A Submarine Journey: The Pyrrole-Imidazole Alkaloids †

Abstract
:1. Introduction
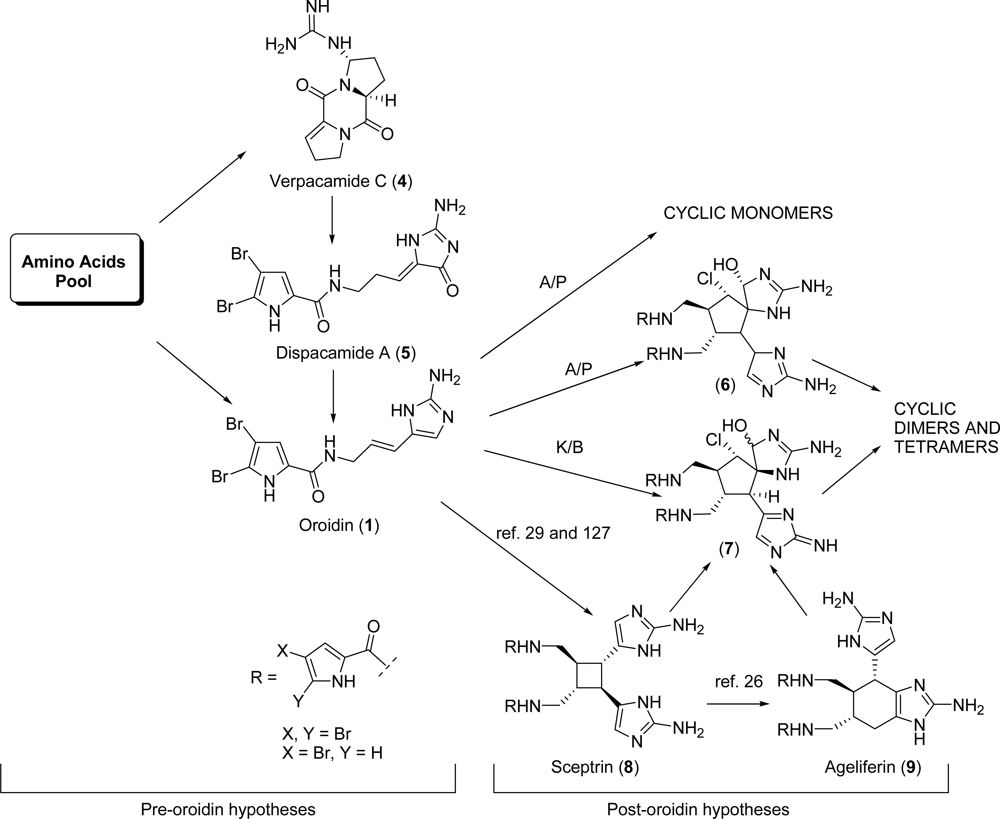
1.1. Pyrrole-Imidazole Alkaloids Sources and Biogenesis
1.2. Pyrrole-Imidazole Alkaloids Ecological Role
1.3. Pyrrole-Imidazole Alkaloids Classification
- Acyclic monomers
- Cyclic monomers
- Acyclic dimers
- Cyclic dimers
- Cyclic tetramers
2. Acyclic Monomers
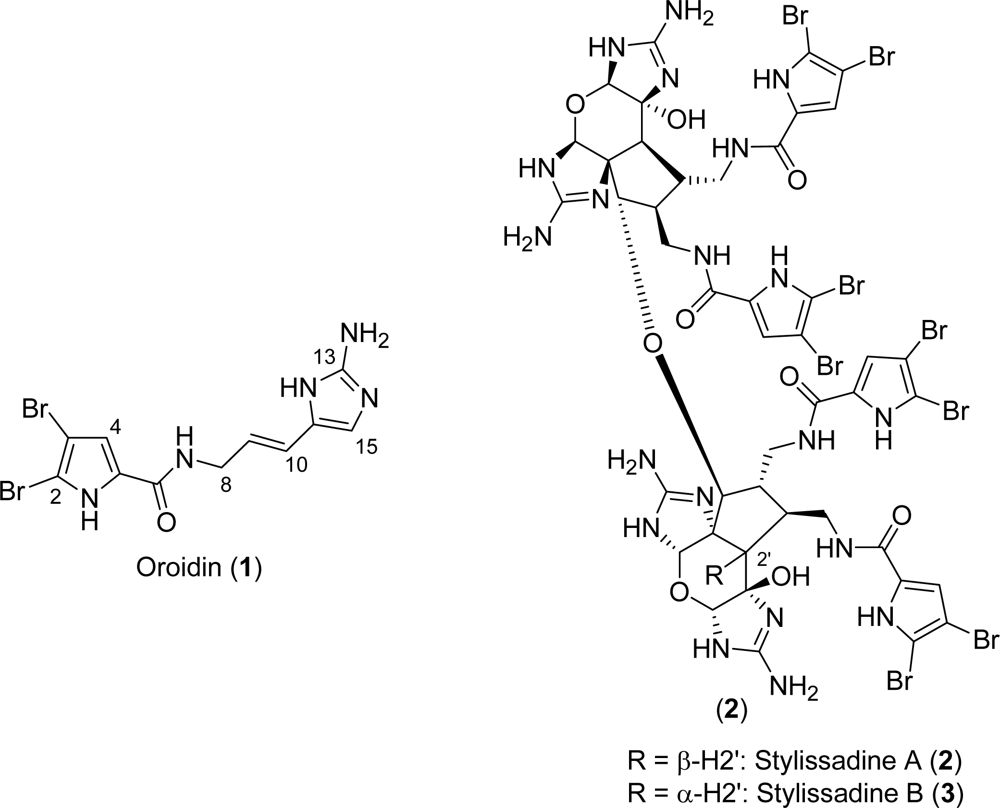
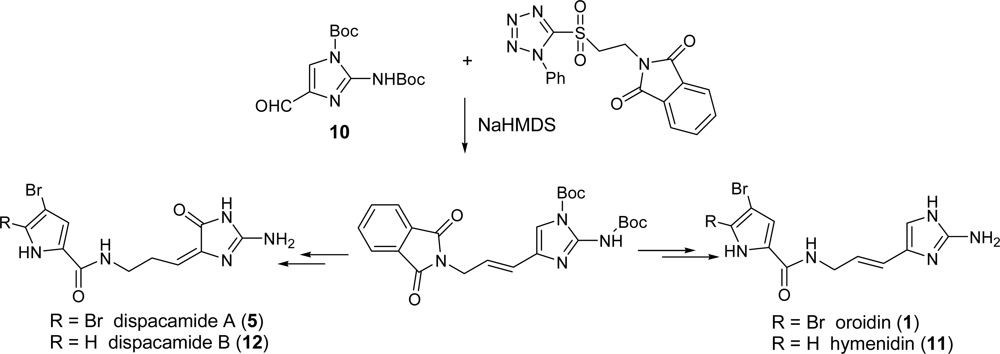
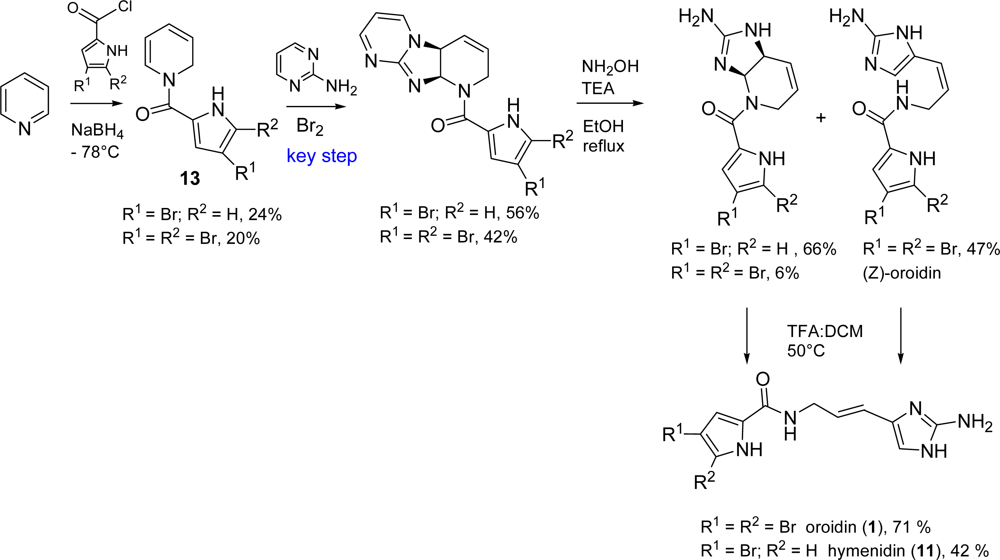
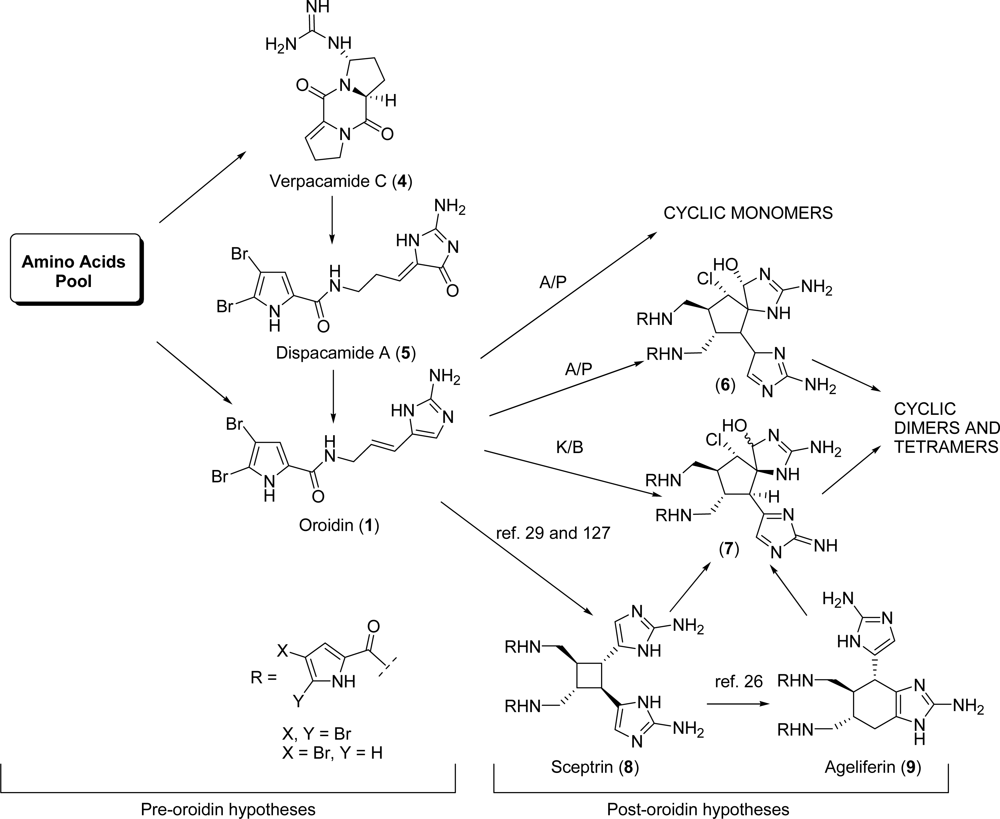
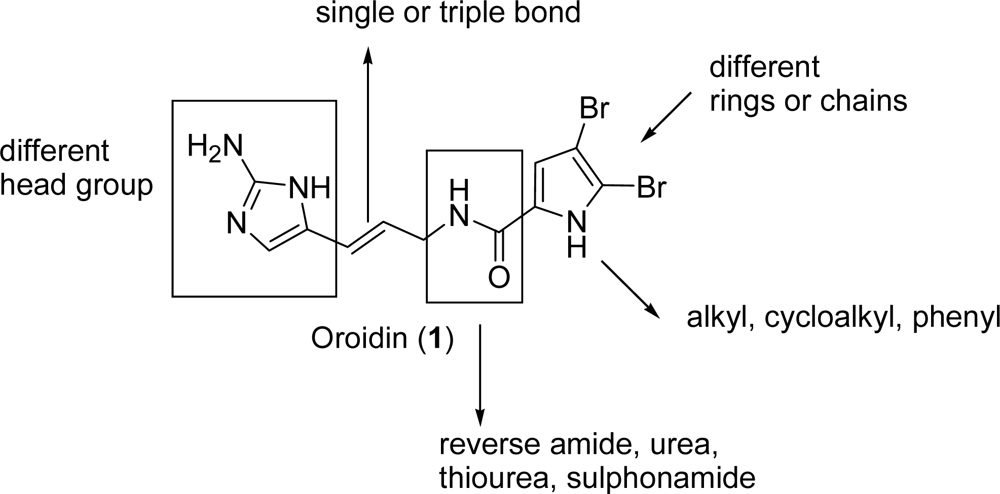
2.1. Oroidin
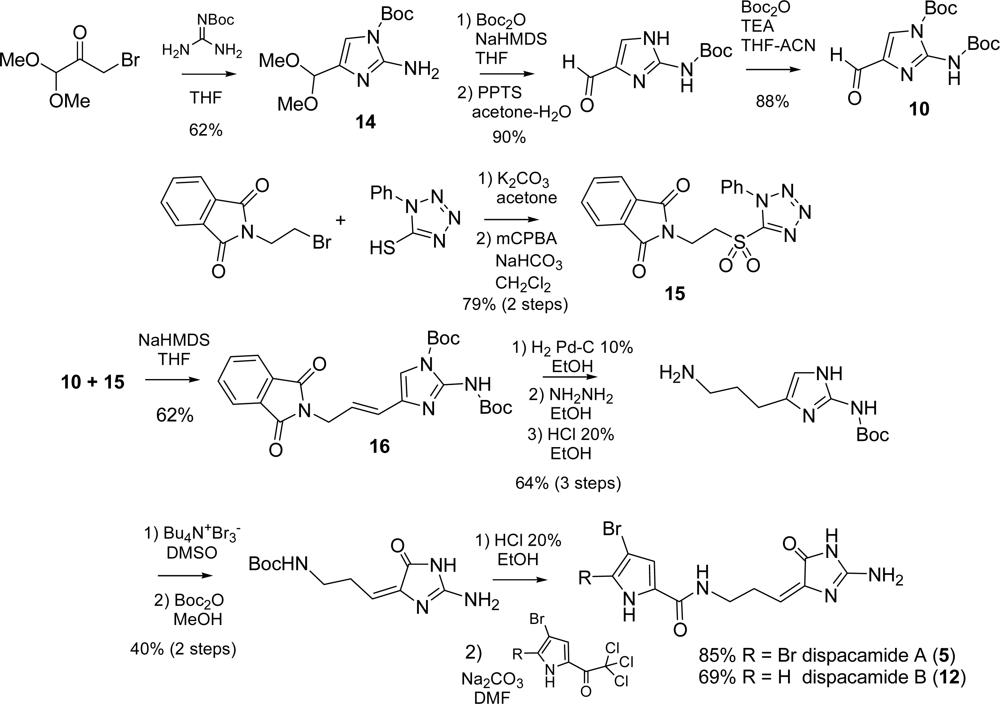
2.2. Dispacamide A
3. Cyclic Monomers
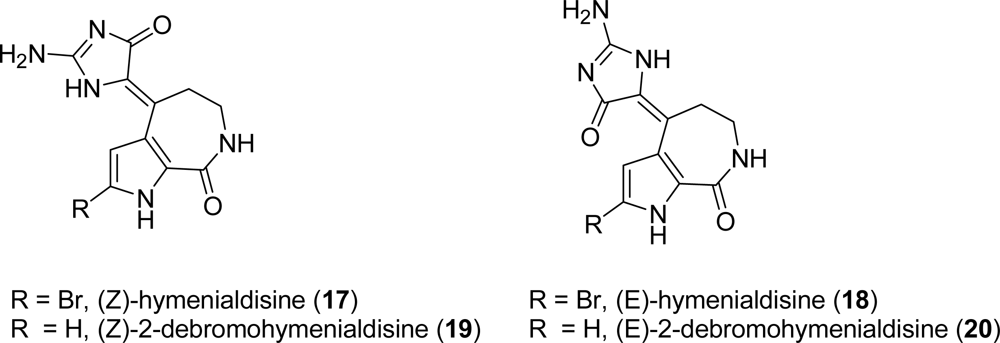
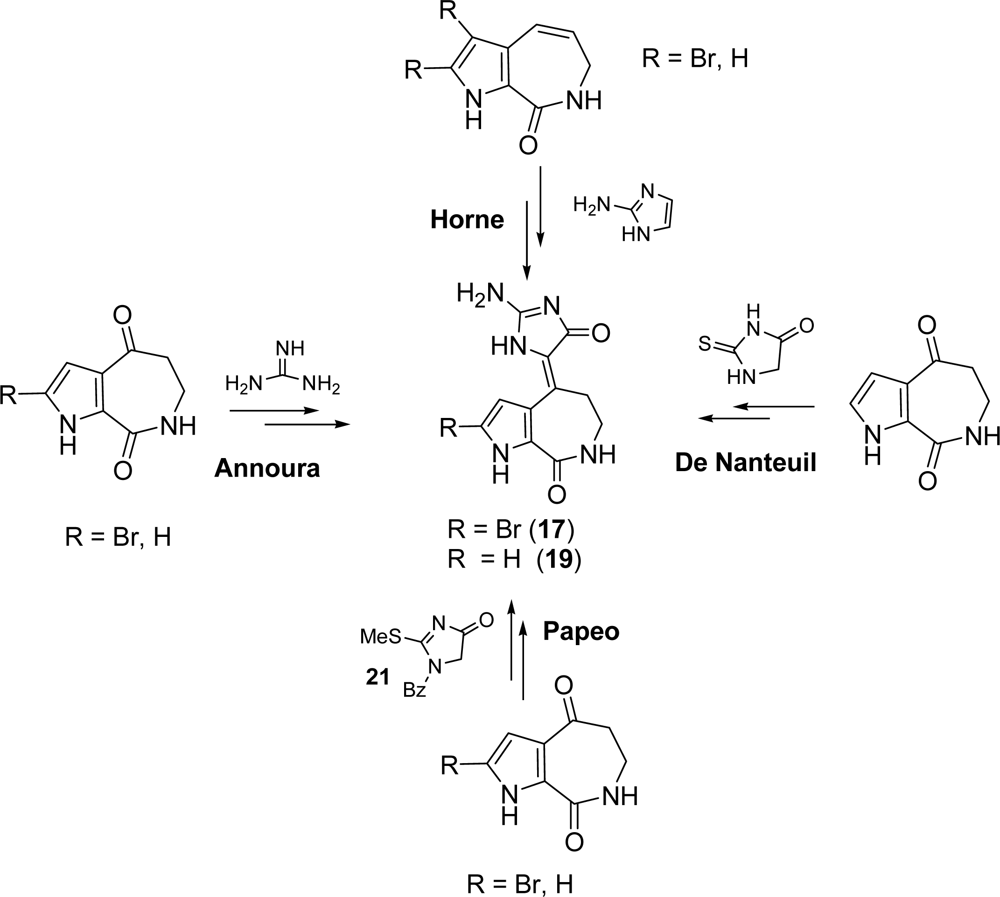
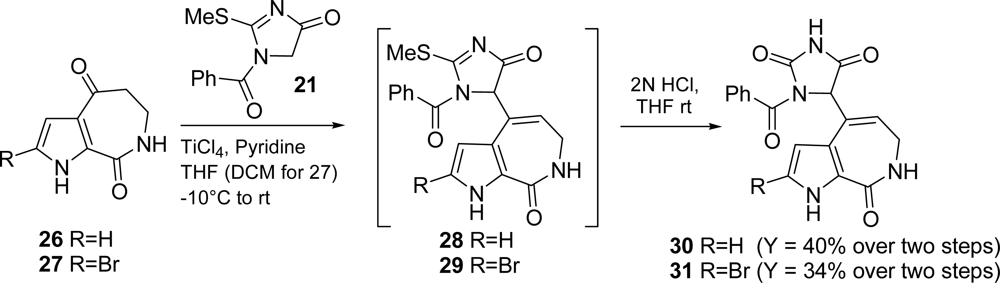
