Generating a Generation of Proteasome Inhibitors: From Microbial Fermentation to Total Synthesis of Salinosporamide A (Marizomib) and Other Salinosporamides

Abstract
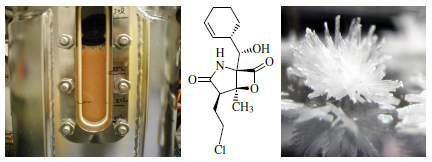
:
1. Introduction
2. Natural and Unnatural Products of S. tropica
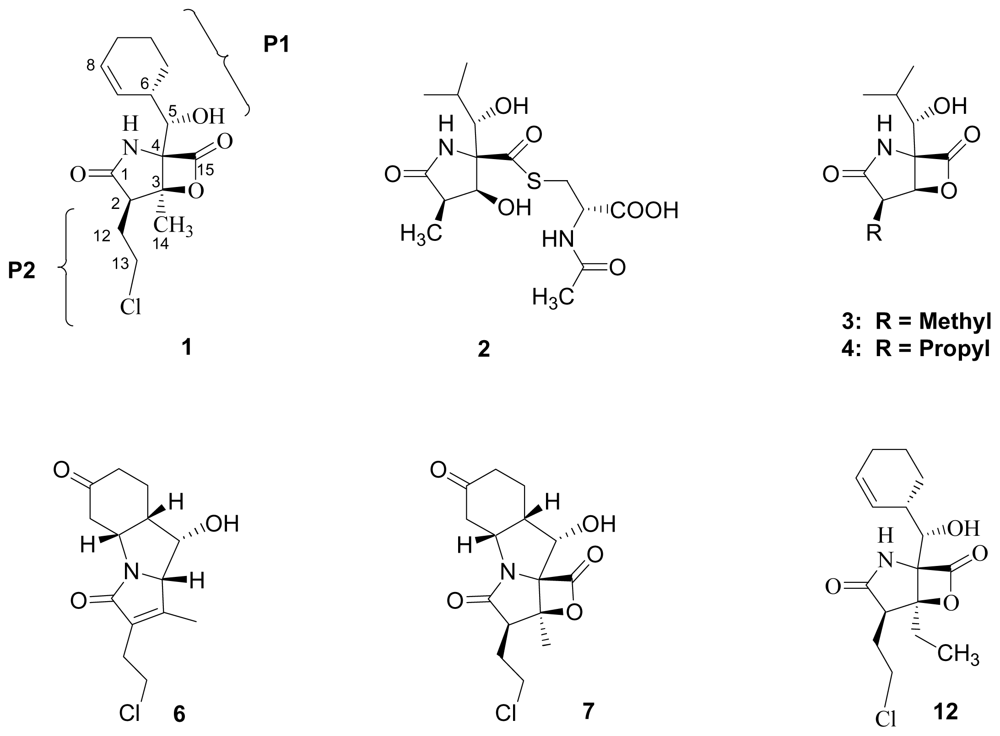
2.1. Natural Products of S. tropica
2.2. Fermentation Optimization of 1 to Clinical Trials Materials
2.3. Products of Precursor-Directed Biosynthesis
2.4. Products of Mutasynthesis
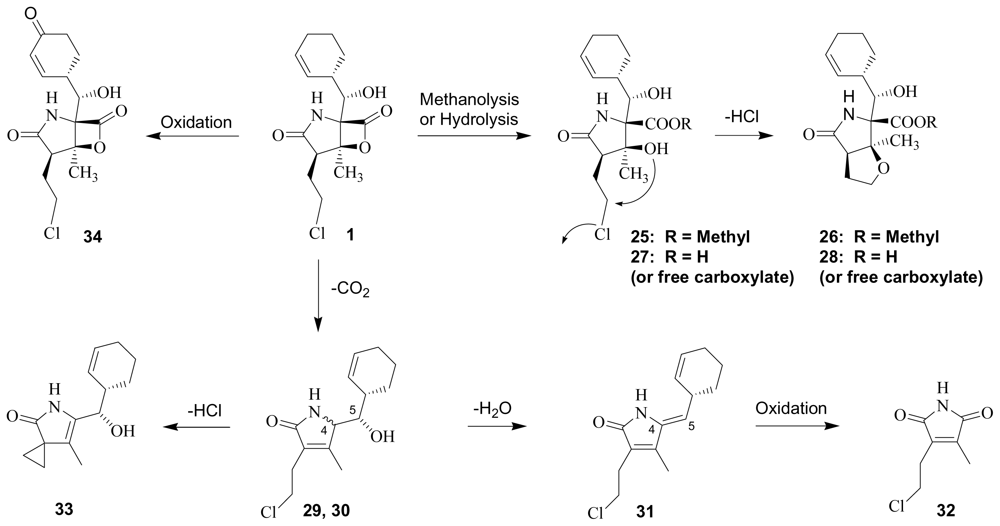
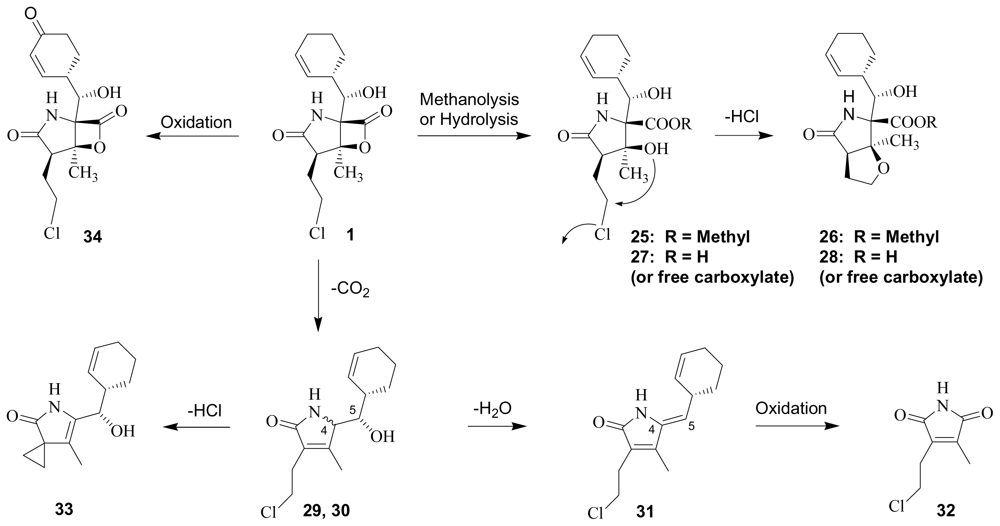
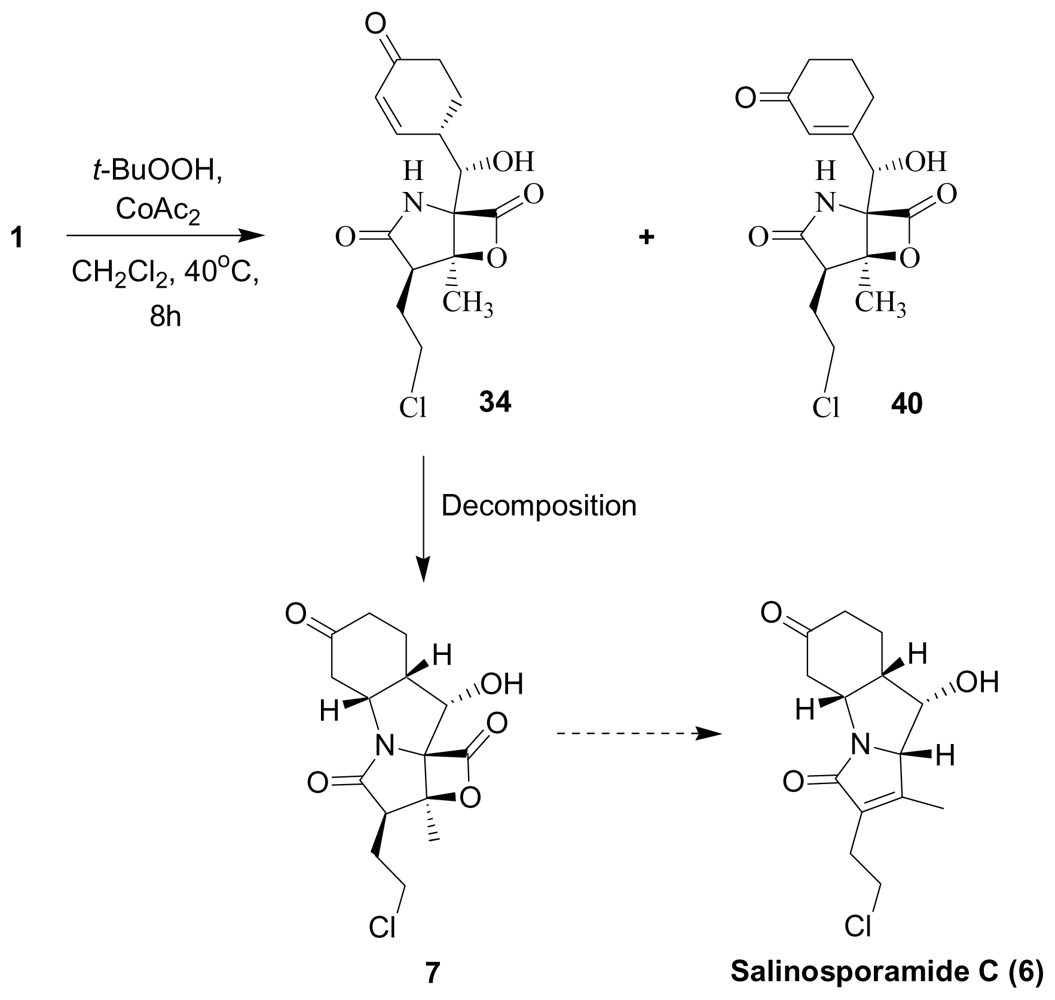
3. Products of Chemical Degradation
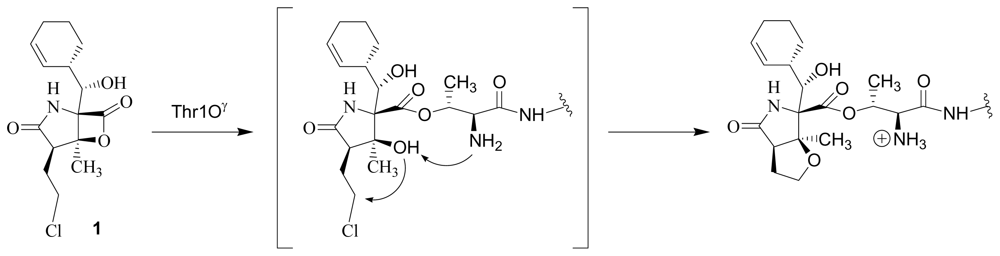
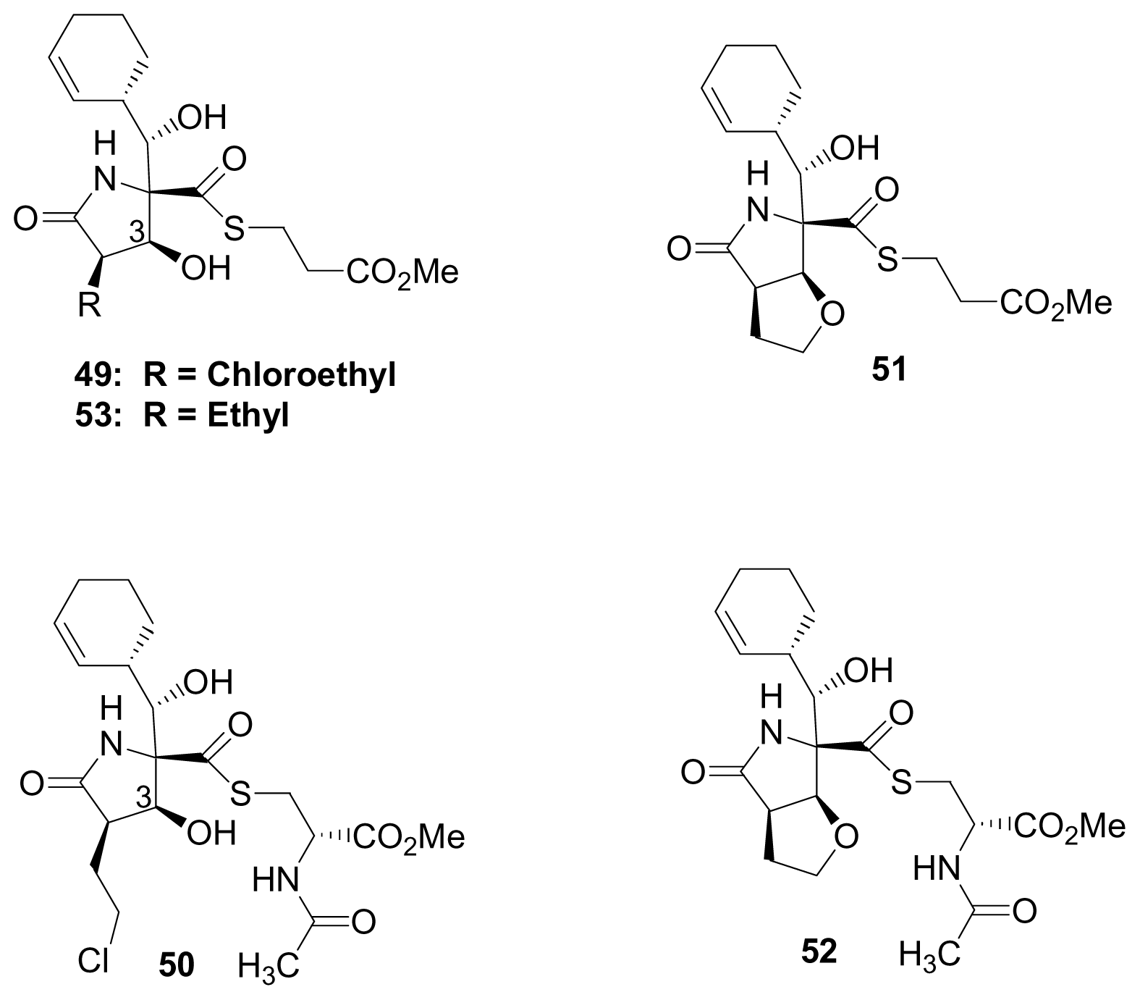
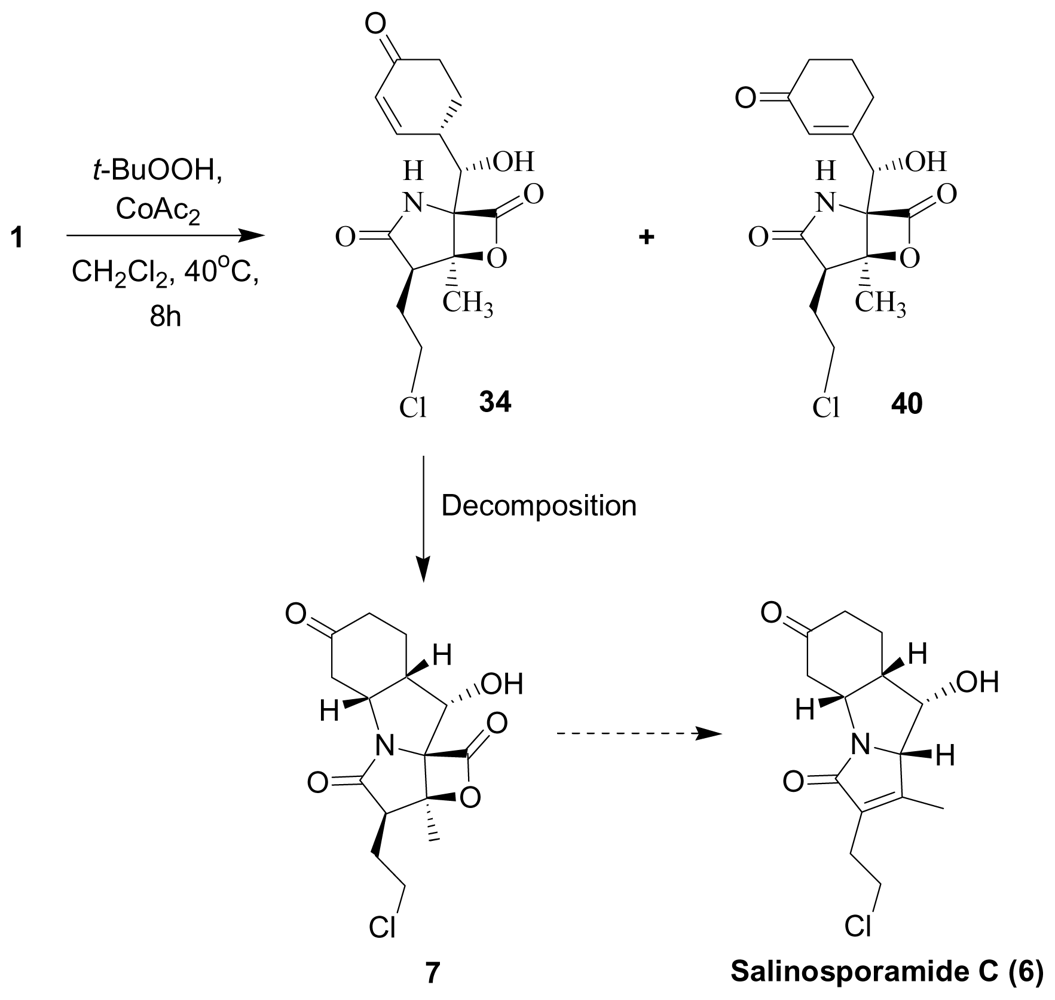
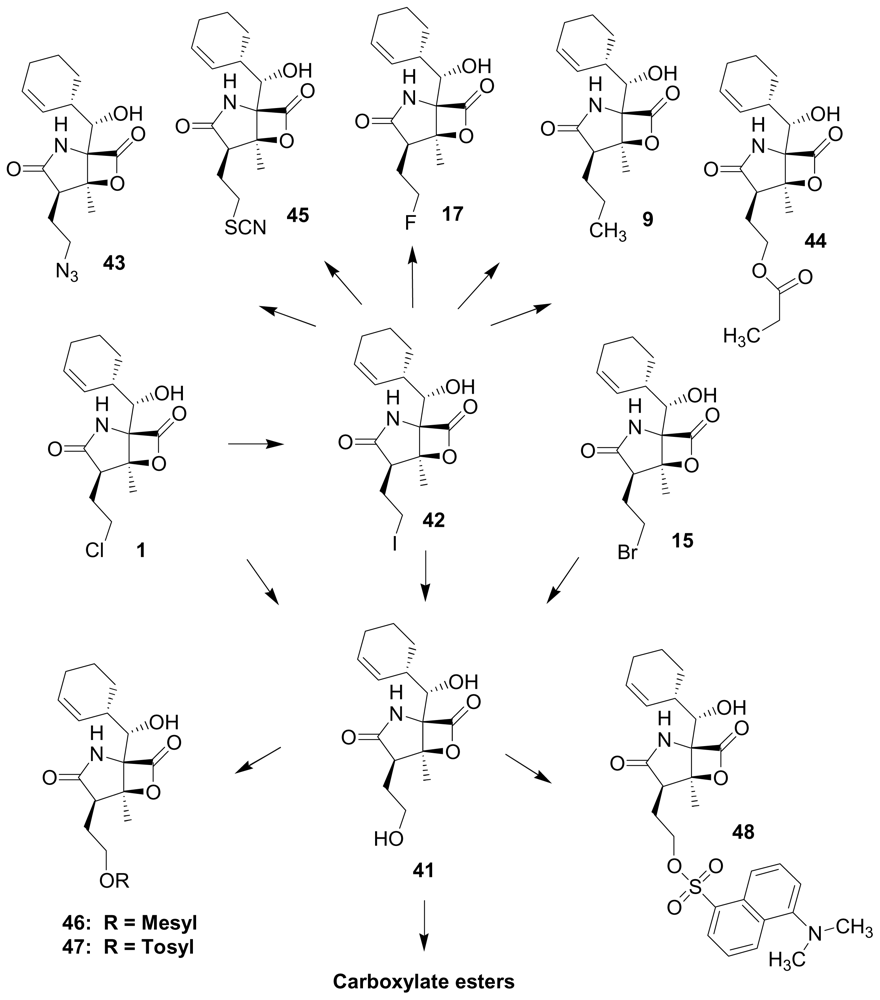
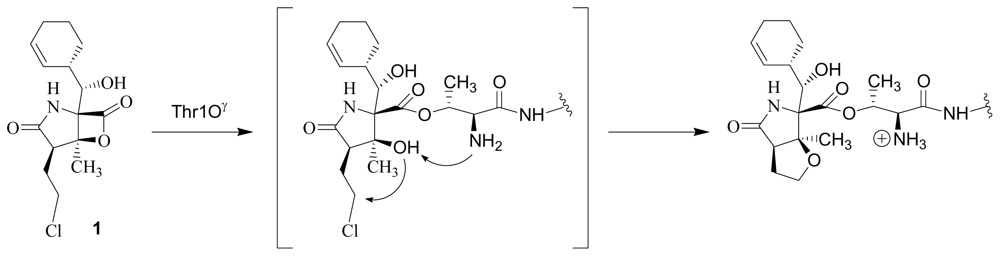
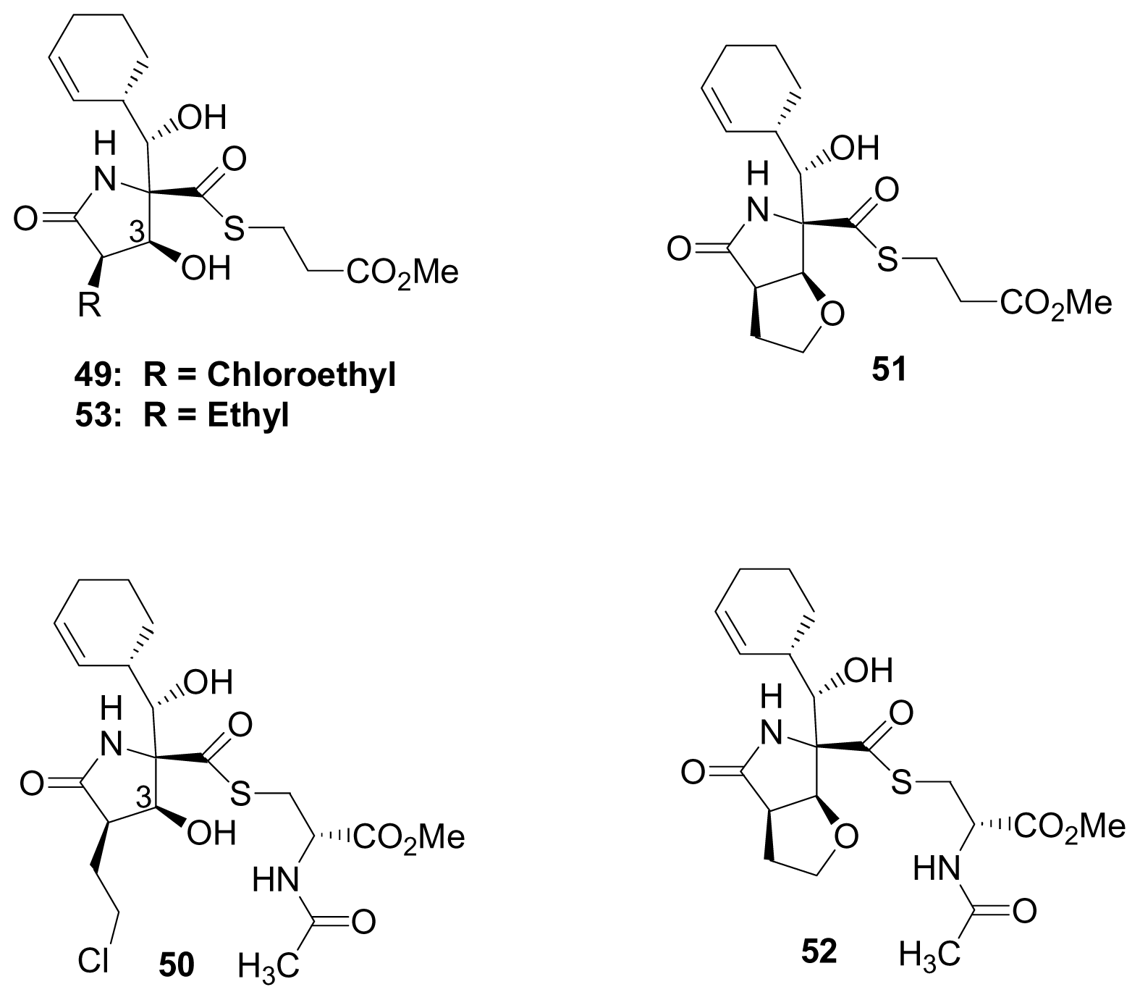
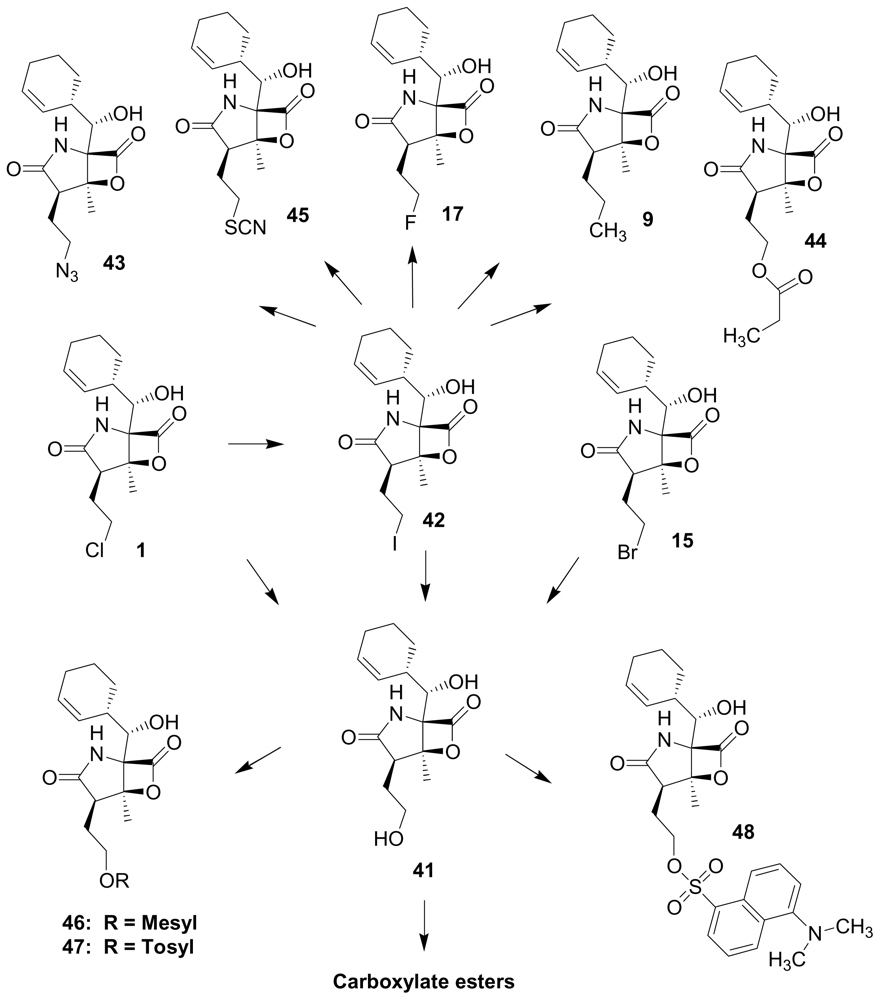
4. Products of Semi-Synthesis
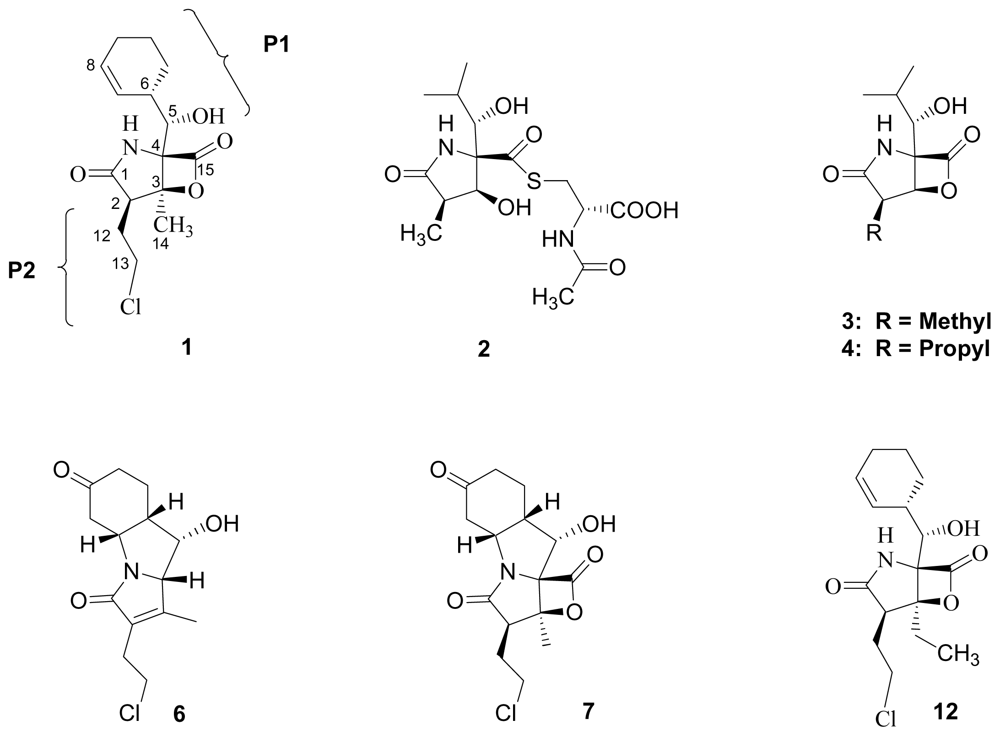
5. Structure-Activity Relationships
5.1. β-Lactone Derivatives
5.2. P1 Analogs
5.3. P2 Analogs
6. Total Synthesis of 1
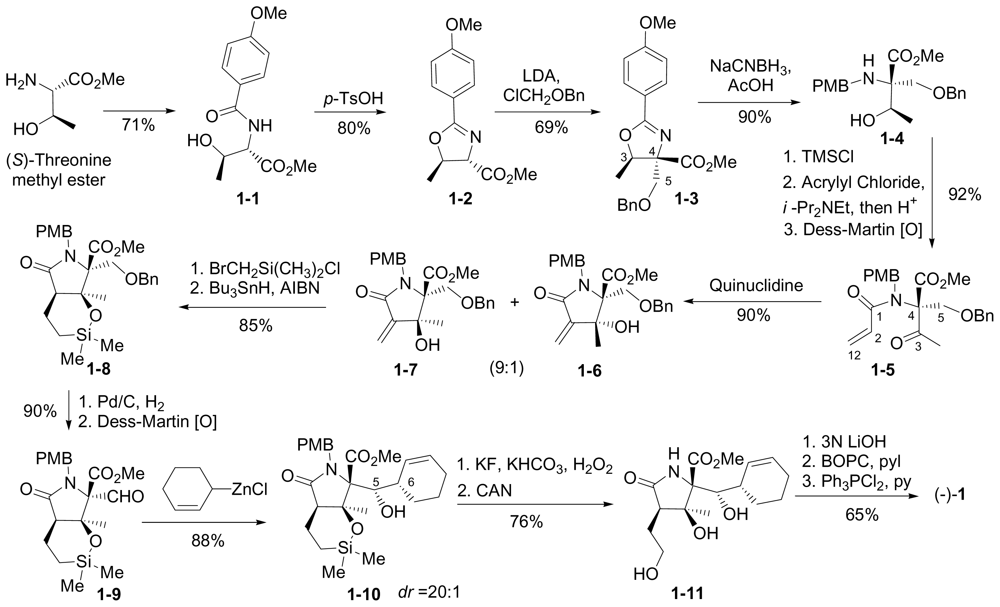
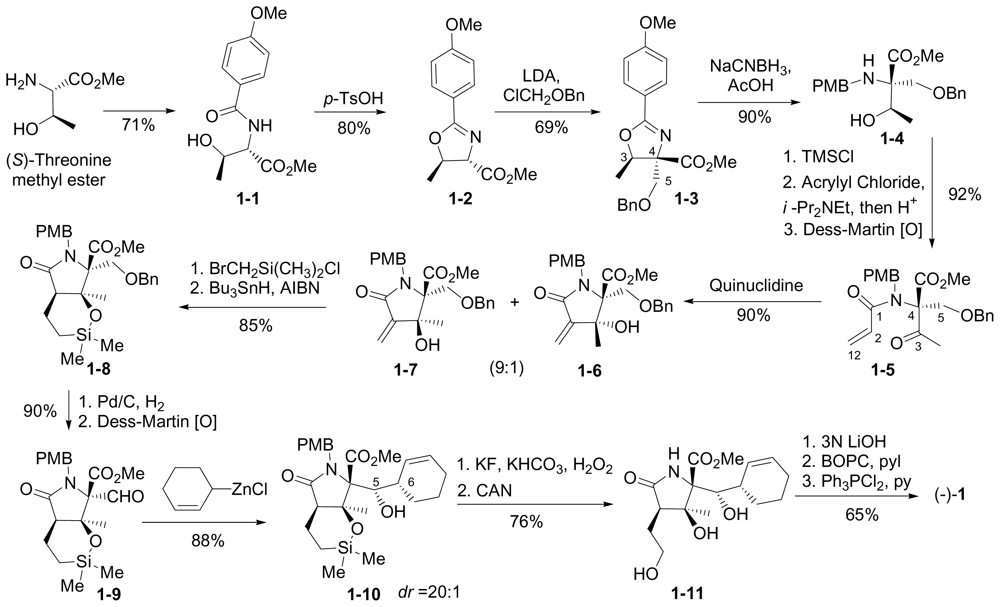
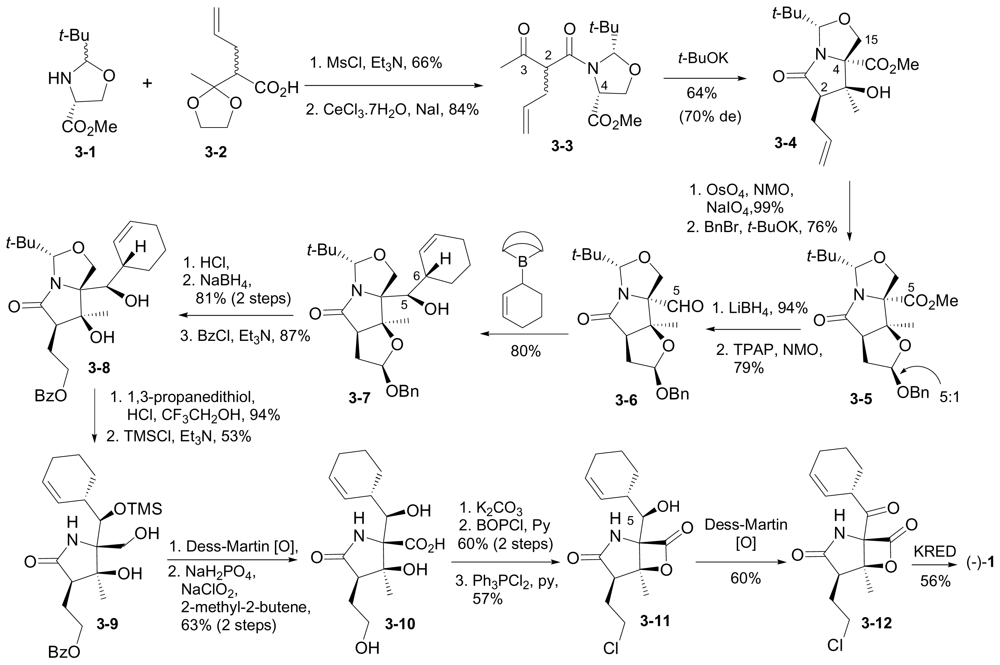
6.1. Enantioselective Synthesis of (-)-1
6.1.1. Corey Enantioselective Synthesis
6.1.2. Danishefsky Enantioselective Synthesis
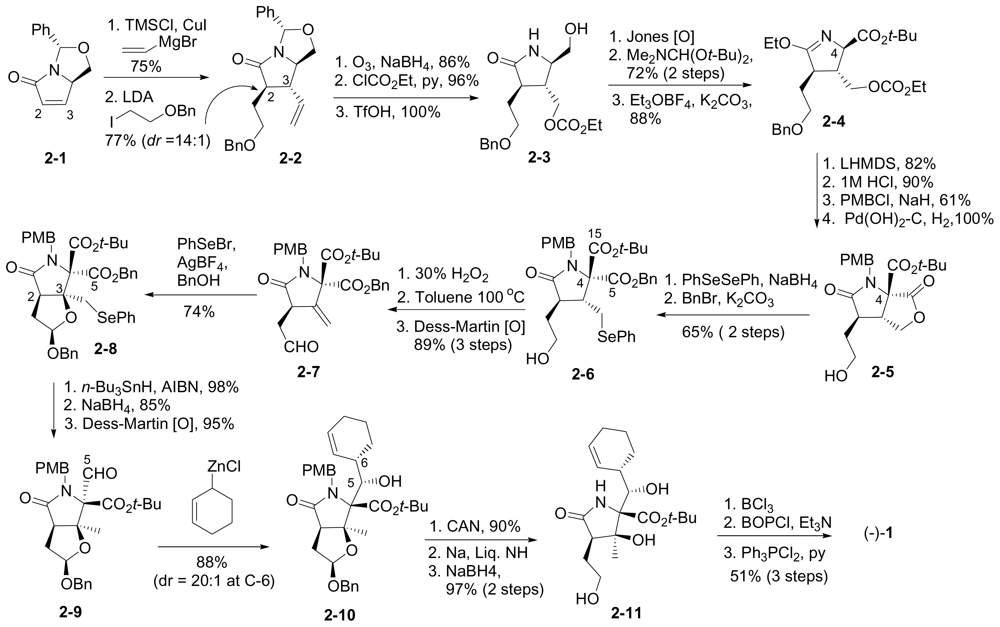
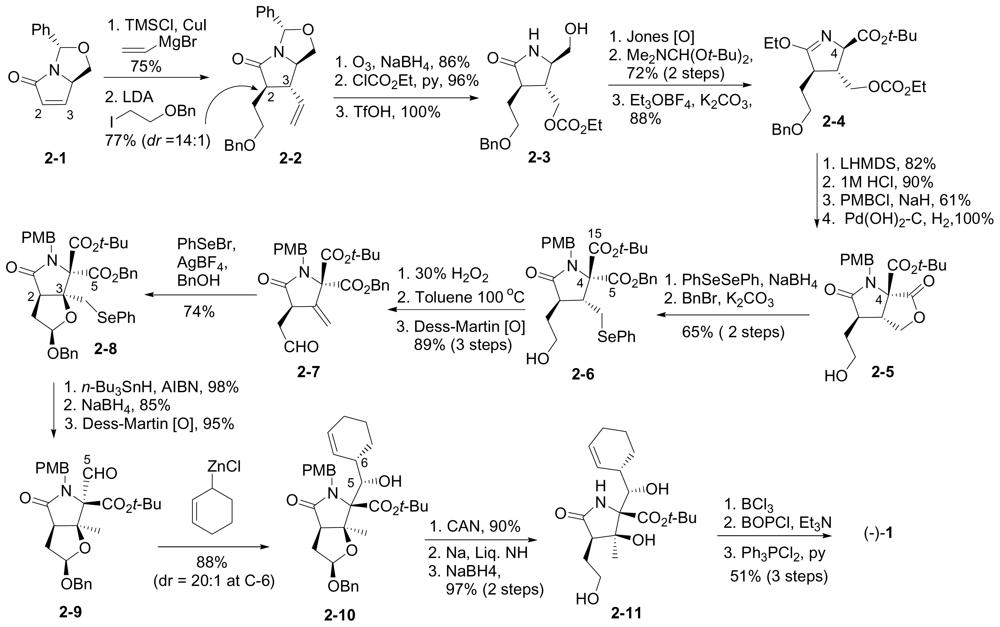
6.1.3. Nereus (Ling) Enantioselective Synthesis
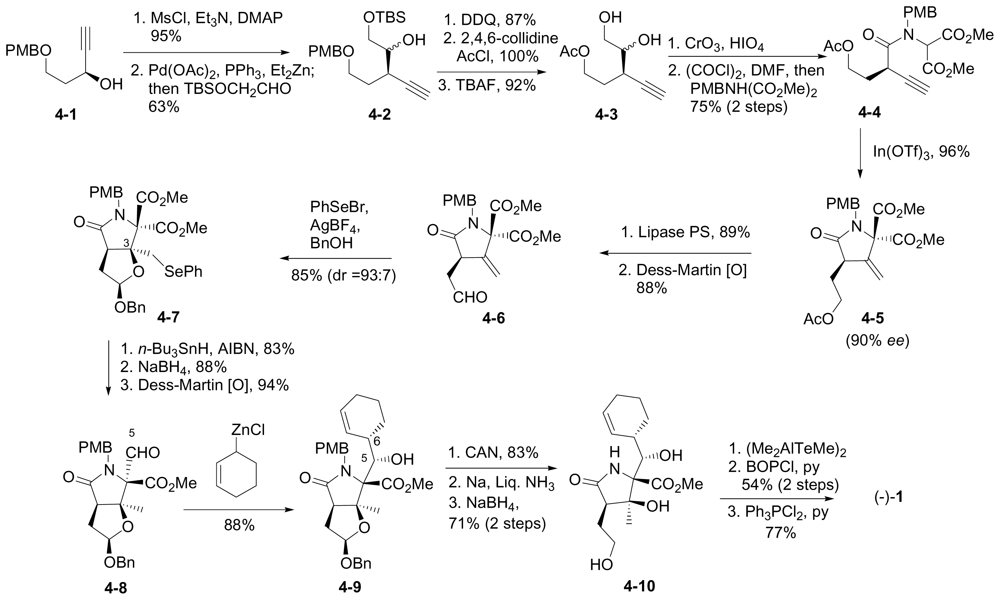
6.1.4. Hatakeyama Enantioselective Synthesis
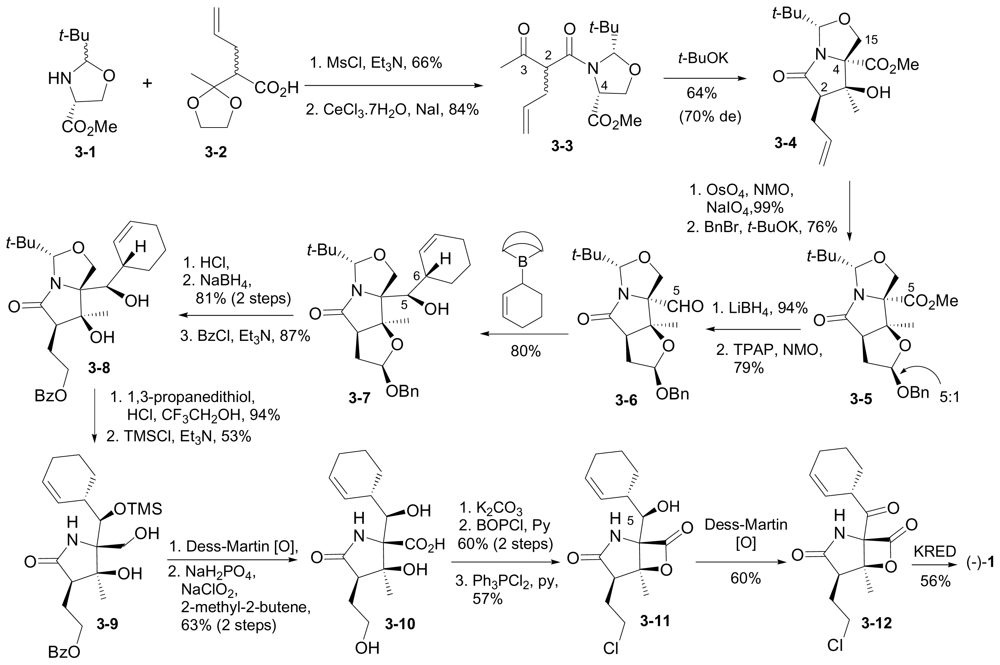
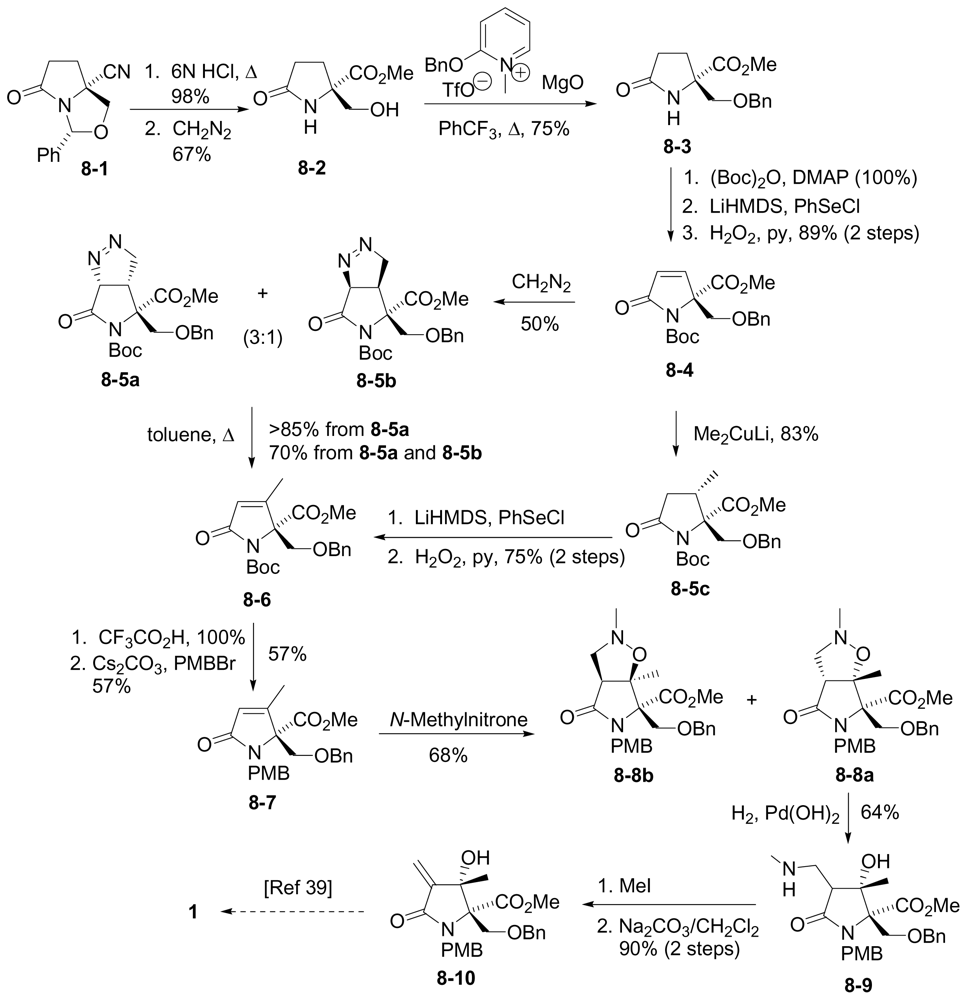
6.1.5. Omura Enantioselective Synthesis
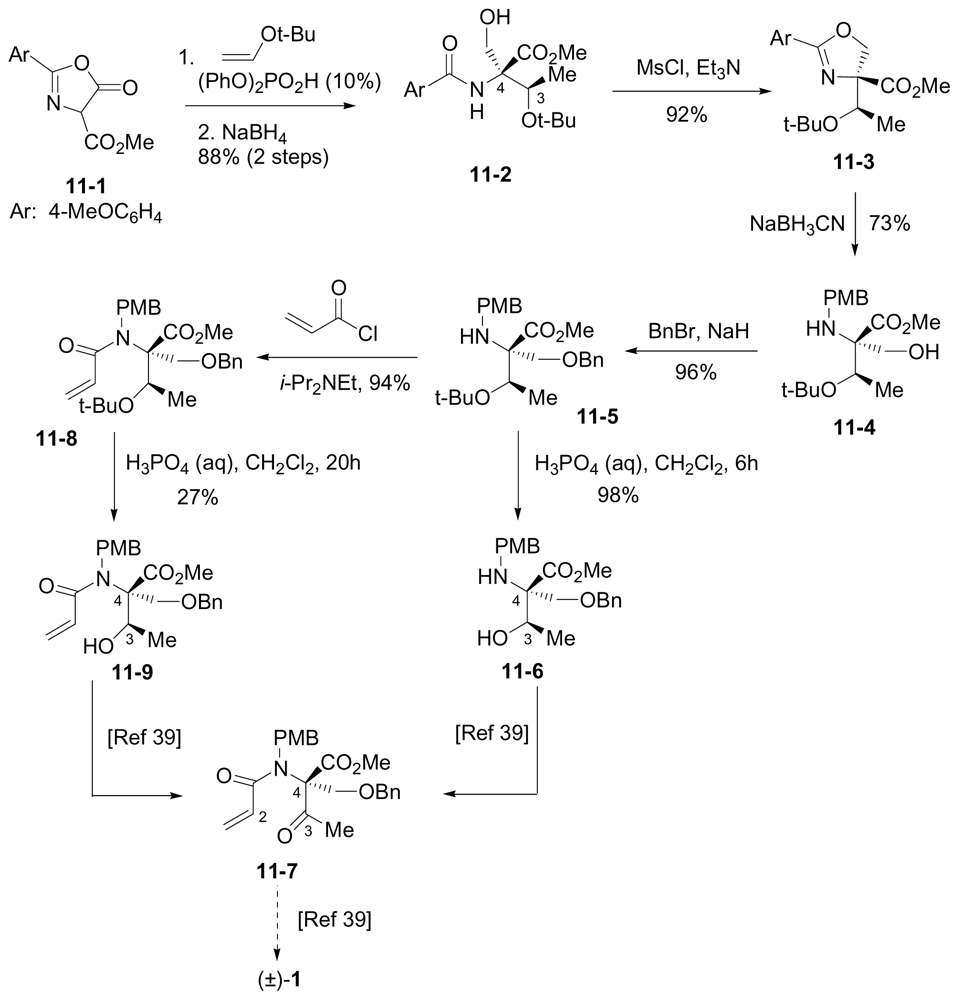
6.2. Racemic Synthesis of (±)-1
6.2.1. Pattenden Racemic Synthesis
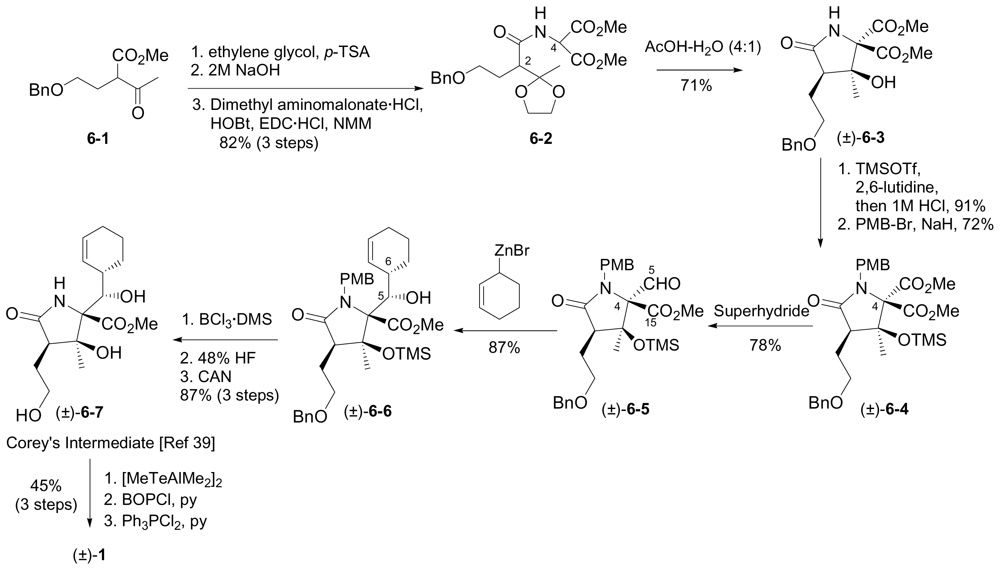
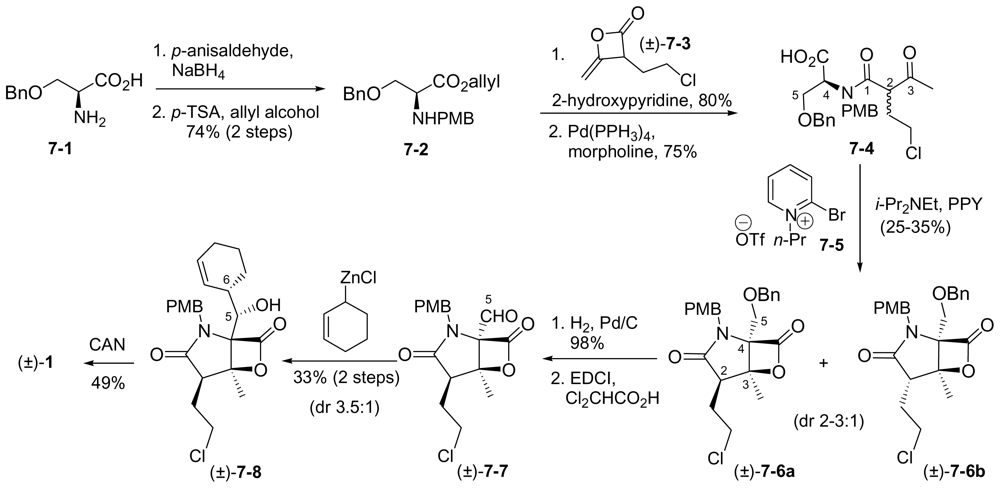
6.2.2. Romo Racemic Synthesis
6.3. Formal Synthesis of 1
6.3.1. Langlois Formal Synthesis
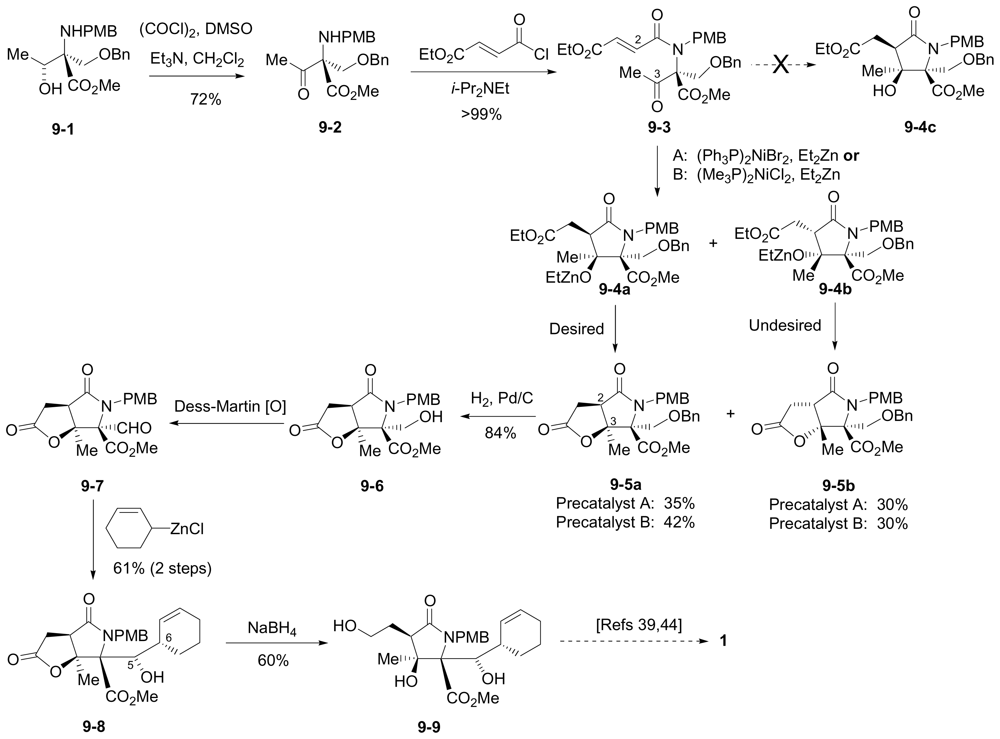
6.3.2. Lam Formal Synthesis
6.3.3. Bode Formal Synthesis
6.3.4. Tepe Formal Synthesis
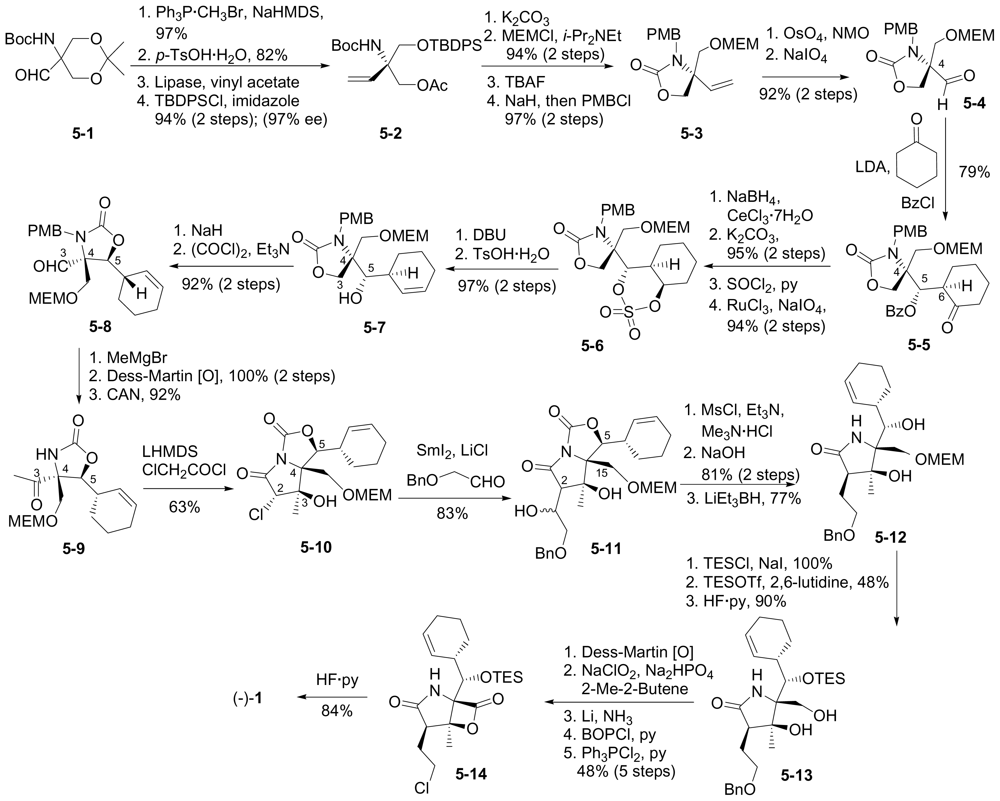
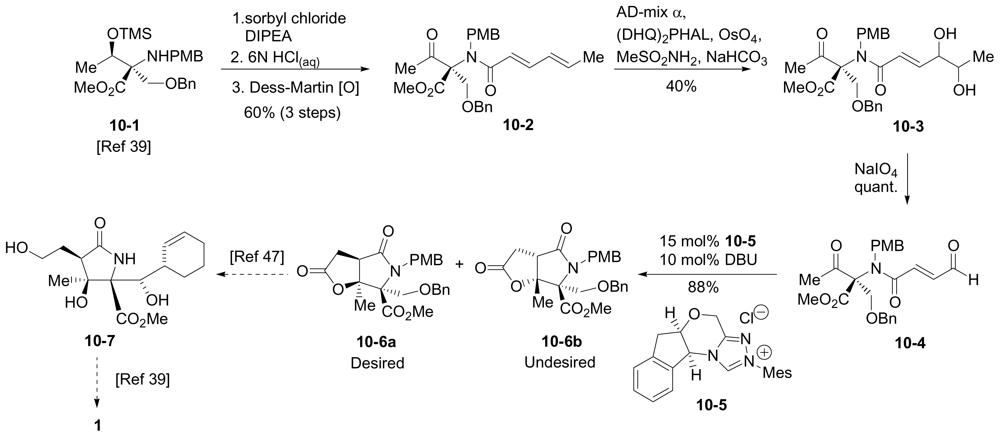
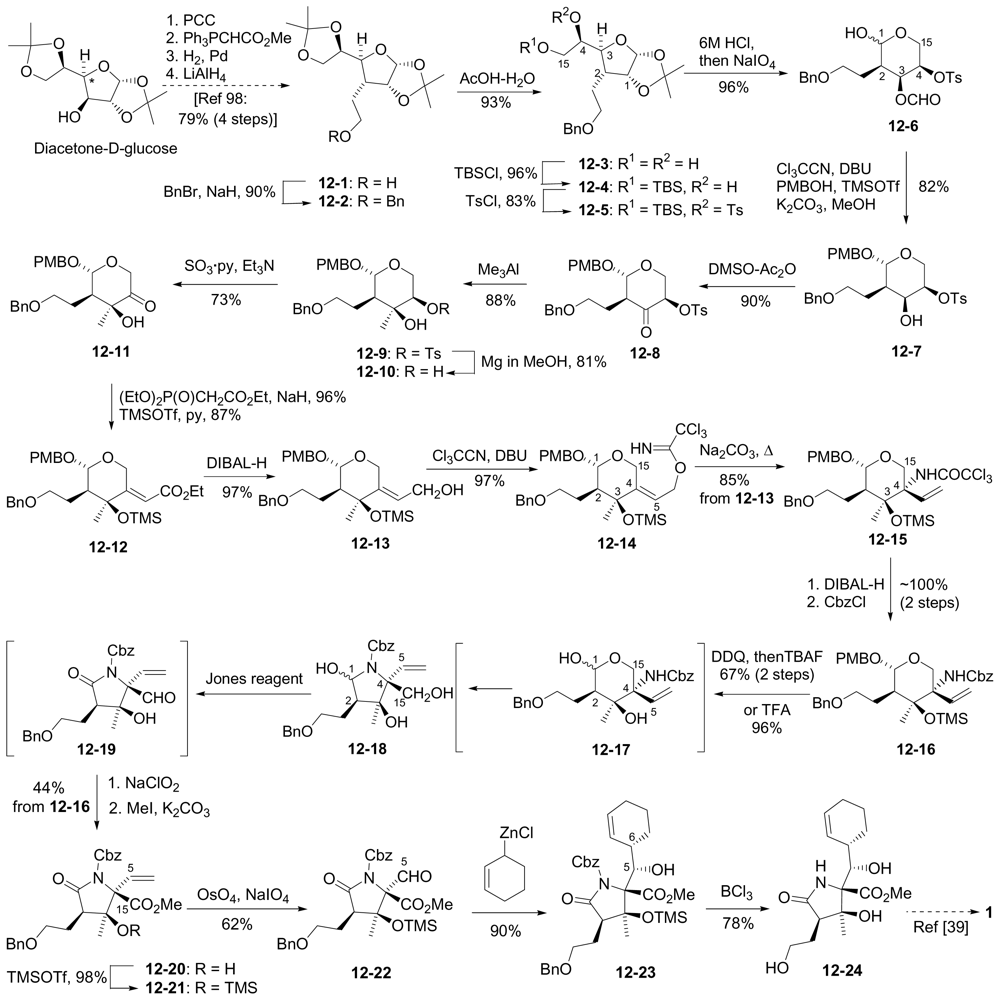
6.3.5. Chida Formal Synthesis
7. Closing Remarks
Acknowledgements
