
New Perspectives in the Chemistry of Marine Pyridoacridine Alkaloids †


Department of Pharmacy—Center for Drug Research, Ludwig-Maximilians University, Butenandtstr. 5-13, 81377 Munich, Germany
*
Author to whom correspondence should be addressed.
†
Dedicated to Prof. Dr. Gunther Seitz, Marburg, on the occasion of his 80th birthday
Mar. Drugs 2016, 14(2), 26; https://doi.org/10.3390/md14020026
Submission received: 28 November 2015
/
Revised: 9 January 2016
/
Accepted: 15 January 2016
/
Published: 26 January 2016
(This article belongs to the Special Issue Synthesis of Antitumor Marine Alkaloids and Related Analogues)
Abstract
:Secondary metabolites from marine organisms are a rich source of novel leads for drug development. Among these natural products, polycyclic aromatic alkaloids of the pyridoacridine type have attracted the highest attention as lead compounds for the development of novel anti-cancer and anti-infective drugs. Numerous sophisticated total syntheses of pyridoacridine alkaloids have been worked out, and many of them have also been extended to the synthesis of libraries of analogues of the alkaloids. This review summarizes the progress in the chemistry of pyridoacridine alkaloids that was made in the last one-and-a-half decades.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
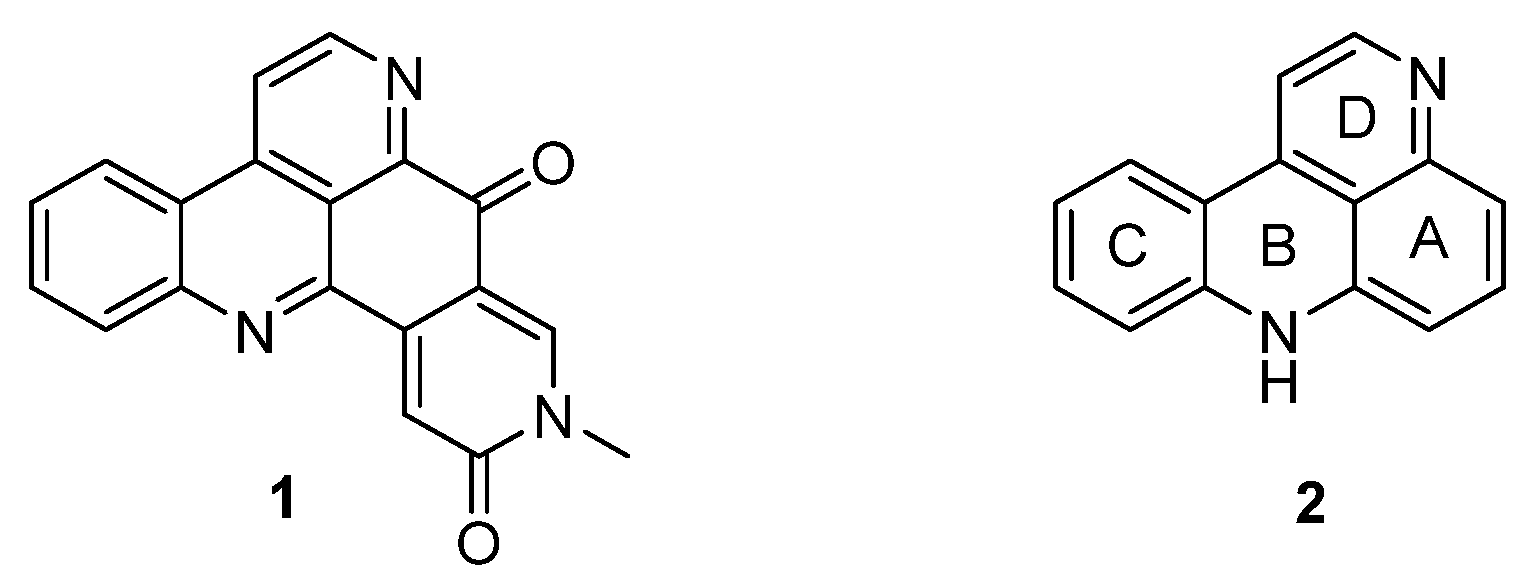
Pyridoacridine alkaloids are a fascinating and emerging class of polycylic alkaloids derived from sessile marine invertebrates such as ascidians, sponges, mollusks, and sea anemones [1]. The era of this alkaloid family started in 1983 with the isolation and identification of amphimedine (1) from a pacific sponge (Amphimedon sp.) by Shoolery [2]. Since then, more than 100 of these polycyclic heteroaromatic natural compounds have been isolated and, according to the biosynthetic considerations of Skyler and Heathcock, a large number of “undiscovered“ alkaloids is assumed [3]. Pyridoacridines are highly colored marine metabolites and are characterized by the 11H-pyrido[4,3,2-mn]acridine skeleton (2) [4] (Figure 1). Besides a few hepta- and octacyclic members, tetra-, penta-, and hexacyclic compounds form the largest subgroup of pyridoacridine alkaloids [3].
Figure 1.
Structure of amphimedine (1) and the 11H-pyrido[4,3,2-mn]acridine scaffold (2).
The pyridoacridines and numerous analogues have been reported to possess manifold biological activities. The outstanding biological effect in this context is beyond doubt their high cytotoxicity. Various mechanisms of action have been described for these anticancer activities. The predominant effects are inhibition of topoisomerase II and the formation of reactive oxygen species (ROS). Furthermore, several pyridoacridines and analogues are associated with antimicrobial, insecticidal and antiparasitic acitivities against Plasmodium, Leishmania, and Trypanosoma species [1]. Consequently, this alkaloid family is of great interest as a source of new lead structures in medicinal chemistry.
The pharmacology of pyridoacridine alkaloids has been reviewed comprehensively very recently [5].
Since only minute amounts of these alkaloids can be obtained from natural sources, total synthesis is an indispensable means for development of drugs from this chemotype. On the other hand, effective strategies for the total syntheses of the alkaloids also open the opportunity for preparing synthetic analogues of the natural products for getting deeper insight into structure-activity relationships and improving pharmacokinetic and pharmacodynamic properties.
2. Ascididemin-Type Pyridoacridines
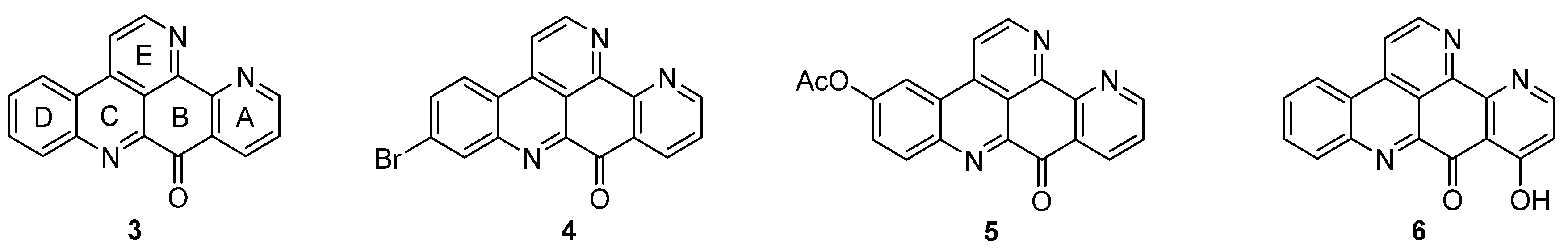
Ascididemin (3) and closely related pyridoacridine alkaloids (bromoleptoclinidinone (4), neocalliactine acetate (5), and 10-hydroxyascididemin (6)) share the same pentacyclic ring system, in which a pyridine ring is annulated to ring A of the 11H-pyrido[4,3,2-mn]acridine scaffold (2) (Figure 2).
Figure 2.
Structures of the ascididemin-type pyridoacridine alkaloids: ascididemin (3), bromoleptoclinidinone (4), neocalliactine acetate (5), 10-hydroxyascididemin (6).
Figure 2.
Structures of the ascididemin-type pyridoacridine alkaloids: ascididemin (3), bromoleptoclinidinone (4), neocalliactine acetate (5), 10-hydroxyascididemin (6).
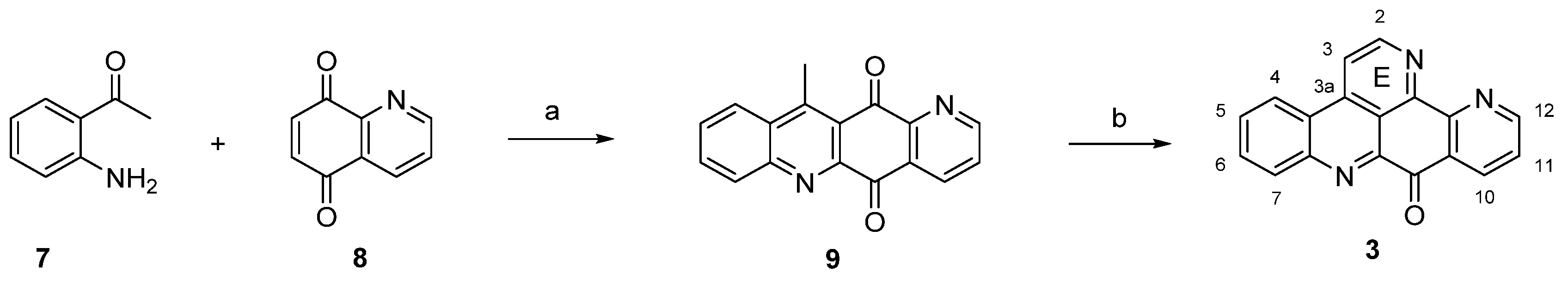
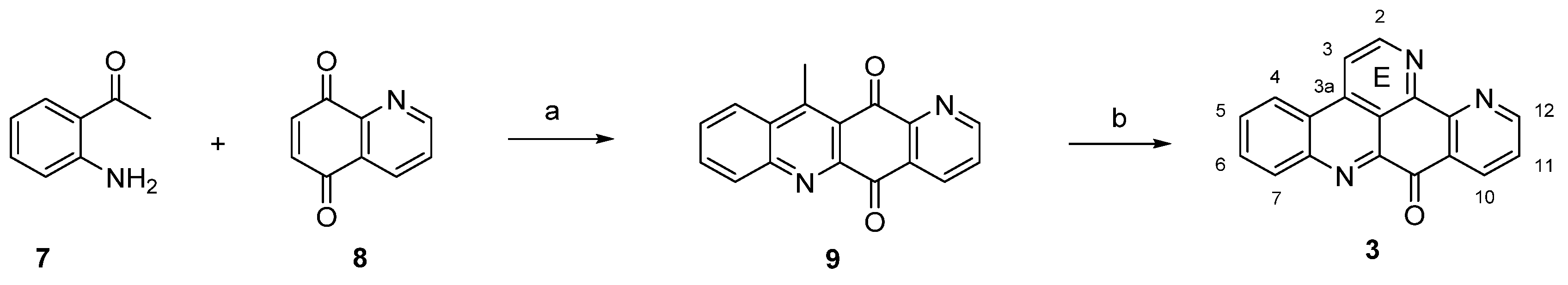
The first and still-leading synthesis of the alkaloid acsididemin (3) was published by Bracher in 1989 [7]. Albeit being presented in numerous reviews before, this synthesis is shown here once again, since its crucial steps have found application in several of the more recent approaches to diverse pyridoacridine alkaloids that will be presented below. The synthesis starts with an oxidative amination of 2-aminoacetophenone (7) and quinoline-5,8-dione (8), followed by an acid-catalyzed cyclization step to give tetracyclic quinone 9. The final annulation of ring E was performed in a one-pot reaction by condensation of the acidic methyl group with N,N-dimethylformamide diethyl acetal, followed by heating with ammonium chloride/glacial acetic acid to give 3 [7] (Scheme 1). This high-yielding four-step approach (43% overall yield) has later been applied to the total synthesis of many related pyridoacridines and analogues thereof by simply using pertinent ring-substituted 2-aminoacetophenones as starting materials [8,9,10,11].
Scheme 1.
First total synthesis of ascididemin (3): (a) CeCl3·7H2O, EtOH, air; then conc. H2SO4/AcOH (73% over two steps); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (59% over two steps).
Scheme 1.
First total synthesis of ascididemin (3): (a) CeCl3·7H2O, EtOH, air; then conc. H2SO4/AcOH (73% over two steps); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (59% over two steps).
The isomer 11 of ascididemin, in which the nitrogen in ring A is shifted from position 13 to 11, was prepared by the Delfourne group [12] in full analogy to Bracher’s method, with a surprisingly regioselective oxidative amination (51% yield) of isoquinoline-5,8-dione (10) and 2-aminoacetophenone (7) as the key step (Scheme 2).
Scheme 2.
Synthesis of ascididemin isomer 11. Reagents and conditions: (a) CeCl3·7H2O, EtOH, air; then conc. H2SO4/AcOH (27% over two steps); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (23% over two steps).
Scheme 2.
Synthesis of ascididemin isomer 11. Reagents and conditions: (a) CeCl3·7H2O, EtOH, air; then conc. H2SO4/AcOH (27% over two steps); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (23% over two steps).
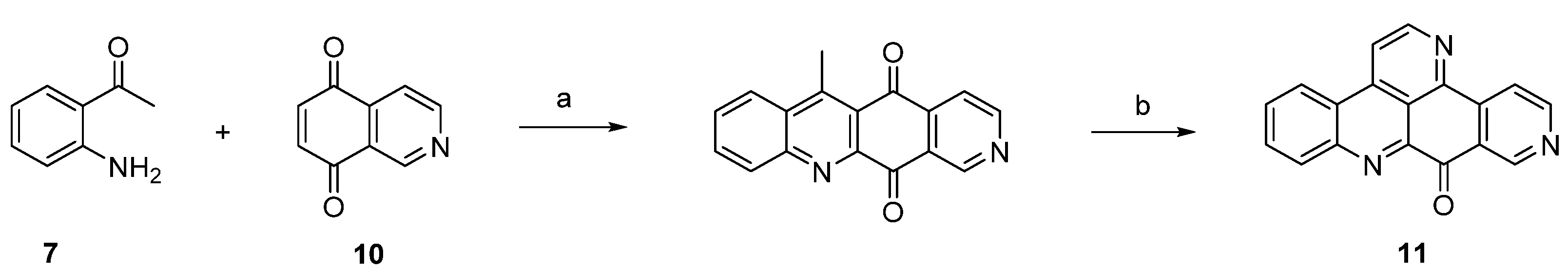
Based on Bracher’s synthetic strategy [7], several modified approaches towards ascididemin-type pyridoacridines have been developed. Most of these syntheses aim at the replacement of the 2-aminoacetophenone building block by more complex 2-substituted anilines, which bear a residue that introduces not only C-3 and C-3a of the final pentacycle (as the acetyl residue does), but contain, in addition, equivalents of the later C-2 an the ring nitrogen of ring E (for numbering of the ring system, see Scheme 1). In earlier approaches, N-trifluoroacetamidokynuramine (bearing a protected 3-aminopropanoyl side-chain) [13] and 2-aminocinnamaldehyde-N,N-dimethylhydrazone [14] were used for this purpose.
In a very recent syntheses of various ascididemin-type alkaloids, consisting of the natural products ascididemin (3), bromoleptoclinidinone (4), neocalliactine acetate (5), 10-hydroxyascididemin (6), as well as two synthetic analogues 5-methoxyascididemin (17) and deazaascididemin (18), Yin et al. introduced a novel C3N unit, the Boc-protected propargylamine group, for this purpose [15].
The required alkyne building blocks 12 were synthesized via Sonogashira reaction of the alkyne N-Boc-propargylamine and a variety of 2-iodoanilines. In analogy to the original synthesis [7], this approach started with an oxidative amination step of alkynylanilines 12 with quinones 13 and 14. The so-obtained arylaminoquinones 15 and 16 were transferred to alkaloids 3–6, and analogues 17 and 18 via a Brønsted acid-promoted tandem annulation in very good yields (Scheme 3) [15].
Scheme 3.
