Monoindole Alkaloids from a Marine Sponge Spongosorites sp.

1
College of Pharmacy, Pusan National University, Busan 609-735, Korea
2
College of Traditional Mongolian Medicine, Inner Mongolia University for the Nationalities, Tongliao Inner Mongolia 028000, China
3
College of Pharmacy, Kyung Hee University, Seoul 130-701, Korea
4
Korea Research Institute of Chemical Technology, Daejon 305-343, Korea
*
Author to whom correspondence should be addressed.
Mar. Drugs 2007, 5(2), 31-39; https://doi.org/10.3390/md502031
Submission received: 5 June 2007
/
Accepted: 23 June 2007
/
Published: 25 June 2007
Abstract
:Seven (1–7) monoindole derivatives were isolated from the MeOH extract of a marine sponge Spongosorites sp. by bioactivity-guided fractionation. The planar structures were established on the basis of NMR and MS spectroscopic analyses. Compounds 1–5 are unique indole pyruvic acid derivatives. Compounds 1–2 and 4–6 are isolated for the first time from a natural source although they were previously reported as synthetic intermediates. Compound 3 was defined as a new compound. Co-occurring bisindoles such as hamacanthins and topsentins might be biosynthesized by condensation of two units of these compounds. The compounds were tested for cytotoxicity against a panel of five human solid tumor cell lines, and compound 7 displayed weak activity.
Introduction
To date, dozens of simple monoindole derivatives were reported from marine sources, such as sponges [1–5], ascidians [6], bryozoans [7], bacteria [8], and fungi [9]. Some of these metabolites were reported to exhibit antibacterial [2,10], antifungal [3], and auxin [4] activities.
In our previous study on cytotoxic compounds from the marine sponge Spongosorites sp., we isolated a series of bisindole alkaloids [11,12]. In our continuing search for cytotoxic metabolites from the same sponge, seven monoindole alkaloids were isolated. Compounds 1–2 and 4–6 were isolated for the first time from a natural source although they were previously reported as synthetic intermediates (Figure 1). Compound 3 was defined as a new compound. Herein we describe the structure elucidation and the biological evaluation of these compounds.