References and Notes
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ 2005, 12, 1178–1190. [Google Scholar]
- Hershko, A; Ciechanover, A. The ubiquitin system. Ann Rev Biochem 1998, 67, 425–479. [Google Scholar]
- Borissenko, L; Groll, M. 20S Proteasome and its inhibitors: Crystallographic knowledge for drug development. Chem Rev 2007, 107, 687–717. [Google Scholar]
- Adams, J. Proteasome Inhibitors in Cancer Therapy; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Orlowski, RZ; Kuhn, DJ. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin Cancer Res 2008, 14, 1649–1657. [Google Scholar]
- Moore, BS; Eustáquio, AS; McGlinchey, RP. Advances in and applications of proteasome inhibitors. Curr Opin Chem Biol 2008, 12, 434–440. [Google Scholar]
- Hillebrand, S; Guth, O; Wiese, W-B; Kunz, K; Ullmann, A; Mattes, A; Schreier, P; Wachendorff-Neumann, U; Kuck, K-H; Lösel, P; Malsam, O; Reinemer, P; Stadler, M; Seip, S; Mayer-Bartschmid, A; Müller, H; Bacon, K. Substituted 2-pyrrolidone derivatives as fungicides and insecticides. Int Publ 2006. WO 2006/005551 A1. [Google Scholar]
- Prudhomme, J; McDaniel, E; Ponts, N; Bertani, S; Fenical, W; Jensen, P; Le Roch, K. Marine actinomycetes: A new source of compounds against the human malaria parasite. PLoS One 2008, 3, e2335. [Google Scholar]
- Skaug, B; Jiang, X; Chen, ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Ann Rev Biochem 2009, 78, 769–796. [Google Scholar]
- Richarson, PG; Barlogie, B; Berenson, J; Singhal, S; Jagannath, S; Irwin, D; Rajkumar, SV; Srkalovic, G; Alsina, M; Alexanian, R; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003, 348, 2609–2617. [Google Scholar]
- Bross, PF; Kane, R; Farrell, AT; Abraham, S; Benson, K; Brower, ME; Bradley, S; Gobburu, JV; Goheer, A; Lee, S-L; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res 2004, 10, 3954–3964. [Google Scholar]
- Chauhan, D; Hideshima, T; Anderson, KC. A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Brit J Cancer 2006, 95, 961–965. [Google Scholar]
- Chauhan, D; Catley, L; Li, G; Podar, K; Hideshima, T; Velankar, M; Mitsiades, C; Mitsiades, N; Yasui, H; Letai, A; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar]
- Stapnes, C; Gjertsen, BT; Reikvam, H; Bruserud, O. Targeted therapy in acute myeloid leukaemia: Current status and future directions. Expert Opin Investig Drugs 2009, 18, 433–455. [Google Scholar]