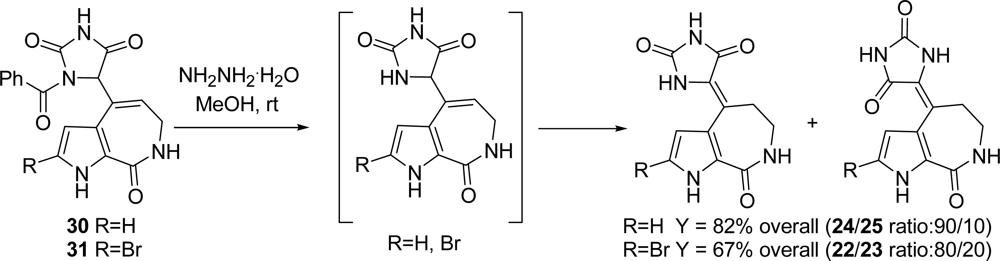
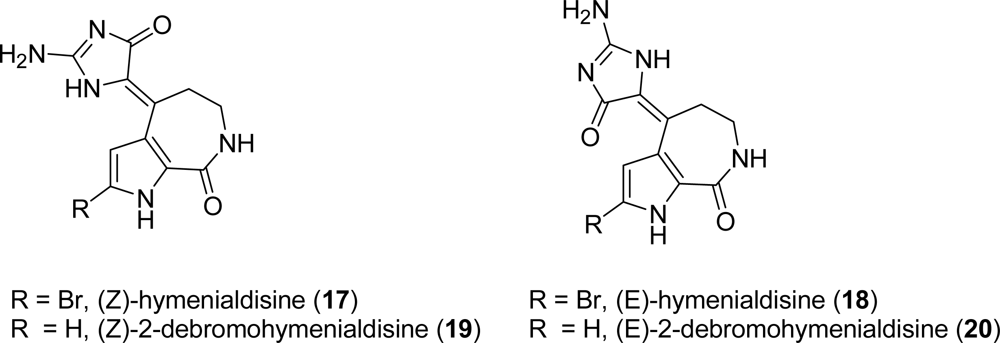
3.1. Hymenialdisine
3.2. Axinohydantoins
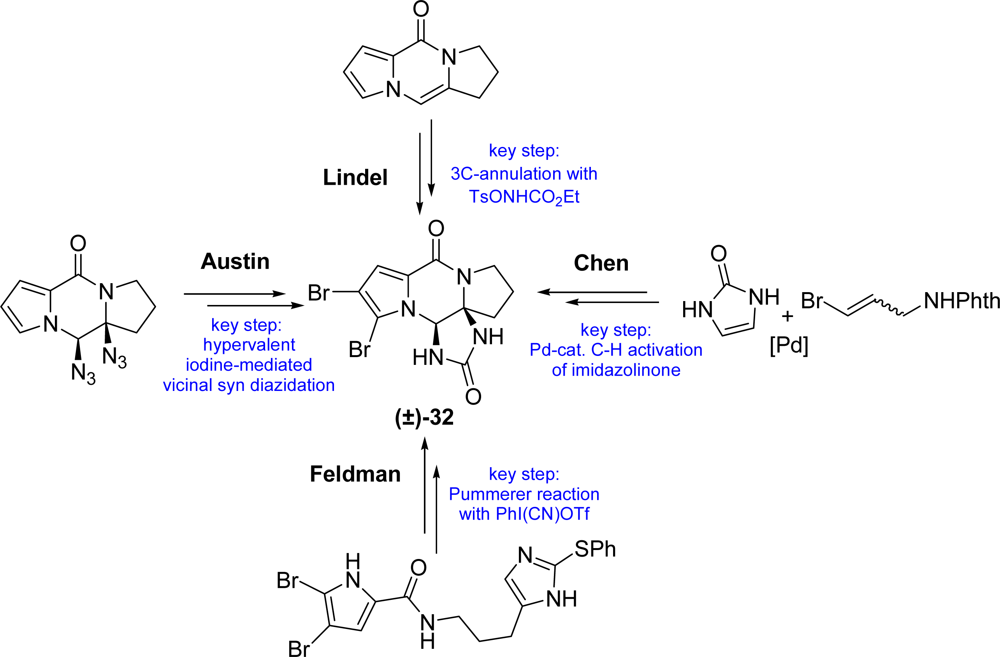
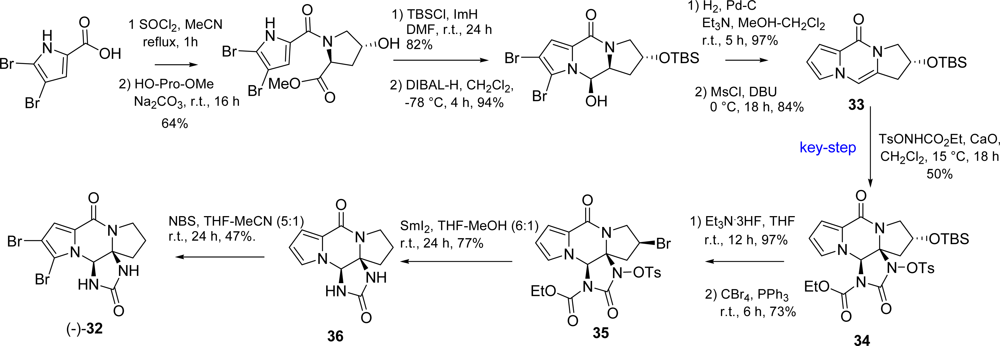
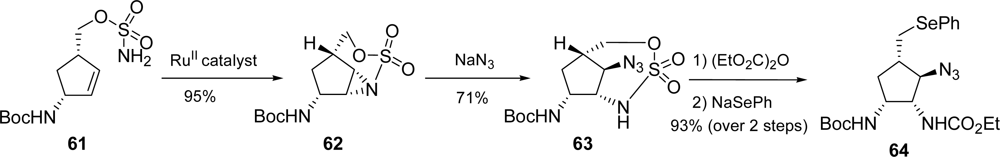
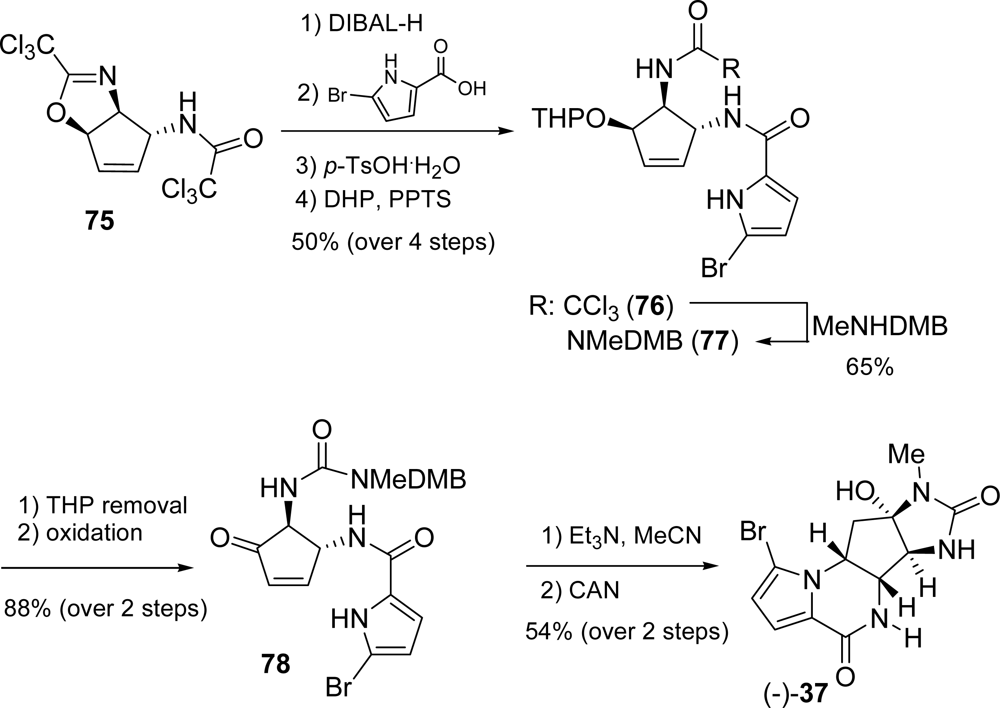
3.3. Dibromophakellstatin
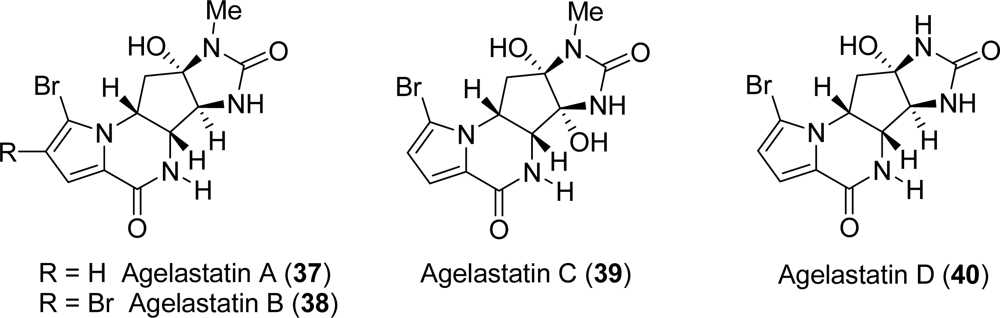
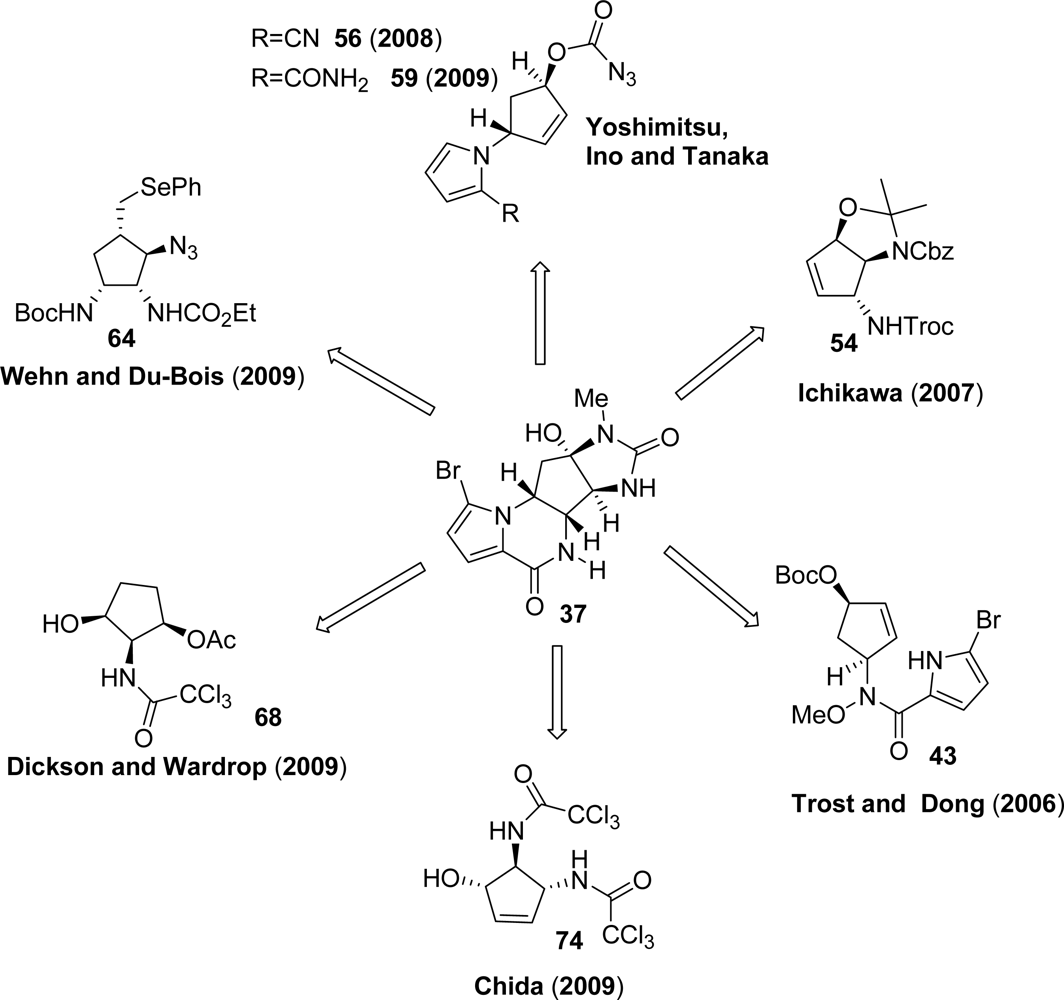
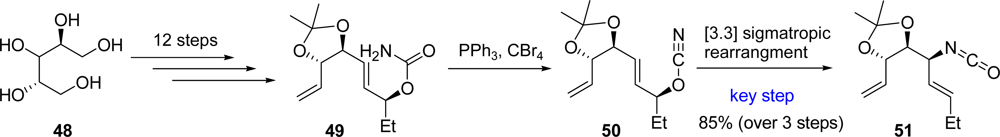
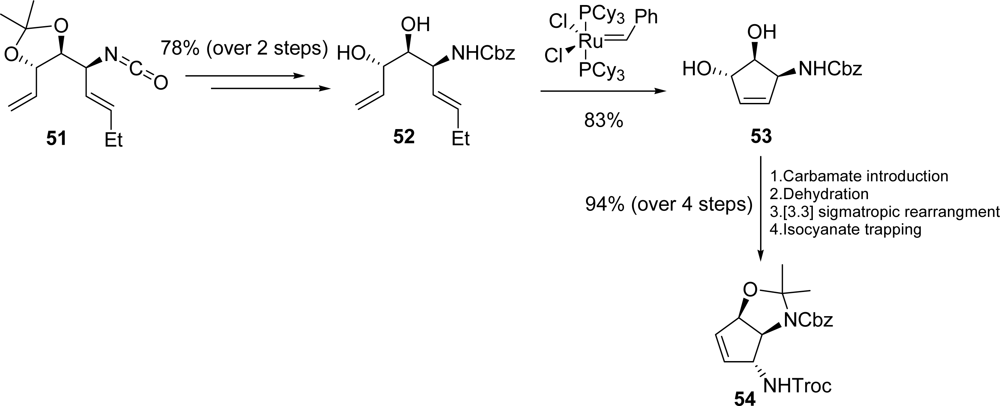
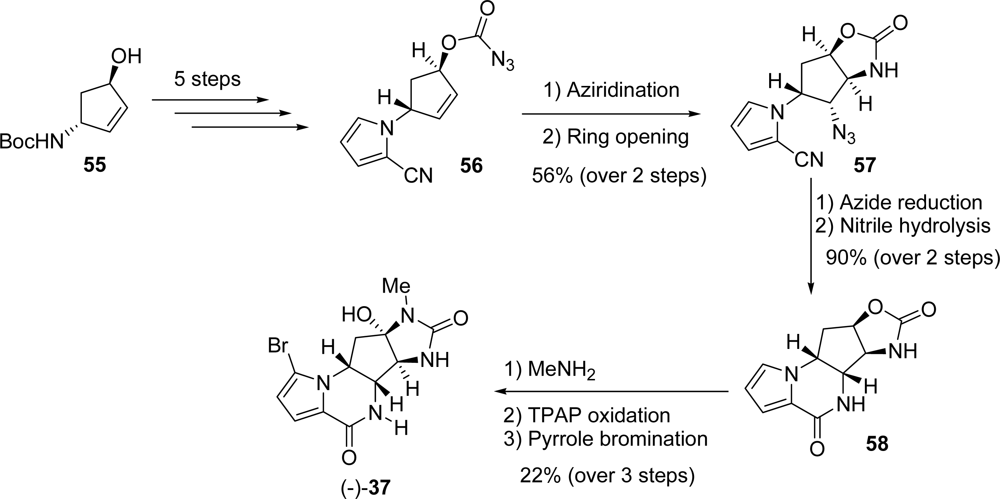
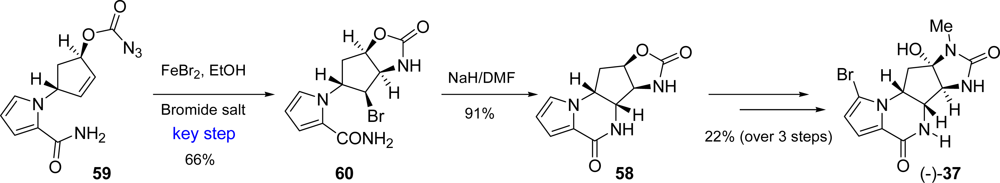
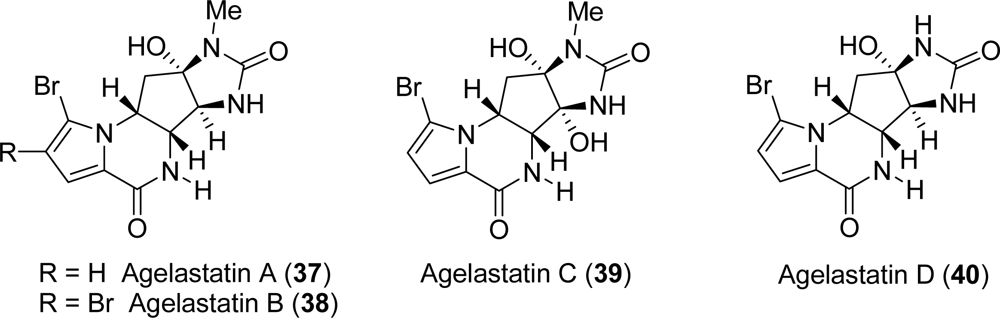
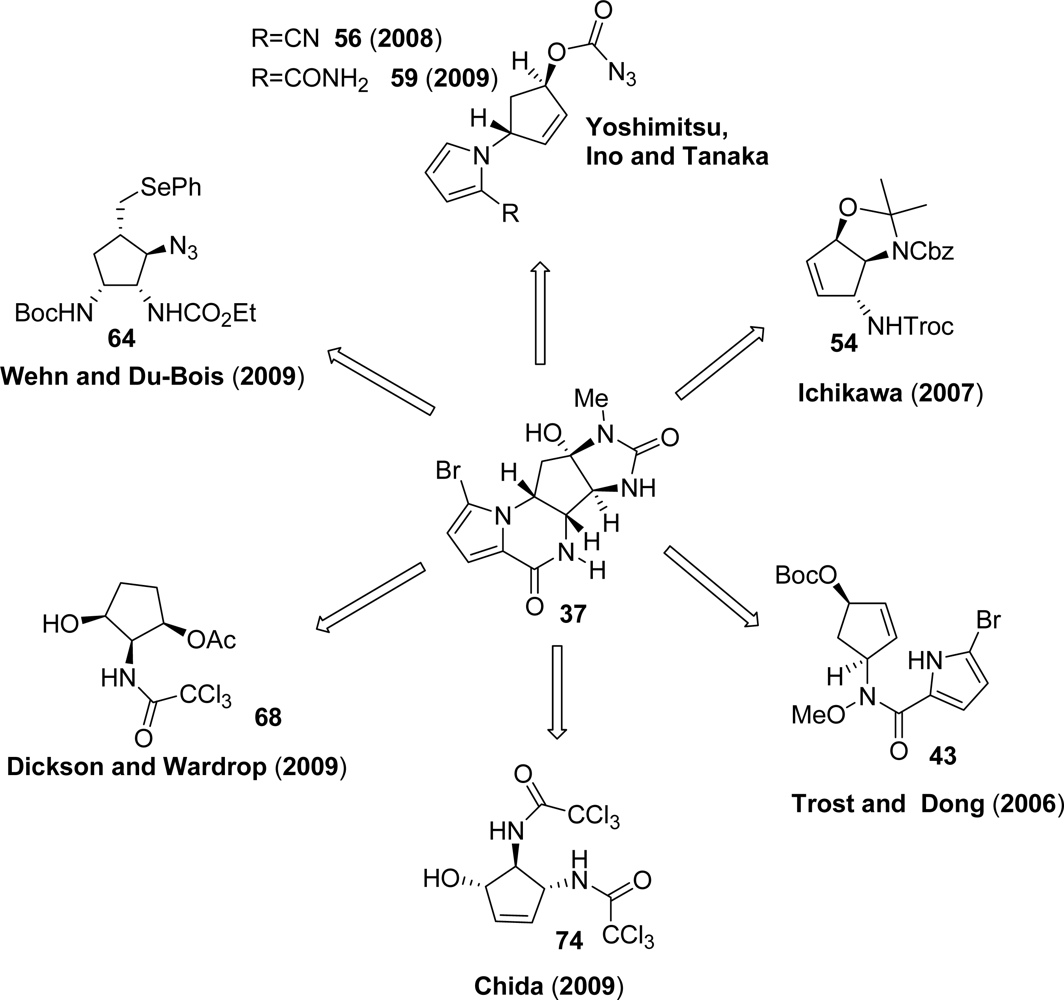
3.4. Agelastatins
4. Acyclic Dimers
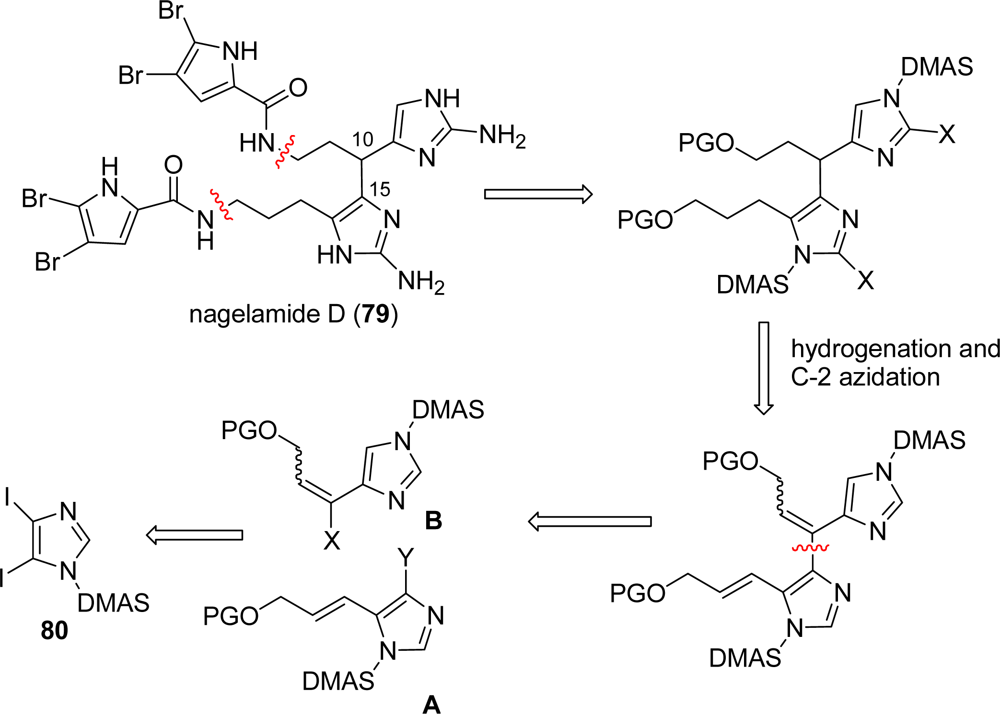
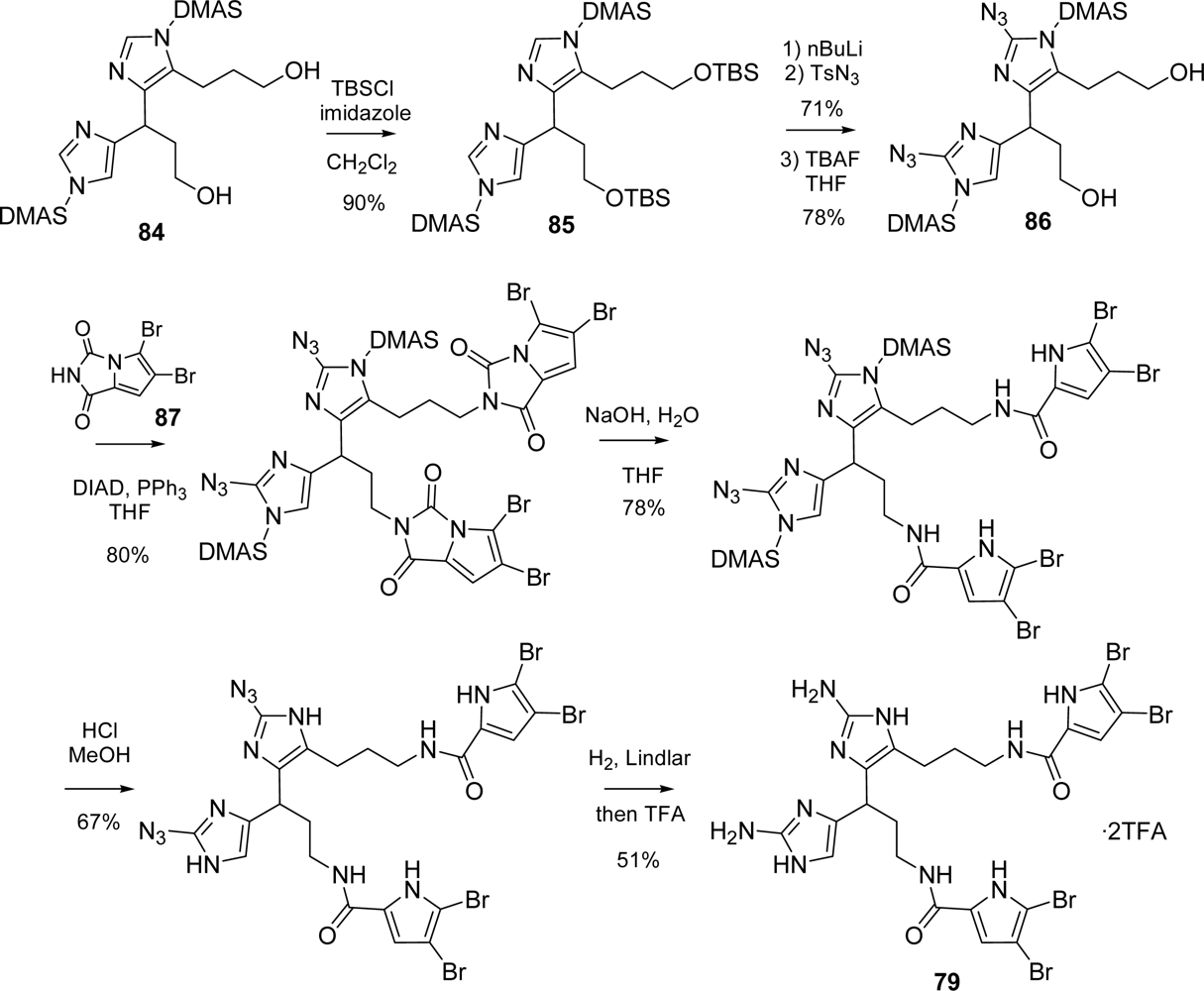
4.1. Nagelamide D
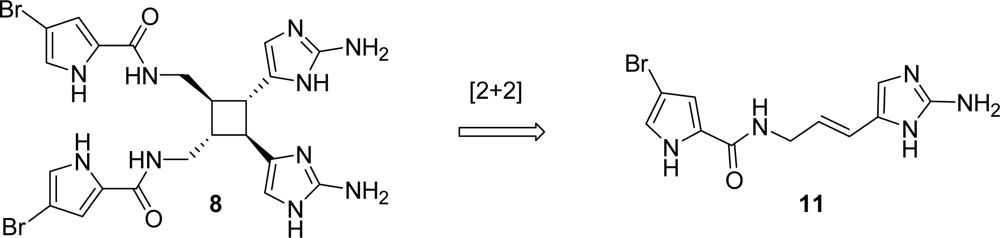