Synthesis of pyridoacridine natural products 3–6 and synthetic ascididemin analogues 5-methoxyascididemin (17) and deazaascididemin (18): (a) CeCl3·7H2O, MeOH, O2 (50%–80%); (b) Fe2(SO4)3, conc. H2SO4/AcOH, O2 (44%–86%).
Scheme 3.
Synthesis of pyridoacridine natural products 3–6 and synthetic ascididemin analogues 5-methoxyascididemin (17) and deazaascididemin (18): (a) CeCl3·7H2O, MeOH, O2 (50%–80%); (b) Fe2(SO4)3, conc. H2SO4/AcOH, O2 (44%–86%).
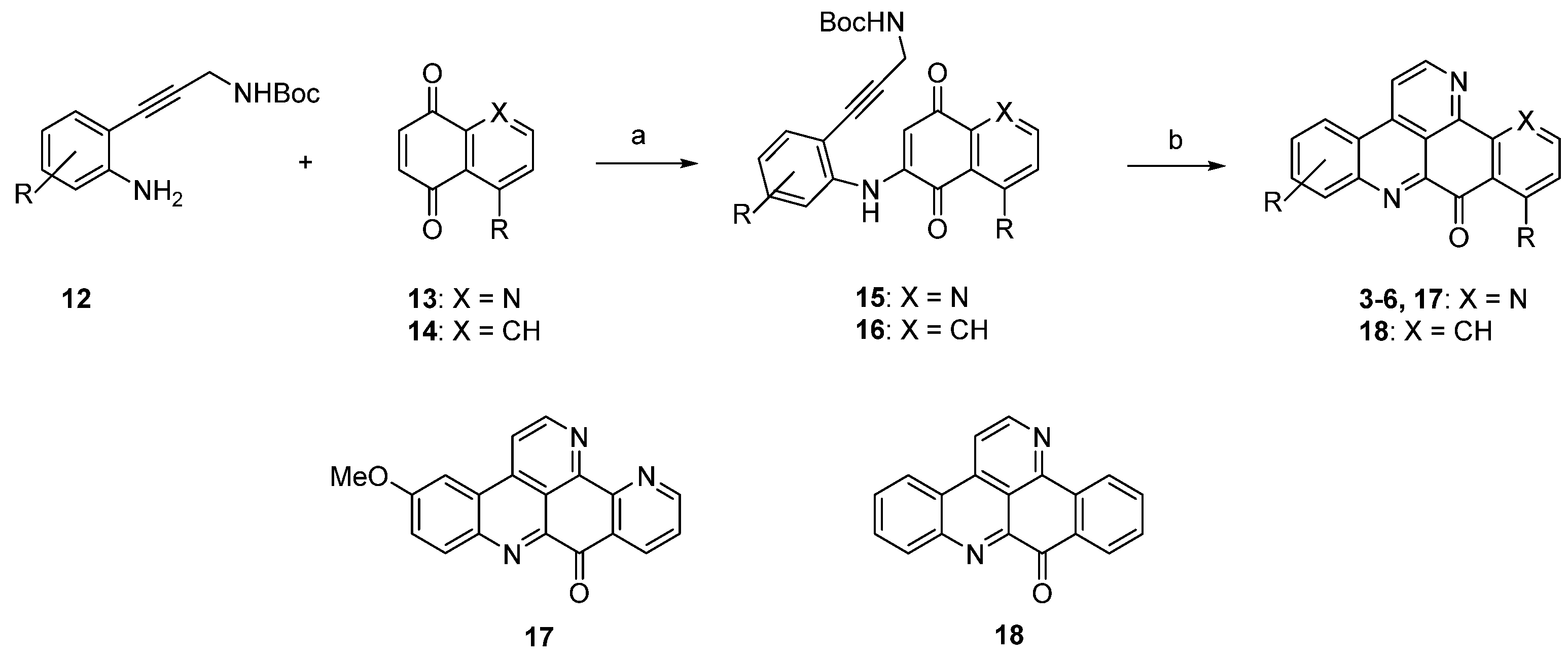
As a number of investigations on the biological activities of ascididemin-type pyridoacridines indicated that cytotoxic activity and selectivity (e.g., against human and protozoal cells) is strongly dependent on the shape of ring A (especially the presence and position of heteroatoms in this ring) [5,16,17,18,19], numerous investigations aimed at working out novel and flexible approaches to ring A analogues of ascididemin (3) were undertaken in the past decade.
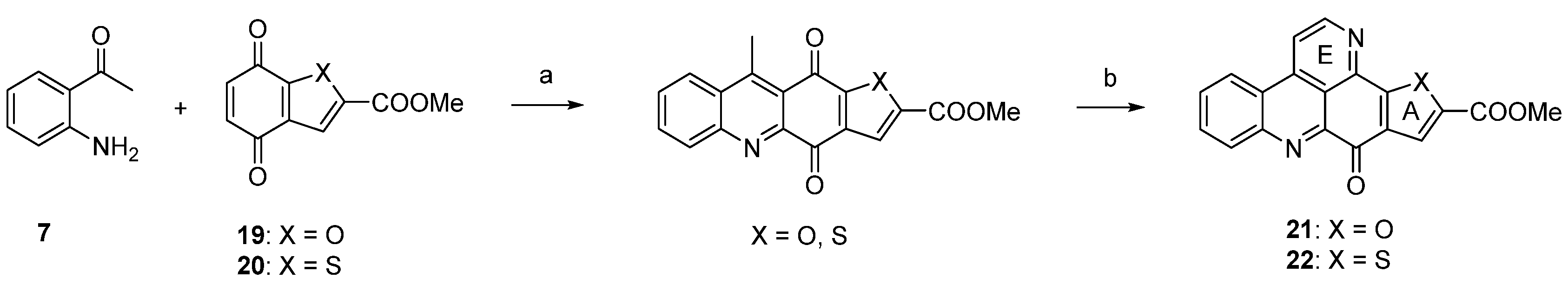
The approach described by Copp in 2010 [18] in general follows Bracher’s strategy [7], but the authors utilized thiophene and furan quinones 19 and 20 instead of quinoline-5,8-dione (8) in order to introduce heteroatom-containing five-membered ring A substitutes. The final annulation of ring E (under concomitant rearomatization) was performed with paraformaldehyde and ammonium chloride to give ascididemin-type pyridoacridines 21 and 22 (Scheme 4) [18].
Scheme 4.
Synthesis of ascididemin-type pyridoacridines 21 and 22 by Copp: (a) CeCl3·7H2O, MeOH, air (22%–92%); then conc. H2SO4/AcOH (81%–94%); (b) NH4Cl, (CH2O)n, AcOH (76%–83%).
Scheme 4.
Synthesis of ascididemin-type pyridoacridines 21 and 22 by Copp: (a) CeCl3·7H2O, MeOH, air (22%–92%); then conc. H2SO4/AcOH (81%–94%); (b) NH4Cl, (CH2O)n, AcOH (76%–83%).
A completely different approach towards ascididemin (3) and its analogues using an anionic ring closure as the key step was published by Kristensen in 2012 [20]. Synthesis of ascididemin (3) started with a Knoevenagel condensation of 2’-fluoroacetophenone (23) and malononitrile. The CH-acidic methyl group in the resulting product 24 was condensed with N,N-dimethylformamide dimethyl acetal to give enamine 25. Upon exposure of 25 to HCl in glacial acetic acid cyclization to the 2-chloropyridine, 26 was accomplished. Negishi cross-coupling with 3-methylpyridin-2-yl zinc bromide under PEPPSI-iPr catalysis gave the bipyridine 27, the central precursor for the anionic cascade. Treating 27 with NaH in DMF under microwave irradiation furnished 9-deoxyascididemin (28) via an anionic ring closure cascade. As 28 proved to be very difficult to handle, the crude intermediate was directly oxidized by bubbling oxygen through the solution. The desired alkaloid 3 was finally isolated in 45% overall yield (Scheme 5). An isomer of the alkaloid, prepared by using 4-methylpyridin-3-yl zinc bromide was obtained in the same manner in comparable overall yield.
Scheme 5.
Synthesis of ascididemin (3) through anionic ring closure: (a) Malononitrile, NH4OAc, toluene/AcOH; (b) dimethylformamide dimethyl acetal, CH2Cl2; (c) HCl gas, AcOH (81% over three steps); (d) 3-methylpyridin-2-ylzinc bromide, PEPPSI-iPr, THF (80%); (e) NaH, DMF; (f) O2 (69% over two steps) [20]; Synthesis of the ring A analogues 18, 30–34; (g) Negishi or Suzuki cross-coupling reactions (44%–88%); (h) NaH, DMPU (19%–29%).
Scheme 5.
Synthesis of ascididemin (3) through anionic ring closure: (a) Malononitrile, NH4OAc, toluene/AcOH; (b) dimethylformamide dimethyl acetal, CH2Cl2; (c) HCl gas, AcOH (81% over three steps); (d) 3-methylpyridin-2-ylzinc bromide, PEPPSI-iPr, THF (80%); (e) NaH, DMF; (f) O2 (69% over two steps) [20]; Synthesis of the ring A analogues 18, 30–34; (g) Negishi or Suzuki cross-coupling reactions (44%–88%); (h) NaH, DMPU (19%–29%).
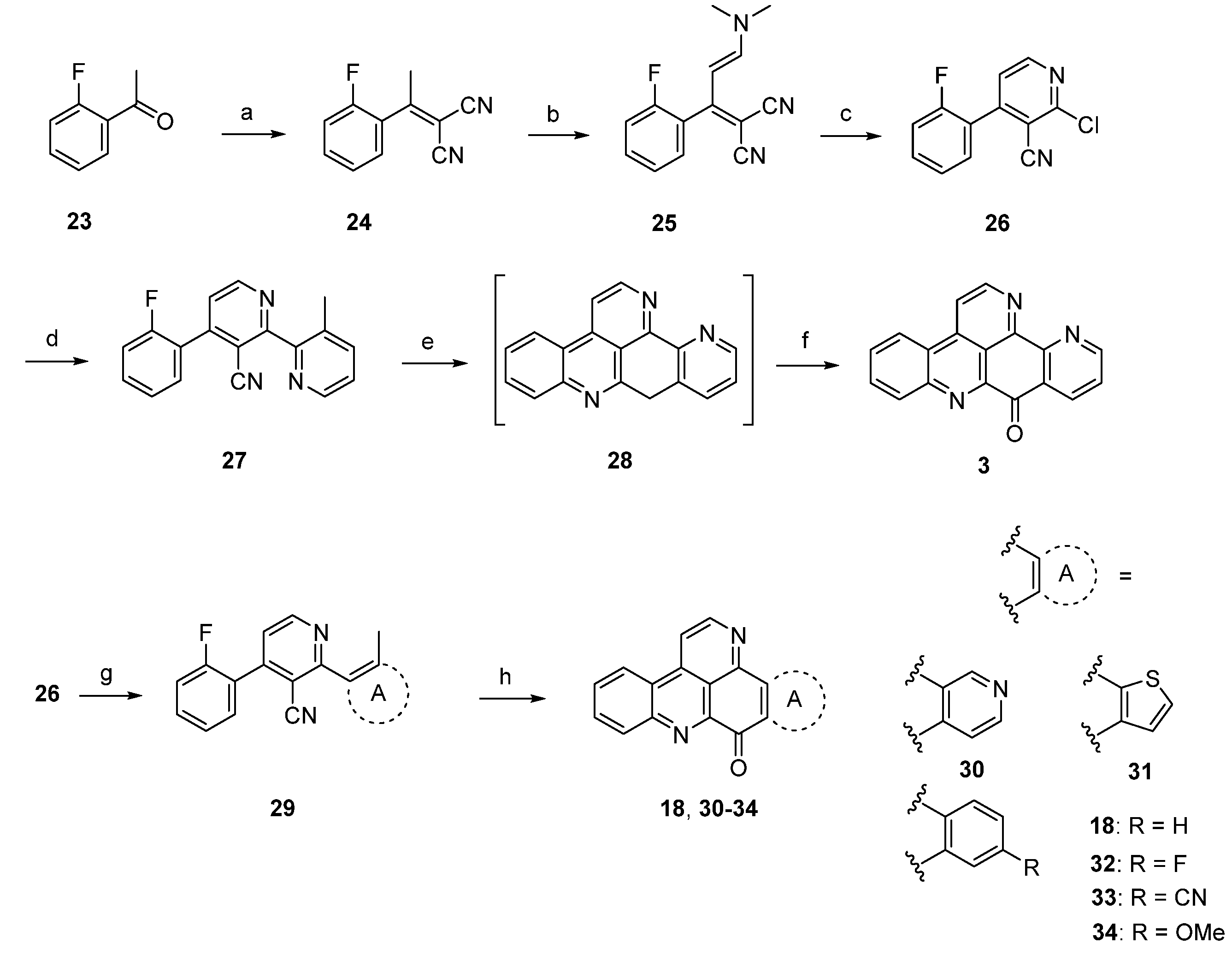
Based on this methodology, several ring A analogues of ascididemin 18, 30–34 were prepared (Scheme 5) [21]. Different ring A equivalents were introduced via Neghishi or Suzuki cross-coupling reactions to give biaryls 29. The following anionic ring closure was achieved here by using NaH in N,N’-dimethylpropylene urea (DMPU). However, the yields of the final cyclization cascade were rather low (12%–29%) here, and several target compounds bearing heteroarenes as ring E equivalents (among them the annulated thiazole kuanoniamine A) could not be obtained at all.