- Feling, RH; Buchanan, GO; Mincer, TJ; Kauffman, CA; Jensen, PR; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew Chem Int Ed 2003, 42, 355–357. [Google Scholar]
- Fenical, WH; Jensen, PR; Palladino, MA; Lam, KS; Lloyd, GK; Potts, BC. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg Med Chem 2009, 17, 2175–2180. [Google Scholar]
- Lam, KS; Lloyd, GK; Neuteboom, STC; Palladino, MA; Sethna, KM; Spear, MA; Potts, BC. Buss, AD, Butler, MS, Eds.; From natural product to clinical trials: NPI-0052 (salinosporamide A), a marine actinomycete-derived anticancer agent. In Natural Products Chemistry for Drug Discovery; Royal Society of Chemistry: Cambridge, UK, 2010; RSC Biomolecular Sciences No. 18; pp. 355–373. [Google Scholar]
- Chauhan, D; Singh, A; Brahmandam, M; Podar, K; Hideshima, T; Richardson, P; Munshi, N; Palladino, MA; Anderson, KC. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 2008, 111, 1654–1664. [Google Scholar]
- Ruiz, S; Krupnik, Y; Keating, M; Chandra, J; Palladino, M; McConkey, D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol Cancer Ther 2006, 5, 1836–1843. [Google Scholar]
- Miller, CP; Ban, K; Dujka, ME; McConkey, DJ; Munsell, M; Palladino, M; Chandra, J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 2007, 110, 267–277. [Google Scholar]
- Miller, CP; Rudra, S; Keating, MJ; Wierda, WG; Palladino, M; Chandra, J. Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: A mechanism for synergy in leukemia cells. Blood 2009, 113, 4289–4299. [Google Scholar]
- Cusack, JC, Jr; Liu, R; Xia, L; Chao, TH; Pien, C; Niu, W; Palombella, VJ; Neuteboom, ST; Palladino, MA. NPI-0052 enhances tumoricidal response to conventional cancer therapy in a colon cancer model. Clin Cancer Res 2006, 12, 6758–6764. [Google Scholar]
- Omura, S; Fujimoto, T; Otoguro, K; Matsuzaki, K; Moriguchi, R; Tanaka, H; Sasaki, Y. Lactacystin, a novel microbial metabolite induces neuritogenesis of neuroblastoma cells. J Antibiot 1991, 44, 113–116. [Google Scholar]
- Omura, S; Matsuzaki, K; Fujimoto, T; Kosuge, K; Furuya, T; Fujita, S; Nakagawa, A. Structure of lactacystin, a new microbial metabolite which induces differentiation of neuroblastoma cells. J Antibiot 1991, 44, 117–118. [Google Scholar]