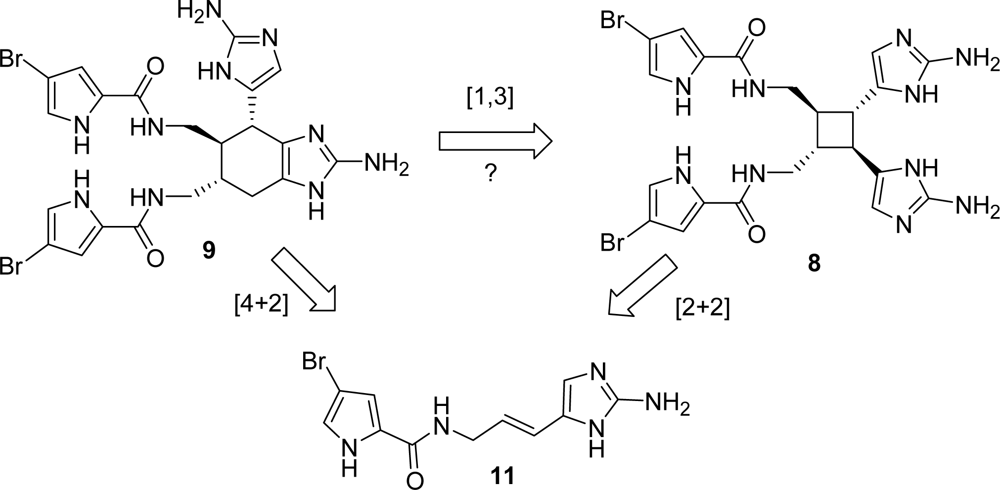
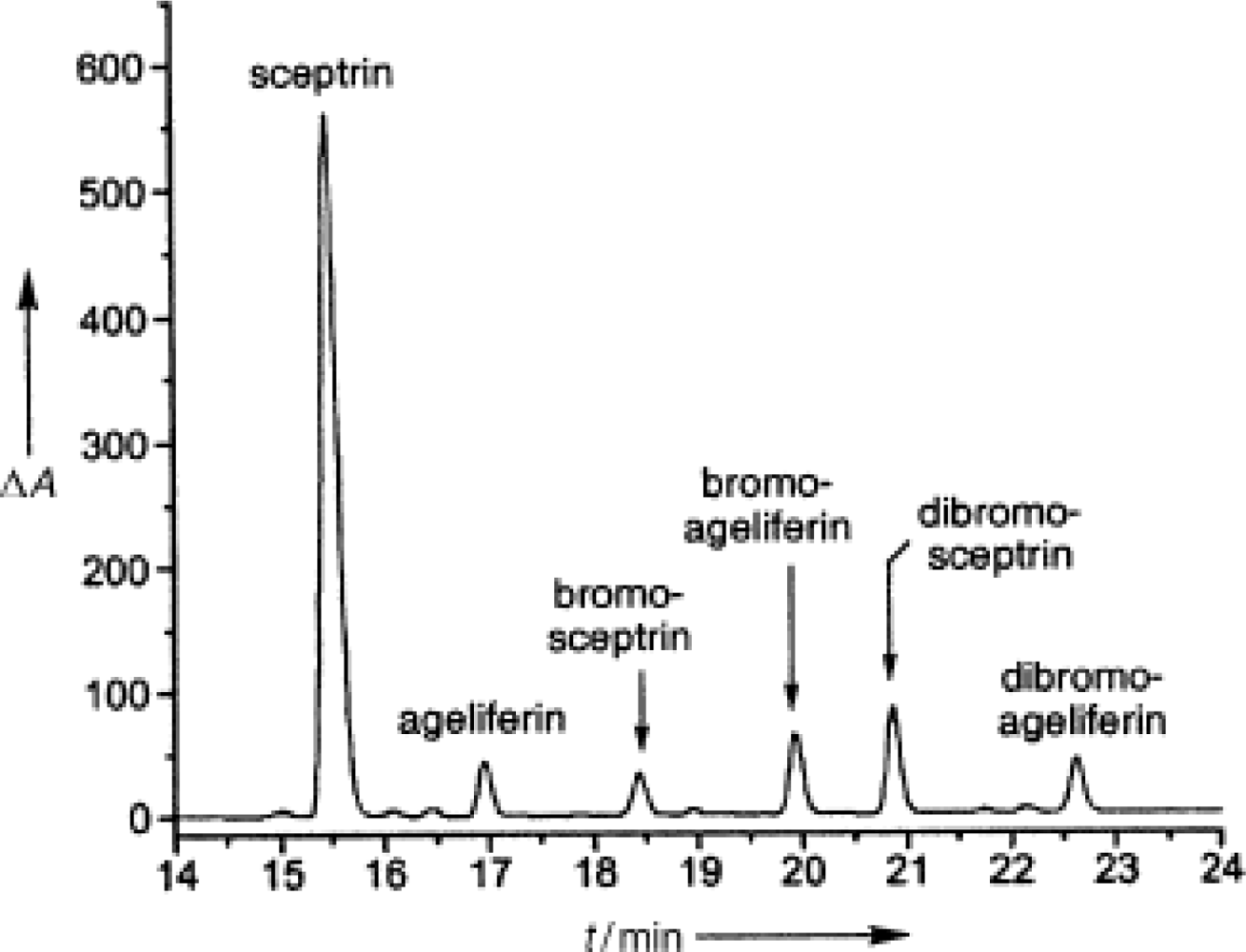
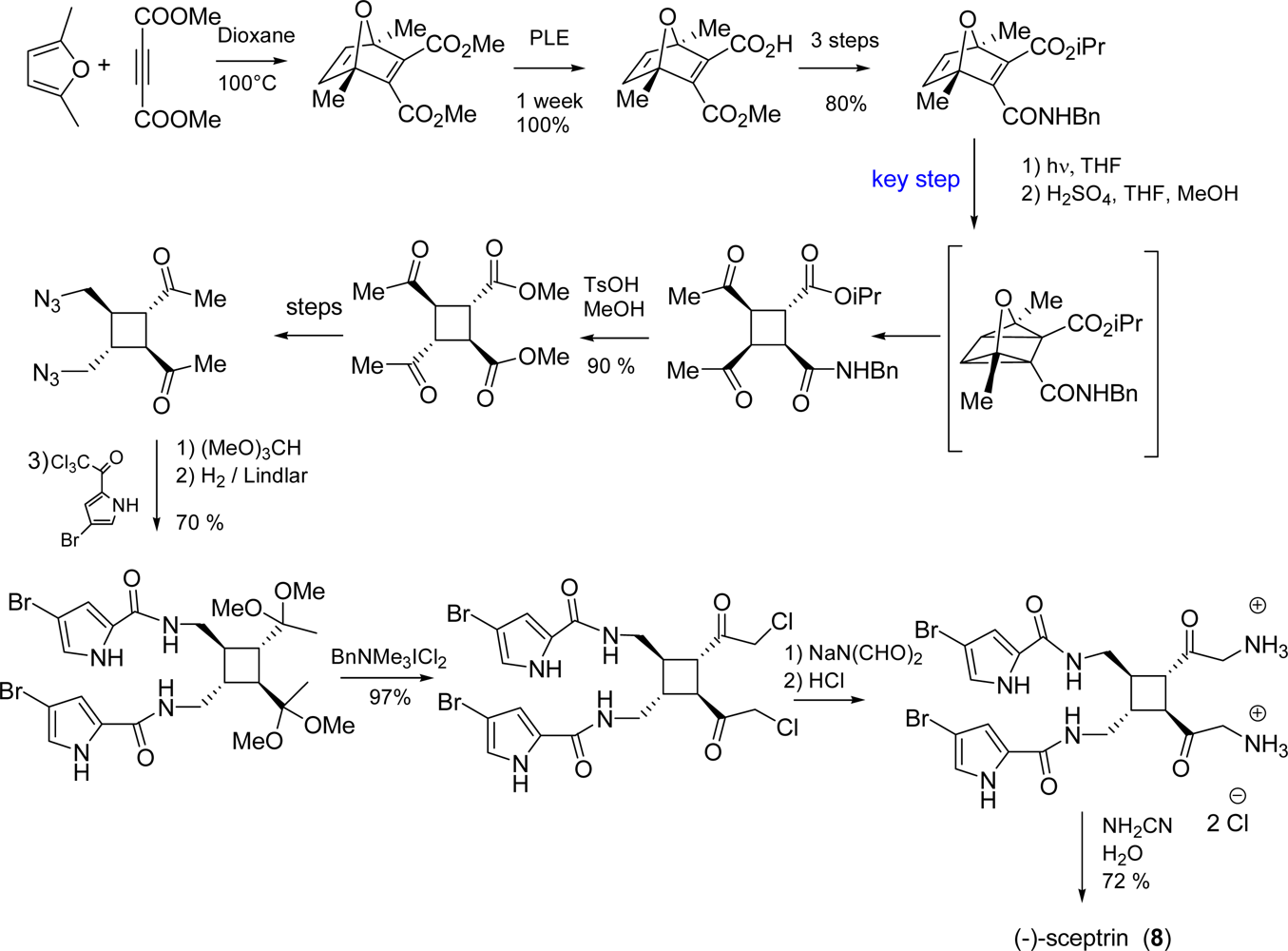
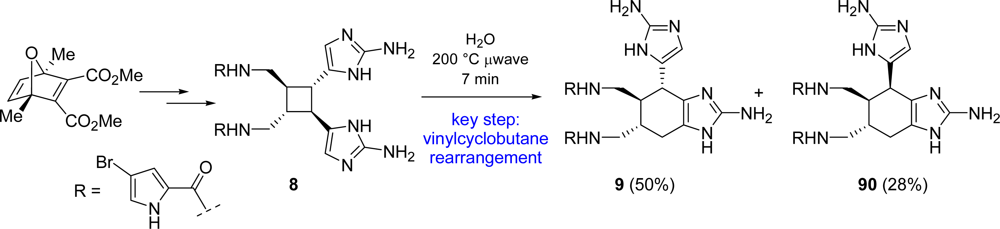
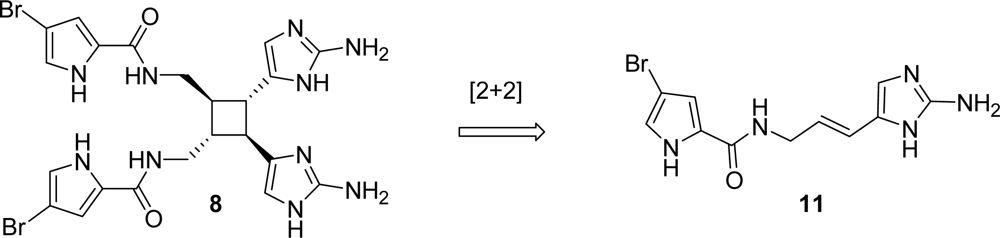
4.2. Sceptrin
- minimal use of protecting groups (benzylamide proved to be crucial for a complete transfer of chirality);
- application of an oxaquadricyclane rearrangement/fragmentation [133] in natural product synthesis to diastereoselectively access the cyclobutane core of sceptrin;
- application of a new chemo- and regioselective halogenation method [134];
- formation of 2-aminoimidazole in mild conditions [135] (these moieties were voluntarily introduced at a later stage of the synthesis because the earlier introduction of this step resulted in compounds intractability).