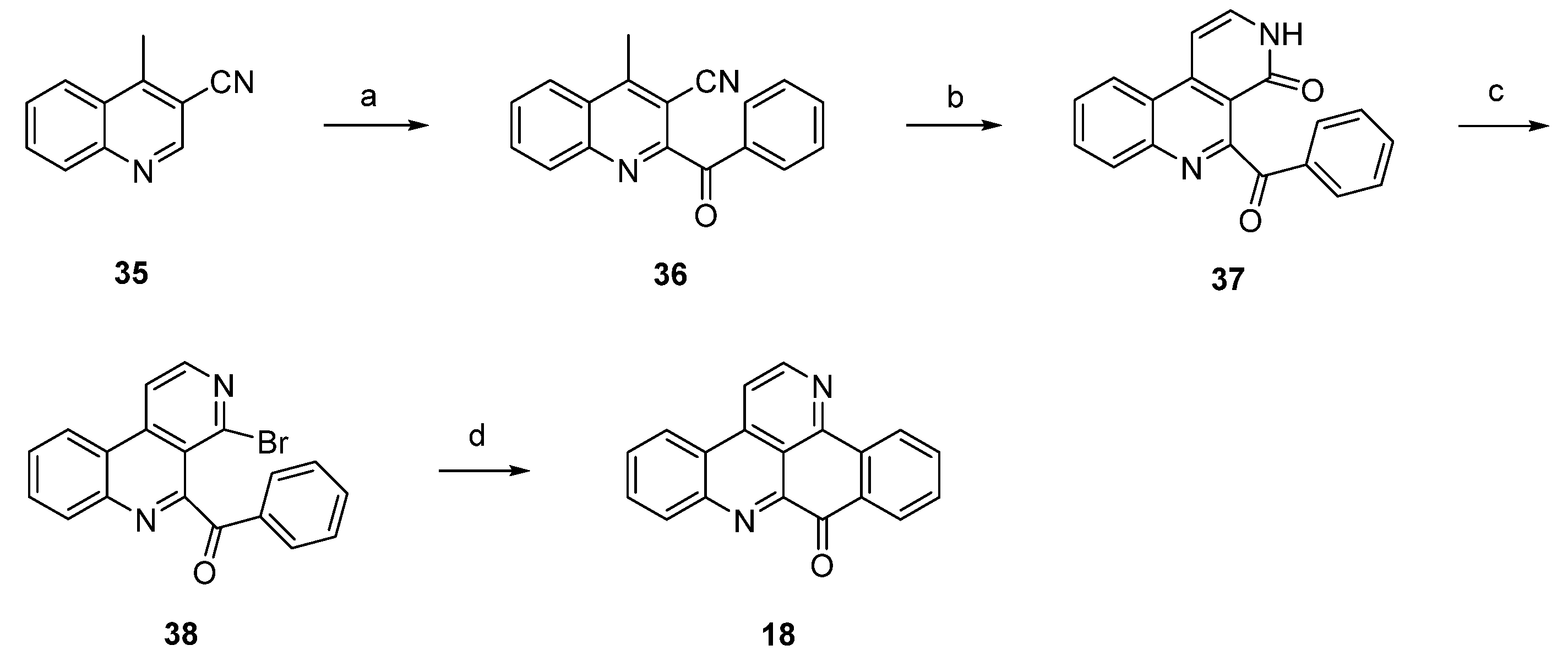
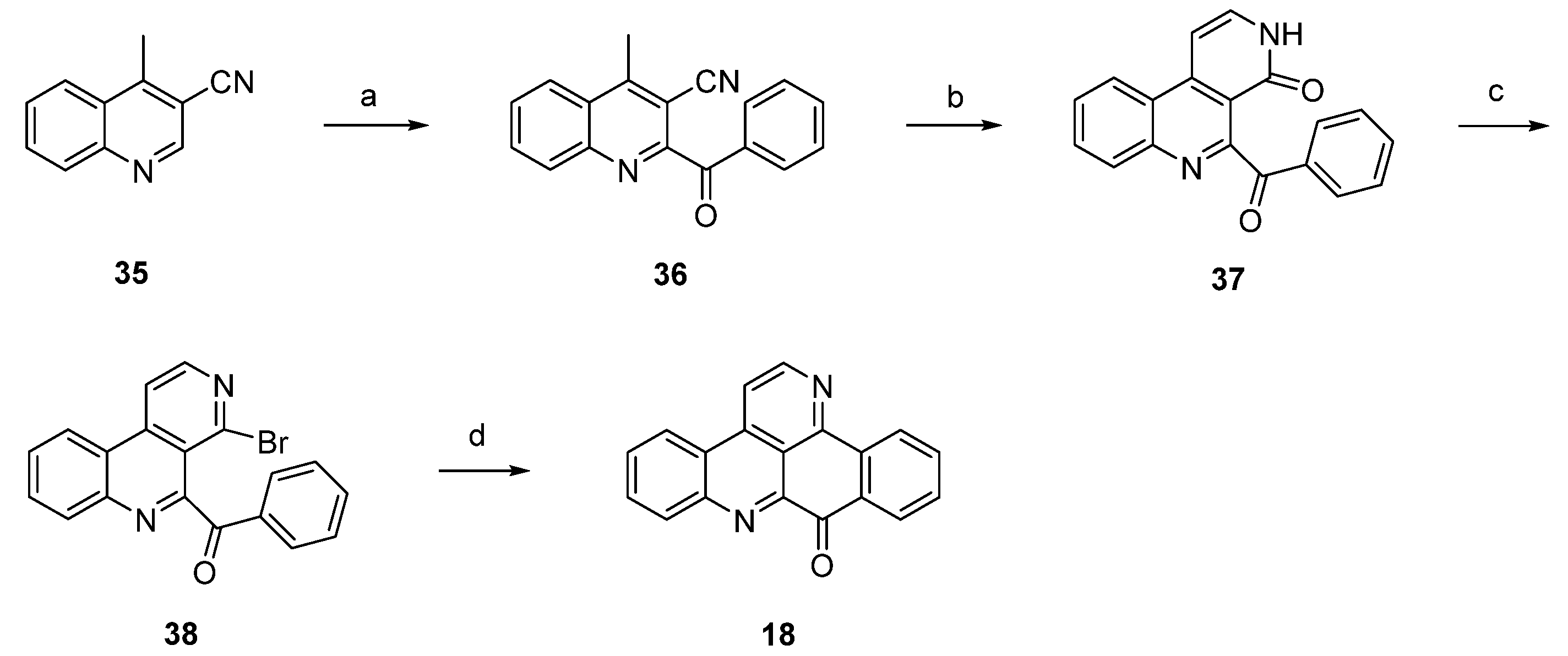
In 2012, Raeder and Bracher published a new synthetic approach to the pyridoacridine ring system involving two radical reactions as the key steps [22]. Readily available quinoline 35 was subjected to Minisci benzoylation with a benzoyl radical generated from benzaldehyde to furnish ketone 36. The initially low yield of this step was increased by replacing sulfuric acid by trifluoroacetic acid and by adding additional amounts of radical starters (iron(II) sulfate, tert-butyl hydroperoxide) after intervals of 45 min. The annulation of the bromopyridine ring, which later forms ring E of the target compound, was accomplished in a two-step protocol. First, 36 was condensed with dimethylformamide diethyl acetal, followed by heating with concentrated sulfuric acid/glacial acetic acid to give pyridone 37. In the second step, 37 was converted to bromo compound 38 by heating with phosphoryl bromide. The final intramolecular cyclization was performed through conversion of the bromopyridine moiety to an aryl radical with tributyltin hydride and azobis(isobutyronitrile) (AIBN) to give deazaascididemin (18) (Scheme 6). Unfortunately, this approach gives only a very low overall yield, and the scope of this route has not been explored.
Scheme 6.
Synthetic approach towards deazaascididemin (18): (a) Benzaldehyde, AcOH, Et3N, H2O, FeSO4, tert-BuOOH (51%); (b) dimethylformamide diethyl acetal, DMF; then H2SO4, AcOH (39%); (c) POBr3, anisole (27%); (d) Bu3SnH, AIBN, toluene (5%).
Scheme 6.
Synthetic approach towards deazaascididemin (18): (a) Benzaldehyde, AcOH, Et3N, H2O, FeSO4, tert-BuOOH (51%); (b) dimethylformamide diethyl acetal, DMF; then H2SO4, AcOH (39%); (c) POBr3, anisole (27%); (d) Bu3SnH, AIBN, toluene (5%).
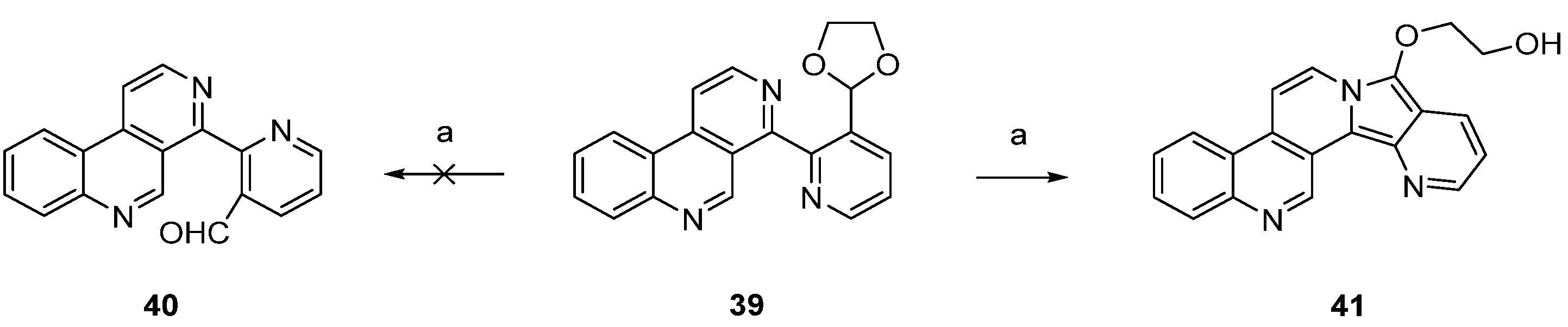
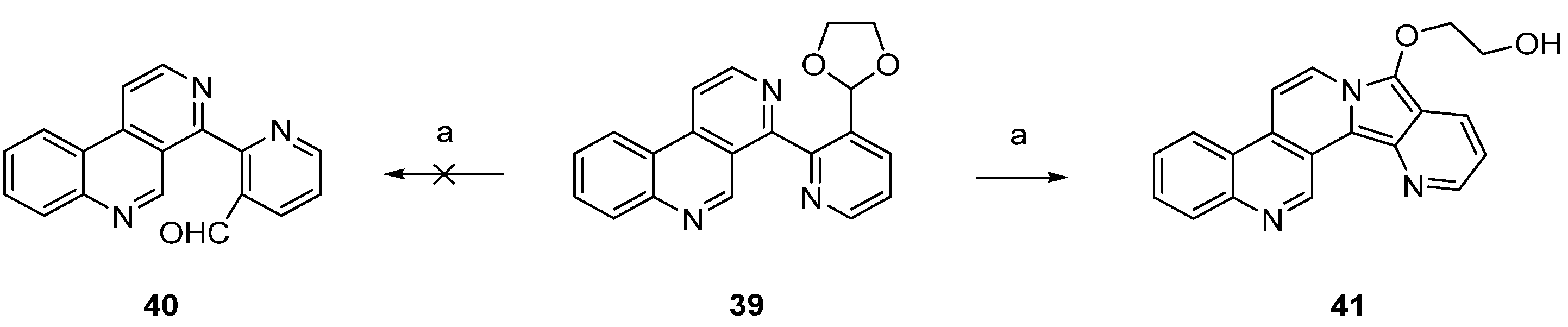
In a related approach, Bracher [23] intended to utilize the Minisci reaction as a final cyclization step for the synthesis of ascididemin (3). However, attempted acidic hydrolysis of dioxolane 39 (to give aldehyde 40 as an acyl radical precursor) resulted in unexpected cyclization to the benzo[f]pyrido[2′,3′:3,4]pyrrolo[2,1-a][2,7]naphthyridine 41 (Scheme 7).
Scheme 7.
Unsuccessful approach to the ascididemin precursor 40 giving benzo[f]pyrido[2′,3′:3,4]pyrrolo[2,1-a][2,7]naphthyridine (41): (a) H2SO4, H2O (43%).
Scheme 7.
Unsuccessful approach to the ascididemin precursor 40 giving benzo[f]pyrido[2′,3′:3,4]pyrrolo[2,1-a][2,7]naphthyridine (41): (a) H2SO4, H2O (43%).
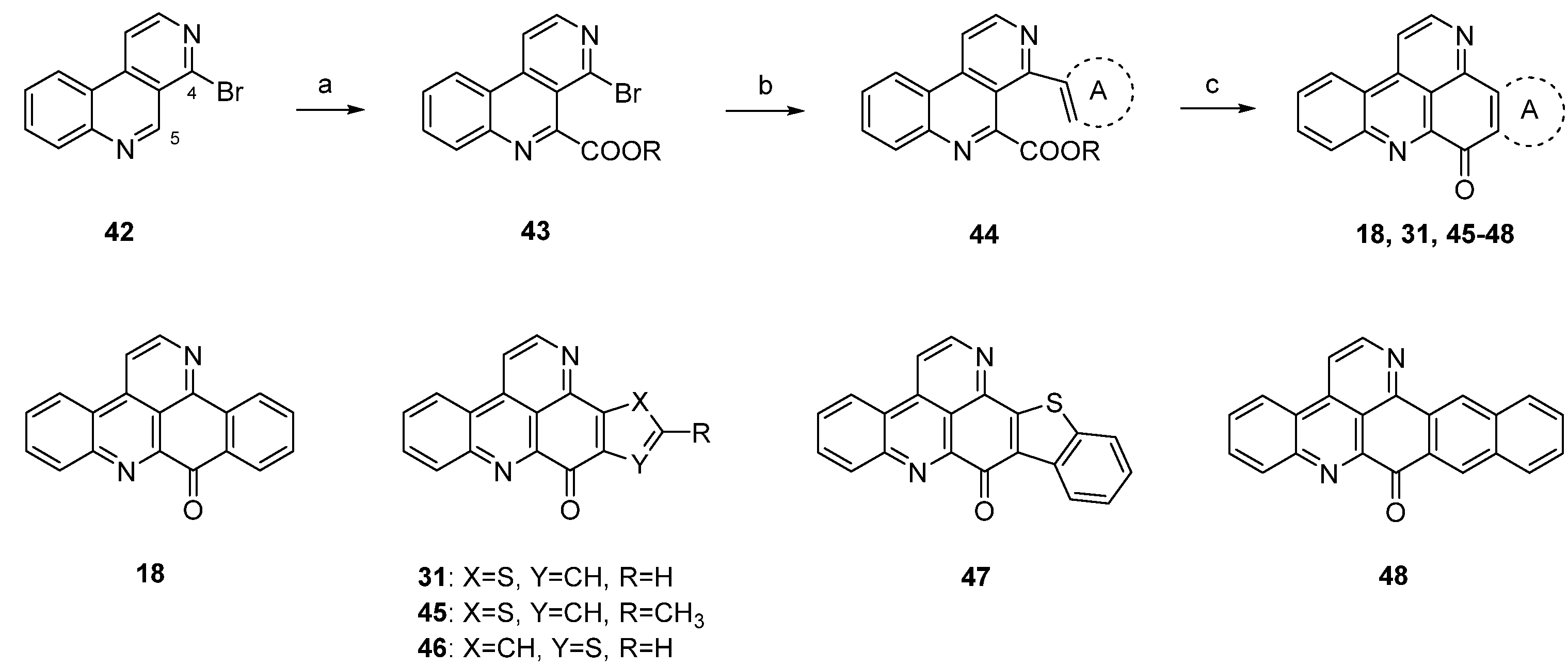
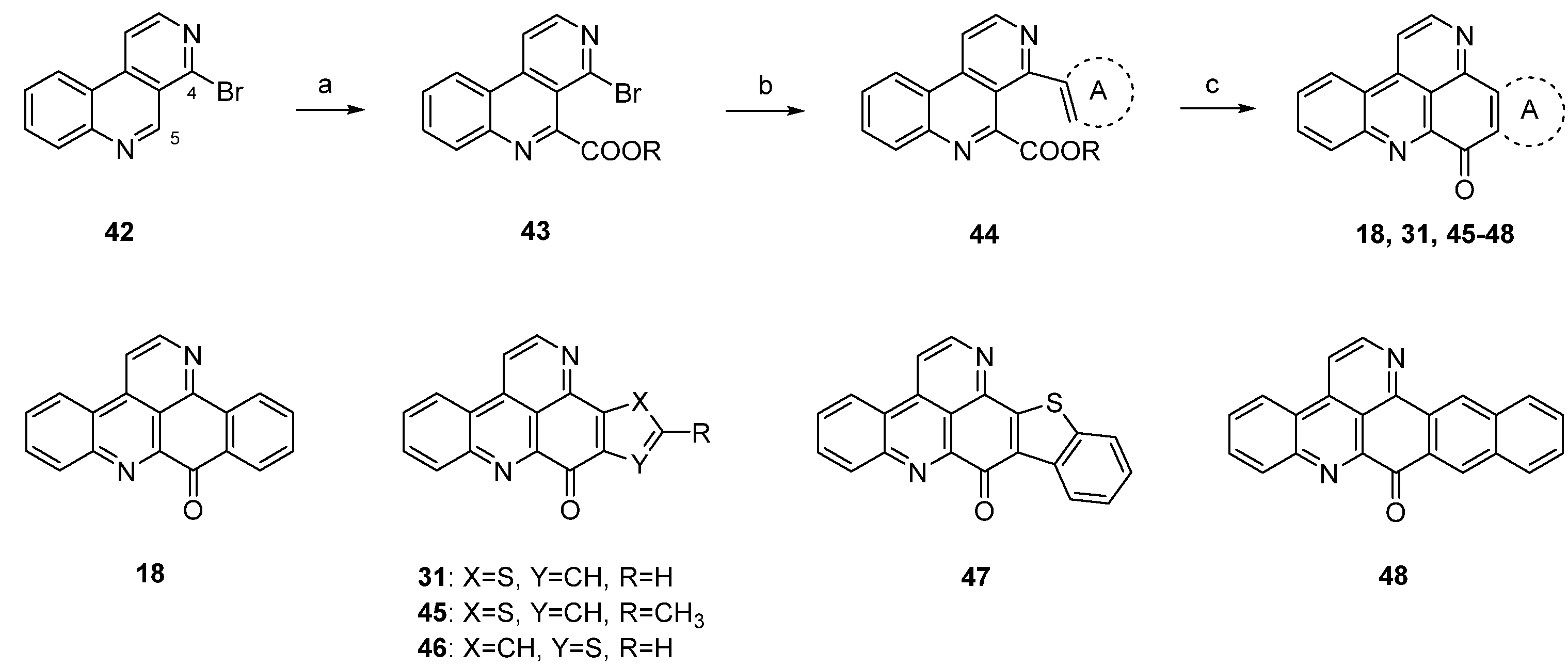
A different synthetic approach to ring A analogues of ascididemin from the Bracher group [24] started with a high-yielding, regioselective Minsici-type homolytic methoxycarbonylation at C-5 of readily available 4-brombenzo[c][2,7]naphthyridine (42), followed by an introduction of ring A equivalents through Suzuki cross-coupling with appropriate (hetero)areneboronic acids. The final intramolecular ring closure was achieved by treating the 4,5-disubstituted benzo[c][2,7]naphthyridines 44 with trifluoromethanesulfonic acid under microwave irradiation. This final superacid-aided Friedel-Crafts-type acylation step furnished pyridoacridines 18, 31, 45–48 in 63%–92% yields (Scheme 8). Significantly lower yields were obtained with the corresponding ethyl esters. However, these cyclization reactions were successful only with electron-rich aryl residues (phenyl, naphthyl, thiophenes) in position 4. Electron-withdrawing substituents (acetyl, chloro, bromo) on a benzene or thiophene ring led to the complete failure of this cyclization step [24].