- Fenteany, G; Standaert, RF; Lane, WS; Choi, S; Corey, EJ; Schreiber, SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar]
- Dick, LR; Cruikshank, AA; Grenier, L; Melandri, FD; Nunes, SL; Stein, RL. Mechanistic studies on the inactivation of the proteasome by lactacystin. J Biol Chem 1996, 271, 7273–7276. [Google Scholar]
- Dick, LR; Cruikshank, AA; Destree, AT; Grenier, L; McCormack, TA; Melandri, FD; Nunes, SL; Palombello, VJ; Parent, LA; Plamondon, L; Stein, RL. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J Biol Chem 1997, 272, 182–188. [Google Scholar]
- Groll, M; Ditzel, L; Löwe, J; Stock, D; Bochtler, M; Bartunik, HD; Huber, R. Structure of 20S proteasome from yeast at 2.4Å resolution. Nature 1997, 386, 463–471. [Google Scholar]
- Corey, EJ; Li, WZ. Total synthesis and biological activity of lactacystin, omuralide, and analogs. Chem Pharm Bull 1999, 47, 1–10. [Google Scholar]
- Masse, CE; Morgan, AJ; Adams, J; Panek, JS. Syntheses and biological evaluation of (+)-lactacystin and analogs. Eur J Org Chem 2000, 2513–2528. [Google Scholar]
- Kang, SH; Kang, SY; Lee, H-S; Buglass, AJ. Total synthesis of natural tert-alkylamino hydroxy carboxylic acids. Chem Rev 2005, 105, 4537–4558. [Google Scholar]
- Shibasaki, M; Kanai, M; Fukuda, N. Total synthesis of lactacystin and salinosporamide A. Chem Asian J 2007, 2, 20–38. [Google Scholar]
- Shah, IM; Lees, KR; Pien, CP; Elliot, PJ. Early clinical experience with the novel proteasome inhibitor PS-519. Br J Clin Pharmacol 2002, 54, 269–276. [Google Scholar]
- Macherla, VR; Mitchell, SS; Manam, RR; Reed, K; Chao, T-H; Nicholson, B; Deyanat-Yazdi, G; Mai, B; Jensen, PR; Fenical, W; et al. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J Med Chem 2005, 48, 3684–3687. [Google Scholar]
- Groll, M; Huber, R; Potts, BCM. Crystal structure of salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of β-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc 2006, 128, 5136–5141. [Google Scholar]
- Millward, M; Spear, MA; Townsend, A; Sweeney, C; Sukumaran, S; Longenecker, A; Palladino, MA; Lloyd, GK; Neuteboom, STC; Price, T. Clinical trial combining proteasome (NPI-0052) and HDAC (vorinostat) inhibition in melanoma, pancreatic and lung cancer. Mol Cancer Ther 2009, 8. Abstr. 107; Meeting Abstract Supplement. [Google Scholar]