Result and discussion
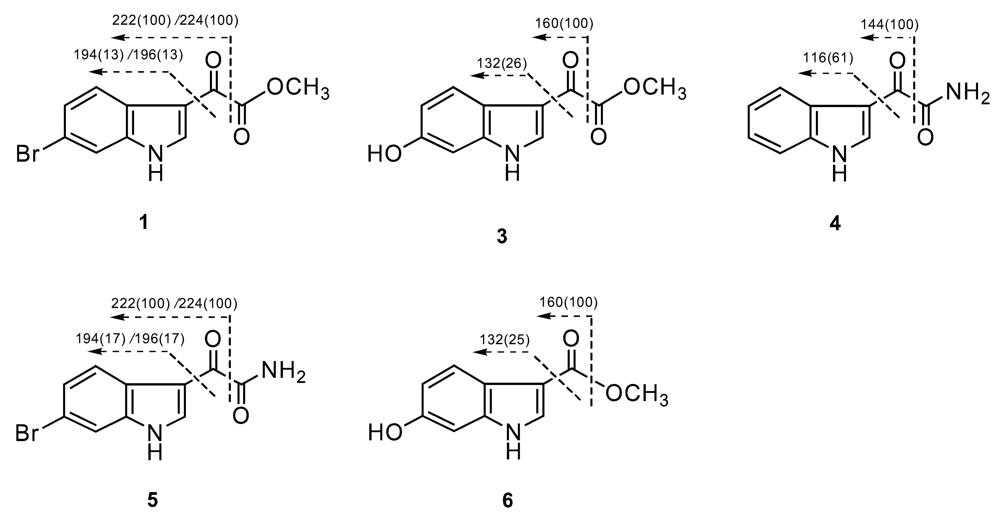
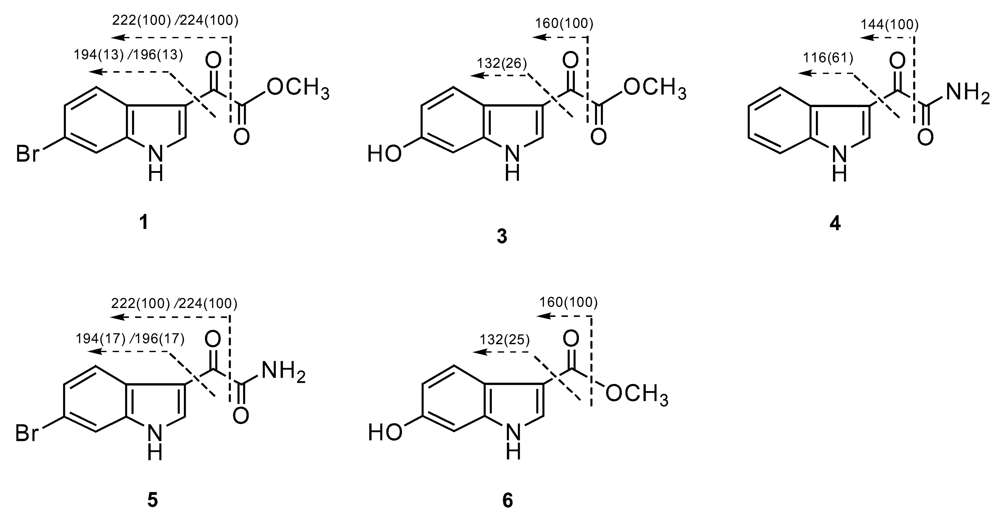
Compound 1 was isolated as a yellow, amorphous powder. The molecular formula was established as C11H8BrNO3 on the basis of the EIMS and NMR data. In the LREIMS of 1, a (M)+ ion cluster was observed at m/z 281/283 in the ratio of 1:1 that is characteristic of a monobrominated compound. The NMR spectrum of 1 were reminiscent of reported indole alkaloids.11,12 Analysis of the 1H, 13C, COSY, HMBC, and HSQC data, along with comparison of chemical shift values with those of known indole alkaloids, allowed us to establish a 6-bromoindol-3-yl residue as a partial structure of 1. The singlet at δH 8.45 (H-2), and a spin system comprised of signals at δH 8.07 (1H, d, J=8.0 Hz, H-4), 7.40 (1H, dd, J=8.0, 2.0 Hz, H-5), and 7.73 (1H, d, J=2.0, H-7) indicated the presence of a 6-bromoindol-3-yl moiety (Table 1). Long-range correlations from H-4 (δH 8.07) to C-3 (δC 112.5) and C-6 (δC 116.2), along with the COSY correlation between H-4 and H-5, and the long-range correlations from H-5 (δH 7.40) to C-3a (δC 124.8) and C-7 (115.5) strongly suggested the presence of a 6-bromoindol-3-yl moiety. The NMR signals at δC 178.2 (C-8), δC 164.0 (C-9), and δH 3.89 (-OCH3, 3H), along with the HMBC correlations of -OCH3/C-9, suggested an oxoacetic acid methyl ester moiety. The EIMS fragments at m/z 194/196, corresponding to C8H5BrN, corroborated the presence of a bromoindole group. These fragments, along with the fragments at m/z 222/224 revealed the presence of a 3-carbonyl-bromoindole group, and established the connectivity between the 6-bromoindole moiety and the oxoacetic acid methyl ester moiety (Figure 2). Therefore, compound 1 was defined as (6-bromo-1H-indol-3-yl) oxoacetic acid methyl ester. Compound 1 was known as an intermediate in the synthesis of some marine natural products, such as didemnimides A and B [13], whereas it has not been reported from a natural source. Pyruvic acid derivatives are unusual natural products, and most of indole pyruvic acid derivatives were isolated from marine sponges [14–16] and ascidians [6].