Scheme 8.
Synthesis of ring A analogues of ascididemin 18, 31, 45–48 through an intramolecular trifluoromethanesulfonic acid-aided Friedel-Crafts-type cyclization step: (a) Methyl pyruvate, H2O2, FeSO4, H2SO4, AcOH; then MnO2, CH2Cl2 (93% over two steps); (b) (hetero)areneboronic acid, Pd(Ph3P)4 (cat.), K2CO3, THF, H2O (41%–75%); (c) CF3-SO3H, microwave irradiation (63%–92%).
Scheme 8.
Synthesis of ring A analogues of ascididemin 18, 31, 45–48 through an intramolecular trifluoromethanesulfonic acid-aided Friedel-Crafts-type cyclization step: (a) Methyl pyruvate, H2O2, FeSO4, H2SO4, AcOH; then MnO2, CH2Cl2 (93% over two steps); (b) (hetero)areneboronic acid, Pd(Ph3P)4 (cat.), K2CO3, THF, H2O (41%–75%); (c) CF3-SO3H, microwave irradiation (63%–92%).
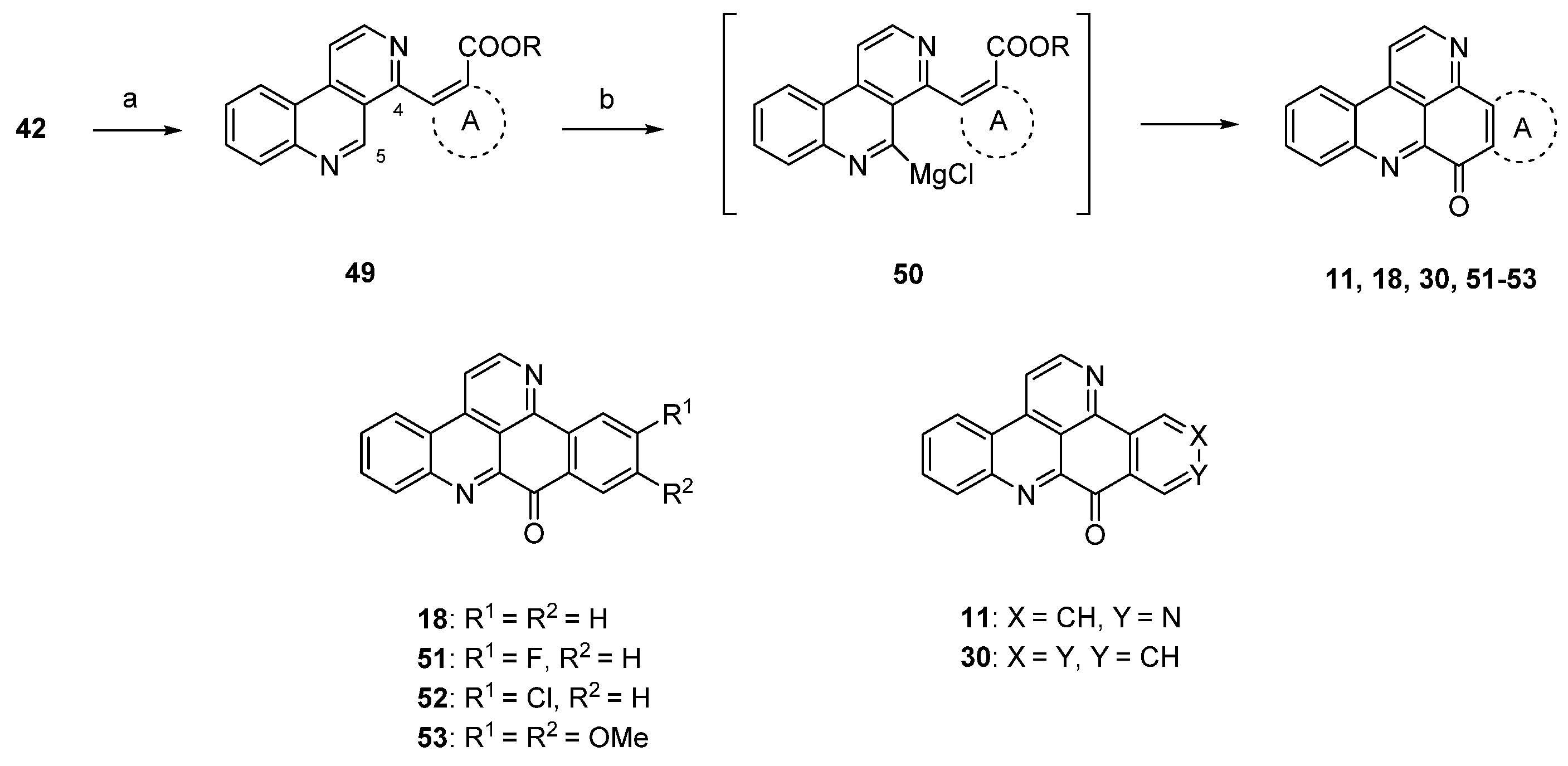
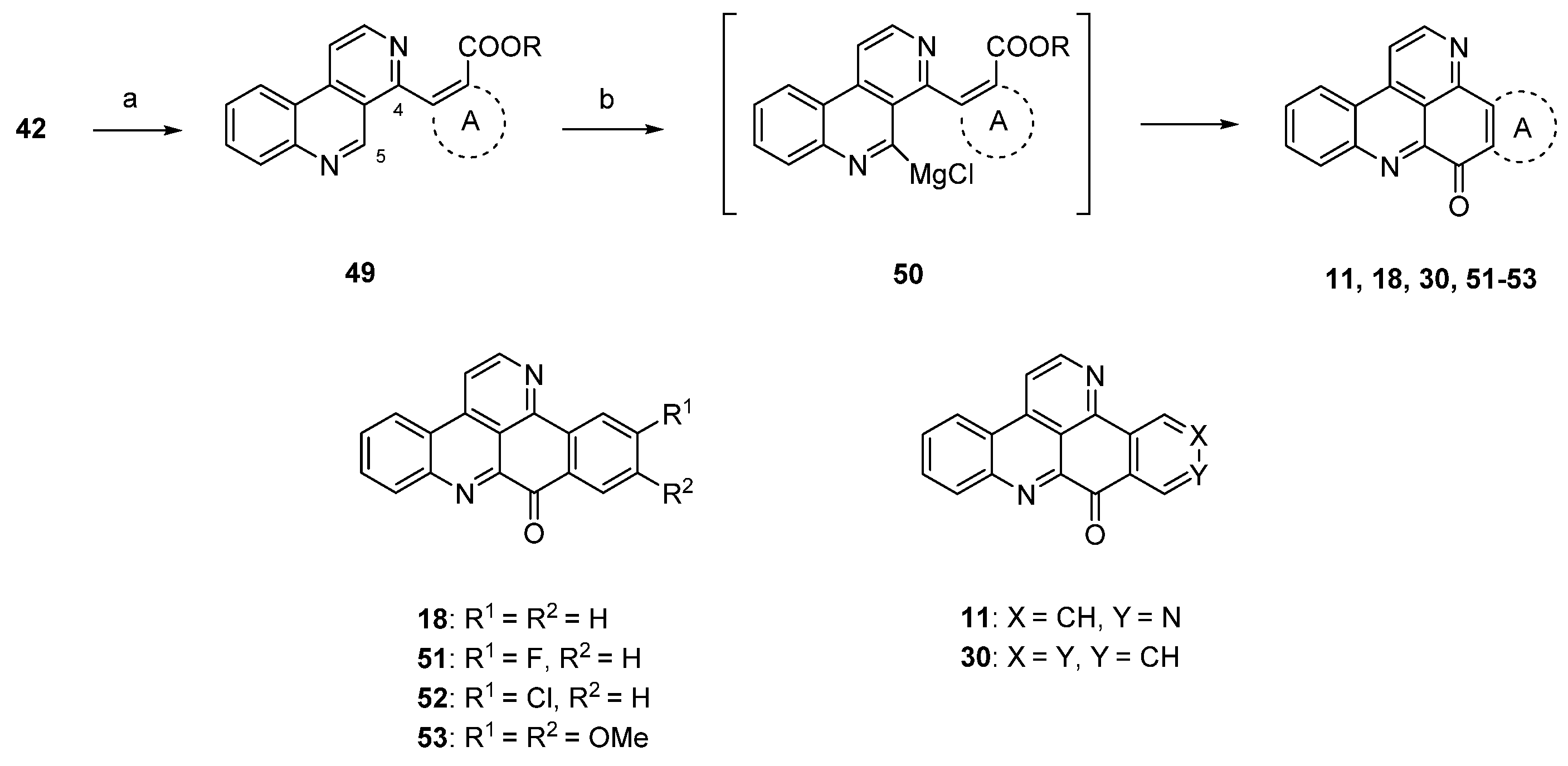
Due to the fact that the above-mentioned protocol [24] only allows for the introduction of electron-rich carbocyclic and heterocyclic ring A substitutes, Plodek et al. developed a new approach to the pyridoacridine ring system in which electron-deficient (hetero)arenes also serve as sources for ring A [25]. The introduction of the ring A scaffolds was achieved through Suzuki cross-coupling of 4-brombenzo[c][2,7]naphthyridine (42) with (hetero)areneboronic acids bearing an ester moiety in the ortho position or through Negishi cross-coupling with pyridylzinc compounds which were obtained by regioselective ring metalation of ethyl nicotinate or ethyl isonicotinate. The resulting 5-substituted benzo[c][2,7]naphthyridines 49 were metalated regioselectively at the peri position (C-5) with Knochel’s TMPMgCl·LiCl (2.2 equivalents), and intramolecular nucleophilic attack of the resulting arylmagnesium species 50 at the ester group furnished ring A analogues 18, 31, 45–48 and isomers 11 and 30 of ascididemin in poor-to-modest yields (Scheme 9) [25]. This key step was inspired by the synthesis of demethyldeoxyamphimedine published by the Bracher group in 2014 [26] (see below).
Scheme 9.
TMPMgCl·LiCl-mediated synthesis of ring A analogues 18, 31, 45–48 and isomers 11 and 30 of ascididemin (3): (a) For Suzuki cross-coupling reactions: areneboronic acid, Pd2(dba)3, P(tBu)3, KF, THF (73%–80%); for Negishi cross-coupling reactions: pyridylzinc compounds, Pd(dba)2, P(2-furyl)3, THF (71%–76%); (b) TMPMgCl·LiCl, THF (27%–39%).
Scheme 9.
TMPMgCl·LiCl-mediated synthesis of ring A analogues 18, 31, 45–48 and isomers 11 and 30 of ascididemin (3): (a) For Suzuki cross-coupling reactions: areneboronic acid, Pd2(dba)3, P(tBu)3, KF, THF (73%–80%); for Negishi cross-coupling reactions: pyridylzinc compounds, Pd(dba)2, P(2-furyl)3, THF (71%–76%); (b) TMPMgCl·LiCl, THF (27%–39%).
The protocol shown in Scheme 9 is the most flexible one for the preparation of ring A analogues of ascididemin (3), which are of special interest for the development of anticancer compounds.
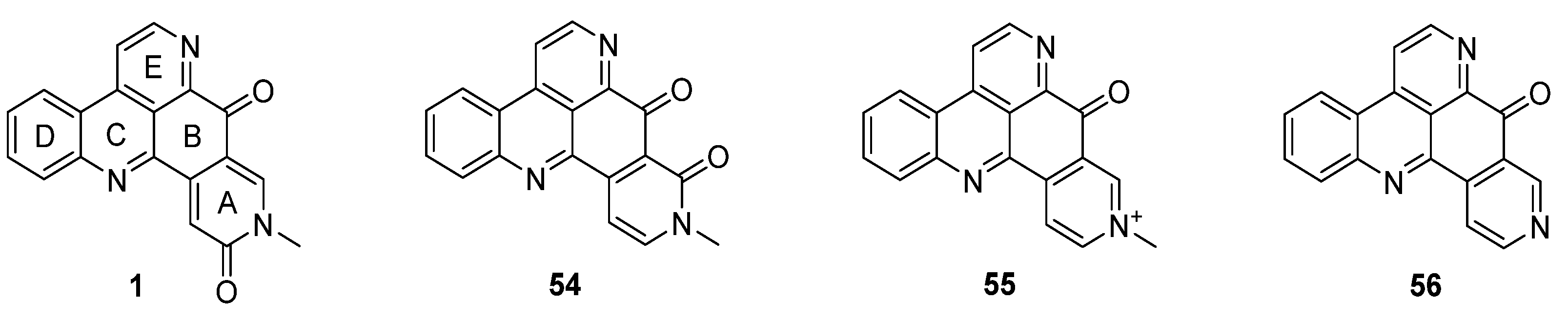
3. Amphimedine-Type Pyridoacridines
Amphimedine-type pyridoacridines consist of the pentacyclic alkaloids amphimedine (1), neoamphimedine (54), deoxyamphimedine (55), and demethyldeoxyamphimedine (56) (Figure 3) [26]. Compared to the ascididemin subclass, rings A and B are connected in a different manner here. While most of the published syntheses of ascididemin-type pyridoacridines are more or less based on Bracher’s synthetic methodology [7], many different and versatile strategies towards the amphimedine scaffold have been developed [6]. During the last 15 years, three approaches towards neoamphimedine (54) and two syntheses of demethyldeoxyamphimedine (56) have been reported.
Figure 3.
Structures of the amphimedine-type pyridoacridine alkaloids: amphimedine (1), neoamphimedine (54), deoxyamphimedine (55) and demethyldeoxyamphimedine (56).
Figure 3.
Structures of the amphimedine-type pyridoacridine alkaloids: amphimedine (1), neoamphimedine (54), deoxyamphimedine (55) and demethyldeoxyamphimedine (56).

Delfourne’s first total synthesis of demethyldeoxyamphimedine (56), which is illustrated in Scheme 10, was published in 2002 [12], years before this compound was identified as a natural product [27]. This approach started with a hetero-Diels-Alder cycloaddition of isoquinoline-5,8-dione (10) and 1-azadieene 57, both of them not commercially available. Besides the desired diazaanthranquinone 58, which was isolated in only 0.8% yield, the poorly separable regioisomer 59 was formed in 1.7% yield. The final ring closure step was accomplished under alkaline conditions to give demethyldeoxyamphimedine (56) in almost quantitative yield. Analogous cyclization of the isomeric intermediate 59 gave the isomer 60 of the alkaloid in 91% yield.
Scheme 10.
Synthesis of demethyldeoxyamphimedine (56): (a) Toluene, N2 atmosphere (0.8%); (b) NaOH, CHCl3 (96%).
Scheme 10.
Synthesis of demethyldeoxyamphimedine (56): (a) Toluene, N2 atmosphere (0.8%); (b) NaOH, CHCl3 (96%).