5. Cyclic Dimers
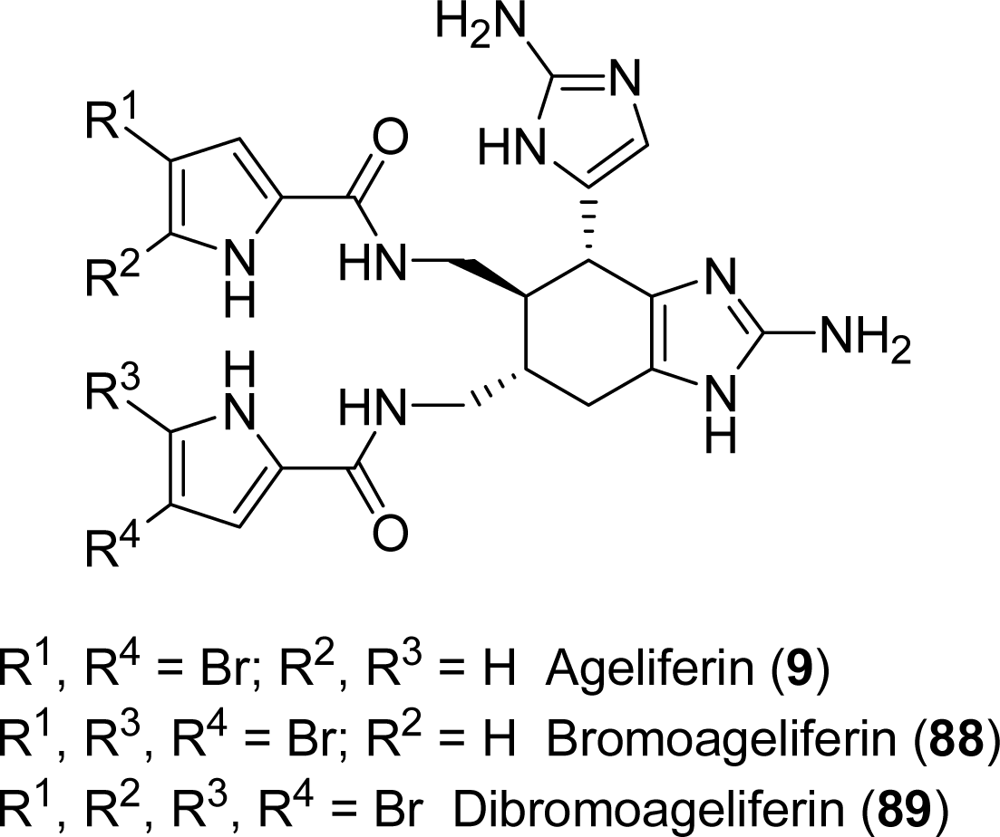
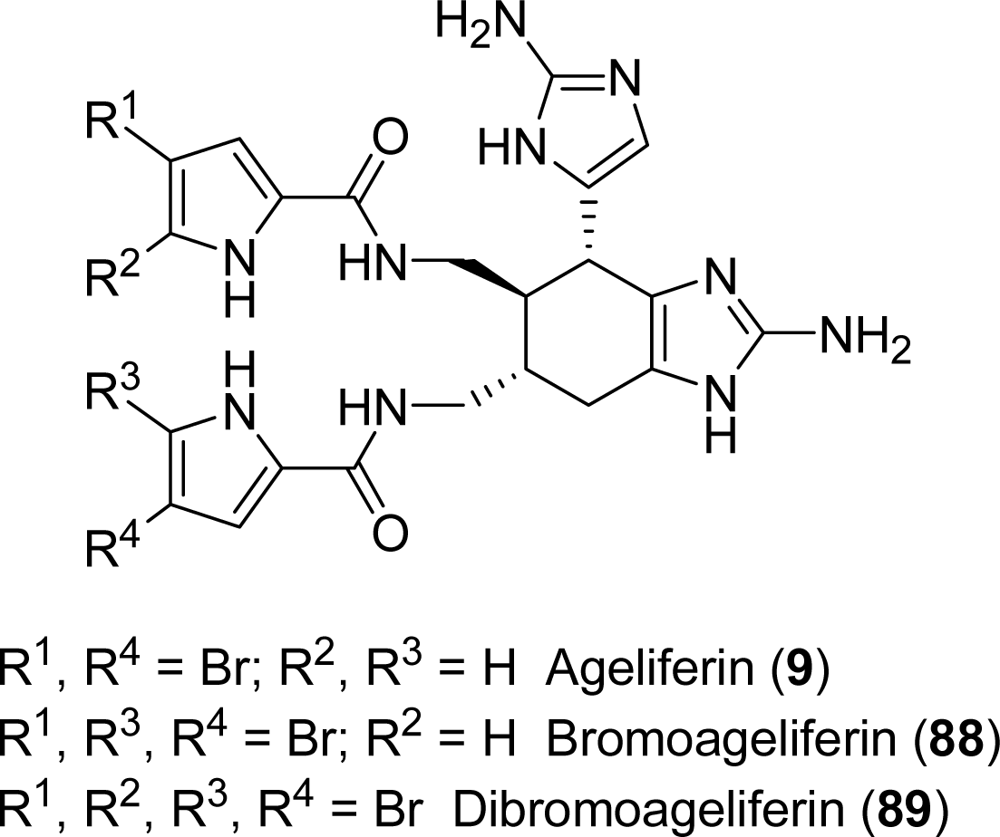
5.1. Ageliferin
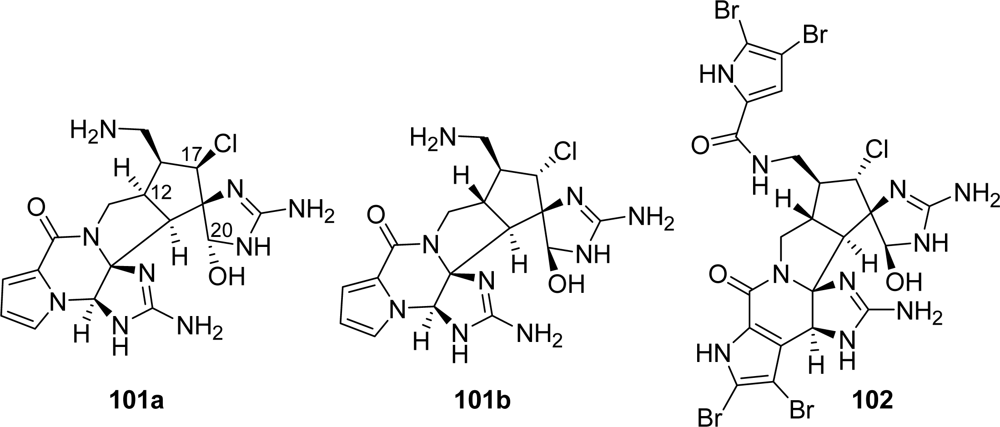
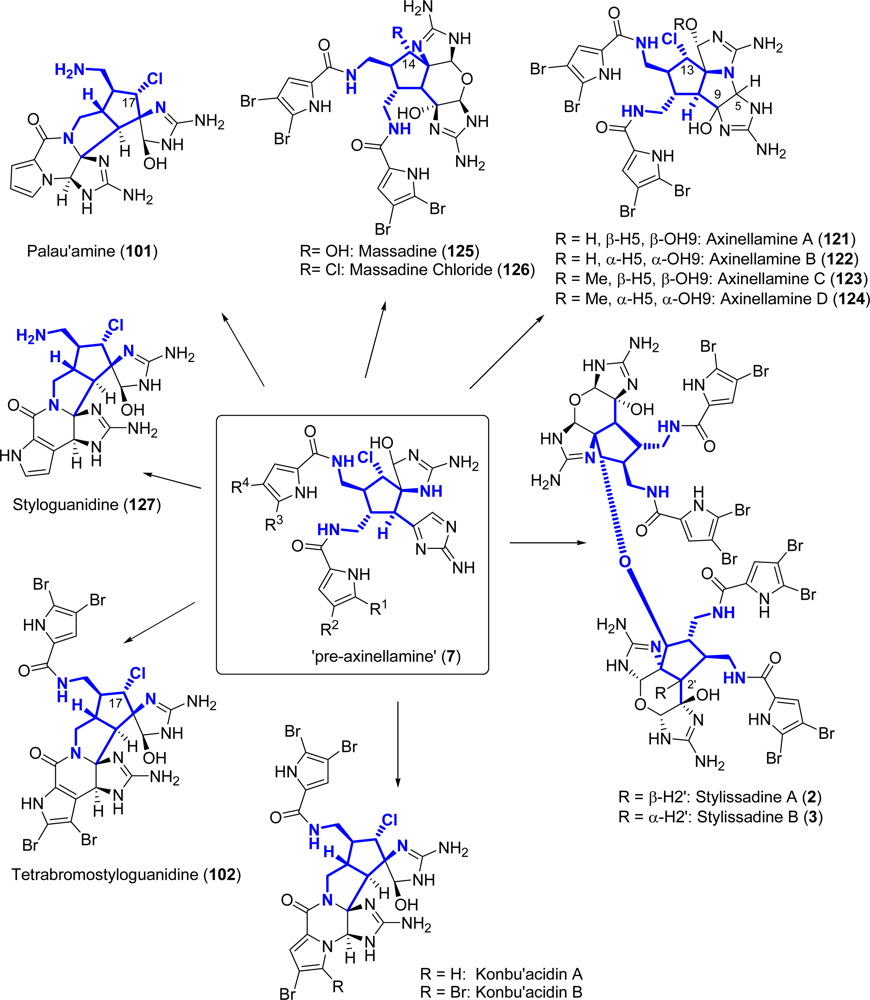
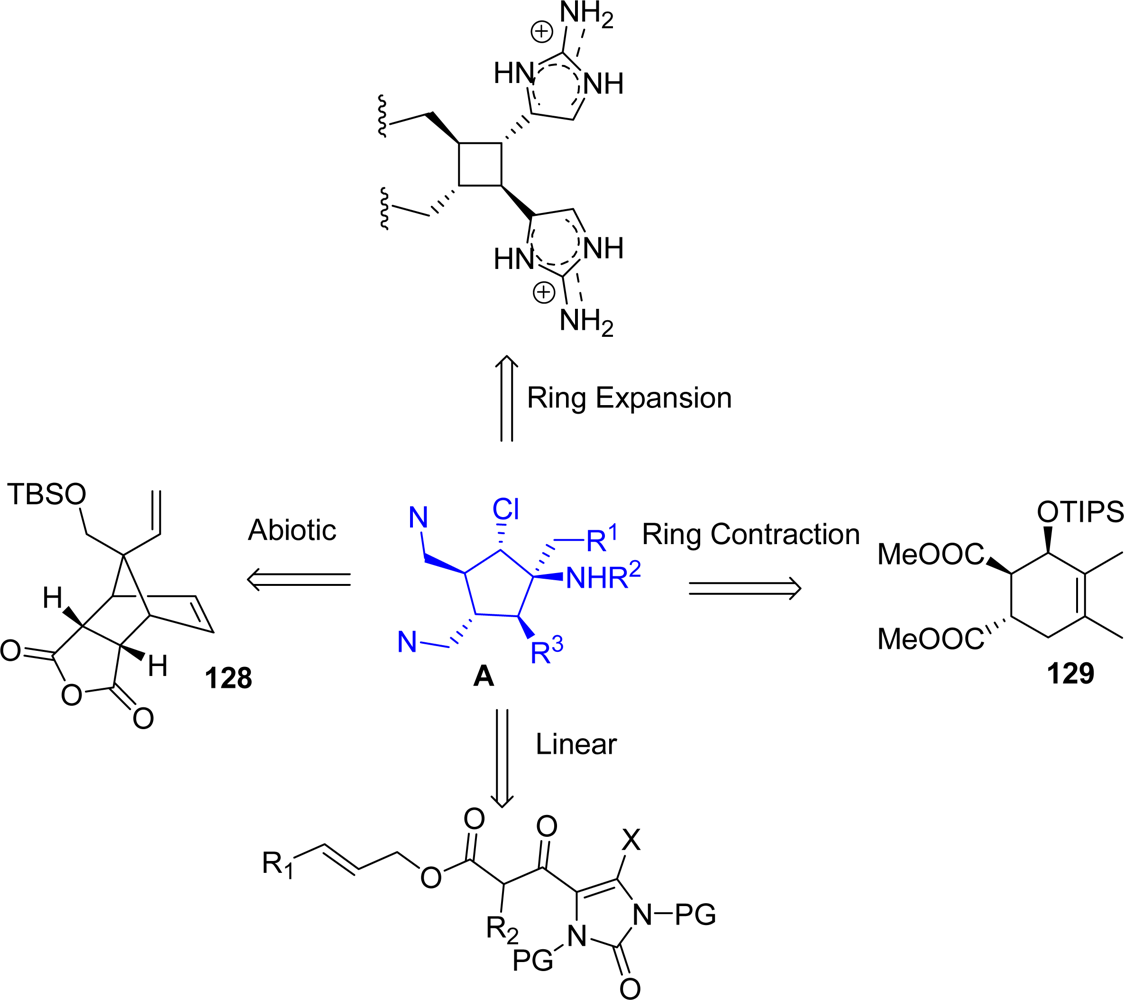
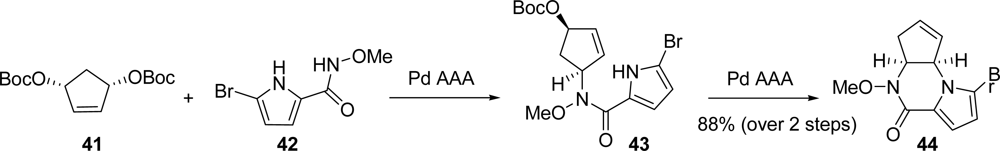
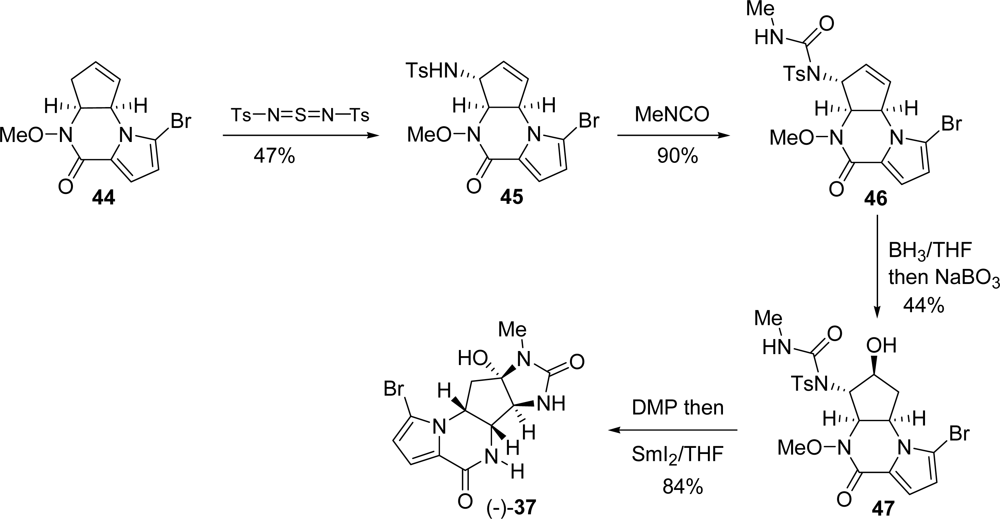
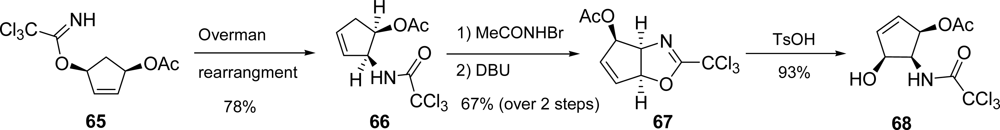
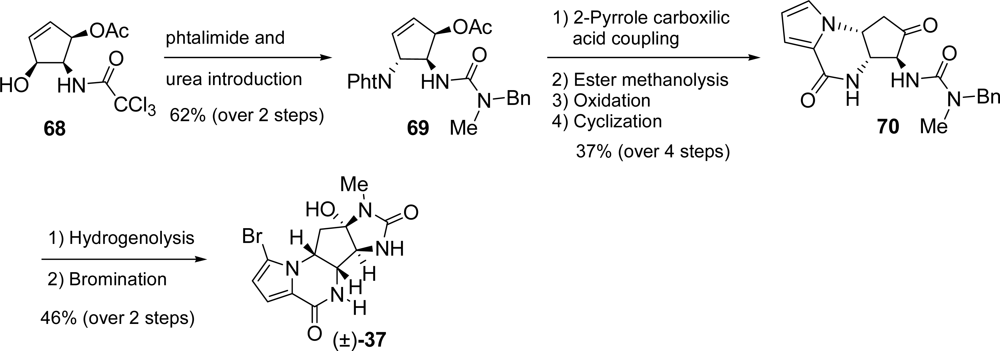
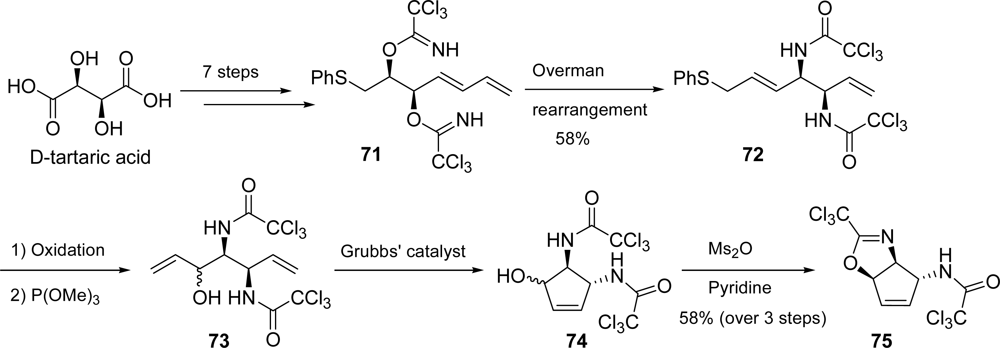
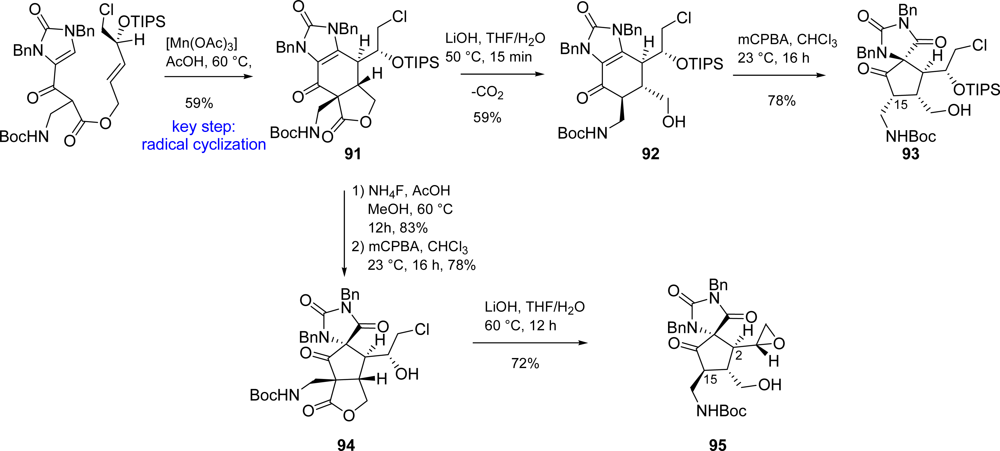
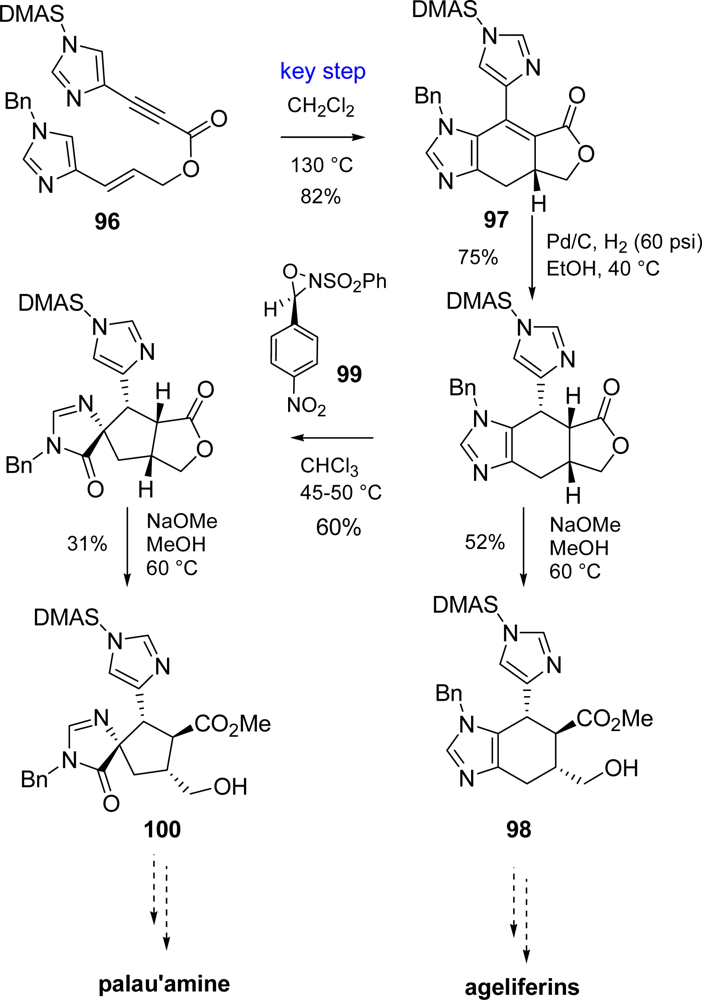
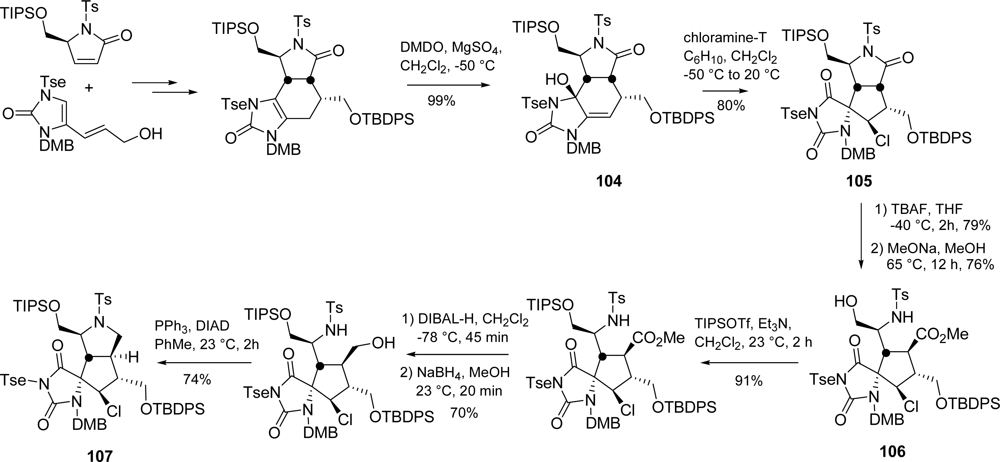
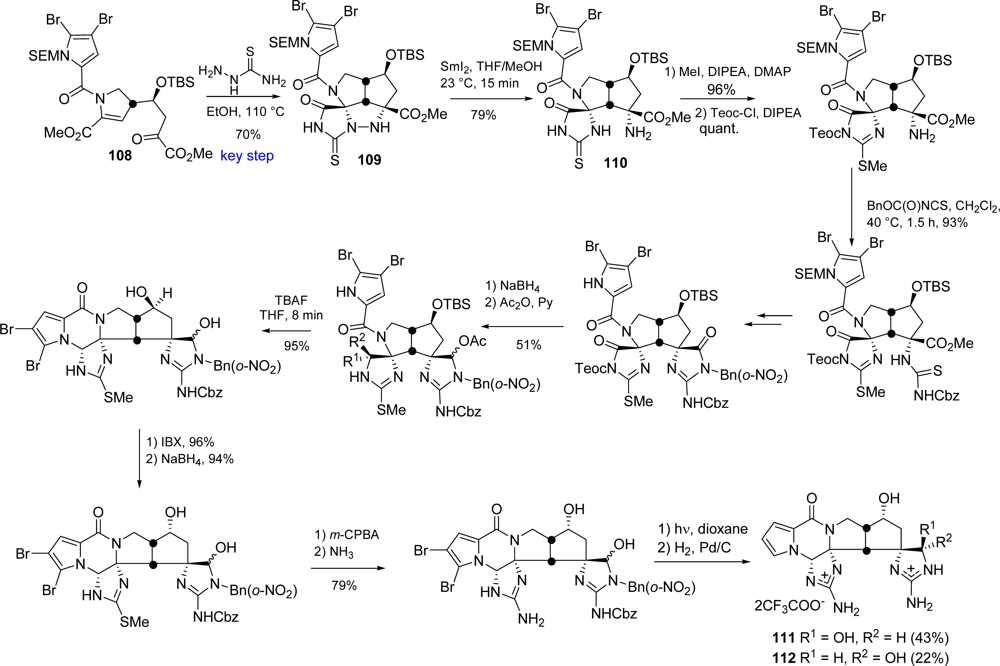
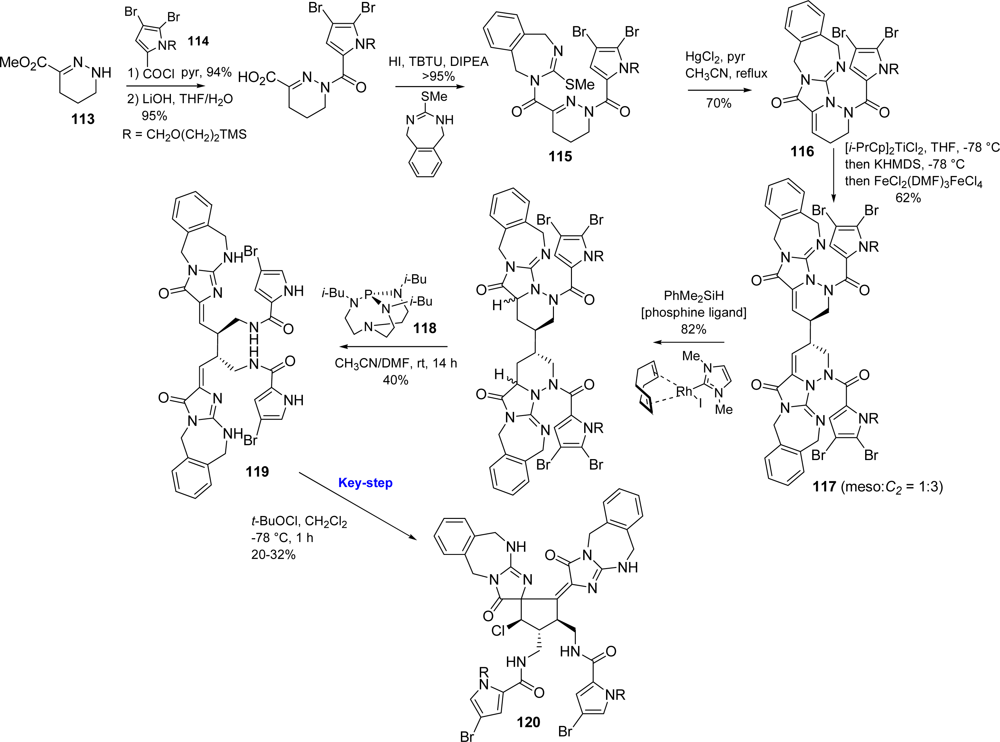
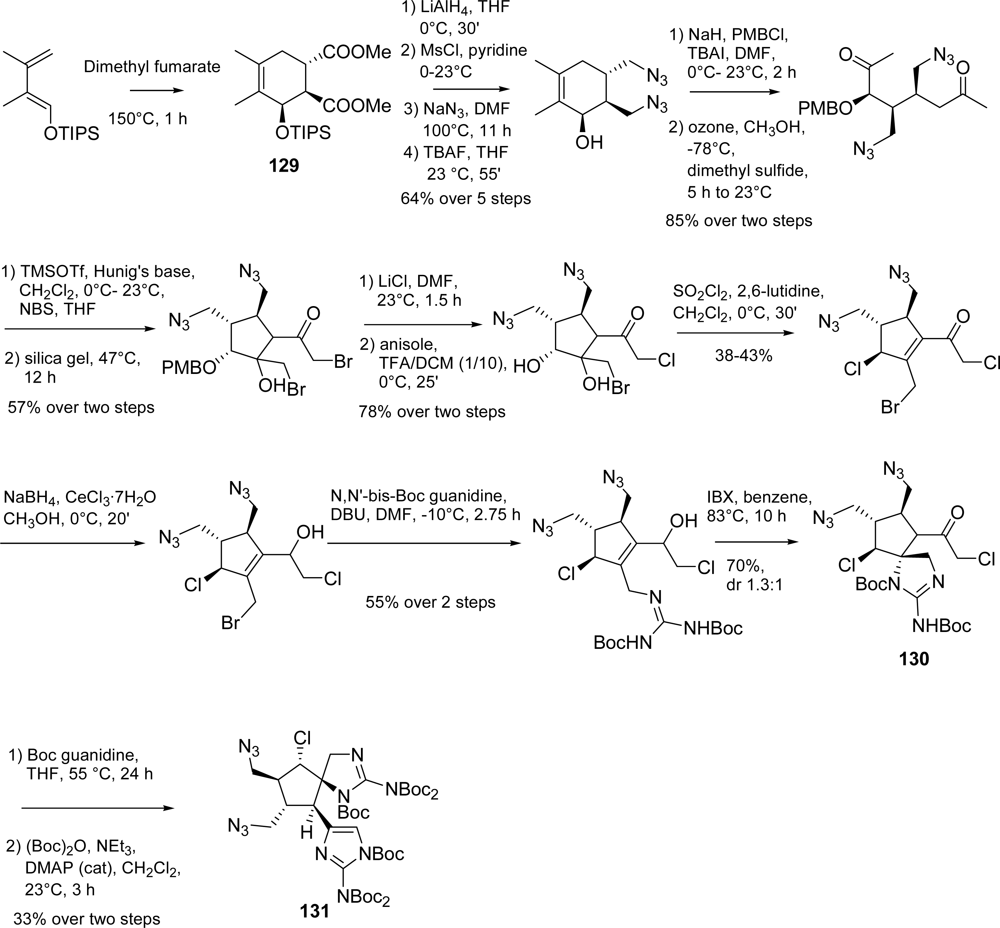
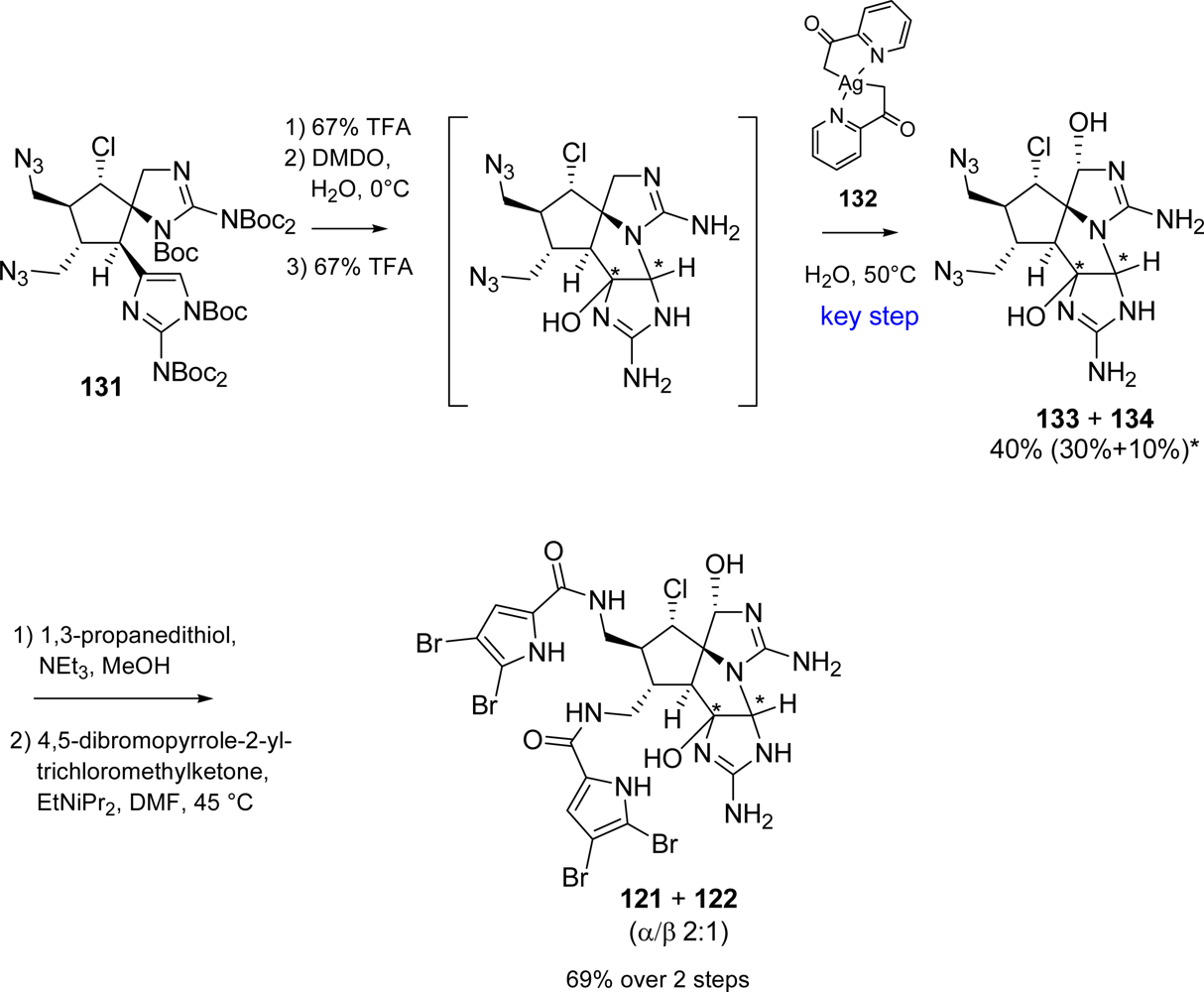
5.2. Palau’amine
5.3. Axinellamines
5.4. Massadines
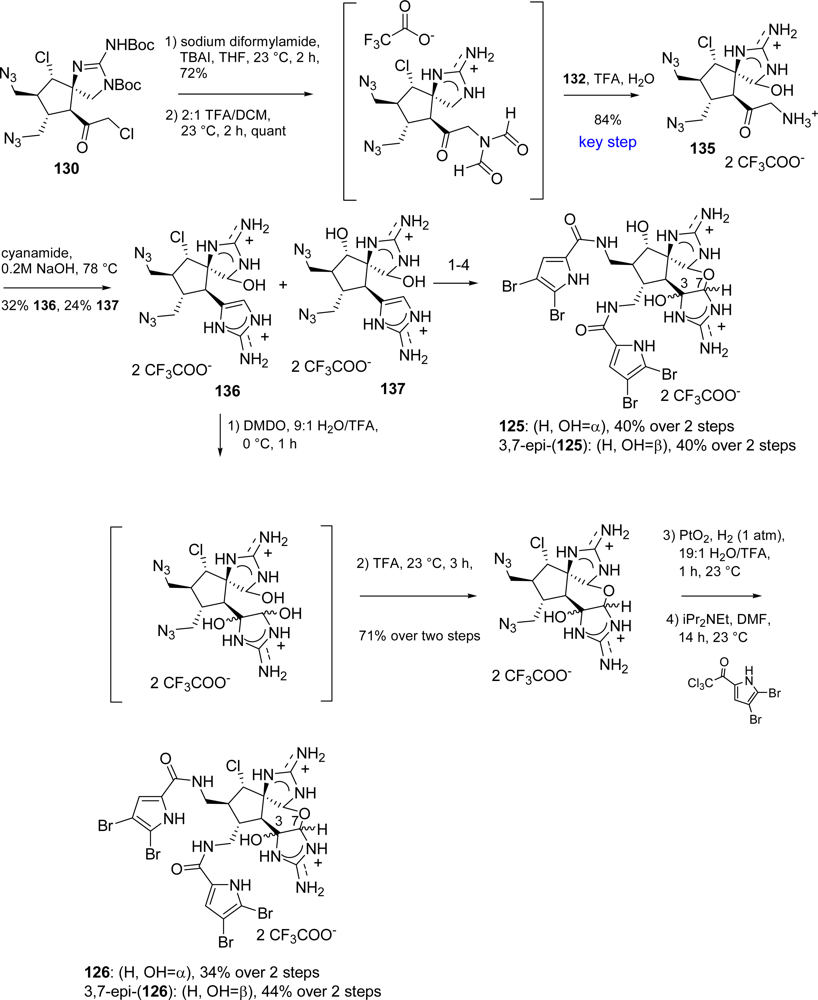
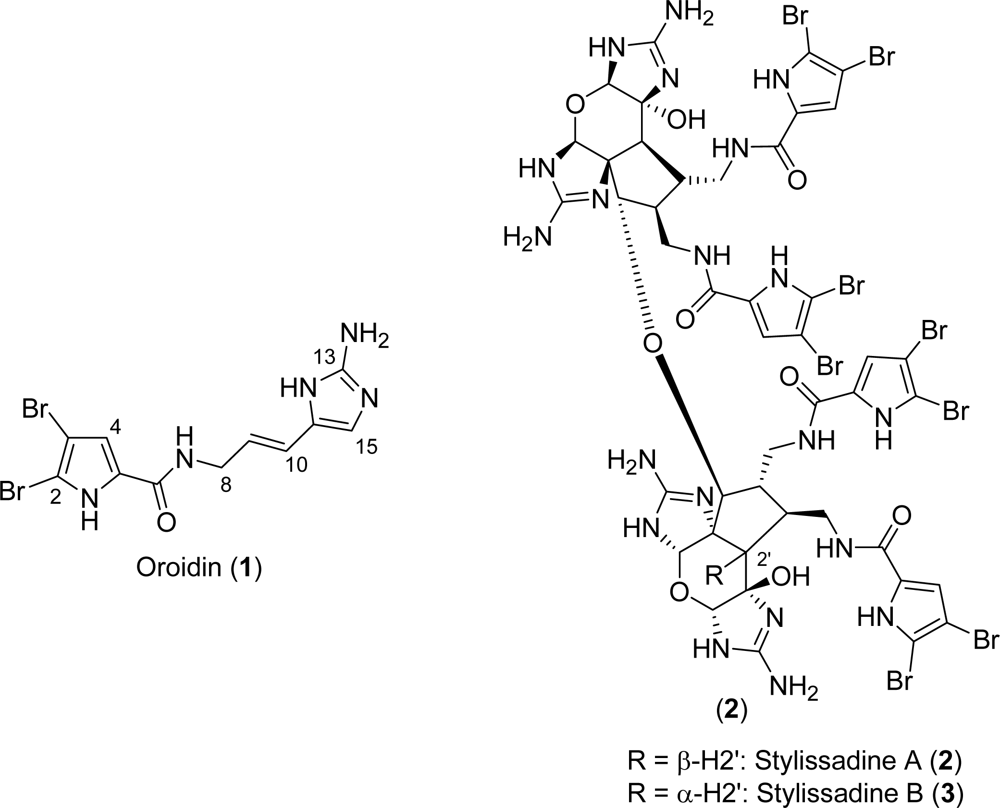
6. Cyclic Tetramers
Stylissadines
7. Others
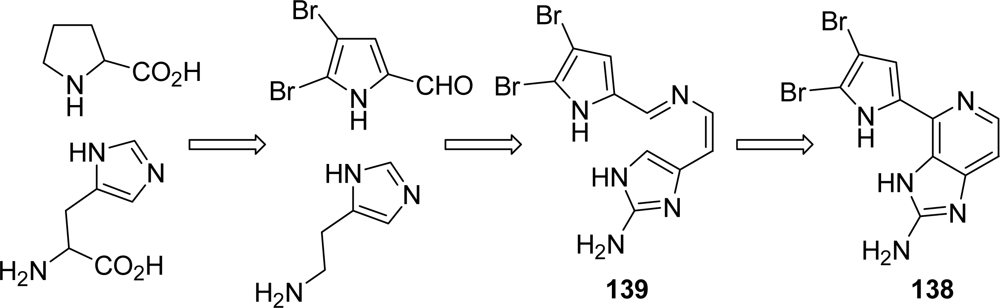
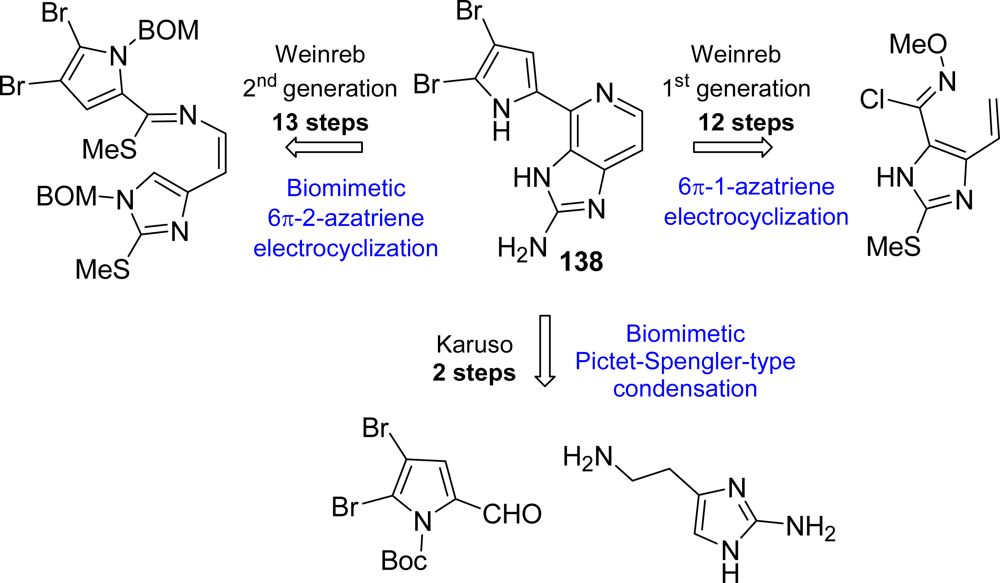
Ageladine A
8. Conclusions
References and Notes
- Wan, Y; Hur, W; Cho, CY; Liu, Y; Adrian, FJ; Lozach, O; Bach, S; Mayer, T; Fabbro, D; Meijer, L; Gray, NS. Synthesis and target identification of hymenialdisine analogs. Chem Biol 2004, 11, 247–259. [Google Scholar]
- Hoffmann, H; Lindel, T. Synthesis of the pyrrole-imidazole alkaloids. Synthesis 2003, 1753–1783. [Google Scholar]
- Jacquot, DEN; Lindel, T. Challenge palau'amine: Current standings. Curr Org Chem 2005, 9, 1551–1565. [Google Scholar]
- Jin, Z. Muscarine, imidazole, oxazole, and thiazole alkaloids. Nat Prod Rep 2005, 22, 196–229. [Google Scholar]
- Jin, Z. Imidazole, oxazole and thiazole alkaloids. Nat Prod Rep 2006, 23, 464–496. [Google Scholar]
- Nakao, Y; Fusetani, N. Enzyme inhibitors from marine invertebrates. J Nat Prod 2007, 70, 689–710. [Google Scholar]
- Weinreb, SM. Some recent advances in the synthesis of polycyclic imidazole-containing marine natural products. Nat Prod Rep 2007, 24, 931–948. [Google Scholar]
- Morris, JC; Phillips, AJ. Marine natural products: Synthetic aspects. Nat Prod Rep 2009, 26, 245–265. [Google Scholar]
- Fattorusso, E; Taglialatela-Scafati, O. Modern Alkaloids, 1st ed; Wyley VCH Verlag GmbH & Co KgaA: Weinheim, Germany, 2008. [Google Scholar]
- Jin, Z. Muscarine, imidazole, oxazole and thiazole alkaloids. Nat Prod Rep 2009, 26, 382–445. [Google Scholar]
- Kitagawa, I; Kobayashi, M; Kitanaka, K; Kido, M; Kyogoku, Y. Marine natural products. XII. On the chemical constituents of the Okinawan marine sponge Hymeniacidon aldis. Chem Pharm Bull 1983, 31, 2321–2328. [Google Scholar]
- Braekman, JC; Daloze, D; Stoller, C; Van Soest, RWM. Chemotaxonomy of Agelas (Porifera: Demospongiae). Biochem Syst Ecol 1992, 20, 417–431. [Google Scholar]
- Richelle-Maurer, E; De Kluijver, MJ; Feio, S; Gaudencio, S; Gaspar, H; Gomez, R; Tavares, R; Van De Vyver, G; van Soest, RWM. Localization and ecological significance of oroidin and sceptrin in the Caribbean sponge Agelas conifera. Biochem Syst Ecol 2003, 31, 1073–1091. [Google Scholar]
- Andrade, P; Willoughby, R; Pomponi, SA; Kerr, RG. Biosynthetic studies of the alkaloid, stevensine, in a cell culture of the marine sponge Teichaxinella morchella. Tetrahedron Lett 1999, 40, 4775–4778. [Google Scholar]
- Assmann, M; Lichte, E; Van Soest, RWM; Köck, M. New bromopyrrole alkaloid from the marine sponge Agelas wiedenmeyeri. Org Lett 1999, 1, 455–457. [Google Scholar]
- Vergne, C; Boury-Esnault, N; Perez, T; Martin, M-T; Adeline, M-T; Dau, ETH; Al Mourabit, A. Verpacamides A-D, a sequence of C11N5 diketopiperazines relating cyclo(Pro-Pro) to cyclo(Pro-Arg), from the marine sponge Axinella vaceleti: Possible biogenetic precursors of pyrrole-2-aminoimidazole alkaloids. Org Lett 2006, 8, 2421–2424. [Google Scholar]
- Travert, N; Al Mourabit, A. A likely biogenetic gateway linking 2-aminoimidazolinone metabolites of sponges to proline: Spontaneous oxidative conversion of the pyrrole-proline-guanidine pseudo-peptide to dispacamide A. J Am Chem Soc 2004, 126, 10252–10253. [Google Scholar]
- Foley, LH; Büchi, G. Biomimetic synthesis of dibromophakellin. J Am Chem Soc 1982, 104, 1776–1777. [Google Scholar]
- Fedoreev, SA; Il'In, SG; Utkina, NK; Maksimov, OB; Reshetnyak, MV; Antipin, MY; Struchkov, YT. The structure of dibromoagelaspongin - A novel bromine-containing guanidine derivative from the marine sponge Agelas sp. Tetrahedron 1989, 45, 3487–3492. [Google Scholar]
- D'Ambrosio, M; Guerriero, A; Debitus, C; Ribes, O; Pusset, J; Leroy, S; Pietra, F. Agelastatin A, a new skeleton cytotoxic alkaloid of the oroidin family. Isolation from the axinellid sponge Agelas dendromorpha of the Coral sea. J. Chem. Soc., Chem. Commun 1993, 1305–1306. [Google Scholar]
- Tsukamoto, S; Kato, H; Hirota, H; Fusetani, N. Mauritiamine, a new antifouling oroidin dimer from the marine sponge Agelas mauritiana. J Nat Prod 1996, 59, 501–503. [Google Scholar]
- Kinnel, RB; Gehrken, H-P; Swali, R; Skoropowski, G; Scheuer, PJ. Palau'amine and its congeners: A family of bioactive bisguanidines from the marine sponge. Stylotella aurantium J Org Chem 1998, 63, 3281–3286. [Google Scholar]
- Garrido-Hernandez, H; Nakadai, M; Vimolratana, M; Li, Q; Doundoulakis, T; Harran, PG. Spirocycloisomerization of tethered alkylidene glycocyamidines: Synthesis of a base template common to the palau'amine family of alkaloids. Angew. Chem., Int. Ed 2005, 44, 765–769. [Google Scholar]
- Faulkner, DJ; Andersen, RJ. Natural products chemistry of the marine environment. Sea 1974, 5, 679–714. [Google Scholar]
- Sharma, G; Magdoff-Fairchild, B. Natural products of marine sponges. 7. The constitution of weakly basic guanidine compounds, dibromophakellin and monobromophakellin. J Org Chem 1977, 42, 4118–4124. [Google Scholar]
- Baran, PS; O'Malley, DP; Zografos, AL. Natural product synthesis: Sceptrin as a potential biosynthetic precursor to complex pyrrole-imidazole alkaloids: The total synthesis of ageliferin. Angew. Chem., Int. Ed 2004, 43, 2674–2677. [Google Scholar]
- Wang, S; Dilley, AS; Poullennec, KG; Romo, D. Planned and unplanned halogenations in route to selected oroidin alkaloids. Tetrahedron 2006, 62, 7155–7161. [Google Scholar]
- Grube, A; Immel, S; Baran, PS; Köck, M. Massadine chloride: A biosynthetic precursor of massadine and stylissadine. Angew. Chem., Int. Ed 2007, 46, 6721–6724. [Google Scholar]
- Al Mourabit, A; Potier, P. Sponge's molecular diversity through the ambivalent reactivity of 2-aminoimidazole: A universal chemical pathway to the oroidin-based pyrrole-imidazole alkaloids and their palau'amine congeners. Eur J Org Chem 2001, 237–243. [Google Scholar]
- Köck, M; Grube, A; Seiple, IB; Baran, PS. The pursuit of palau'amine. Angew. Chem., Int. Ed 2007, 46, 6586–6594. [Google Scholar]
- Van Pee, K-H. Microbial biosynthesis of halometabolites. Arch Microbiol 2001, 175, 250–258. [Google Scholar]
- Butler, A; Carter-Franklin, JN. The role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products. Nat Prod Rep 2004, 21, 180–188. [Google Scholar]
- Dong, C; Flecks, S; Unversucht, S; Haupt, C; van Pee, K-H; Naismith, JH. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. [Google Scholar]
- Nishimura, S; Matsunaga, S; Shibazaki, M; Suzuki, K; Furihata, K; Van Soest, RWM; Fusetani, N. Massadine, a novel geranylgeranyltransferase type I inhibitor from the marine sponge Stylissa aff. massa. Org. Lett 2003, 5, 2255–2257. [Google Scholar]
- Jacquot, DEN; Mayer, P; Lindel, T. Chiroptical analysis of marine sponge alkaloids sharing the pyrrolopyrazinone core. Chem Eur J 2004, 10, 1141–1148. [Google Scholar]