Compound 2 was isolated as a yellow, amorphous powder. The molecular formula was established as C11H9NO3 on the basis of the FABMS and NMR data. In the LRFABMS of 2, a (M + H)+ ion was observed at m/z 204. The main difference from compound 1 was lack of bromine atom on the indole ring. Therefore, compound 2 was defined as (1H-indol-3-yl) oxoacetic acid methyl ester. Compound 2 was known as an intermediate in the synthesis of natural products, such as didemnimides A and B [13], rebeccamycin, and 11-dechlororebeccamycin [17], whereas it has not been reported as a natural product.
Compound 3 was isolated as a yellow, amorphous powder. The molecular formula was established as C11H9NO4 on the basis of the EIMS and NMR data. In the LREIMS of 1, a (M)+ ion was observed at m/z 219. The main difference from compound 2 was an additional hydroxyl group on the indole ring. A singlet at δH 8.22 (1H, s, H-2), and a spin system comprised of signals at δH 7.82 (1H, d, J=8.0, H-4), 6.74 (1H, dd, J=8.0, 2.0, H-5), and 6.87 (1H, d, J=2.0, H-7), were observed in 1H NMR spectrum. The HMBC correlations from H-2 (δH 8.22), H-5 (δH 6.74), and H-7 (δH 6.87) to C-3a (δC 118.5), from H-2 to C-3 (δC 112.5) and C-7a (δC 138.5), and from H-5 (δH 6.74) to C-6 (δC 154.4), indicated the presence of a 6-hydroxyindol-3-yl moiety. The EIMS fragments at m/z 132 and 160 corroborated the proposed structure (Figure 2). Therefore, compound 3 was defined as (6-hydroxy-1H-indol-3-yl) oxoacetic acid methyl ester. To the best of our knowledge, compound 3 has not been reported previously either from a natural source or as a synthetic product.
Compound 4 was isolated as a white, amorphous powder. The molecular formula was established as C10H8N2O2 on the basis of the EIMS and NMR data. In the LREIMS of 3, a (M)+ ion was observed at m/z 188. The main difference from compound 2 was the presence of an oxoacetamide moiety instead of the oxoacetic acid methyl ester moiety. The 13C signals at δC 182.9 (C-8) and δC 165.9 (C-9), the 1H singlets at δH 8.06 and δH 7.69 (each 1H, -NH2) (Tables 1 and 2), along with the long-range correlation between -NH2 (δH 7.69) and C-8 (δC 182.9), established an oxoacetamide moiety. The EIMS fragments at m/z 116 and 144 revealed the presence of a 3-carbonylindole group, and established the connectivity between the oxoacetamide moiety and the indole moiety (Figure 2). Thus, compound 4 was defined as (1H-indol-3-yl) oxoacetamide, which was also known as an intermediate in the synthesis of some marine natural products, such as arborescidines [18] and dihydrohamacanthins [19], but has not been isolated previously from a natural source.
Compound 5 was isolated as a yellow, amorphous powder. The molecular formula was established as C10H7BrN2O2 on the basis of the EIMS and NMR data. In the EIMS data of 5, a (M)+ ion cluster was observed at m/z 266/268. The main difference from compound 4 was an additional bromine atom on the indole ring. The fragments at m/z 194/196 and 222/224 revealed the presence of 3-carbonyl-bromoindole group (Figure 2). Therefore, compound 5 was defined as (6-bromo-1H-indol-3-yl) oxoacetamide, which was also reported as an intermediate in the synthesis of some natural products, such as arborescidines [18], dihydrohamacanthins [19], but has not been isolated from a natural source.