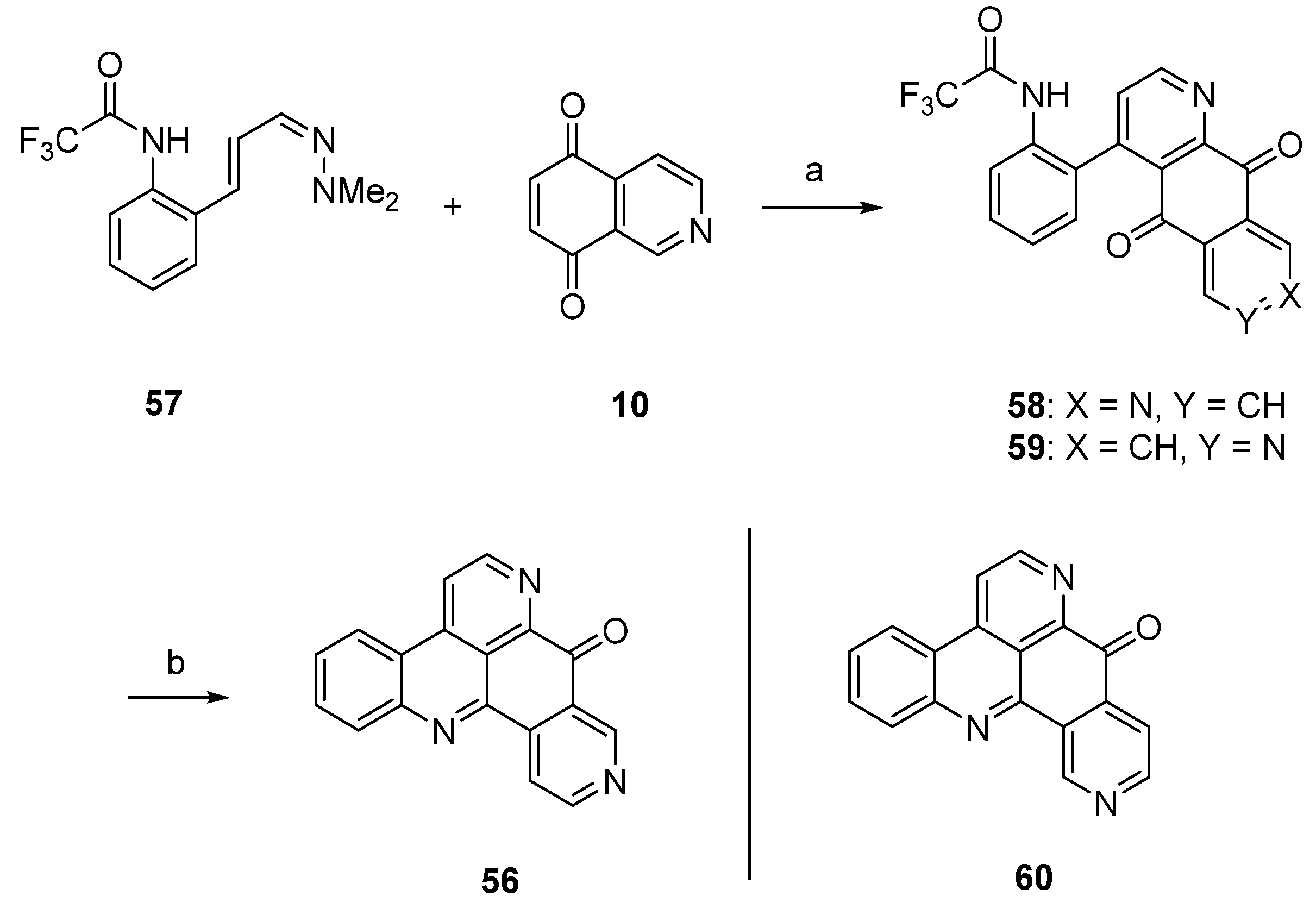
A highly effective total synthesis of demethyldeoxyamphimedine (56) was reported by Melzer et al. in 2014 [26]. Regioselective direct metalation of ethyl nicotinate (61) at C-4 using TMPMgCl·BF3·LiCl and subsequent transmetalation with ZnCl2 gave pyridylzinc compound 62, which was directly subjected to a Negishi cross-coupling reaction with 2-iodoaniline. The resulting biaryl (not shown in Scheme 11) underwent spontaneous lactamization to give benzo[c][2,7]naphthyridin-5(6H)-one (63). This lactam was converted to 5-bromobenzo[c][2,7]naphthyridine (64) with phosphoryl bromide. In another Negishi cross-coupling reaction, tricyclus 64 was coupled with organozinc intermediate 62 to give biaryl 65. The final ring closure step was achieved via direct regioselective metalation at C-4 of 65 utilizing 2.2 equivalents of Knochel’s TMPMgCl·LiCl, followed by intramolecular trapping of the ester group to give alkaloid 56 in 6.4% total yield. Thus, the authors prepared demethyldeoxyamphimedine (56) by using only two commercial building blocks, ethyl nicotinate and 2-iodoaniline (Scheme 11) [26].
Scheme 11.
Synthesis of demethyldeoxyamphimedine (56) by Melzer et al: (a) TMPMgCl·BF3·LiCl, THF; then ZnCl2; (b) 2-iodoaniline, Pd(dba)2, P(2-furyl)3, THF (50% over two steps); (c) POBr3 (59%); (d) pyridylzinc compound 62, Pd(dba)2, P(2-furyl)3, THF (78%); (e) TMPMgCl·LiCl, THF (28%).
Scheme 11.
Synthesis of demethyldeoxyamphimedine (56) by Melzer et al: (a) TMPMgCl·BF3·LiCl, THF; then ZnCl2; (b) 2-iodoaniline, Pd(dba)2, P(2-furyl)3, THF (50% over two steps); (c) POBr3 (59%); (d) pyridylzinc compound 62, Pd(dba)2, P(2-furyl)3, THF (78%); (e) TMPMgCl·LiCl, THF (28%).
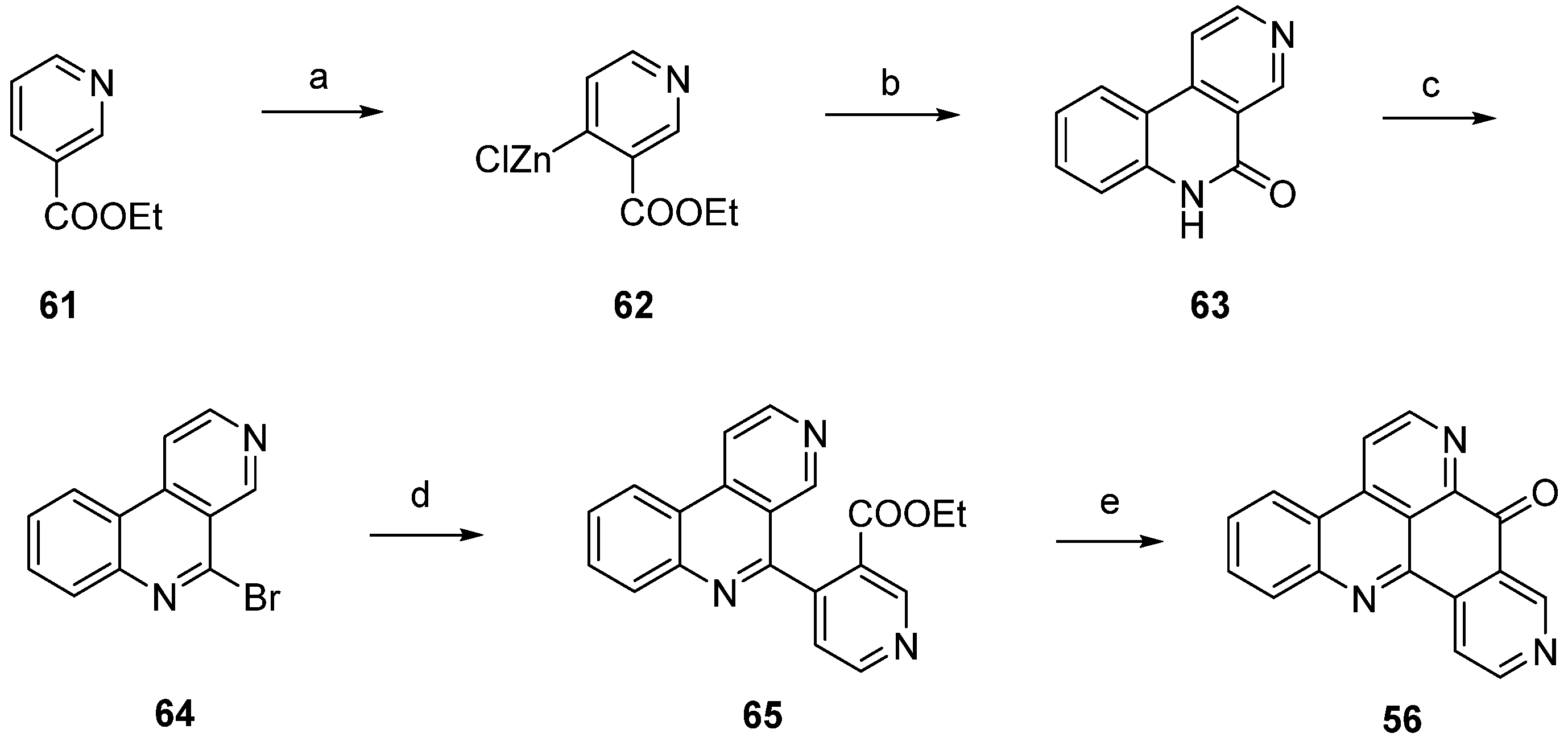
The first total synthesis of neoamphimedine (54) was reported by Ireland in 2007 [28]. This approach started with O-methylation of dinitrophenol 66 with diazomethane, followed by selective reduction of one of the two nitro groups with 10% palladium on carbon and subsequent acetylation of the so-obtained amine (not shown in Scheme 12) with acetic anhydride to afford acetanilide 67. Catalytic reduction and following conversion with ethyl (2-nitrobenzoyl)acetate furnished β-keto amide 68 in very good yields. Treatment of 68 with polyphosphoric acid gave quinolone 69 in a Knorr cyclization reaction. Quinoline 70 was obtained in a two-step protocol through treating 69 with trifluoromethanesulfonic anhydride and subsequent hydrogenolysis of the resulting triflate ester. The following transformation of the 7-acetylamino group of 70 into a nitrile was accomplished under Sandmeyer conditions after acidic hydrolysis. Next, the nitrile of 71 was hydrolyzed by heating with concentrated sulfuric acid to give the carboxylic acid 72, and further amidated with N-methylamino acetaldehyde dimethylacetal to give carboxamide 73. Pomeranz-Fritsch–type cyclization with sulfuric acid gave oxo-diazaanthracene 74. The desired alkaloid 54 was obtained by catalytic reduction of the nitro group of 74, subsequent CAN oxidation to a tricyclic quinone, followed by intramolecular condensation with the amino group in a total yield of 2% over 12 steps (Scheme 12) [28].
Based on this methodology, LaBarbera published an improved total synthesis of neoamphimedine (54) [29]. The central quinolone intermediate 77 was synthesized in three steps, starting from nitrobenzoate 75. Catalytic hydrogenation of 75 gave the aniline 76, which was converted to 77 by treatment with Meldrum’s acid and trimethyl orthoformate. The following thermal ring closure gave 4-quinolone 78 with an overall yield of 78%. Treatment of 78 with trifluoromethanesulfonic anhydride furnished triflate ester 79, which was smoothly converted to the biaryl 81 in a Stille cross-coupling reaction with readily available trimethyl-(2-nitrophenyl)stannane (80). Next, alkaline hydrolysis of the methyl ester group gave carboxyclic acid 72. The following steps were performed as described above [28] and afforded neoamphimedine (54) in 25% overall yield (Scheme 13) [29].
Scheme 12.
Ireland’s synthesis of neoamphimedine (54): (a) CH2N2; then 10% Pd/C in cyclohexene/EtOH; then AcOH/Ac2O (77% over two steps); (b) 10% Pd/C in cyclohexene/EtOH; then ethyl (2-nitrobenzoyl)acetate, m-xylenes (96%); (c) PPA (52%); (d) Tf2O, CH2Cl2, Et3N; then formic acid, Et3N, DMF, Pd(OAc)2, dppf (61% over two steps); (e) AcOH, H2O, H2SO4; then NaNO2; then CuCN (50%); (f) H2SO4 (80%); (g) N-methylamino acetaldehyde dimethyl acetal, EDC, CH2Cl2 (87%); (h) H2SO4 (43%); (i) 10% Pd/C, cyclohexene/EtOH; then CAN (30%).
Scheme 12.
Ireland’s synthesis of neoamphimedine (54): (a) CH2N2; then 10% Pd/C in cyclohexene/EtOH; then AcOH/Ac2O (77% over two steps); (b) 10% Pd/C in cyclohexene/EtOH; then ethyl (2-nitrobenzoyl)acetate, m-xylenes (96%); (c) PPA (52%); (d) Tf2O, CH2Cl2, Et3N; then formic acid, Et3N, DMF, Pd(OAc)2, dppf (61% over two steps); (e) AcOH, H2O, H2SO4; then NaNO2; then CuCN (50%); (f) H2SO4 (80%); (g) N-methylamino acetaldehyde dimethyl acetal, EDC, CH2Cl2 (87%); (h) H2SO4 (43%); (i) 10% Pd/C, cyclohexene/EtOH; then CAN (30%).
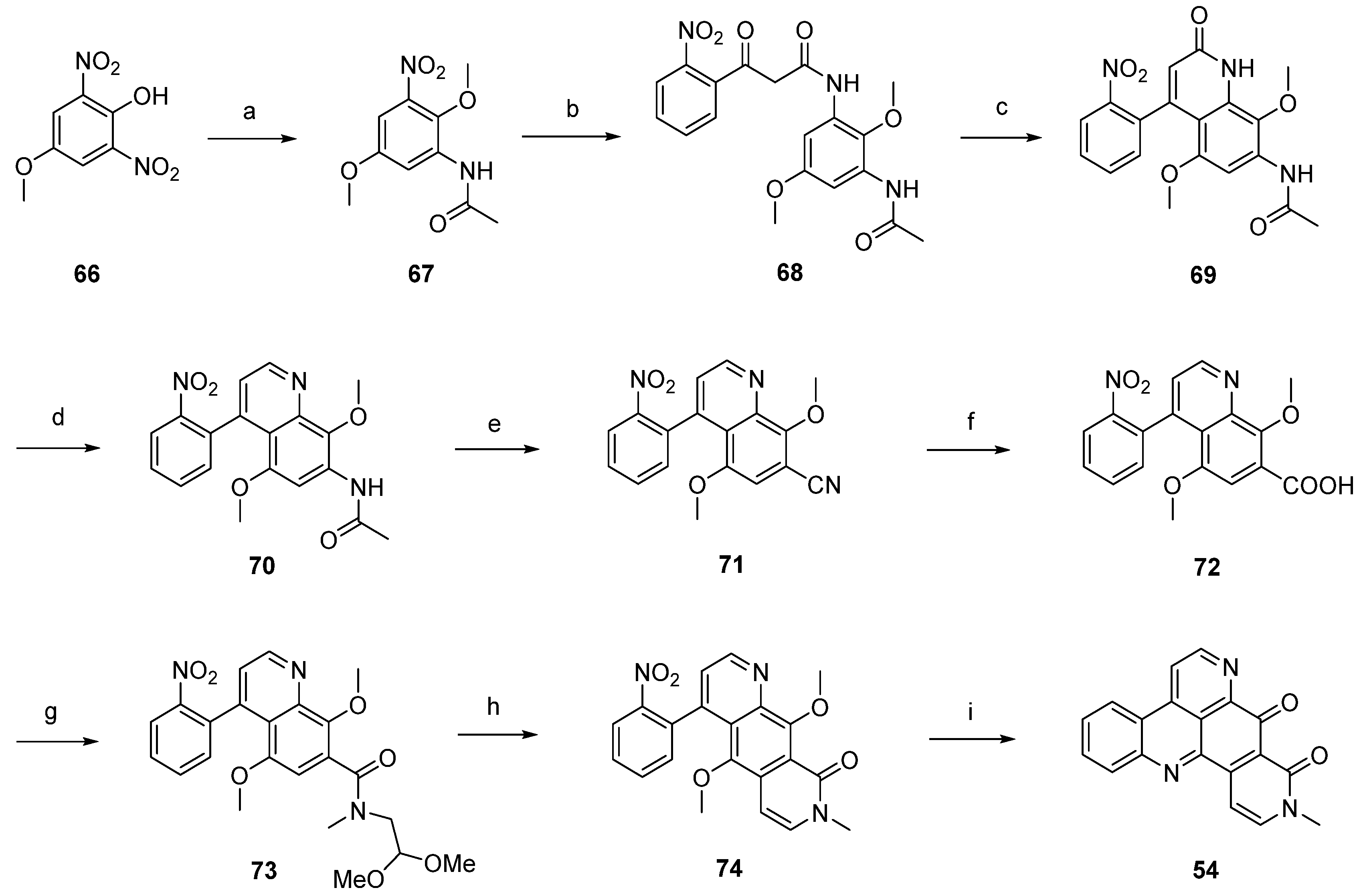
Scheme 13.
