan, E.; Nigam, N.; Roy, P.; George, J.; Shukla, Y. Induction of apoptosis by lupeol in human epidermoid carcinoma A431 cells through regulation of mitochondrial, Akt/PKB and NFkappaB signaling pathways. Cancer Biol. Ther 2009, 8, 1632–1639. [Google Scholar]
- Shin, D.Y.; Kim, G.Y.; Kim, N.D.; Jung, J.H.; Kim, H.S.; Choi, Y.H. Induction of apoptosis by pectenotoxin-2 is mediated with the induction of DR4/DR5, Egr-1 and NAG-1, activation of caspases and modulation of the Bcl-2 family in p53-deficient Hep3B hepatocellular carcinoma cells. Oncol. Rep 2008, 19, 517–526. [Google Scholar]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther 2002, 1, 639–649. [Google Scholar]
- Chae, H.D.; Choi, T.S.; Kim, B.M.; Jung, J.H.; Bang, Y.J.; Shin, D.Y. Oocyte-based screening of cytokinesis inhibitors and identification of pectenotoxin-2 that induces Bim/Bax-mediated apoptosis in p53-deficient tumors. Oncogene 2005, 24, 4813–4819. [Google Scholar]
- Bunch, J.T.; Bae, N.S.; Leonardi, J.; Baumann, P. Distinct requirements for Pot1 in limiting telomere length and maintaining chromosome stability. Mol. Cell. Biol 2005, 25, 5567–5578. [Google Scholar]
- Philippi, C.; Loretz, B.; Schaefer, U.F.; Lehr, C.M. Telomerase as an emerging target to fight cancer—Opportunities and challenges for nanomedicine. J. Control. Releases 2010, 146, 228–240. [Google Scholar]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar]
- Cong, Y.S.; Wen, J.; Bacchetti, S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum. Mol. Genet 1999, 8, 137–142. [Google Scholar]
- Kim, M.O.; Moon, D.O.; Kang, S.H.; Heo, M.S.; Choi, Y.H.; Jung, J.H.; Lee, J.D.; Kim, G.Y. Pectenotoxin-2 represses telomerase activity in human leukemia cells through suppression of hTERT gene expression and Akt-dependent hTERT phosphorylation. FEBS Lett 2008, 582, 3263–3269. [Google Scholar]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem 1999, 274, 13085–13090. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, G.-Y.; Kim, W.-J.; Choi, Y.H. Pectenotoxin-2 from Marine Sponges: A Potential Anti-Cancer Agent—A Review. Mar. Drugs 2011, 9, 2176-2187. https://doi.org/10.3390/md9112176
Kim G-Y, Kim W-J, Choi YH. Pectenotoxin-2 from Marine Sponges: A Potential Anti-Cancer Agent—A Review. Marine Drugs. 2011; 9(11):2176-2187. https://doi.org/10.3390/md9112176
Chicago/Turabian StyleKim, Gi-Young, Wun-Jae Kim, and Yung Hyun Choi. 2011. "Pectenotoxin-2 from Marine Sponges: A Potential Anti-Cancer Agent—A Review" Marine Drugs 9, no. 11: 2176-2187. https://doi.org/10.3390/md9112176