- Fattorusso, E; Taglialatela-Scafati, O. Two novel pyrrole-imidazole alkaloids from the Mediterranean sponge. Agelas oroides Tetrahedron Lett 2000, 41, 9917–9922. [Google Scholar]
- D'Ambrosio, M; Guerriero, A; Ripamonti, M; Debitus, C; Waikedre, J; Pietra, F. The active centers of agelastatin A, a strongly cytotoxic alkaloid of the Coral Sea axinellid sponge Agelas dendromorpha, as determined by comparative bioassays with semisynthetic derivatives. Helv Chim Acta 1996, 79, 727–735. [Google Scholar]
- Chanas, B; Pawlik, JR; Lindel, T; Fenical, W. Chemical defense of the Caribbean sponge Agelas clathrodes (Schmidt). J Exp Mar Biol Ecol 1997, 208, 185–196. [Google Scholar]
- Wilson, DM; Puyana, M; Fenical, W; Pawlik, JR. Chemical defense of the Caribbean reef sponge Axinella corrugata against predatory fishes. J Chem Ecol 1999, 25, 2811–2823. [Google Scholar]
- Lindel, T; Hoffmann, H; Hochgurtel, M; Pawlik, JR. Structure-activity relationship of inhibition of fish feeding by sponge-derived and synthetic pyrrole-imidazole alkaloids. J Chem Ecol 2000, 26, 1477–1496. [Google Scholar]
- Assmann, M; Van Soest, RWM; Köck, M. New antifeedant bromopyrrole alkaloid from the Caribbean sponge Stylissa caribica. J Nat Prod 2001, 64, 1345–1347. [Google Scholar]
- Assmann, M; Lichte, E; Pawlik, JR; Köck, M. Chemical defenses of the Caribbean sponges Agelas wiedenmayeri and Agelas conifera. Mar Ecol Prog Ser 2000, 207, 255–262. [Google Scholar]
- Bickmeyer, U; Drechsler, C; Köck, M; Assmann, M. Brominated pyrrole alkaloids from marine Agelas sponges reduce depolarization-induced cellular calcium elevation. Toxicon 2004, 44, 45–51. [Google Scholar]
- Bickmeyer, U. Bromoageliferin and dibromoageliferin, secondary metabolites from the marine sponge Agelas conifera, inhibit voltage-operated, but not store-operated calcium entry in PC12 cells. Toxicon 2005, 45, 627–632. [Google Scholar]
- De Nanteuil, G; Ahond, A; Guilhem, J; Poupat, C; Tran Huu Dau, E; Potier, P; Pusset, M; Pusset, J; Laboute, P. Marine invertebrates from the New Caledonian lagoon. V. Isolation and identification of metabolites of a new species of sponge Pseudaxinyssa cantharella. Tetrahedron 1985, 41, 6019–6033. [Google Scholar]
- Fedoreev, SA; Utkina, NK; Il'In, SG; Reshetnyak, MV; Maksimov, OB. The structure of dibromoisophakellin from the marine sponge. Acanthella carteri Tetrahedron Lett 1986, 27, 3177–3180. [Google Scholar]
- Forenza, S; Minale, L; Riccio, R; Fattorusso, E. New bromopyrrole derivatives from the sponge Agelas oroides. J Chem Soc D 1971, 1129–1130. [Google Scholar]
- De Nanteuil, G; Ahond, A; Poupat, C; Thoison, O; Potier, P. Synthesis of oroidine. Bull Soc Chim Fr 1986, 813–816. [Google Scholar]
- Daninos-Zeghal, S; Al Mourabit, A; Ahond, A; Poupat, C; Potier, P. Synthesis of marine 2-aminoimidazole metabolites: Hymenidin, oroidin, and keramadine. Tetrahedron 1997, 53, 7605–7614. [Google Scholar]
- Berree, F; Girard-Le Bleis, P; Carboni, B. Synthesis of the marine sponge alkaloid oroidin and its analogues via Suzuki cross-coupling reactions. Tetrahedron Lett 2002, 43, 4935–4938. [Google Scholar]
- Olofson, A; Yakushijin, K; Horne, DA. Synthesis of C11N5 marine alkaloids oroidin, clathrodin, and dispacamides. Preparation and transformation of 2-amino-4,5-dialkoxy-4,5-dihydroimidazoline from 2-aminoimidazoles. J Org Chem 1998, 63, 1248–1253. [Google Scholar]
- Little, TL; Webber, SE. A simple and practical synthesis of 2-aminoimidazoles. J Org Chem 1994, 59, 7299–7305. [Google Scholar]
- Ando, N; Terashima, S. A novel synthesis of the 2-aminoimidazol-4-carbaldehyde derivatives, versatile synthetic intermediates for 2-aminoimidazole alkaloids. Synlett 2006, 2836–2840. [Google Scholar]
- Schroif-Gregoire, C; Travert, N; Zaparucha, A; Al Mourabit, A. Direct access to marine pyrrole-2-aminoimidazoles, oroidin, and derivatives, via new acyl-1,2-dihydropyridine intermediates. Org Lett 2006, 8, 2961–2964. [Google Scholar]
- Yamada, A; Kitamura, H; Yamaguchi, K; Fukuzawa, S; Kamijima, C; Yazawa, K; Kuramoto, M; Wang, G-Y-S; Fujitani, Y; Uemura, D. Development of chemical substances regulating biofilm formation. Bull Chem Soc Jpn 1997, 70, 3061–3069. [Google Scholar]
- Richards, JJ; Huigens, RW, III; Ballard, TE; Basso, A; Cavanagh, J; Melander, C. Inhibition and dispersion of proteobacterial biofilms. Chem Commun 2008, 1698–1700. [Google Scholar]
- Donlan, RM; Costerton, JW. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 2002, 15, 167–193. [Google Scholar]
- Musk, DJ, Jr; Hergenrother, PJ. Chemical countermeasures for the control of bacterial biofilms: Effective compounds and promising targets. Curr Med Chem 2006, 13, 2163–2177. [Google Scholar]
- Huigens, RW, III; Richards, JJ; Parise, G; Ballard, TE; Zeng, W; Deora, R; Melander, C. Inhibition of Pseudomonas aeruginosa biofilm formation with bromoageliferin analogues. J Am Chem Soc 2007, 129, 6966–6967. [Google Scholar]
- Richards, JJ; Ballard, TE; Melander, C. Inhibition and dispersion of Pseudomonas aeruginosa biofilms with reverse amide 2-aminoimidazole oroidin analogs. Org Biomol Chem 2008, 6, 1356–1363. [Google Scholar]
- Ballard, TE; Richards, JJ; Wolfe, AL; Melander, C. Synthesis and antibiofilm activity of a second-generation reverse-amide oroidin library: A structure-activity relationship study. Chem Eur J 2008, 14, 10745–10761. [Google Scholar]
- Richards, JJ; Reed, CS; Melander, C. Effects of N-pyrrole substitution on the anti-biofilm activities of oroidin derivatives against Acinetobacter baumannii. Bioorg Med Chem Lett 2008, 18, 4325–4327. [Google Scholar]
- Richards, JJ; Reyes, S; Stowe, SD; Tucker, AT; Ballard, TE; Mathies, LD; Cavanagh, J; Melander, C. Amide isosteres of oroidin: Assessment of antibiofilm activity and C. elegans toxicity. J Med Chem 2009, 52, 4582–4585. [Google Scholar]
- Richards, JJ; Ballard, TE; Huigens, RW, III; Melander, C. Synthesis and screening of an oroidin library against Pseudomonas aeruginosa biofilms. ChemBioChem 2008, 9, 1267–1279. [Google Scholar]
- Cafieri, F; Fattorusso, E; Mangoni, A; Taglialatela-Scafati, O. Dispacamides, anti-histamine alkaloids from Caribbean Agelas sponges. Tetrahedron Lett 1996, 37, 3587–3590. [Google Scholar]
- Lindel, T; Hoffmann, H. Synthesis of dispacamide from the marine sponge Agelas dispar. Tetrahedron Lett 1997, 38, 8935–8938. [Google Scholar]
- Fresneda, PM; Molina, P; Sanz, MA. A convergent approach to midpacamide and dispacamide pyrrole-imidazole marine alkaloids. Tetrahedron Lett 2001, 42, 851–854. [Google Scholar]
- Meijer, L; Thunnissen, AMWH; White, AW; Garnier, M; Nikolic, M; Tsai, LH; Walter, J; Cleverley, KE; Salinas, PC; Wu, YZ; Biernat, J; Mandelkow, EM; Kim, SH; Pettit, GR. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol 2000, 7, 51–63. [Google Scholar]
- Nguyen, TNT; Tepe, JJ. Preparation of hymenialdisine, analogues and their evaluation as kinase inhibitors. Curr Med Chem 2009, 16, 3122–3143. [Google Scholar]
- Sharma, GM; Buyer, JS; Pomerantz, MW. Characterization of a yellow compound isolated from the marine sponge Phakellia flabellata. J Chem Soc, Chem Commun 1980, 435–436. [Google Scholar]
- Cimino, G; De Rosa, S; De Stefano, S; Mazzarella, L; Puliti, R; Sodano, G. Isolation and X-ray crystal structure of a novel bromo compound from two marine sponges. Tetrahedron Lett 1982, 23, 767–768. [Google Scholar]
- Mattia, CA; Mazzarella, L; Puliti, R. 4-(2-Amino-4-oxo-2-imidazolin-5-ylidene)-2-bromo-4,5,6,7-tetrahydropyrrolo[2,3-c]azepin-8-one methanol solvate: A new bromo compound from the sponge Acanthella Aurantiaca. 1982, 2513–2515. [Google Scholar]