Improved synthesis of neoamphimedine (54): (a) Pd/C, H2, MeOH; (b) Meldrum’s acid, trimethyl orthoformate (90% over two steps); (c) Ph2O, reflux (87%); (d) Tf2O, DMAP, 2,6-lutidine, CH2Cl2 (92%); (e) trimethyl-(2-nitrophenyl)stannane (80), CuI, Pd(OAc)2, dppe, DMF (83%); (f) LiOH; following steps, see Scheme 12.
Scheme 13.
Improved synthesis of neoamphimedine (54): (a) Pd/C, H2, MeOH; (b) Meldrum’s acid, trimethyl orthoformate (90% over two steps); (c) Ph2O, reflux (87%); (d) Tf2O, DMAP, 2,6-lutidine, CH2Cl2 (92%); (e) trimethyl-(2-nitrophenyl)stannane (80), CuI, Pd(OAc)2, dppe, DMF (83%); (f) LiOH; following steps, see Scheme 12.
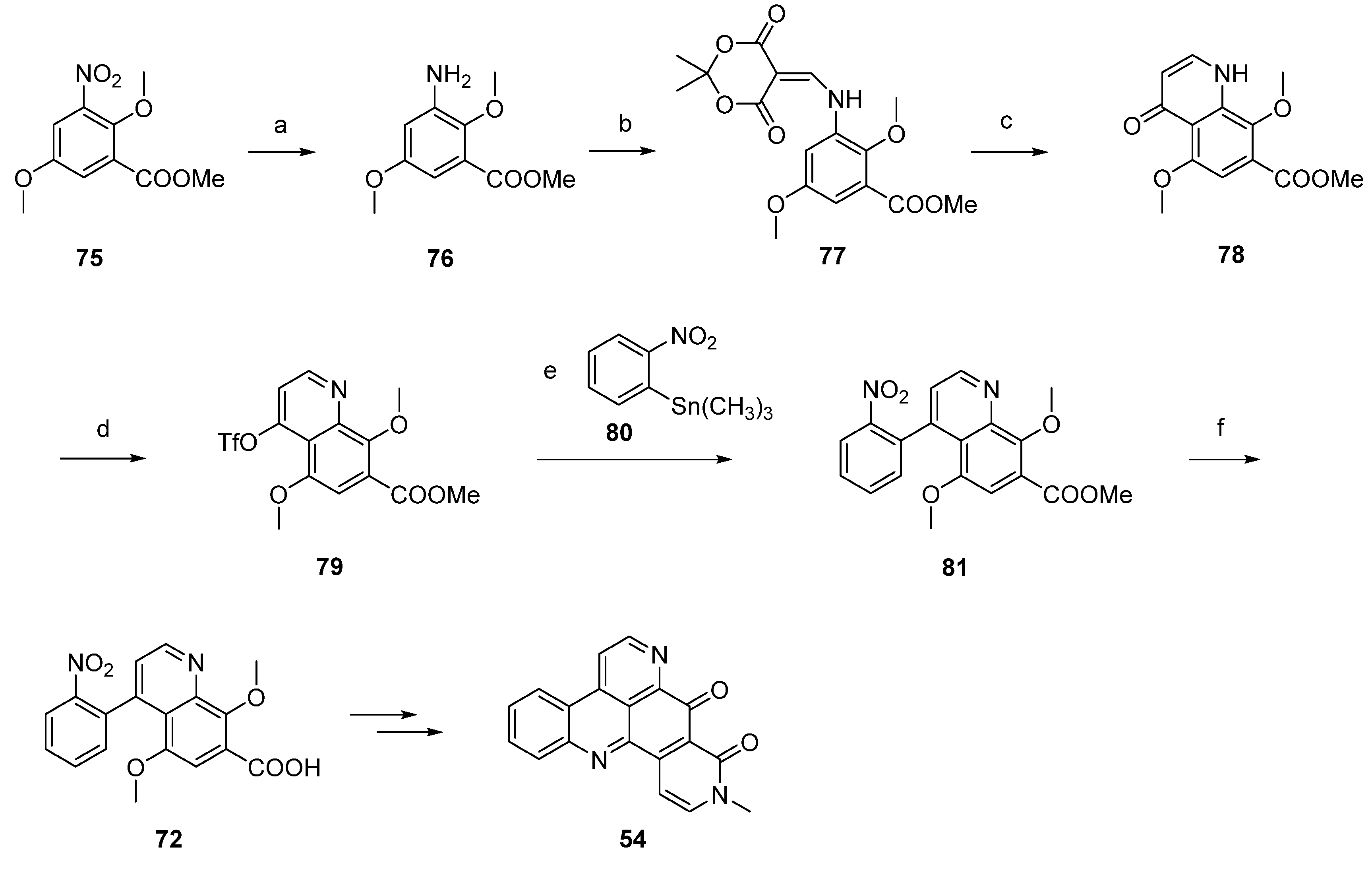
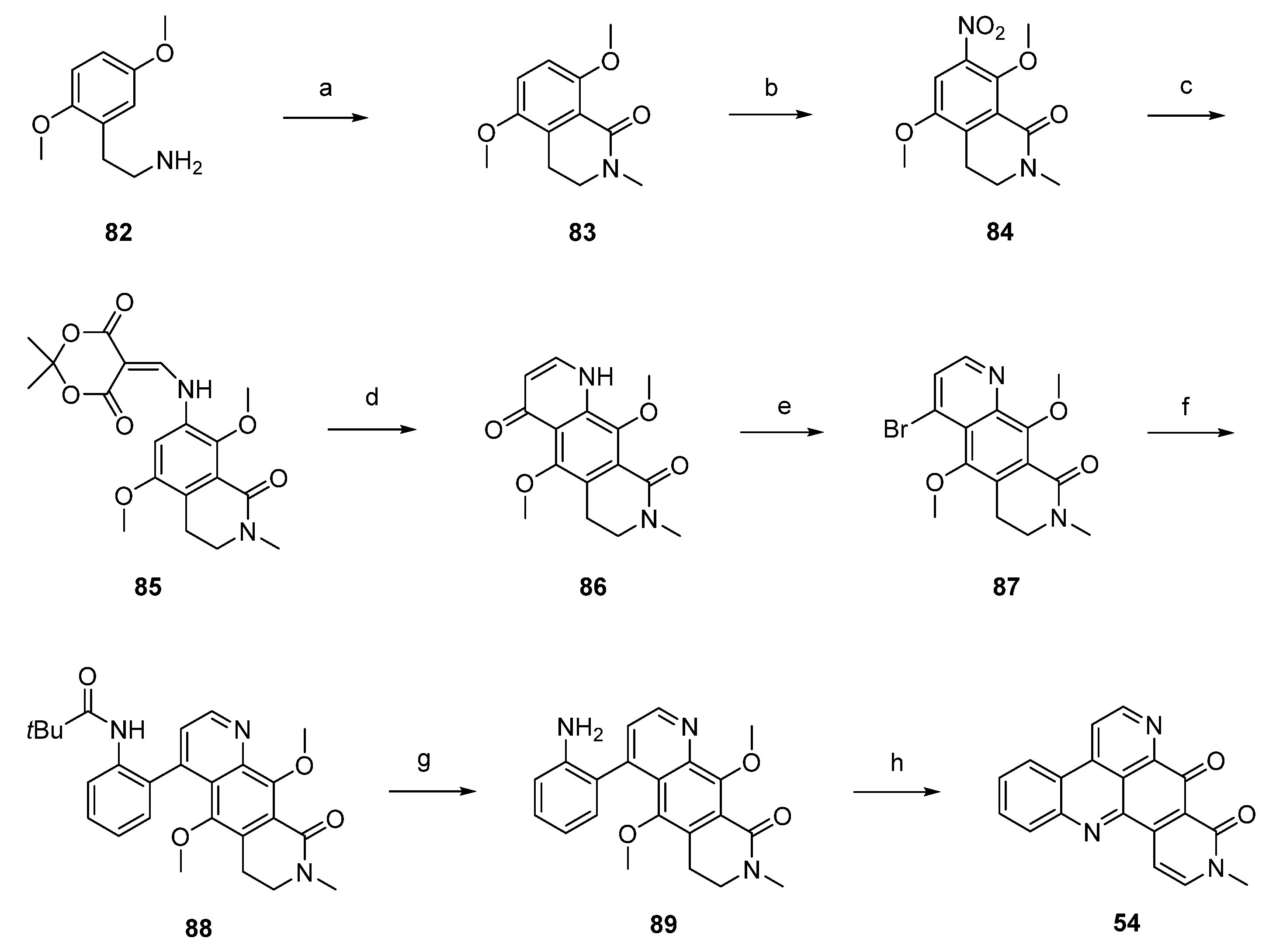
Another synthetic approach to neoamphimedine (54) was reported by Nakahara et al. in 2012 [30]. Commercially available 2,5-dimethoxyphenethylamine (82) was N-acylated with ethyl chloroformate and triethylamine. N-Methylation of the so-obtained N-carbethoxy derivative (not shown in Scheme 14) was performed with methyl iodide and sodium hydride. Subsequent Bischler-Napieralski–type cyclization with triflic anhydride and N,N-dimethylaminopyridine (DMAP) furnished quinolone 83 in 74% overall yield. Following regioselective nitration of 83 with cupric nitrate trihydrate in acetic anhydride afforded 84 in excellent yield. Catalytic hydrogenation of the nitro group utilizing 10% palladium on carbon and subsequent reaction with Meldrum’s acid and trimethyl orthoformate yielded enamine 85, which was cyclized to acridinedione 86 by heating in diphenyl ether. Subsequent bromination with phosphoryl bromide afforded 87, and Suzuki cross-coupling with 2-(pivaloylamino)phenylboronic acid gave intermediate 88 in high yield. Acid-mediated removal of the pivaloyl protective group gave amine 89. Conversion to the alkaloid 54 was performed in two steps: O-demethylation with BBr3 gave a hydroquinone intermediate which underwent cyclization and dehydrogenation in the dihydropyridone ring upon oxidation with CAN (31% yield over both steps). Using nitric acid as the oxidant led to a significant loss in yield (6%). The total yield of neoamphimedine (54) over 12 steps was 6% (Scheme 14) [30].
Scheme 14.
Synthesis of neoamphimedine (54): (a) ClCO2Et, Et3N, THF; then MeI, NaH, THF; then DMAP, (CF3SO2)O, CH2Cl2 (74% over three steps); (b) Cu(NO2)2∙3H2O, Ac2O (96%); (c) Pd/C, H2, MeOH; then Meldrum’s acid, trimethyl orthoformate (81% over two steps); (d) Ph2O, reflux (83%); (e) POBr3, THF (70%); (f) 2-(pivaloylamino)phenylboronic acid, Pd(PPh3)4, K2CO3, H2O, EtOH, toluene (90%); (g) H2SO4/H2O (64%); (h) BBr3, CH2Cl2; then CAN/H2O, MeCN (31%).
Scheme 14.
Synthesis of neoamphimedine (54): (a) ClCO2Et, Et3N, THF; then MeI, NaH, THF; then DMAP, (CF3SO2)O, CH2Cl2 (74% over three steps); (b) Cu(NO2)2∙3H2O, Ac2O (96%); (c) Pd/C, H2, MeOH; then Meldrum’s acid, trimethyl orthoformate (81% over two steps); (d) Ph2O, reflux (83%); (e) POBr3, THF (70%); (f) 2-(pivaloylamino)phenylboronic acid, Pd(PPh3)4, K2CO3, H2O, EtOH, toluene (90%); (g) H2SO4/H2O (64%); (h) BBr3, CH2Cl2; then CAN/H2O, MeCN (31%).
4. Eilatin-Type Pyridoacridines
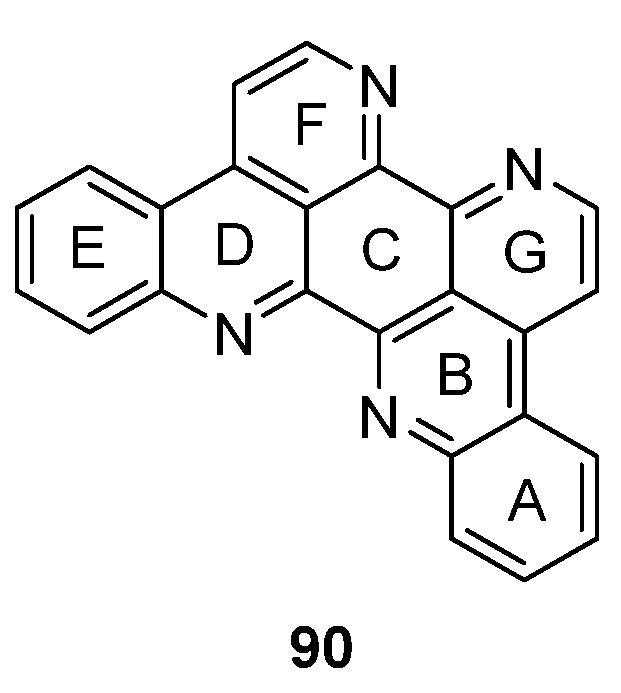
The symmetrical alkaloid eilatin (90, Figure 4) is the only known heptacyclic member of the pyridoacridine alkaloid class and was isolated in 1988 by Rudi et al. from the tunicate Eudistoma sp. [31]. Since synthetic approaches towards eilatin (90) have not been reviewed before, this chapter will close this gap and gives a short overview on all published syntheses since its isolation.
Figure 4.
Structure of eilatin (90).