Compound 6 was isolated as colorless oil. The molecular formula was established as C10H9NO3 on the basis of the EIMS and NMR data. In the LREIMS of 6, a [M]+ ion was observed at m/z 191. Analysis of the 1H, 13C, COSY, HMBC, and HSQC data, allowed us to establish a 6-hydroxyindol residue as a partial structure of 6. The long-range correlation from H-2 (δH 7.86, 1H, s) and -OCH3 (δH 3.76, 3H, s) to C-8 (δC 164.8) established the presence of a formic acid methyl ester and the connectivity between the 6-hydroxyindol moiety and the carboxylic acid methyl ester. The EIMS fragments at m/z 132 and 160 corroborated the proposed structure (Figure 1). Therefore, compound 6 was defined as (6-hydroxy-1H-indol-3-yl) carboxylic acid methyl ester, which was known as an intermediate in the organic synthesis of a 5-HT4 receptor antagonist [20], but has not been reported from a natural source.
Compound 7 was also isolated as a yellow, amorphous powder. According to the MS and NMR data of 7, the main difference from 6 was lack of a hydroxyl group in the indole moiety. The MS and NMR data of 7 matched well with reported data [8], and was identified as (1H-indol-3-yl) carboxylic acid methyl ester which was previously reported from marine-derived bacteria [8] and fungi [21], and red alga [22], with cytotoxicity against K562 human chronic leukemia (MIC s 14.0 μg/mL) [21].
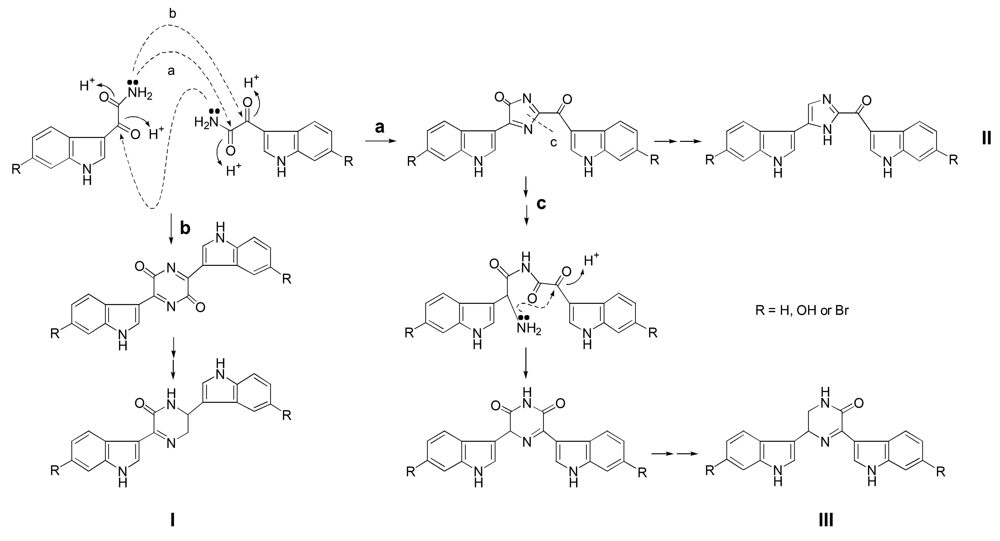
It is expected that (1H-indol-3-yl) oxoacetamide derivatives serve as intermediate for the biogenesis of co-occurring bisindole alkaloids, topsentins and hamacanthins [11,12] (Scheme 1). Schiff base formation between amino and carbonyl groups may (either via a or b) leads to the genesis of hamacanthin A (I) and topsentin (II) skeletons. Cleavage of the C–N bond (c) in the topsentin skeleton, and successive Schiff base formation between newly generated amino group and the intact carbonyl group may lead to a genesis of hamacanthin B skeleton (III).