- Williams, DH; Faulkner, DJ. Isomers and tautomers of hymenialdisine and debromohymenialdisine. Nat Prod Lett 1996, 9, 57–64. [Google Scholar]
- Annoura, H; Tatsuoka, T. Total syntheses of hymenialdisine and debromohymenialdisine: Stereospecific construction of the 2-amino-4-oxo-2-imidazolin-5(Z)-disubstituted ylidene ring system. Tetrahedron Lett 1995, 36, 413–416. [Google Scholar]
- Xu, Y-Z; Yakushijin, K; Horne, DA. Synthesis of C11N5 marine sponge alkaloids: (±)-Hymenin, stevensine, hymenialdisine, and debromohymenialdisine. J Org Chem 1997, 62, 456–464. [Google Scholar]
- Sosa, ACB; Yakushijin, K; Horne, DA. A practical synthesis of (Z)-debromohymenialdisine. J Org Chem 2000, 65, 610–611. [Google Scholar]
- Portevin, B; Golsteyn, RM; Pierre, A; De Nanteuil, G. An expeditious multigram preparation of the marine protein kinase inhibitor debromohymenialdisine. Tetrahedron Lett 2003, 44, 9263–9265. [Google Scholar]
- Papeo, G; Posteri, H; Borghi, D; Varasi, M. A new glycocyamidine ring precursor: Syntheses of (Z)-hymenialdisine, (Z)-2-debromohymenialdisine, and (±)-endo-2-debromohymenialdisine. Org Lett 2005, 7, 5641–5644. [Google Scholar]
- Endo, M; Nakagawa, M; Hamamoto, Y; Ishihama, M. Pharmacologically active substances from southern pacific marine invertebrates. Pure Appl Chem 1986, 58, 387–394. [Google Scholar]
- He, Q; Chen, W; Qin, Y. Synthesis of 2-substituted endo-hymenialdisine derivatives. Tetrahedron Lett 2007, 48, 1899–1901. [Google Scholar]
- Chacun-Lefevre, L; Joseph, B; Merour, J-Y. Synthesis and reactivity of azepino[3,4-b]indol-5-yl trifluoromethanesulfonate. Tetrahedron 2000, 56, 4491–4499. [Google Scholar]
- Kaiser, HM; Zenz, I; Lo, WF; Spannenberg, A; Schroeder, K; Jiao, H; Goerdes, D; Beller, M; Tse, MK. Preparation of novel unsymmetrical bisindoles under solvent-free conditions: Synthesis, crystal structures, and mechanistic aspects. J Org Chem 2007, 72, 8847–8858. [Google Scholar]
- Mangu, N; Kaiser, HM; Kar, A; Spannenberg, A; Beller, M; Tse, MK. Synthesis of novel hymenialdisine analogues using solvent-free and silica gel-promoted ring opening of epoxides. Tetrahedron 2008, 64, 7171–7177. [Google Scholar]
- Parmentier, J-G; Portevin, B; Golsteyn, RM; Pierre, A; Hickman, J; Gloanec, P; De Nanteuil, G. Synthesis and CHK1 inhibitory potency of hymenialdisine analogues. Bioorg Med Chem Lett 2009, 19, 841–844. [Google Scholar]
- Pettit, GR; Herald, CL; Leet, JE; Gupta, R; Schaufelberger, DE; Bates, RB; Clewlow, PJ; Doubek, DL; Manfredi, KP; Rützler, K; Schmidt, JM; Tackett, LP; Ward, FB; Bruck, M; Camou, F. Antineoplastic agents. 168. Isolation and structure of axinohydantoin. Can J Chem 1990, 68, 1621–1624. [Google Scholar]
- Groszek, G; Kantoci, D; Pettit, GR. The isolation and structure elucidation of debromoaxinohydantoin. Liebigs Ann 1995, 715–716. [Google Scholar]
- Patil, AD; Freyer, AJ; Killmer, L; Hofmann, G; Johnson, RK. (Z)-Axinohydantoin and debromo-(Z)-axinohydantoin from the sponge Stylotella aurantium. Inhibitors of protein kinase C. Nat Prod Lett 1997, 9, 201–207. [Google Scholar]
- Inaba, K; Sato, H; Tsuda, M; Kobayashi, JI. Spongiacidins A-D, new bromopyrrole alkaloids from Hymeniacidon sponge. J Nat Prod 1998, 61, 693–695. [Google Scholar]
- Sosa, ACB; Yakushijin, K; Horne, DA. Synthesis of axinohydantoins. J Org Chem 2002, 67, 4498–4500. [Google Scholar]
- Tutino, F; Posteri, H; Borghi, D; Quartieri, F; Mongelli, N; Papeo, G. Stereoselective synthesis of (Z)-axino- and (Z)-debromoaxinohydantoin. Tetrahedron 2009, 65, 2372–2376. [Google Scholar]
- Boyd, MR; Pettit, GR; McNulty, J; Herald, DL; Doubek, DL; Chapuis, J-C; Schmidt, JM; Tackett, LP. Antineoplastic agents. 362. Isolation and X-ray crystal structure of dibromophakellstatin from the Indian Ocean sponge. Phakellia mauritiana J Nat Prod 1997, 60, 180–183. [Google Scholar]
- Jacquot, DEN; Zoellinger, M; Lindel, T. Total synthesis of the marine natural product rac-dibromophakellstatin. Angew. Chem., Int. Ed 2005, 44, 2295–2298. [Google Scholar]
- Zoellinger, M; Mayer, P; Lindel, T. Total synthesis of the cytostatic marine natural product dibromophakellstatin via three-component imidazolidinone anellation. J Org Chem 2006, 71, 9431–9439. [Google Scholar]
- Feldman, KS; Skoumbourdis, AP. Extending Pummerer reaction chemistry. Synthesis of (±)-dibromophakellstatin by oxidative cyclization of an imidazole derivative. Org Lett 2005, 7, 929–931. [Google Scholar]
- Feldman, KS; Skoumbourdis, AP; Fodor, MD. Extending Pummerer reaction chemistry. Synthesis studies in the phakellin alkaloid area. J Org Chem 2007, 72, 8076–8086. [Google Scholar]
- Chung, R; Yu, E; Incarvito, CD; Austin, DJ. Hypervalent iodine-mediated vicinal syn diazidation: Application to the total synthesis of (±)-dibromophakellstatin. Org Lett 2004, 6, 3881–3884. [Google Scholar]
- Lu, J; Tan, X; Chen, C. Palladium-catalyzed direct functionalization of imidazolinone: Synthesis of dibromophakellstatin. J Am Chem Soc 2007, 129, 7768–7769. [Google Scholar]
- Zoellinger, M; Mayer, P; Lindel, T. Enantioselective total synthesis of (−)-dibromophakellstatin. Synlett 2007, 2756–2758. [Google Scholar]
- Zoellinger, M; Kelter, G; Fiebig, H-H; Lindel, T. Antitumor activity of the marine natural product dibromophakellstatin in vitro. Bioorg. Med. Chem. Lett 2007, 17, 346–349. [Google Scholar]
- D'Ambrosio, M; Guerriero, A; Chiasera, G; Pietra, F. Conformational preferences and absolute configuration of agelastatin A, a cytotoxic alkaloid of the axinellid sponge Agelas dendromorpha from the Coral Sea, via combined molecular modeling, NMR, and exciton splitting for diamide and hydroxyamide derivatives. Helv Chim Acta 1994, 77, 1895–1902. [Google Scholar]
- Hong, TW; Jimenez, DR; Molinski, TF. Agelastatins C and D, new pentacyclic bromopyrroles from the sponge Cymbastela sp., and potent arthropod toxicity of (−)-agelastatin A. J Nat Prod 1998, 61, 158–161. [Google Scholar]
- Pettit, GR; Ducki, S; Herald, DL; Doubek, DL; Schmidt, JM; Chapuis, J-C. Antineoplastic agents. 470. Absolute configuration of the marine sponge bromopyrrole agelastatin A. Oncol Res 2005, 15, 11–20. [Google Scholar]
- Hale, KJ; Domostoj, MM; El-Tanani, M; Campbell, FC; Mason, CK. Strategies and Tactics in Organic Synthesis; Harmata, M, Ed.; Academic Press: London, UK, 2005. [Google Scholar]
- Mason, CK; Mcfarlane, S; Johnston, PG; Crowe, P; Erwin, PJ; Domostoj, MM; Campbell, FC; Manaviazar, S; Hale, KJ; El-Tanani, M. Agelastatin A: A novel inhibitor of osteopontin-mediated adhesion, invasion, and colony formation. Mol Cancer Ther 2008, 7, 548–558. [Google Scholar]
- Longley, DB; Johnston, PG. Molecular mechanisms of drug resistance. J Pathol 2005, 205, 275–292. [Google Scholar]
- Hale, KJ; Domostoj, MM; Tocher, DA; Irving, E; Scheinmann, F. Enantiospecific formal total synthesis of the tumor and GSK-3β inhibiting alkaloid, (−)-agelastatin A. Org Lett 2003, 5, 2927–2930. [Google Scholar]
- Stien, D; Anderson, GT; Chase, CE; Koh, Y-H; Weinreb, SM. Total synthesis of the antitumor marine sponge alkaloid agelastatin A. J Am Chem Soc 1999, 121, 9574–9579. [Google Scholar]
- Feldman, KS; Saunders, JC. Alkynyliodonium salts in organic synthesis. Application to the total synthesis of (−)-agelastatin A and (−)-agelastatin B. J Am Chem Soc 2002, 124, 9060–9061. [Google Scholar]
- Feldman, KS; Saunders, JC; Wrobleski, ML. Alkynyliodonium salts in organic synthesis. Development of a unified strategy for the syntheses of (−)-agelastatin A and (−)-agelastatin B. J Org Chem 2002, 67, 7096–7109. [Google Scholar]