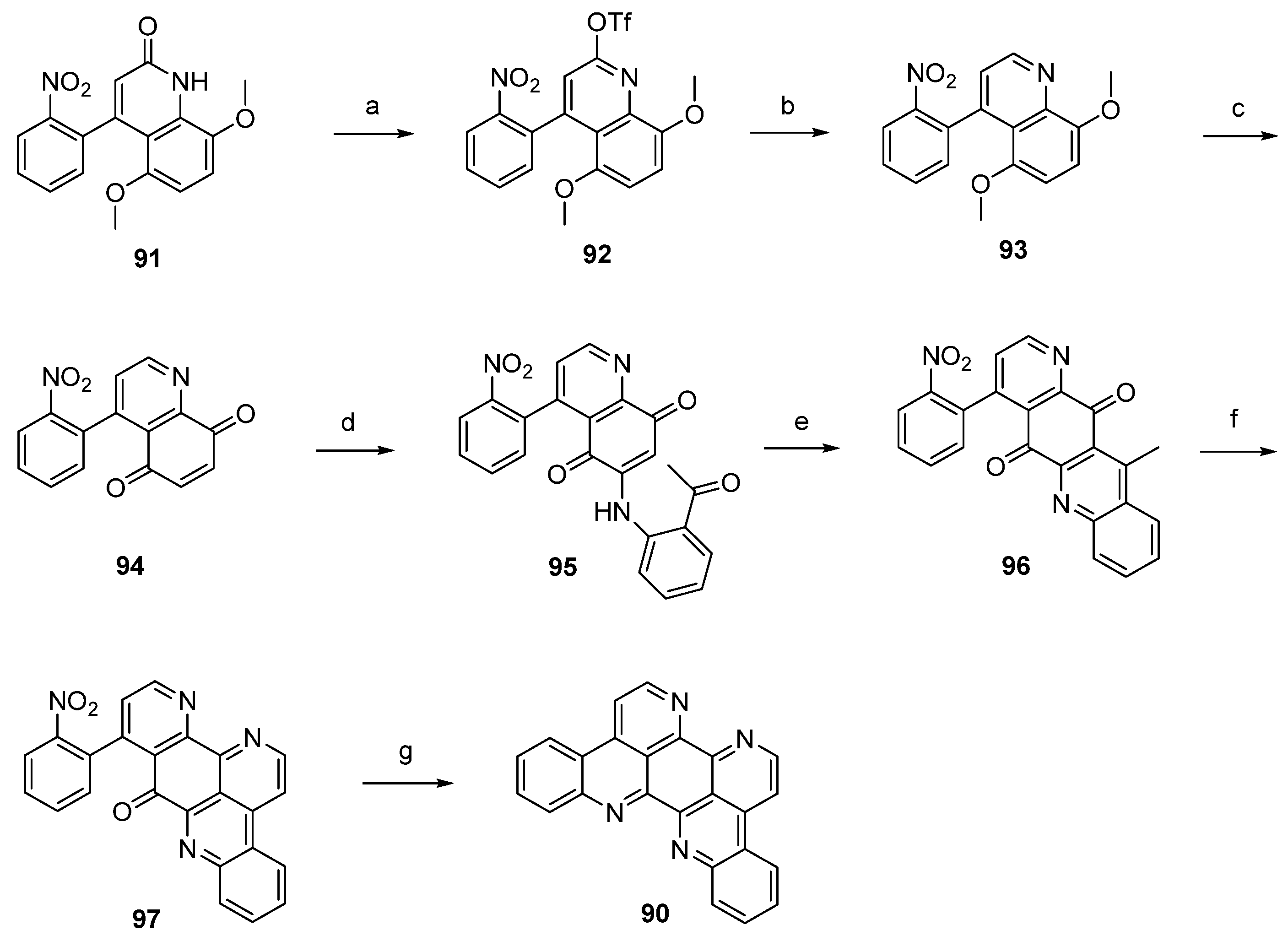
The first total synthesis of eilatin (90), which is illustrated in Scheme 15, was reported by the Kubo group in 1993 [32]. The alkaloid was obtained in seven steps (overall yield 13%) from intermediate 91, which had been developed before as an intermediate for a total synthesis of amphimedine (1) [33]. Arylquinolone 91 was converted to the aryl triflate 92 with trifluoromethanesulfonic anhydride, and subsequent palladium-catalyzed reductive removal of the sulfonyloxy group with triethylammonium formate furnished 4-arylquinoline 93. Following oxidative demethylation of 93 using CAN gave p-quinone 94. The next three steps follow Bracher’s ascididemine synthesis (see Scheme 1), thus arylaminoquinoline-5,8-dione 95 was prepared by regioselective oxidative amination of p-quinone 94 with 2-aminoacetophenone in the presence of CeCl3. Next, an acid-catalyzed cyclization step was performed furnishing tetracyclic quinone 96, and the annulation of ring G was achieved using condensation with N,N-dimethylformamide diethyl acetal followed by ammonium chloride treatment to afford pentacyclus 97. The final ring closure step was accomplished through catalytic hydrogenation of the nitro compound 97 with 10% palladium on carbon and spontaneous cyclocondensation to give eilatin (90) (Scheme 15) [32].
Scheme 15.
Synthesis of eilatin (90): (a) Tf2O, CH2Cl2, Et3N (93%); (b) formic acid, Et3N, Pd(OAc)2, dppf, DMF (87%); (c) CAN, acetonitrile/H2O (60%); (d) CeCl3·7H2O, 2-aminoacetophenone, EtOH (54%); (e) conc. H2SO4/AcOH (83%); (f) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (75% over two steps); (g) 10% Pd/C, EtOH (85%).
Scheme 15.
Synthesis of eilatin (90): (a) Tf2O, CH2Cl2, Et3N (93%); (b) formic acid, Et3N, Pd(OAc)2, dppf, DMF (87%); (c) CAN, acetonitrile/H2O (60%); (d) CeCl3·7H2O, 2-aminoacetophenone, EtOH (54%); (e) conc. H2SO4/AcOH (83%); (f) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (75% over two steps); (g) 10% Pd/C, EtOH (85%).
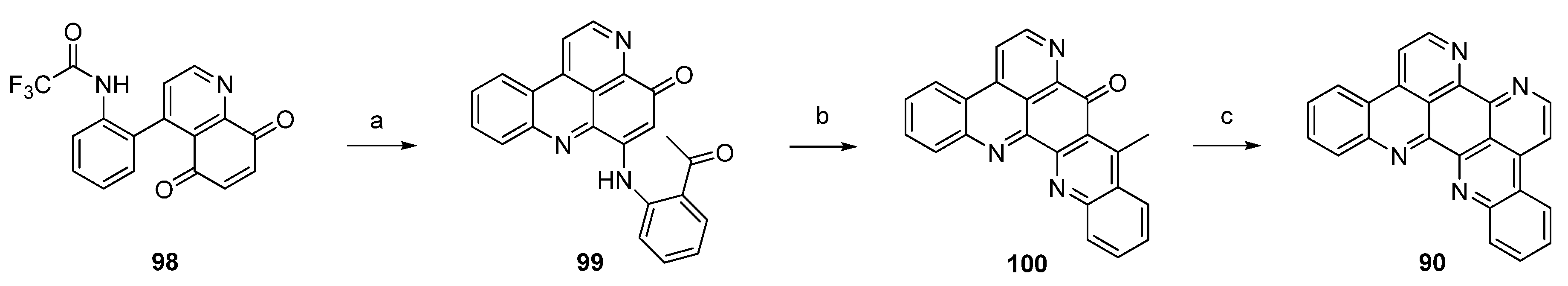
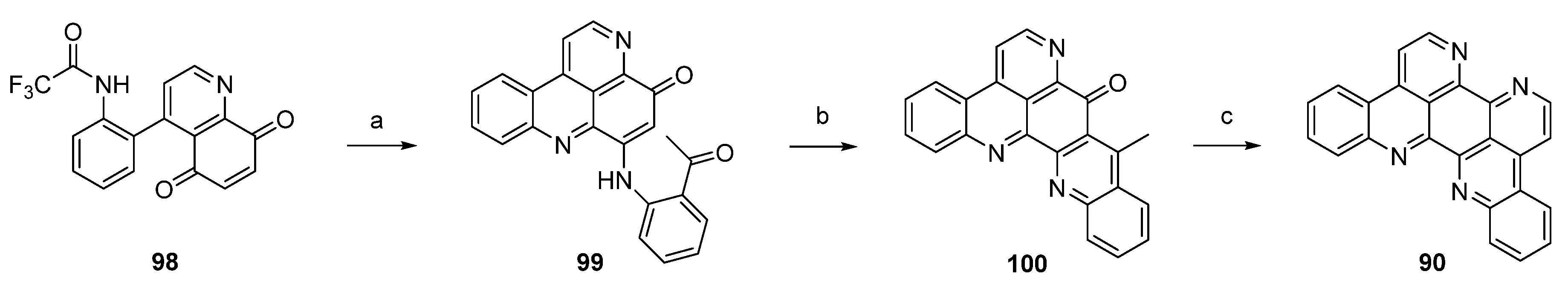
In a closely related approach, the same authors synthesized eilatin (90) starting from quinolone-5,8-dione 98 bearing a trifluoroacetyl-protected amino group at the phenyl ring. Regioselective CeCl3-catalyzed oxidative amination of 98 and 2-aminoacetophenone furnished, obviously under spontaneous N-deprotection/cyclocondensation, the tetracyclic aminoquinone 99. After an acid-catalyzed cyclization step, which afforded hexacyclus 100, eilatin (90) was obtained in a one-pot annulation of ring G using N,N-dimethylformamide diethyl acetal and ammonium chloride (Scheme 16) [32].
Scheme 16.
Synthesis of eilatin (90): (a) CeCl3·7H2O, 2-aminoacetophenone, EtOH (52%); (b) conc. H2SO4/AcOH (75%); (c) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (41% over two steps).
Scheme 16.
Synthesis of eilatin (90): (a) CeCl3·7H2O, 2-aminoacetophenone, EtOH (52%); (b) conc. H2SO4/AcOH (75%); (c) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl, AcOH (41% over two steps).
Kashman’s biomimetic approach towards eilatin (90) suggests that kynuramine and o-benzoquinone (or the corresponding catechol, 102), both natural products, can be considered as potential biosynthetic precursors of this alkaloid. In the first step, the monoprotected trifluoroacetyl kynuramine 101 was reacted with catechol (102) under oxidative conditions to give compound 103 in very low yield. Treatment of 103 with ammonia in methanol and DMAP directly furnished alkaloid 90. An alternative route towards eilatin (90) was accomplished by the treatment of 103 first with BF3 etherate. The so-obtained pentacyclus 104 was converted to eilatin (90) under alkaline conditions (NH3-MeOH) (Scheme 17) [34]. No yields were given for the final steps in both approaches.
Scheme 17.
Biomimetic synthesis of eilatin (90): (a) NaIO3, EtOH (15%); (b) BF3·OEt2, CH2Cl2 (no yield given); (c) NH3, MeOH (no yield given) [34].
Scheme 17.
Biomimetic synthesis of eilatin (90): (a) NaIO3, EtOH (15%); (b) BF3·OEt2, CH2Cl2 (no yield given); (c) NH3, MeOH (no yield given) [34].
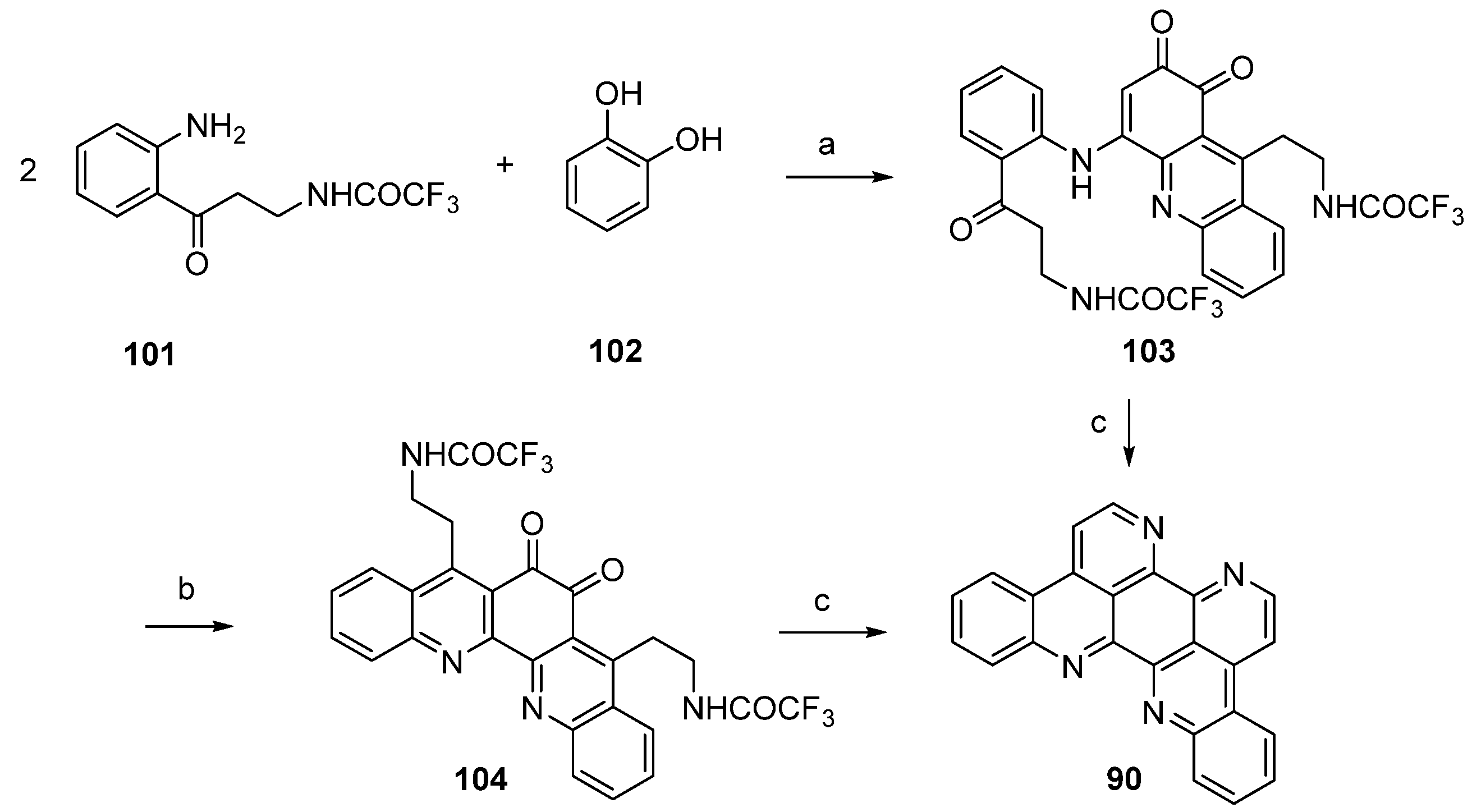
In this and an accompanying paper [35] the Kashman group described several unsuccessful approaches to eilatin (90). One of these attempts is illustrated in Scheme 18. In a conversion closely related to the one described in Scheme 17, compound 105 was prepared from 2-aminoacteophenone (7) and catechol (102) under oxidative conditions. Ring closure was accomplished by treating 105 with BF3-etherate and this furnished the symmetrical pentacyclic dibenzo-1,10-phenanthroline-5,6-dione 106 in high yield. A completion of this synthesis (annulation of rings F and G of eilatin) has not yet been published.
Scheme 18.
Synthesis of postulated eilatin precursor 106: (a) NaIO3, EtOH (60%); (b) BF3·OEt2, CH2Cl2 (70%).
Scheme 18.
Synthesis of postulated eilatin precursor 106: (a) NaIO3, EtOH (60%); (b) BF3·OEt2, CH2Cl2 (70%).
The synthetic strategy employed by Glazer and Tor [36] for the preparation of a RuII-eilatin complex started with “pre-eilatin” (107), a formal seco analogue of the alkaloid, which was obtained in 54% yield by Pd-catalyzed homo-coupling of 4-bromobenzo[c][2,7]naphthyridine (42). Treatment of “pre-eilatin” (107) with Ru(bpy)2Cl2·5H2O in ethylene glycol and water gave the dark-red RuII complex 108 ([Ru(bpy)2(pre-eilatin)]2+), which was converted into the deep-green [Ru(bpy)2(eilatin)]2+ complex 109 by exposure to palladium on carbon in ethylene glycol and acetone at elevated temperatures in almost quantitative yield. Separation of free eilatin from the complex 109 is not mentioned in the publication. It is noteworthy that treatment of the free “pre-eilatin” (107) ligand with Pd/C did not yield eilatin (90) (Scheme 19). This approach is quite short, but affords stoichiometric amounts of the expensive Ru complex for the key step.
Scheme 19.
Synthesis of RuII-eilatin complex 109: (a) Pd(OAc)2, Bu4NBr, K2CO3, iPrOH, DMF (b) Ru(bpy)2Cl2·5H2O, ethylene glycol, water (62%); (c) Pd/C, ethylene glycol-acetone (97%).
Scheme 19.
Synthesis of RuII-eilatin complex 109: (a) Pd(OAc)2, Bu4NBr, K2CO3, iPrOH, DMF (b) Ru(bpy)2Cl2·5H2O, ethylene glycol, water (62%); (c) Pd/C, ethylene glycol-acetone (97%).