Compounds 1, 2, and 4–7 were evaluated for cytotoxicity against a panel of five human solid tumor cell lines. Compound 7 showed weak cytotoxicity to human lung cancer, human ovarian cancer, human skin cancer, human CNS cancer, and human colon cancer with ED50 values 24.1, 13.4, 15.2, 26.2, and 4.85 μg/mL, respectively, while other compounds did not show significant activity (ED50>30μg/mL). The ED50 values of doxorubicin against these tumor cell lines in the same experiment were 0.02, 0.14, 0.03, 0.04, and 0.10μg/mL, respectively.
Experimental
General Experimental Procedures
1H and 13C NMR spectra were recorded on a Varian Unity 300 and Varian INOVA 500 instruments. Chemical shifts were reported with reference to the respective residual solvent or deuterated solvent peaks (δH 2.5 and δC 39.5 for DMSO-d6). FABMS data were obtained on a JEOL JMS SX-102A; EIMS data were obtained on a Shimadzu QP5050. HPLC was performed with an YMC ODS-H80 column (250 – 10 mm i.d., 4 μm, 80 Å) and C18-5E Shodex packed column (250 – 10 mm i.d., 5 μm, 100 Å) using a Shodex RI-71 detector.
Animal Material
The sponges were collected by hand using SCUBA (20 m depth) in October 2002, off the coast of Jeju Island, Korea. The collected sample was a loose association of two sponges Spongosorites sp. and Halichondria sp. The two sponges were separated and only Spongosorites sp. was subjected to chemical analysis. The morphology of the sponge was described elsewhere [11]. A voucher specimen (registry No. Spo. 44) is deposited at the Natural History Museum, Hannam University. Korea.
Extraction and Isolation
Evaluation was performed at Korea Research Institute of Chemical Technology. The frozen sponge (0.8 kg) was chopped into small pieces and extracted with MeOH at room temperature. The MeOH extract showed significant toxicity to brine shrimp larvae (LD50 23.7 μg/mL). The MeOH extract was partitioned between CH2Cl2 and water. The CH2Cl2 layer was further partitioned between aqueous MeOH and n-hexane. Aqueous MeOH fraction was subjected to a reversed-phase flash column chromatography (YMC Gel ODS-A, 60 Å, 230 mesh) with a stepped gradient solvent system of 60 to 100% MeOH/H2O to afford 16 fractions. Fraction 2 (0.80 g), one of the bioactive fractions (LD50 33.9 μg/mL), was subjected to a reversed-phase HPLC (YMC ODS-H80 column) eluting with 75% MeOH to afford 13 sub-fractions. Compound 1 (0.95 mg) was obtained by separation of the sub-fraction 2–8 on a reversed-phase HPLC eluting with 58% MeCN. Compound 2 (2.2 mg) was obtained by separation of the sub-fraction 2-2 on a reversed-phase HPLC eluting with 35% MeCN. The sub-fraction 2-1 was subjected to successive reversed-phase HPLC (YMC ODS-H80 column) eluting with 38% MeCN, and further purification with 43% MeCN (C18-5E Shodex packed column) to afford compounds 3 (0.4 mg), 4 (0.78 mg) and 6 (0.62 mg). Compounds 5 (1.2 mg) and 7 (3.3 mg) were obtained by separation of sub-fractions 2–5 and 2–4, respectively, on a reversed-phase HPLC (Shodex C18 M10E column) eluting with 42% MeCN.
(6-Bromo-1H-indol-3-yl) oxoacetic acid methyl ester (1): yellow amorphous powder; 1H NMR data, see Table 1; 13C NMR data, see Table 2; LREIMS m/z 281/283 (M)+.
(1H-Indol-3-yl)oxoacetic acid methyl ester (2): yellow amorphous powder; IR (film) νmax 3206 (br), 1727, 1615 cm−1; UV (MeOH) λmax (log ∈) 362 (3.11), 262 (3.03) nm; 1H NMR data, see Table 1; 13C NMR data, see Table 2; LRFABMS m/z 204 (M + H)+.