- Townsend, AR; Millward, M; Price, T; Mainwaring, P; Spencer, A; Longenecker, A; Palladino, MA; Lloyd, GK; Spear, MA; Padrik, P. Clinical trial of NPI-0052 in advanced malignancies including lymphoma and leukemia (Advanced Malignancies Arm). J Clin Oncol 2009, 27, 15s. [Google Scholar]
- Hamlin, PA; Aghajanian, C; Younes, A; Hong, DS; Palladino, MA; Longenecker, AM; Lloyd, GK; Hannah, AL; Spear, MA; Kurzrock, R. First-in-human phase 1 study of the novel structure proteasome inhibitor NPI-0052. J Clin Oncol 2009, 27, 15s. [Google Scholar]
- Reddy, LR; Saravanan, P; Corey, EJ. A simple stereocontrolled synthesis of salinosporamide A. J Am Chem Soc 2004, 126, 6230–6231. [Google Scholar]
- Endo, A; Danishefsky, SJ. Total synthesis of salinosporamide A. J Am Chem Soc 2005, 127, 8298–8299. [Google Scholar]
- Ling, T; Macherla, VR; Manam, RR; McArthur, KA; Potts, BCM. Enantioselective total synthesis of (-)-salinosporamide A (NPI-0052). Org Lett 2007, 9, 2289–2292. [Google Scholar]
- Takahashi, K; Midori, M; Kawano, K; Ishihara, J; Hatakeyama, S. Entry to heterocycles based on indium-catalyzed Conia-ene reactions: Asymmetric synthesis of (-)-salinosporamide A. Ang Chem Int Ed 2008, 47, 6244–6246. [Google Scholar]
- Fukuda, T; Sugiyama, K; Arima, S; Harigaya, Y; Nagamitsu, T; Omura, S. Total synthesis of salinosporamide A. Org Lett 2008, 10, 4239–4242. [Google Scholar]
- Mulholland, NP; Pattenden, G; Walters, IAS. A concise total synthesis of salinosporamide A. Org Biomol Chem 2006, 4, 2845–2846. [Google Scholar]
- Ma, G; Nguyen, H; Romo, D. Concise total synthesis of (±)-salinosporamide A, (±)-cinnabaramide A, and derivatives via a bis-cyclization process; implications for a biosynthetic pathway? Org Lett 2007, 9, 2143–2146. [Google Scholar]
- Caubert, V; Masse, J; Retailleau, P; Langlois, N. Stereoselective formal synthesis of the potent proteasome inhibitor: Salinosporamide A. Tetrahedron Lett 2007. [Google Scholar]
- Margalef, IV; Rupnicki, L; Lam, HW. Formal synthesis of salinosporamide A using a nickel-catalyzed reductive aldol cyclization-lactonization as a key step. Tetrahedron 2008, 64, 7896–7901. [Google Scholar]
- Struble, JR; Bode, JW. Formal synthesis of salinosporamide A via NHC-catalyzed intramolecular lactonization. Tetrahedron 2009, 65, 4957–4967. [Google Scholar]