- Domostoj, MM; Irving, E; Scheinmann, F; Hale, KJ. New total synthesis of the marine antitumor alkaloid (−)-agelastatin A. Org Lett 2004, 6, 2615–2618. [Google Scholar]
- Davis, FA; Deng, J. Asymmetric total synthesis of (−)-agelastatin A using sulfinimine (N-sulfinyl imine) derived methodologies. Org Lett 2005, 7, 621–623. [Google Scholar]
- Nicolaou, KC; Vourloumis, D; Winssinger, N; Baran, PS. The art and science of total synthesis at the dawn of the twenty-first century. Angew. Chem., Int. Ed 2000, 39, 44–122. [Google Scholar]
- Trost, BM; Crawley, ML. Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem Rev 2003, 103, 2921–2943. [Google Scholar]
- Trost, BM; Dong, G. New class of nucleophiles for palladium-catalyzed asymmetric allylic alkylation. Total synthesis of agelastatin A. J Am Chem Soc 2006, 128, 6054–6055. [Google Scholar]
- Trost, BM; Dong, G. A stereodivergent strategy to both product enantiomers from the same enantiomer of a stereoinducing catalyst: Agelastatin A. Chem Eur J 2009, 15, 6910–6919. [Google Scholar]
- Ichikawa, Y; Yamaoka, T; Nakano, K; Kotsuki, H. Synthesis of (−)-agelastatin A by [3.3] sigmatropic rearrangement of allyl cyanate. Org Lett 2007, 9, 2989–2992. [Google Scholar]
- Yoshimitsu, T; Ino, T; Tanaka, T. Total synthesis of (−)-agelastatin A. Org Lett 2008, 10, 5457–5460. [Google Scholar]
- Yoshimitsu, T; Ino, T; Futamura, N; Kamon, T; Tanaka, T. Total synthesis of the β-catenin inhibitor, (−)-agelastatin A: A second-generation approach based on radical aminobromination. Org Lett 2009, 11, 3402–3405. [Google Scholar]
- Wehn, PM; Du Bois, J. A stereoselective synthesis of the bromopyrrole natural product (−)-agelastatin A. Angew. Chem., Int. Ed 2009, 48, 3802–3805. [Google Scholar]
- Dickson, DP; Wardrop, DJ. Total synthesis of (±)-agelastatin A, a potent inhibitor of osteopontin-mediated neoplastic transformations. Org Lett 2009, 11, 1341–1344. [Google Scholar]
- Hama, N; Matsuda, T; Sato, T; Chida, N. Total synthesis of (−)-agelastatin A: The application of a sequential sigmatropic rearrangement. Org Lett 2009, 11, 2687–2690. [Google Scholar]
- Endo, T; Tsuda, M; Okada, T; Mitsuhashi, S; Shima, H; Kikuchi, K; Mikami, Y; Fromont, J; Kobayashi, JI. Nagelamides A-H, new dimeric bromopyrrole alkaloids from marine sponge Agelas species. J Nat Prod 2004, 67, 1262–1267. [Google Scholar]
- Bhandari, MR; Sivappa, R; Lovely, CJ. Total synthesis of the putative structure of nagelamide D. Org Lett 2009, 11, 1535–1538. [Google Scholar]
- Lovely, CJ; Du, H; Sivappa, R; Bhandari, MR; He, Y; Dias, HVR. Preparation and Diels-Alder chemistry of 4-vinylimidazoles. J Org Chem 2007, 72, 3741–3749. [Google Scholar]
- Mee, SPH; Lee, V; Baldwin, JE. Significant enhancement of the Stille reaction with a new combination of reagents-copper(I) iodide with cesium fluoride. Chem Eur J 2005, 11, 3294–3308. [Google Scholar]
- Usami, Y. Recent synthetic studies leading to structural revisions of marine natural products. Mar Drugs 2009, 7, 314–330. [Google Scholar]
- Walker, RP; Faulkner, DJ; Van Engen, D; Clardy, J. Sceptrin, an antimicrobial agent from the sponge Agelas sceptrum. J Am Chem Soc 1981, 103, 6772–6773. [Google Scholar]
- D'Auria, M; Racioppi, R. Photochemical dimerization of esters of urocanic acid. J Photochem Photobiol 1998, 112, 145–148. [Google Scholar]
- Baran, PS; Zografos, AL; O'Malley, DP. Short total synthesis of (±)-sceptrin. J Am Chem Soc 2004, 126, 3726–3727. [Google Scholar]
- Birman, VB; Jiang, X-T. Synthesis of sceptrin alkaloids. Org Lett 2004, 6, 2369–2371. [Google Scholar]
- Baran, PS; Li, K; O'Malley, DP; Mitsos, C. Short, enantioselective total synthesis of sceptrin and ageliferin by programmed oxaquadricyclane fragmentation. Angew. Chem., Int. Ed 2006, 45, 249–252. [Google Scholar]
- O'Malley, DP; Li, K; Maue, M; Zografos, AL; Baran, PS. Total synthesis of dimeric pyrrole-imidazole alkaloids: Sceptrin, ageliferin, nagelamide E, oxysceptrin, nakamuric acid, and the axinellamine carbon skeleton. J Am Chem Soc 2007, 129, 4762–4775. [Google Scholar]
- Nelsen, SF; Calabrese, JC. Nucleophilic cleavage of quadricyclene-2,3-dicarboxylate derivatives by iodide. J Am Chem Soc 1973, 95, 8385–8389. [Google Scholar]
- Kajigaeshi, S; Kakinami, T; Moriwaki, M; Fujisaki, S; Maeno, K; Okamoto, T. Halogenation using quaternary ammonium polyhalides. Part X. α-Chlorination of aromatic acetyl derivatives with benzyltrimethylammonium dichloroiodate. Synthesis 1988, 545–546. [Google Scholar]
- Lancini, GC; Lazzari, E; Arioli, V; Bellani, P. Synthesis and relationship between structure and activity of 2-nitroimidazole derivatives. J Med Chem 1969, 12, 775–780. [Google Scholar]
- Keifer, PA; Schwartz, RE; Koker, MES; Hughes, RG, Jr; Rittschof, D; Rinehart, KL. Bioactive bromopyrrole metabolites from the Caribbean sponge Agelas conifera. J Org Chem 1991, 56, 2965–2975. [Google Scholar]
- Cafieri, F; Carnuccio, R; Fattorusso, E; Taglialatela-Scafati, O; Vallefuoco, T. Anti-histaminic activity of bromopyrrole alkaloids isolated from Caribbean Agelas sponges. Bioorg Med Chem Lett 1997, 7, 2283–2288. [Google Scholar]
- Shen, X; Perry, TL; Dunbar, CD; Kelly-Borges, M; Hamann, MT. Debromosceptrin, an alkaloid from the Caribbean sponge Agelas conifera. J Nat Prod 1998, 61, 1302–1303. [Google Scholar]
- Rosa, R; Silva, W; Escalona De Motta, G; Rodriguez, AD; Morales, JJ; Ortiz, M. Antimuscarinic activity of a family of C11N5 compounds isolated from Agelas sponges. Experientia 1992, 48, 885–887. [Google Scholar]
- Vassas, A; Bourdy, G; Paillard, JJ; Lavayre, J; Pais, M; Quirion, JC; Debitus, C. Naturally occurring somatostatin and vasoactive intestinal peptide inhibitors. Isolation of alkaloids from two marine sponges. Planta Med 1996, 62, 28–30. [Google Scholar]
- Rodriguez, AD; Lear, MJ; La Clair, JJ. Identification of the binding of sceptrin to MreB via a bidirectional affinity protocol. J Am Chem Soc 2008, 130, 7256–7258. [Google Scholar]
- Gitai, Z; Dye, NA; Reisenauer, A; Wachi, M; Shapiro, L. MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 2005, 120, 329–341. [Google Scholar]
- Rinehart, KL. Biologically active marine natural products. Pure Appl Chem 1989, 61, 525–528. [Google Scholar]
- Kobayashi, J; Tsuda, M; Murayama, T; Nakamura, H; Ohizumi, Y; Ishibashi, M; Iwamura, M; Ohta, T; Nozoe, S. Ageliferins, potent actomyosin ATPase activators from the Okinawan marine sponge Agelas sp. Tetrahedron 1990, 46, 5579–5586. [Google Scholar]
- Assmann, M; Köck, M. Bromosceptrin, an alkaloid from the marine sponge Agelas conifera. Z Naturforsch, C J Biosci 2002, 57, 157–160. [Google Scholar]
- Northrop, BH; O'Malley, DP; Zografos, AL; Baran, PS; Houk, KN. Mechanism of the vinylcyclobutane rearrangement of sceptrin to ageliferin and nagelamide E. Angew. Chem., Int. Ed 2006, 45, 4126–4130. [Google Scholar]
- Tan, X; Chen, C. Regiocontrol in MnIII-mediated oxidative heterobicyclizations: Access to the core skeletons of oroidin dimers. Angew. Chem., Int. Ed 2006, 45, 4345–4348. [Google Scholar]
- Sivappa, R; Mukherjee, S; Dias, HVR; Lovely, CJ. Studies toward the total synthesis of the oroidin dimers. Org Biomol Chem 2009, 7, 3215–3218. [Google Scholar]
- Kinnel, RB; Gehrken, HP; Scheuer, PJ. Palau'amine: A cytotoxic and immunosuppressive hexacyclic bisguanidine antibiotic from the sponge Stylotella agminata. J Am Chem Soc 1993, 115, 3376–3377. [Google Scholar]
- Kobayashi, H; Kitamura, K; Nagai, K; Nakao, Y; Fusetani, N; Van Soest, RWM; Matsunaga, S. Carteramine A, an inhibitor of neutrophil chemotaxis, from the marine sponge Stylissa carteri. Tetrahedron Lett 2007, 48, 2127–2129. [Google Scholar]