(6-Hydroxy-1H-indol-3-yl) oxoacetic acid methyl ester (3): yellow amorphous powder; 1H NMR data, see Table 1; 13C NMR data, see Table 2; LREIMS m/z 219 (M)+.
(1H-Indol-3-yl) oxoacetamide (4): white amorphous powder; 1H NMR data, see Table 1; 13C NMR data, see Table 2; LREIMS m/z 188 (M)+.
(6-Bromo-1H-indol-3-yl) oxoacetamide (5): yellow amorphous powder; IR (film) νmax 3386, 3211, 1663, 1591, 1572, 1407 cm−1; UV (MeOH) λmax (log ∈) 320 (2.61), 275 (2.75), 258 (2.73), 212 (3.21) nm; 1H NMR data, see Table 1; 13C NMR data, see Table 2; LREIMS m/z 266/268 (M)+.
(6-Hydroxy-1H-indol-3-yl) carboxylic acid methyl ester (6): colorless oil; 1H NMR data, see Table 1; 13C NMR data, see Table 2; LREIMS m/z 191 (M)+.
(1H-Indol-3-yl) carboxylic acid methyl ester (7): yellow amorphous powder; IR (film) νmax 3255 (br), 1693, 1620, 1591, 1531, 1444, 1197 cm−1; UV (MeOH) λmax (log ∈) 349 (2.62), 240 (2.75); LREIMS m/z 175 (M)+.
Evaluation of Cytotoxicity
Acknowledgments
This study was supported by a grant from the Korea Research Foundation (2003-041-E 00331). The authors thank Prof. C. J. Sim, Hannam University, for the taxonomical work on the sponge.
- Samples Availability: Not available.
References
- Kobayashi, J; Murayama, T; Ishibashi, M; Kosuge, S; Takamatsu, M; Ohizumi, Y; Kobayashi, H; Ohta, T; Nozoe, S; Sasaki, T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Kyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar]
- Segraves, NL; Crews, P. Investigation of brominated tryptophan alkaloids from two Thorectidae sponges: Thorectandra and Smenospongia. J Nat Prod 2005, 68, 1484–1488. [Google Scholar]
- Li, H; Matsunaga, S; Fusetani, N. Bioactive marine meabolites. Part 52. Simple antifungal metabolites from a marine sponge, Halichondria sp. Comp Biochem Phys B: Biochem Mol Bio 1994, 2, 261–264. [Google Scholar]
- Rasmussen, T; Christophersen, C; Nielsen, PH; Rajagopal, R. Auxin activity of brominated indoles from the marine sponge Pseudosuberites hyalinus. J Mar Biotechnol 1995, 2, 167–169. [Google Scholar]
- Kobayashi, J; Cheng, J; Yamamura, S; Sasaki, T; Ohizumi, Y. Penaresin, a new sarcoplasmic reticulum Ca-inducer from the Okinawan marine sponge Penares sp Heterocycles 1990, 31, 2205–2208.