- Mosey, RA; Tepe, JJ. New synthetic route to access (±) salinosporamide A via an oxazolone-mediated ene-type reaction. Tetrahedron Lett 2009, 50, 295–297. [Google Scholar]
- Momose, T; Kaiya, Y; Hasegawa, J; Sato, T; Chida, N. Formal synthesis of salinosporamide A starting from D-glucose. Synthesis 2009, 17, 2983–2991. [Google Scholar]
- Williams, PG; Buchanan, GO; Feling, RH; Kauffman, CA; Jensen, PR; Fenical, W. New cytotoxic salinosporamides from the marine actinomycete Salinispora tropica. J Org Chem 2005, 70, 6196–6203. [Google Scholar]
- Reed, KA; Manam, RR; Mitchell, SS; Xu, J; Teisan, S; Chao, T-H; Deyanat-Yazdi, G; Neuteboom, STC; Lam, KS; Potts, BCM. Salinosporamides D-J from the marine actinomycete Salinospora tropica, bromosalinosporamide, and thioester derivatives are potent inhibitors of the 20S proteasome. J Nat Prod 2007, 70, 269–276. [Google Scholar]
- Manam, RR; Macherla, VR; Tsueng, G; Dring, CW; Weiss, J; Neuteboom, ST; Lam, KS; Potts, BC. Antiprotealide is a natural product. J Nat Prod 2009, 72, 295–297. [Google Scholar]
- Lam, KS; Tsueng, G; McArthur, KA; Mitchell, SS; Potts, BCM; Xu, J. Effects of halogens on the production of salinosporamides by the obligate marine actinomycete Salinispora tropica. J Antibiot 2007, 60, 13–19. [Google Scholar]
- Tsueng, G; McArthur, KA; Potts, BCM; Lam, KS. Unique butyric acid incorporation patterns for salinosporamides A and B reveal distinct biosynthetic origins. Appl Microbiol Biotechnol 2007, 75, 999–1005. [Google Scholar]
- Beer, LL; Moore, BS. Biosynthetic convergence of salinosporamides A and B in the marine actinomycete Salinospora tropica. Org Lett 2007, 9, 845–848. [Google Scholar]
- Tsueng, G; Teisan, S; Lam, KS. Defined salt formulations for the growth of Salinispora tropica strain NPS21184 and the production of salinosporamide A (NPI-0052) and related analogs. Appl Microbiol Biotechnol 2008, 78, 827–832. [Google Scholar]
- Tsueng, G; Lam, KS. Effect of cobalt and vitamin B12 on the production of salinosporamides by Salinispora tropica. J Antibiot 2009, 62, 213–216. [Google Scholar]
- Potts, BC; Macherla, VR; Mitchell, SS; Manam, RR; Reed, KA; Lam, KS; Neuteboom, STC; Chao, TH; Nicholson, B; Billstrom, C. [3.2.0]Heterocyclic compounds and methods of using the same WO 2006/028525 A2. 2006.