- Grube, A; Köck, M. Structural assignment of tetrabromostyloguanidine: Does the relative configuration of the palau'amines need revision? Angew Chem, Int Ed 2007, 46, 2320–2324. [Google Scholar]
- Buchanan, MS; Carroll, AR; Quinn, RJ. Revised structure of palau'amine. Tetrahedron Lett 2007, 48, 4573–4574. [Google Scholar]
- O'Malley, DP; Yamaguchi, J; Young, IS; Seiple, IB; Baran, PS. Total synthesis of (±)-axinellamines A and B. Angew. Chem., Int. Ed 2008, 47, 3581–3583. [Google Scholar]
- Su, S; Seiple, IB; Young, IS; Baran, PS. Total syntheses of (±)-massadine and massadine chloride. J Am Chem Soc 2008, 130, 16490–16491. [Google Scholar]
- Lovely, CJ; Du, H; He, Y; Dias, HVR. Oxidative rearrangement of imidazoles with dimethyldioxirane. Org Lett 2004, 6, 735–738. [Google Scholar]
- Dilley, AS; Romo, D. Enantioselective strategy to the spirocyclic core of palau'amine and related bisguanidine marine alkaloids. Org Lett 2001, 3, 1535–1538. [Google Scholar]
- Dransfield, PJ; Dilley, AS; Wang, S; Romo, D. A unified synthetic strategy toward oroidin-derived alkaloids premised on a biosynthetic proposal. Tetrahedron 2006, 62, 5223–5247. [Google Scholar]
- Zancanella, MA; Romo, D. Facile synthesis of the trans-fused azabicyclo[3.3.0]octane core of the palau'amines and the tricyclic core of the axinellamines from a common intermediate. Org Lett 2008, 10, 3685–3688. [Google Scholar]
- Wang, S; Romo, D. Enantioselective synthesis of (+)-monobromophakellin and (+)-phakellin: A concise phakellin annulation strategy applicable to palau'amine. Angew. Chem., Int. Ed 2008, 47, 1284–1286. [Google Scholar]
- Katz, JD; Overman, LE. Studies towards the total synthesis of palau'amine. Formation of 4,5-dihydropyrrole-2-carboxylate intermediates by alkene-enamide ring-closing metathesis. Tetrahedron 2004, 60, 9559–9568. [Google Scholar]
- Lanman, BA; Overman, LE; Paulini, R; White, NS. On the structure of palau'amine: Evidence for the revised relative configuration from chemical synthesis. J Am Chem Soc 2007, 129, 12896–12900. [Google Scholar]
- Li, Q; Hurley, P; Ding, H; Roberts, AG; Akella, R; Harran, PG. Exploring symmetry-based logic for a synthesis of palau'amine. J Org Chem 2009, 74, 5909–5919. [Google Scholar]
- Urban, S; Leone, PDA; Carroll, AR; Fechner, GA; Smith, J; Hooper, JNA; Quinn, RJ. Axinellamines A-D, novel imidazo-azolo-imidazole alkaloids from the Australian marine sponge Axinella sp. J Org Chem 1999, 64, 731–735. [Google Scholar]
- Arndt, H-D; Riedrich, M. Synthesis of marine alkaloids from the oroidin family. Angew. Chem., Int. Ed 2008, 47, 4785–4788. [Google Scholar]
- Starr, JT; Koch, G; Carreira, EM. Enantioselective synthesis of the cyclopentyl core of the axinellamines. J Am Chem Soc 2000, 122, 8793–8794. [Google Scholar]
- Dransfield, PJ; Wang, S; Dilley, A; Romo, D. Highly regioselective Diels-Alder reactions toward oroidin alkaloids: Use of a tosylvinyl moiety as a nitrogen masking group with adjustable electronics. Org Lett 2005, 7, 1679–1682. [Google Scholar]
- Kato, T; Shizuri, Y; Izumida, H; Yokoyama, A; Endo, M. Styloguanidines, new chitinase inhibitors from the marine sponge Stylotella aurantium. Tetrahedron Lett 1995, 36, 2133–2136. [Google Scholar]
- Poullennec, KG; Romo, D. Enantioselective total synthesis of (+)-dibromophakellstatin. J Am Chem Soc 2003, 125, 6344–6345. [Google Scholar]
- Du, H; He, Y; Rasapalli, S; Lovely, CJ. New methods of imidazole functionalization - from imidazole to marine alkaloids. Synlett 2006, 965–992. [Google Scholar]
- Sivappa, R; Hernandez, NM; He, Y; Lovely, CJ. Studies toward the total synthesis of axinellamine and massadine. Org Lett 2007, 9, 3861–3864. [Google Scholar]
- Yamaguchi, J; Seiple, IB; Young, IS; O'Malley, DP; Maue, M; Baran, PS. Synthesis of 1,9-dideoxy-pre-axinellamine. Angew. Chem., Int. Ed 2008, 47, 3578–3580. [Google Scholar]
- Belanger, G; Hong, F-T; Overman, LE; Rogers, BN; Tellew, JE; Trenkle, WC. Stereocontrolled synthesis of triazacyclopenta[cd]pentalenes by intramolecular 1,3-dipolar cycloaddition reactions of azomethine imines. J Org Chem 2002, 67, 7880–7883. [Google Scholar]
- Lanman, BA; Overman, LE. Evaluation of strategies for the synthesis of the guanidine hemiaminal portion of palau'amine. Heterocycles 2006, 70, 557–570. [Google Scholar]
- König, SG; Miller, SM; Leonard, KA; Loewe, RS; Chen, BC; Austin, DJ. A Transient N-O-linked Pauson-Khand strategy for the synthesis of the deschloro carbocyclic core of the palau'amines and styloguanidines. Org Lett 2003, 5, 2203–2206. [Google Scholar]
- Grube, A; Köck, M. Stylissadines A and B: The first tetrameric pyrrole-imidazole alkaloids. Org Lett 2006, 8, 4675–4678. [Google Scholar]
- Buchanan, MS; Carroll, AR; Addepalli, R; Avery, VM; Hooper, JNA; Quinn, RJ. Natural products, stylissadines A and B, specific antagonists of the P2X7 receptor, an important inflammatory target. J Org Chem 2007, 72, 2309–2317. [Google Scholar]
- Fujita, M; Nakao, Y; Matsunaga, S; Seiki, M; Itoh, Y; Yamashita, J; Van Soest, RWM; Fusetani, N. Ageladine A: An antiangiogenic matrix metalloproteinase inhibitor from the marine sponge. Agelas nakamurai J Am Chem Soc 2003, 125, 15700–15701. [Google Scholar]
- Meketa, ML; Weinreb, SM. Total synthesis of ageladine A, an angiogenesis inhibitor from the marine sponge Agelas nakamurai. Org Lett 2006, 8, 1443–1446. [Google Scholar]
- Meketa, ML; Weinreb, SM; Nakao, Y; Fusetani, N. Application of a 6π-1-azatriene electrocyclization strategy to the total synthesis of the marine sponge metabolite ageladine A and biological evaluation of synthetic analogues. J Org Chem 2007, 72, 4892–4899. [Google Scholar]
- Meketa, ML; Weinreb, SM. A new total synthesis of the zinc matrixmetalloproteinase inhibitor ageladine A featuring a biogenetically patterned 6π-2-azatriene electrocyclization. Org Lett 2007, 9, 853–855. [Google Scholar]
- Meketa, ML; Weinreb, SM. A convergent total synthesis of the marine sponge alkaloid ageladine A via a strategic 6π-2-azatriene electrocyclization. Tetrahedron 2007, 63, 9112–9119. [Google Scholar]
- Shengule, SR; Karuso, P. Concise total synthesis of the marine natural product ageladine A. Org Lett 2006, 8, 4083–4084. [Google Scholar]
- Ando, N; Terashima, S. Synthesis and matrix metalloproteinase (MMP)-12 inhibitory activity of ageladine A and its analogs. Bioorg Med Chem Lett 2007, 17, 4495–4499. [Google Scholar]
- Bickmeyer, U; Grube, A; Klings, K-W; Köck, M. Ageladine A, a pyrrole-imidazole alkaloid from marine sponges, is a pH sensitive membrane permeable dye. Biochem Biophys Res Commun 2008, 373, 419–422. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | IC50 (nM) | Enzyme | IC50 (nM) |
|---|---|---|---|
| CDK1/cyclin B | 22 | Erk 1 | 470 |
| CDK2/cyclin A | 70 | Erk 2 | 2000 |
| CDK2/cyclin E | 40 | c-raf | >10,000 |
| CDK3/cyclin E | 100 | MAPKK | 1200 |
| CDK4/cyclin D1 | 600 | GSK-3β | 10 |
| CDK5/p25 | 28 | CK1 | 35 |
| CDK6/cyclin D2 | 700 | CK2 | 7000 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Forte, B.; Malgesini, B.; Piutti, C.; Quartieri, F.; Scolaro, A.; Papeo, G. A Submarine Journey: The Pyrrole-Imidazole Alkaloids. Mar. Drugs 2009, 7, 705-753. https://doi.org/10.3390/md7040705
Forte B, Malgesini B, Piutti C, Quartieri F, Scolaro A, Papeo G. A Submarine Journey: The Pyrrole-Imidazole Alkaloids. Marine Drugs. 2009; 7(4):705-753. https://doi.org/10.3390/md7040705
Chicago/Turabian StyleForte, Barbara, Beatrice Malgesini, Claudia Piutti, Francesca Quartieri, Alessandra Scolaro, and Gianluca Papeo. 2009. "A Submarine Journey: The Pyrrole-Imidazole Alkaloids" Marine Drugs 7, no. 4: 705-753. https://doi.org/10.3390/md7040705