- Lindquist, N; Fenical, W. Polyandrocarpamides A–D, novel metabolites from the marine ascidian Polyandrocarpa sp. Tetrahedron Lett 1990, 31, 2521–2524. [Google Scholar]
- Peters, L; König, GM; Terlau, H; Wright, AD. Four new bromotryptamine derivatives from the marine bryozoan Flustra foliacea. J Nat Prod 2002, 65, 1633–1637. [Google Scholar]
- Zheng, L; Yan, X; Xu, J; Chen, H; Lin, W. Hymeniacidon perleve associated bioactive bacterium Psedomonas sp. NJ6-3-1. Appl Biochem Micro 2005, 41, 29–33. [Google Scholar]
- Li, Y; Li, X; Kim, D; Choi, H; Son, B. Indolyl alkaloid derivatives, Nb-acetyltrptamine and oxaline from a marine-derived fungus. Arch Pharm Res 2003, 26, 21–23. [Google Scholar]
- Van Lear, GE; Morton, GO; Fulmor, W. New antibacterial bromoindole metabolites from the marine sponge Polyfibrospongia maynardii. Tetrahedron Lett 1973, 4, 299–300. [Google Scholar]
- Bao, B; Sun, Q; Yao, X; Hong, J; Lee, CO; Sim, CJ; Im, KS; Jung, JH. Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J Nat Prod 2005, 68, 711–715. [Google Scholar]
- Bao, B; Sun, Q; Yao, X; Hong, J; Lee, CO; Cho, HY; Im, KS; Jung, JH. Bisindole alkaloids of the topsentin and hamacanthin classes from a marine sponge Spongosorites sp. J Nat Prod 2007, 70, 2–8. [Google Scholar]
- Hughes, TV; Cava, MP. Total synthesis of didemnimide A and B. Tetrahedron Lett 1998, 39, 9629–9630. [Google Scholar]
- Jimènez, C; Quiñoà, E; Adamczeski, M; Hunter, LM; Crews, P. Novel sponge-derived amino acids. 12. Tryptophan-derived pigments and accompanying sesterterpenes from Fascaplysinopis reticulata. J Org Chem 1991, 56, 3403–3410. [Google Scholar]
- Dumdei, E; Andersen, RJ. Igzamide, a metabolite of the marine sponge Plocamissma Igzo. J Nat Prod 1993, 56, 792–794. [Google Scholar]
- Bokesch, HR; Pannell, LK; McKee, TC; Boyd, MR. Coscinamides A, B and C, three new bis indole alkaloids from the marine sponge Coscinoderma sp. Tetrahedron Lett 2000, 41, 6305–6308. [Google Scholar]
- Faul, MM; Winneroski, LL; Krumrich, CA. Synthesis of rebeccamycin and 11-dechlororebeccamycin. J Org Chem 1999, 64, 2465–2470. [Google Scholar]
- Santos, LS; Pilli, RA; Rawal, VH. Enantioselective total syntheses of (+)-arborescidine A, (–)-arborescidine B, and (–)-arborescidine C. J Org Chem 2004, 69, 1283–1289. [Google Scholar]
- Miyake, FY; Yakushijin, K; Horne, DA. Synthesis of marine sponge bisindole alkaloids dihydrohamacanthins. Org Lett 2002, 4, 941–943. [Google Scholar]
- Fedouloff, M; Hossner, F; Voyle, M; Ranson, J; Powles, J; Riley, G; Sanger, G. Synthesis and pharmacological activity of metabolites of the 5-HT4 receptor antagonist SB-207266. Bioorg Med Chem 2001, 9, 2119–2128. [Google Scholar]
- Hu, S; Tan, R; Hong, K; Yu, Z; Zhu, H. Methyl indole-3-carboxylate. Acta Cryst 2005, E61, 1654–1656. [Google Scholar]
- Bano, S; Ahmad, VU; Perveen, S; Bano, N; Shafiuddin; Shameel, M. Marine natural products; II. Chemical constituents of red alga Botryocladia Leptopoda. Planta Med 1987, 53, 117–118. [Google Scholar]
Figure 1.
Seven (1–7) monoindole derivatives were isolated from the MeOH extract of a marine sponge Spongosorites sp.
Figure 1.
Seven (1–7) monoindole derivatives were isolated from the MeOH extract of a marine sponge Spongosorites sp.
Figure 2.
Key fragmentations of [M]+ ions of 1 and 3–6 in LREIMS (relative intensity in parentheses).
Figure 2.
Key fragmentations of [M]+ ions of 1 and 3–6 in LREIMS (relative intensity in parentheses).
Scheme 1.
Hypothetical biogenesis of topsentins and hamacanthins.