- Manam, RR; Macherla, VR; Potts, BCM. Stereoselective enzymatic reduction of keto-salinosporamide to (-)-salinosporamide A (NPI-0052). Tetrahedron Lett 2007, 48, 2537–2540. [Google Scholar]
- Manam, RR; McArthur, KA; Chao, TH; Weiss, J; Ali, JA; Palombella, VJ; Groll, M; Lloyd, GK; Palladino, MA; Neuteboom, STC; et al. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. J Med Chem 2008, 51, 6711–6724. [Google Scholar]
- Udwary, DW; Zeigler, L; Asolkar, RN; Singan, V; Lapidus, A; Fenical, W; Jensen, PR; Moore, BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinospora tropica. Proc Natl Acad Sci USA 2007, 104, 10376–10381. [Google Scholar]
- Eustáquio, AS; Moore, BS. Mutasynthesis of fluorosalinosporamide, a potent and reversible inhibitor of the proteasome. Angew Chem Int Ed 2008, 47, 3936–3938. [Google Scholar]
- McGlinchey, RP; Nett, M; Eustáquito, AS; Asolkar, RN; Fenical, W; Moore, BS. Engineered biosynthesis of antiprotealide and other unnatural salinosporamide proteasome inhibitors. J Am Chem Soc 2008, 130, 7822–7823. [Google Scholar]
- Nett, M; Gulder, TAM; Kale, AJ; Hughes, CC; Moore, BS. Function-oriented biosynthesis of β-lactone proteasome inhibitors in Salinispora tropica. J Med Chem 2009, 52, 6163–6167. [Google Scholar]
- Eustáquio, AS; O’Hagen, D; Moore, BS. Engineering fluorometabolite production: Fluorinase expression in Salinispora tropica yields fluorosalinosporamide. J Nat Prod 2010. [Google Scholar] [CrossRef]
- Groll, M; McArthur, KA; Macherla, VR; Manam, RR; Potts, BC. Snapshots of the fluorosalinosporamide/20S complex offer mechanistic insights for fine tuning proteasome inhibition. J Med Chem 2009, 52, 5420–5428. [Google Scholar]
- Eustáquio, AS; Pojer, F; Noel, JP; Moore, BS. Discovery and characterization of a marine bacterial SAM-dependent chlorinase. Nat Chem Biol 2008, 4, 69–74. [Google Scholar]
- Mincer, TJ; Jensen, PR; Kauffman, CA; Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl Environ Microbiol 2002, 68, 5005–5011. [Google Scholar]
- Maldonado, L; Fenical, W; Jensen, PR; Kauffman, CK; Mincer, TJ; Ward, AC; Bull, AT; Goodfellow, M. Salinispora arenicola gen. nov., sp. nov. and Salinispora tropica sp. nov., obligate marine actinomycetes belonging to the family Micromonosporaceae. Int J Syst Evol Microbiol 2005, 55, 1759–1766. [Google Scholar]
- Reddy, LR; Fournier, JF; Reddy, BVS; Corey, EJ. An efficient, stereocontrolled synthesis of a potent omuralide-salinosporin hybrid for selective proteasome inhibition. J Am Chem Soc 2005, 127, 8974–8976. [Google Scholar]
- Reddy, LR; Fournier, J-F; Reddy, BVS; Corey, EJ. New synthetic route for the enantioselective total synthesis of salinosporamide A and biologically active analogues. Org Lett 2005, 7, 2699–2701. [Google Scholar]
- Lee, WW; Benitez, A; Goodman, L; Baker, BR. Potential anticancer agents. XL. Synthesis of the β-anomer of 9-(D-arabinofuranosyl)-adenine. J Am Chem Soc 1960, 82, 2648–2649. [Google Scholar]
- Howells, JD; Ryder, A. (Parke, Davis and Co.) Fermentation process for 9-(β-D-arabinofuranosyl)adenine. US Patent 3,616,208, 26 October 1971. [Google Scholar]
- Stadler, M; Bitzer, J; Mayer-Bartschmid, A; Müller, H; Benet-Buchholz, J; Gantner, F; Tichy, HV; Reinemer, P; Bacon, KB. Cinnabaramides A-G: Analogues of lactacystin and salinosporamide from a terrestrial streptomycete. J Nat Prod 2007, 70, 246–252. [Google Scholar]
- Lam, KS; Gustavson, DR; Veitch, JA; Forenza, S. Effect of cerulenin on the production of esperamicin A1 by Actinomadura verrucosospora. J Ind Microbiol 1993, 12, 99–102. [Google Scholar]
- Lam, KS; Veitch, JA; Lowe, SE; Forenza, S. Effect of neutral resins on the production of dynemicins by Micromonospora cherisina. J Ind Microbiol 1995, 15, 453–456. [Google Scholar]
- Woo, EJ; Starks, CM; Carney, JR; Arslanian, R; Cadapan, L; Zavala, S; Licari, P. Migrastatin and a new compound, isomigrastatin, from Streptomyces platensis. J Antibiot 2002, 55, 141–146. [Google Scholar]
- Frykman, S; Tsuruta, H; Galazzo, J; Licari, P. Characterization of product capture resin during microbial cultivations. J Ind Microbiol Biotechnol 2006, 33, 445–453. [Google Scholar]
- Denora, N; Potts, BCM; Stella, VJ. A mechanistic and kinetic study of the β-lactone hydrolysis of salinosporamide A (NPI-0052), a novel proteasome inhibitor. J Pharm Sci 2007, 96, 2037–2047. [Google Scholar]
- Tsueng, G; Lam, KS. Stabilization effect of resin on the production of potent proteasome inhibitor NPI-0052 during submerged fermentation of Salinispora tropica. J Antibiot 2007, 60, 469–472. [Google Scholar]
- Sedriks, JA. Corrosions of Stainless Steels, 2nd ed; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Thiericke, R; Rohn, J. Biological variation of microbial metabolites by precursor-directed biosynthesis. Nat Prod Rep 1993, 10, 265–289. [Google Scholar]
- Ritacco, FV; Graziani, EI; Summers, MY; Zabriskie, TM; Yu, K; Bernan, VS; Carter, GT; Greenstein, M. Production of novel rapamycin analogs by precursor-directed biosynthesis. Appl Environ Microbiol 2005, 71, 1971–1976. [Google Scholar]
- Moran, S; Rai, DK; Clark, BR; Murphy, CD. Precursor-directed biosynthesis of fluorinated iturin A in Bacillus spp. Org Biomol Chem 2009, 7, 644–646. [Google Scholar]
- Güschow, S; Rackham, EJ; Elkins, B; Newill, PL; Hill, LM; Gross, RJ. New apacidamycin antibiotics through precursor-directed biosynthesis. ChemBioChem 2009, 10, 355–360. [Google Scholar]
- Amir-Heidari, B; Thirlway, J; Micklefield, J. Auxotrophic-precursor directed biosynthesis of nonribosomal lipopeptides with modified tryptophan residues. Org Biomol Chem 2008, 6, 975–978. [Google Scholar]
- Stirrett, K; Denoya, C; Westpheling, J. Branched-chain amino acid catabolism provides precursors for the type II polyketide antibiotic, actinorhodin, via pathways that are nutrient dependent. J Ind Microbiol Biotechnol 2009, 36, 129–137. [Google Scholar]
- Nagaoka, K; Demain, AL. Mutational biosynthesis of a new antibiotic, streptomutin A, by an idiotroph of Streptomyces griseus. J Antibiot 1975, 28, 627–635. [Google Scholar]
- Rinehart, KL, Jr; Stroshane, RM. Biosynthesis of aminocyclitol antibiotics. J Antibiot 1976, 29, 319–353. [Google Scholar]
- Levengood, MR; Knerr, PJ; Oman, TJ; van der Donk, WA. In vitro mutasynthesis of lantibiotic analogues containing nonproteinogenic amino acids. J Am Chem Soc 2009, 131, 12024–12025. [Google Scholar]
- Eichner, S; Floss, HG; Sasse, F; Kirschning, A. New, highly active nonbenzoquinone geldanamycin derivatives by using mutasynthesis. ChemBioChem 2009, 10, 1801–1805. [Google Scholar]
- Heide, L. Genetic engineering of antibiotic biosynthesis for the generation of new aminocoumarins. Biotechnol Adv 2009, 27, 1006–1014. [Google Scholar]
- Gupta, S; Lakshmanan, V; Kim, BS; Fecik, R; Reynolds, KA. Generation of novel pikromycin antibiotic products through mutasynthesis. ChemBioChem 2008, 9, 1609–1616. [Google Scholar]
- Donadio, S; Sosio, M. Biosynthesis of glycopeptides: Prospects for improved antibacterials. Curr Top Med Chem 2008, 8, 654–666. [Google Scholar]
- Tannhauser, P; Pratt, RJ; Jensen, EV. The preparation of 21-flurosteroids. J Am Chem Soc 1956, 78, 2658–2659. [Google Scholar]
- Ando, T; Cork, DG; Fujita, M; Kimura, T; Tatsuno, T. Silver fluoride supported on calicium fluoride. Improved fluorination and halofluorination reactions. Chem Lett 1988, 1877–1878. [Google Scholar]
- Fenteany, G; Schreiber, SL. Lactacystin, proteasome function, and cell fate. J Biol Chem 1998, 273, 8545–8548. [Google Scholar]
- Seebach, D; Sting, AR; Hoffman, M. Self-regeneration of stereocenters (SRS)–applications, limitations, and abandonment of a synthetic principle. Angew Chem Int Ed 1996, 35, 2708–2748. [Google Scholar]
- Andrews, MD; Brewster, AG; Moloney, MG. Highly functionalized pyroglutamates by intramolecular aldol reactions: Towards the pyroglutamate skeleton of oxazolomycin. Synlett 1996, 612–614. [Google Scholar]
- Kramer, GW; Brown, HC. Organoboranes: XIX. The preparation and some unusual chemistry of b-allyl derivatives of 9-borabicyclo[3.3.1]nonane. J Organomet Chem 1977, 132, 9–27. [Google Scholar]
- Conia, JM; Le Perchec, P. The thermal cyclisation of unsaturated carbonyl compounds. Synthesis 1975, 1, 1–19. [Google Scholar]
- Hatakeyama, S. Indium-catalyzed Conia-ene reaction for alkaloid synthesis. Pure Appl Chem 2009, 81, 217–226. [Google Scholar]