{kind=link}
{kind=link}
{kind=link}
| position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 12.19 | 11.52 | ||||
| (br s) | (br s) | |||||
| 2 | 8.45 | 8.44 | 8.22 | 8.69 | 8.68 | 7.86 |
| (d, J=2.0 Hz) | (s) | (s) | (s) | (s) | (s) | |
| 4 | 8.07 | 8.16 | 7.82 | 8.22 | 8.12 | 7.74 |
| (d, J=8.0 Hz) | (d, J=7.0 Hz) | (d, J=8.0 Hz) | (d, J=6.0 Hz) | (d, J=8.5 Hz) | (d, J=8.5 Hz) | |
| 5 | 7.40 | 7.27 | 6.74 | 7.25 | 7.36 | 6.68 |
| (dd, J=8.0, 2.0 Hz) | (t, J=7.0 Hz) | (dd, J=8.0, 2.0 Hz) | (t, J=6.0 Hz) | (dd, J=8.5, 2.0 Hz) | (dd, J=8.5, 2.0 Hz) | |
| 6 | 7.30 | 7.25 | ||||
| (t, J=7.0 Hz) | (t, J=6.0 Hz) | |||||
| 7 | 7.73 | 7.55 | 6.87 | 7.52 | 7.70 | 6.81 |
| (d, J=2.0 Hz) | (d, J=7.0 Hz) | (d, J=2.0 Hz) | (d, J=6.0 Hz) | (d, J=2.0 Hz) | (d, J=2.0 Hz) | |
| -OCH3 | 3.89 | 3.90 (s) | 3.87 | 3.76 | ||
| (s) | (s) | (s) | ||||
| -NH2 | 8.06 | 8.05 | ||||
| (br s) | (br s) | |||||
| 7.69 | 7.67 | |||||
| (br s) | (br s) | |||||
| -OH | 9.17 | |||||
| (br s) |
| position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 2 | 139.5 | 136.8 | 134.5 | 138.1 | 140.2 | 130.6 |
| 3 | 112.5 | 112.7 | 112.5 | 112.0 | 112.0 | 106.3 |
| 3a | 124.8 | 125.5 | 118.5 | 126.1 | 125.6 | 118.7 |
| 4 | 122.5 | 121.1 | 121.4 | 121.2 | 122.8 | 120.8 |
| 5 | 125.3 | 122.8 | 112.2 | 122.4 | 125.0 | 111.6 |
| 6 | 116.2 | 123.8 | 154.4 | 123.3 | 115.6 | 153.7 |
| 7 | 115.5 | 112.4 | 97.7 | 112.4 | 115.6 | 97.2 |
| 7a | 138.6 | 138.4 | 138.5 | 136.2 | 140.0 | 137.4 |
| 8 | 178.2 | 178.6 | a | 182.9 | 180.0 | 164.8 |
| 9 | 164.0 | 164.9 | 164.4 | 165.9 | 165.9 | |
| -OCH3 | 52.4 | 52.5 | 51.9 | 50.4 |
aThe carbonyl carbon signal was not detected due to low concentration of the NMR sample.
Share and Cite
MDPI and ACS Style
Bao, B.; Zhang, P.; Lee, Y.; Hong, J.; Lee, C.-O.; Jung, J.H. Monoindole Alkaloids from a Marine Sponge Spongosorites sp.. Mar. Drugs 2007, 5, 31-39. https://doi.org/10.3390/md502031
AMA Style
Bao B, Zhang P, Lee Y, Hong J, Lee C-O, Jung JH. Monoindole Alkaloids from a Marine Sponge Spongosorites sp.. Marine Drugs. 2007; 5(2):31-39. https://doi.org/10.3390/md502031
Chicago/Turabian StyleBao, Baoquan, Ping Zhang, Yoonmi Lee, Jongki Hong, Chong-O. Lee, and Jee H. Jung. 2007. "Monoindole Alkaloids from a Marine Sponge Spongosorites sp." Marine Drugs 5, no. 2: 31-39. https://doi.org/10.3390/md502031