- Mulholland, NP; Pattenden, G; Walters, IAS. A concise and straightforward total synthesis of (±)-salinosporamide A, based on a biosynthetic model. Org Biomol Chem 2008, 6, 2782–2789. [Google Scholar]
- Henry-Riyad, H; Lee, CS; Purohit, VC; Romo, D. Bicyclic- and tricyclic-beta-lactones via organonucleophile-promoted bis-cyclizations of keto acids: Enantioselective synthesis of (+)-dihydroplakevulin. Org Lett 2006, 8, 4363–4366. [Google Scholar]
- Caubert, V; Langlois, N. Studies toward the synthesis of salinosporamide A, a potent proteasome inhibitor. Tetrahedron Lett 2006, 47, 4473–4475. [Google Scholar]
- Langlois, N; Le Nguyen, BK. Diastereoselective syntheses of deoxydysibetaine, dysibetaine and its 4-epimer. J Org Chem 2004, 69, 7558–7564. [Google Scholar]
- Joensuu, PM; Murray, GJ; Fordyce, EAF; Luebbers, T; Lam, HW. Selective nickel-catalyzed reductive aldol cyclizations using diethylzinc as the stoichiometric reductant: Scope and mechanistic insight. J Am Chem Soc 2008, 130, 7328–7338. [Google Scholar]
- Hewlett, NM; Hupp, CD; Tepe, JJ. Reactivity of oxazol-5-(4H)-ones and their application toward natural product synthesis. Synthesis 2009, 17, 2825–2839. [Google Scholar]
- Chida, N; Takeoka, J; Ando, K; Tsutsumi, N; Ogawa, S. Stereoselective total synthesis of (+)-lactacystin from D-glucose. Tetrahedron 1997, 53, 16287–16298. [Google Scholar]
- Fleet, GWJ; James, K; Lunn, RJ; Matthews, CJ. An enantiospecific synthesis of S-quinuclidinol from D-glucose. Tetrahedron Lett 1986, 27, 3057–3058. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Str # | Compound Name(s) | P2 | CT-L (IC50, nM)a | Source/Method of Production |
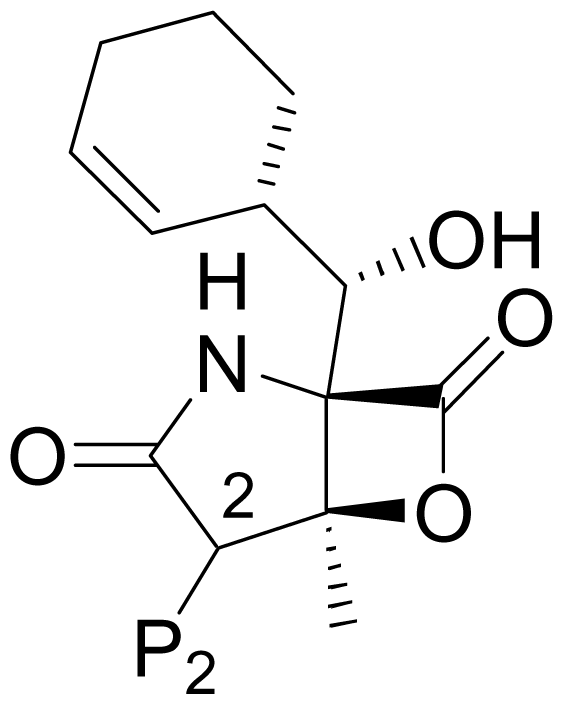
| 1 | Salinosporamide A NPI-0052 | 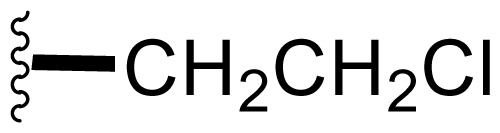 | 2.5 ± 1.2 [61] 2.0 ± 0.3 [61]b 3.5 ± 0.3 [13]c | Natural Product [15] Total Synthesis [39–45] Formal Synthesis [46–50] |
| 5 | Salinosporamide B NPI-0047 | 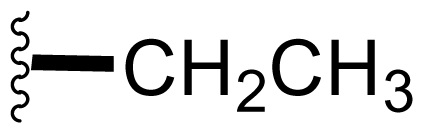 | 26 ± 6.7 [61] | Natural Product [51] Directed Biosynthesis [55,56,Table 4] Modified Media [54,58,Table 4] |
| 8 | Salinosporamide D | 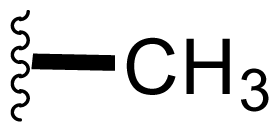 | 7.5 ± 0.6 [61] | Natural Product [52] Directed Biosynthesis [Table 4] Modified Media[Table 4] |
| 9 | Salinosporamide E | 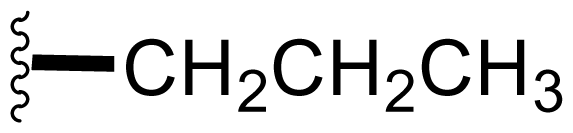 | 24 ± 5 [34] | Natural Product [52] Semi-synthesis [34] Directed Biosynthesis ± Modified Media [Table 4] |
| 10 | Salinosporamide F |  | 330 ± 20 [34] | Natural Product [34,52] |
| 11 | Salinosporamide G |  | 3200, 3300 [59] | Natural Product [52] |
| 16 | Salinosporamide H | 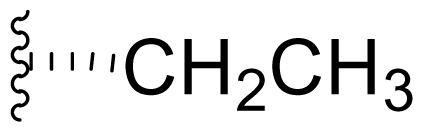 | 1400 [59] | Byproduct of directed biosynthesis of 15 [52] |
| 15 | Bromosalinosporamide | 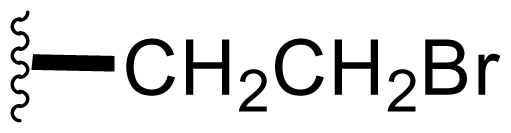 | 2.6 ± 0.4 [34] | Directed Biosynthesis [34,52,54] + Modified Media [Table 4] |
| 42 | Iodosalinosporamide | 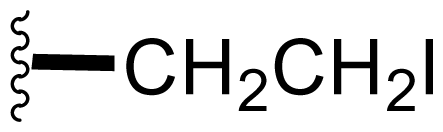 | 2.8 ± 0.5 [34] | Semi-synthesis [34] |
| 17 | Fluorosalinosporamide | 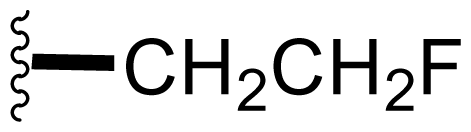 | 9.2 ± 10.2 [61] 1.5 ± 0.05 [63]b | Semi-synthesis [61] Mutasynthesis [63,66] Directed Biosynthesis in Modified Media [Table 4] |
| 43 | Azidosalinosporamide |  | 7.7 ± 1.5 [34] | Semi-synthesis [34] |
| 45 | Thiocyano-salinosporamide | 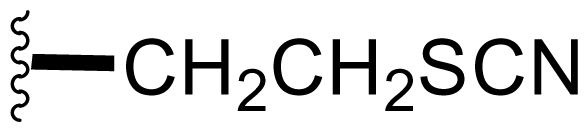 | 3.4 ± 0.2 [59] | Semi-synthesis [59] |
| 41 | Hydroxy-salinosporamide |  | 14 ± 1.5 [61] | Semi-synthesis [34,61] |
| 44 | 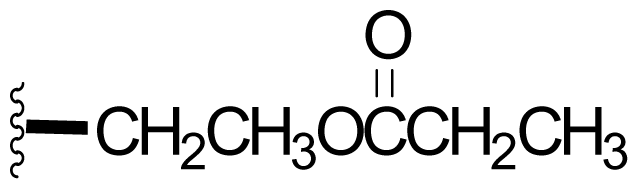 | 7,3 [59] | Semi-synthesis [59] | |
| 46 | Mesylsalinosporamide |  | 4.3 ± 0.8 [61] | Semi-synthesis [61] |
| 47 | Tosylsalinosporamide | 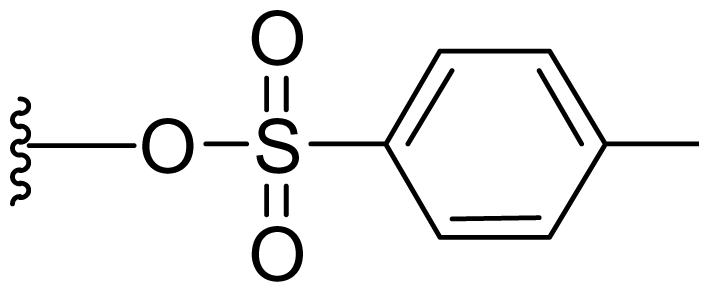 | 2.5 ± 0.4 [61] | Semi-synthesis [61] Total synthesis [61] |
| 48 | Dansylsalinosporamide | 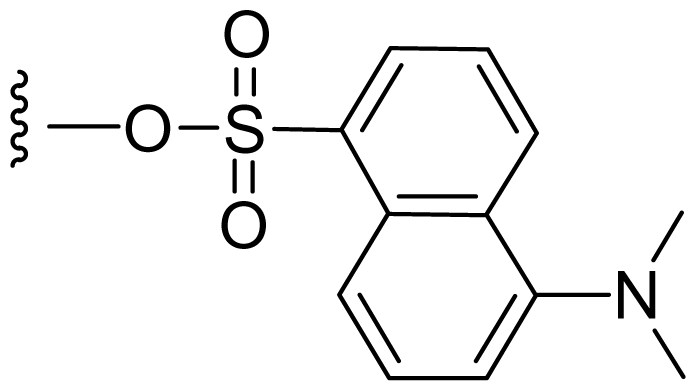 | 3.0 ± 0.5 [61] | Semi-synthesis [61] |
 | ||||
|---|---|---|---|---|
| Str # | Compound Name(s) | P1 | CT-L (IC50, nM)a | Source/Method of Production |
| 1 | Salinosporamide A NPI-0052 |  | 2.5 ± 1.2 [61] 2.0 ± 0.3 [61]b 3.5 ± 0.3 [13]c | Natural Product [15] Total Synthesis [39–45] Formal Synthesis [46–50] |
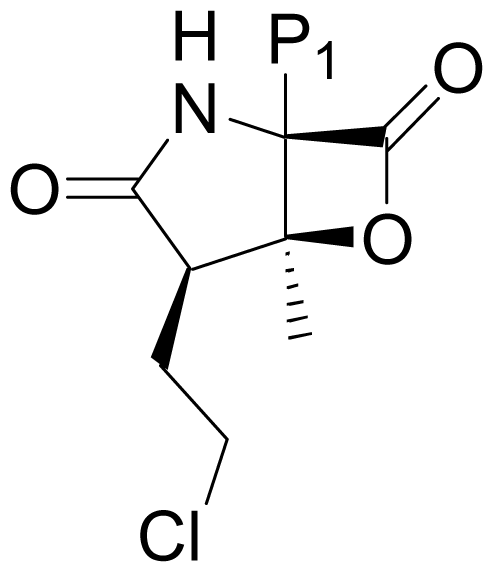
| 13 | Salinosporamide J |  | 52 ± 2 [52] | Natural Product [52] |
| 36 | C-5-epi-salinosporamide | 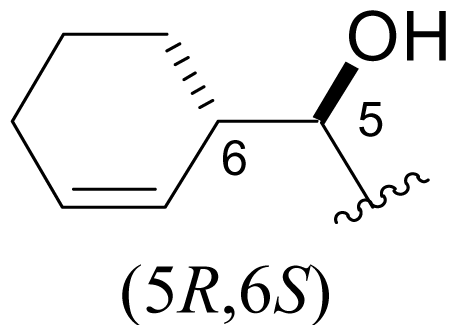 | > 20000 [34] | Semi-synthesis [34,60] Total synthesis [41] |
| 35 | Keto-salinosporamide | 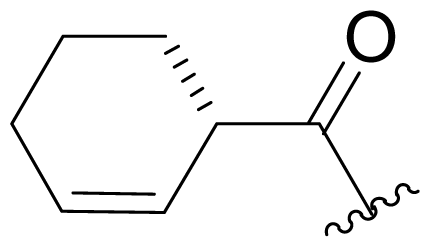 | 8200 ± 600 [34] | Semi-synthesis [34,60] Total synthesis [41] |
| 18 | NPI-2056 Salinosporamide X1 | 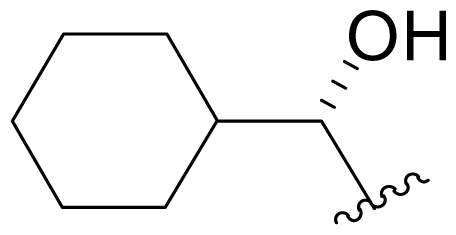 | 20 ± 3 [34] 27.5 ± 3.7 [65]b | Semi-synthesis [34] Mutasynthesis [64,65] |
| 19 | Salinosporamide X7 | 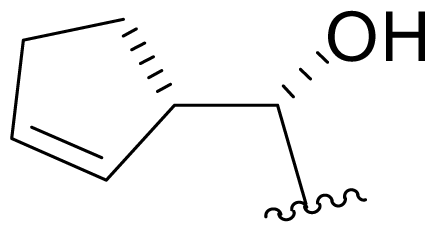 | 2.2 ± 0.1 [65]b | Mutasynthesis [65] |
| 20 | Salinosporamide X2 |  | 9.3 ± 1.6 [65]b | Mutasynthesis [64,65] |
| 21 | Salinosporamide X3 | 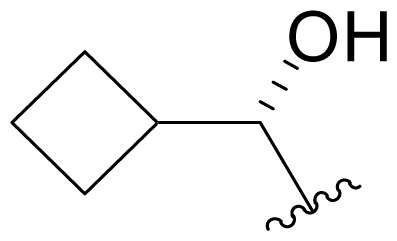 | 93.4 ± 4.3 [65]b | Mutasynthesis [65] |
| 14 | Antiprotealide |  | 31 ± 5 [53]d 27 ± 2 [53]e 101 ± 15 [65]b | Natural Product [53] Total Synthesis [71,72] Mutasynthesis [64,65] Directed Biosynthesis [Table 4] |
| 22 | Salinosporamide X5 | 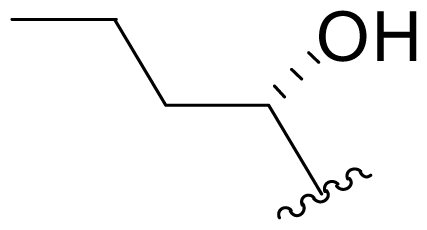 | 245 ± 38 [65]b | Mutasynthesis [65] |
| 23 | Salinosporamide X6 |  | 132 ± 19 [65]b | Mutasynthesis [65] |
| 24 | Salinosporamide X4 |  | 1029 ± 419 [65]b | Mutasynthesis [65] |
| 37 | NPI-2060 | 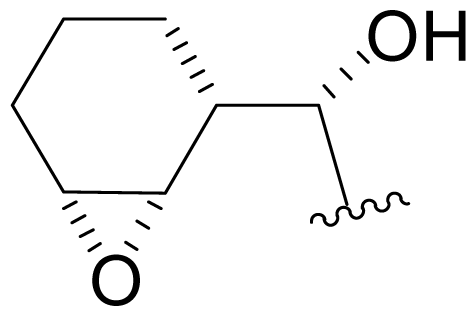 | 6.3 ± 0.6 [34] | Semi-synthesis [34] |
| 38 | NPI-2061 | 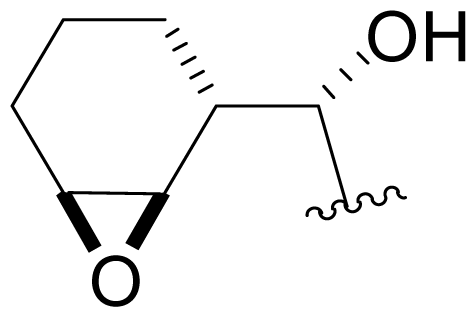 | 91 ± 8 [34] | Semi-synthesis [34] |
| 39 | NPI-2064 |  | 8200 ± 3000 [34] | Semi-synthesis [34] |
| 34 | NPI-2157 | 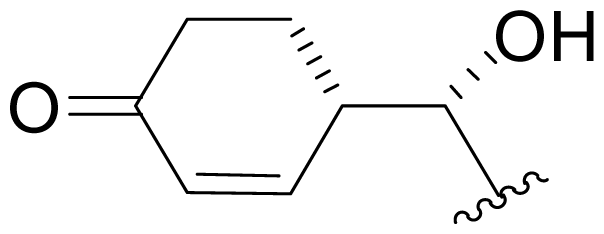 | NR | Degradation [Scheme 1] Semi-synthesis [Scheme 2] |
| 40 | NPI-2167 | 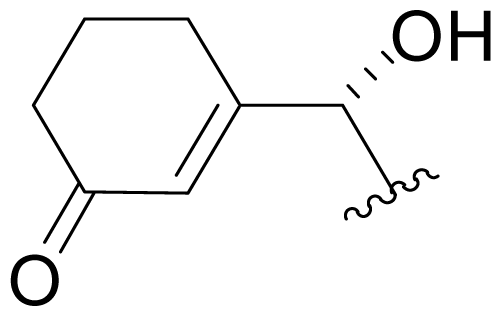 | NR | Semi-synthesis [Scheme 2] |
| Improvement step | Improvement parameters | Shake flask (mg/L) | Fermentor (mg/L) |
|---|---|---|---|
| – | Original condition | 4 | – |
| 1 | Effect of resins | 70 | 25 |
| 2 | Length and timing of seed, production and resin addition cycle | 120 | 120 |
| 3 | Media formulation | 220 | 220 |
| 4 | Single colony isolation | 330 | 330 |
| 5 | Statistical design media optimization | 450 | 360 |
| Condition | Precursor | 1 Chloro | 15 Bromo | 17 Fluoro | 14 Antiprotealide | 5 Ethyl | 8 Methyl | 9 Propyl |
|---|---|---|---|---|---|---|---|---|
| 1 [53] | None (NaCl-based medium) | 277 | 0 | 0 | 3.0 | 4.4 | 0.15 | 0.11 |
| 2 [54] | NaBr (NaBr-based medium) | 1.2 | 19.4 | 0 | 0 | 80.3 | 0 | 0 |
| 3 | None (Na2SO4-based medium) | 53 | 0 | 0 | ND | 7.3 | 0.34 | 0.18 |
| 4 | 1.5% NaBr (Na2SO4-based medium) | 18.7 | 73.3 | 0 | 0 | 22.3 | 0 | 0 |
| 5 | 0.025% 5′-FDA (Na2SO4-based medium) | 48.1 | 0 | 55.8 | ND | 2.9 | Trace | Trace |
| 6 | 1% Valerate (Na2SO4-based medium) | 45 | 0 | 0 | ND | 8.3 | 0.12 | 145 |
| 7 | 1% Propionate (NaCl-based medium) | 222 | 0 | 0 | 0 | 5.2 | 4.81 | 0 |
| 8 [55] | 1% Butyrate (NaCl-based medium) | 189 | 0 | 0 | ND | 19.0 | 0 | 0 |
| 9 | 1% Valerate (NaCl-based medium) | 131 | 0 | 0 | ND | 5.0 | 0.15 | 121 |
| 10 | 1% Valine (NaCl-based medium) | 177 | 0 | 0 | 0 | 16.7 | 0 | 0 |
| 11 | 1% Leucine (NaCl-based medium) | 112 | 0 | 0 | 11 | 11 | 0.43 | 0.23 |
| 12 | 1% Isoleucine (NaCl-based medium) | 71 | 0 | 0 | 0 | 6.6 | 4.63 | 0 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Potts, B.C.; Lam, K.S. Generating a Generation of Proteasome Inhibitors: From Microbial Fermentation to Total Synthesis of Salinosporamide A (Marizomib) and Other Salinosporamides. Mar. Drugs 2010, 8, 835-880. https://doi.org/10.3390/md8040835
Potts BC, Lam KS. Generating a Generation of Proteasome Inhibitors: From Microbial Fermentation to Total Synthesis of Salinosporamide A (Marizomib) and Other Salinosporamides. Marine Drugs. 2010; 8(4):835-880. https://doi.org/10.3390/md8040835
Chicago/Turabian StylePotts, Barbara C., and Kin S. Lam. 2010. "Generating a Generation of Proteasome Inhibitors: From Microbial Fermentation to Total Synthesis of Salinosporamide A (Marizomib) and Other Salinosporamides" Marine Drugs 8, no. 4: 835-880. https://doi.org/10.3390/md8040835
APA StylePotts, B. C., & Lam, K. S. (2010). Generating a Generation of Proteasome Inhibitors: From Microbial Fermentation to Total Synthesis of Salinosporamide A (Marizomib) and Other Salinosporamides. Marine Drugs, 8(4), 835-880. https://doi.org/10.3390/md8040835