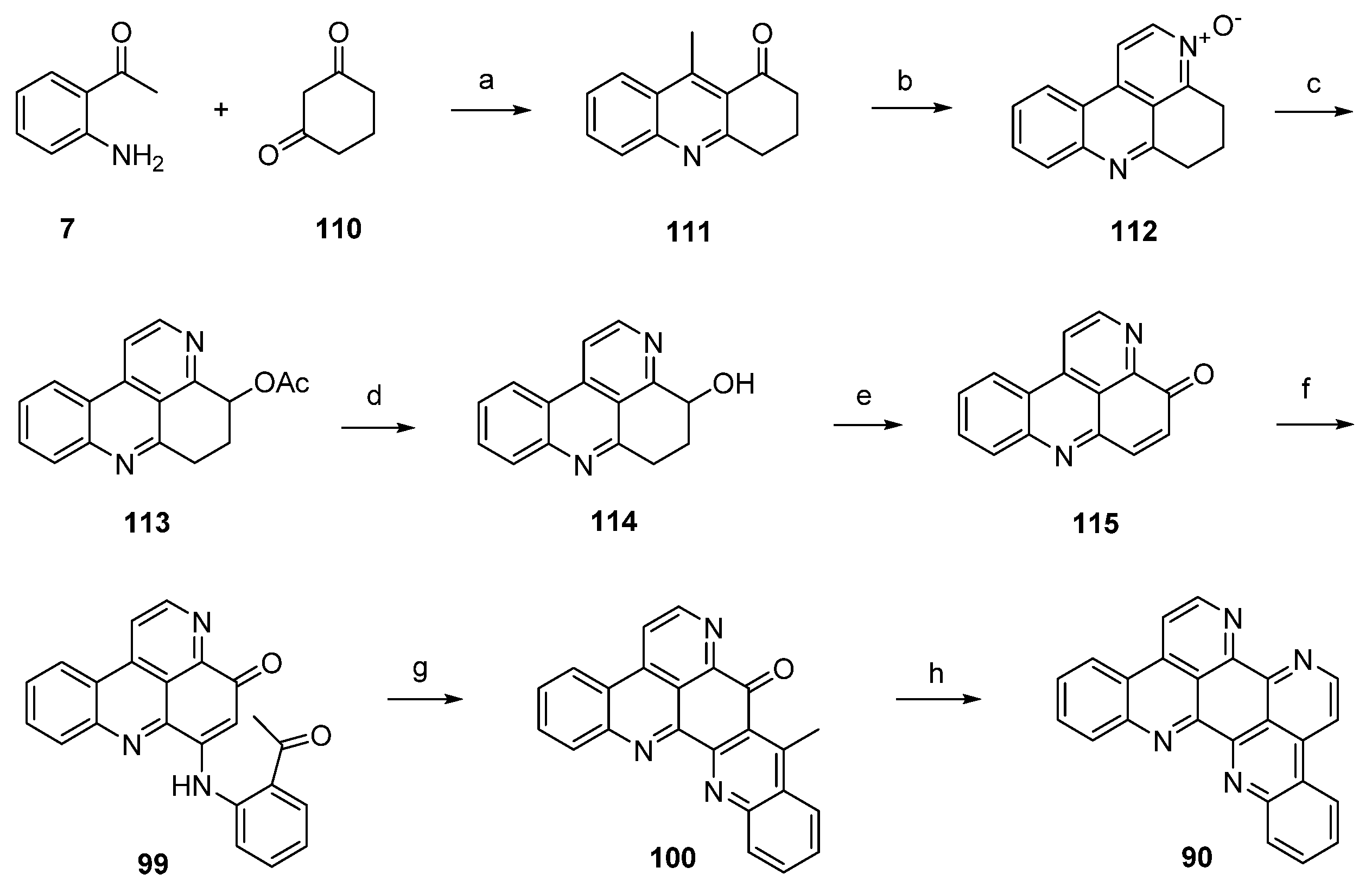
A divergent synthesis leading to both eilatin (90) and its isomer isoeilatin (119) was published by Plodek and Bracher in 2015 [37]. This approach started with a Friedländer reaction of 2-aminoacetophenone (7) and 1,3-cyclohexanedione (110) to afford acridone 111. Following one-pot annulation of a pyridine-N-oxide ring was accomplished by condensation of the acidic methyl group of 111 with N,N-dimethylformamide diethyl acetal under controlled conditions (55 °C) and subsequent ring closure with hydroxylamine hydrochloride to give N-oxide 112. In a Boekelheide rearrangement, this N-oxide was converted to the acetoxy compound 113 by heating with acetic anhydride. Alkaline ester hydrolysis of 113 afforded alcohol 114 in 84% yield. Subsequent oxidation of this alcohol under mild conditions with MnO2 under concomitant ring dehydrogenation furnished pyridoacridone 115 in 66% yield. Further conversion to eilatin (90) was performed in analogy to Bracher’s total synthesis of ascididemin (3) [7]. Thus, regioselective oxidative amination of 115 with 2-aminoacetophenone under CeCl3 catalysis gave arylamino derivative 99. Next, acid-catalyzed cyclization furnished hexacyclic compound 100 in almost quantitative yield. The final annulation of ring G was performed in a one-pot reaction by condensation of the acidic methyl group with N,N-dimethylformamide diethyl acetal, followed by heating with ammonium chloride/glacial acetic acid to give 90. Hence, alkaloid 90 was synthesized in seven steps with an overall yield of 6.9% from only two building blocks (1,3-cyclohexanedione, 2-aminoacetophenone) (Scheme 20) [37].
Scheme 20.
Synthesis of eilatin (90): (a) Reflux (54%); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH2OH·HCl (41% over two steps); (c) acetic anhydride (66%); (d) 10% NaOH in H2O/MeOH (84%); (e) MnO2 (66%); (f) 2-aminoacetophenone, CeCl3·7H2O (74%); (g) 10% H2SO4 in AcOH (96%); (h) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl (65% over two steps).
Scheme 20.
Synthesis of eilatin (90): (a) Reflux (54%); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH2OH·HCl (41% over two steps); (c) acetic anhydride (66%); (d) 10% NaOH in H2O/MeOH (84%); (e) MnO2 (66%); (f) 2-aminoacetophenone, CeCl3·7H2O (74%); (g) 10% H2SO4 in AcOH (96%); (h) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl (65% over two steps).
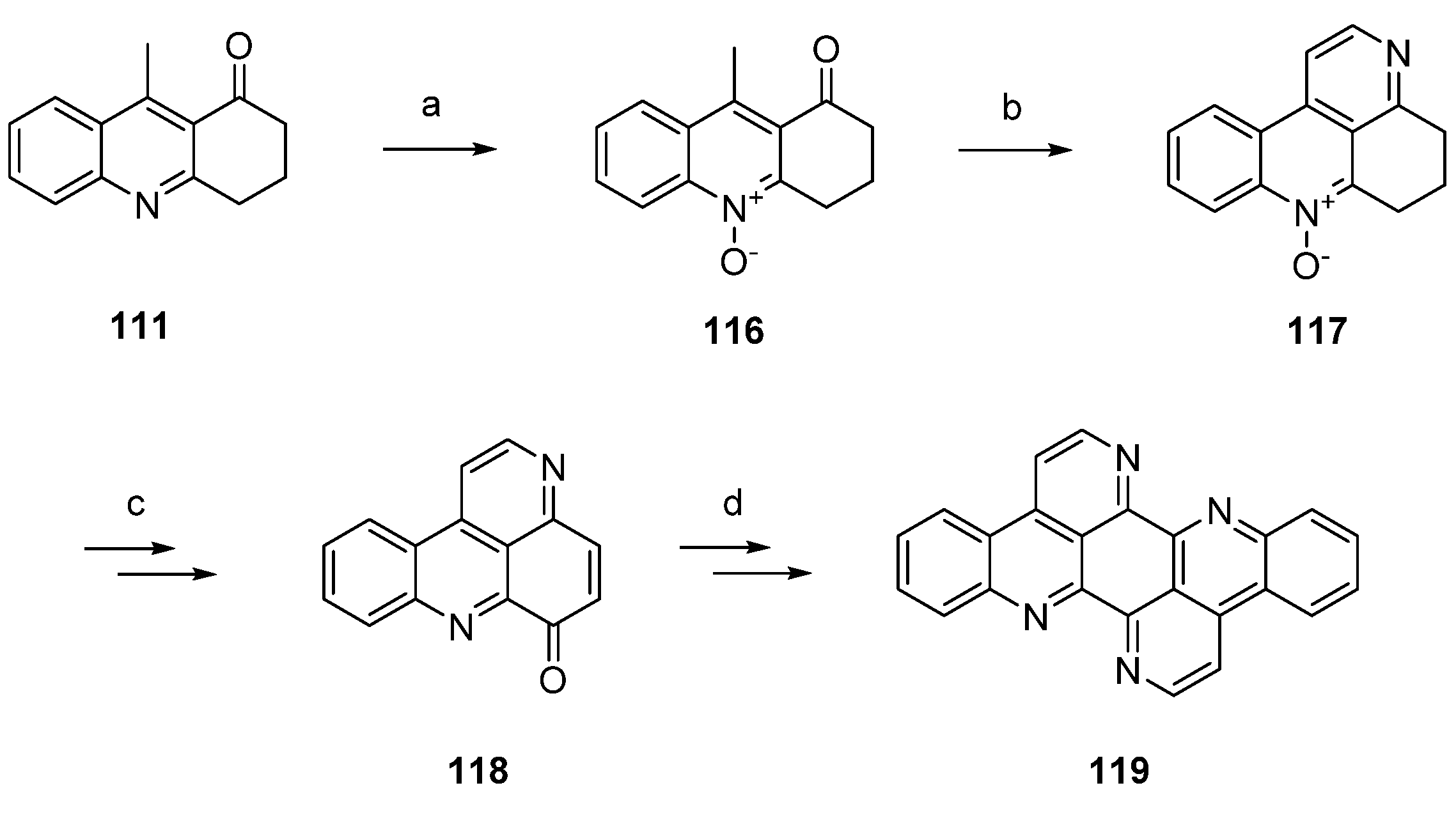
Isoeilatin (119), a synthetic isomer of 90, was prepared from the same building blocks by using different reaction conditions. In this synthesis, acridone 111 was first treated with meta-chloroperoxybenzoic acid (mCPBA) to furnish N-oxide 116. An annulation of a pyridine ring through condensation with N,N-dimethylformamide diethyl acetal and subsequent ring closure with ammonium chloride gave tetracyclic compound 117, an isomer of the N-oxide 112 utilized in the synthesis of eilatin (90), in 38% yield. In analogy to the above-mentioned protocol, N-oxide 117 was subjected to Boekelheide rearrangement, ester hydrolysis and oxidation with MnO2 to furnish pyridoacridone 118. The following steps (oxidative amination, acid-catalyzed cyclization, annulation of the seventh ring using N,N-dimethylformamide diethyl acetal and ammonium chloride) were performed in full analogy to the above-mentioned total synthesis of eilatin (90) [37]. Isoeilatin (119) was obtained in eight steps with 5.1% total yield (Scheme 21) [37].
Scheme 21.
Synthesis of isoeilatin (119): (a) mCPBA, CH2Cl2 (88%); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl (38% over two steps); (c) acetic anhydride (80%); then 10% NaOH in H2O/MeOH; then MnO2, toluene (50% over two steps); (d) 2-aminoacetophenone, CeCl3·7H2O; then 10% H2SO4 in AcOH; then N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl (38% over four steps).
Scheme 21.
Synthesis of isoeilatin (119): (a) mCPBA, CH2Cl2 (88%); (b) N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl (38% over two steps); (c) acetic anhydride (80%); then 10% NaOH in H2O/MeOH; then MnO2, toluene (50% over two steps); (d) 2-aminoacetophenone, CeCl3·7H2O; then 10% H2SO4 in AcOH; then N,N-dimethylformamide diethyl acetal, DMF; then NH4Cl (38% over four steps).
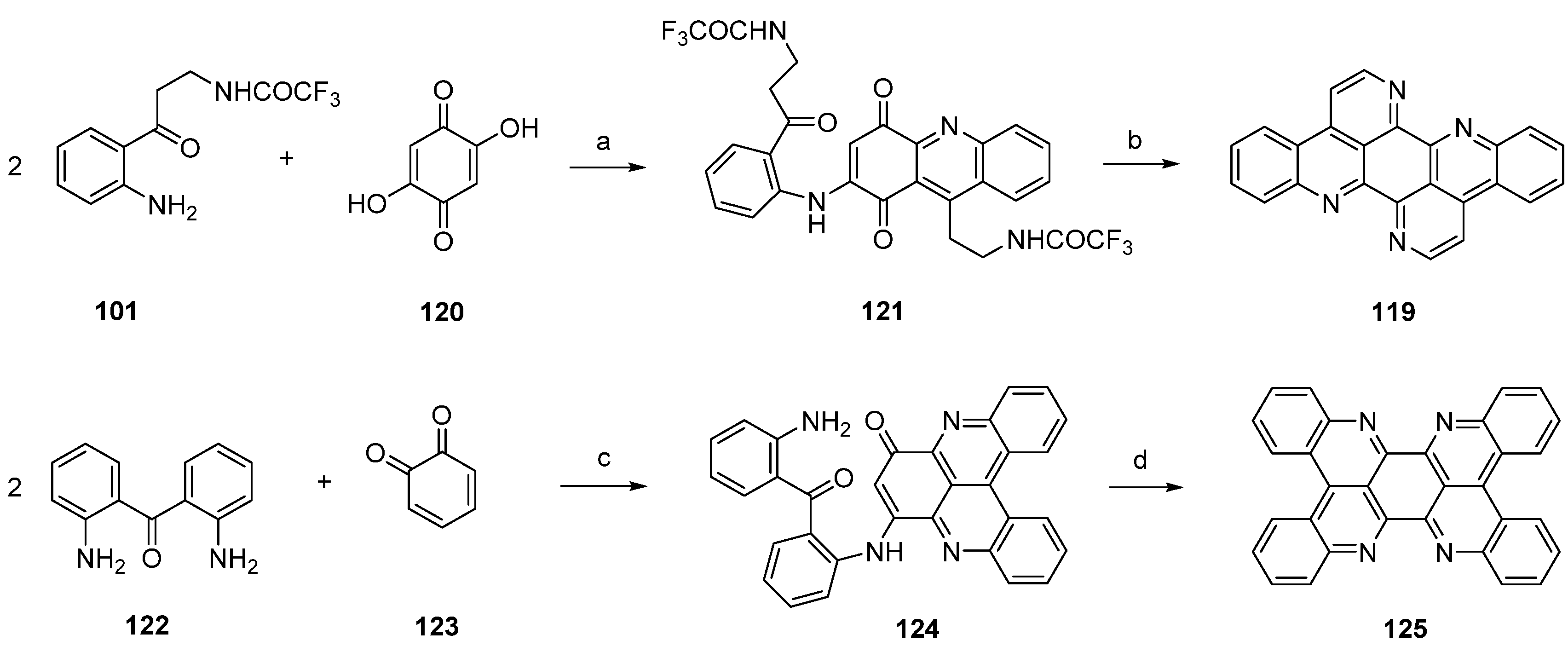
Another approach towards isoeilatin (119) was reported by Kashman [38] in a paper published in 1994. Reaction of monoprotected trifluoroacetyl kynuramine 101 and 2,5-dihydroxy-1,4-cyclohexanedione (120) under acidic conditions (AcOH/Et3N) furnished isoeilatin precursor 121 in 7% yield. Following treatment of with ammonia in methanol provided isoeilatin (119) (Scheme 22).
The authors also described the synthesis of a dibenzo analogue of eilatin (125) in this publication [38]. For this purpose, 2,2′-diaminobenzophenone (122) was reacted with o-benzoquinone (123) in the presence of CeCl3 to give compound 124. Subsequent treatment with acid (AcOH/H2SO4/Et3N) of 124 afforded the highly symmetrical “dibenzoeilatin” (125) (Scheme 22) [38].
Scheme 22.
Synthesis of isoeilatin (119): (a) AcOH/Et3N (7%); (b) NH3 in methanol (43%). Synthesis of dibenzoeilatin (125); (c) CeCl3·7H2O, EtOH (36%); (d) AcOH/H2SO4/Et3N (no yield given).
Scheme 22.
Synthesis of isoeilatin (119): (a) AcOH/Et3N (7%); (b) NH3 in methanol (43%). Synthesis of dibenzoeilatin (125); (c) CeCl3·7H2O, EtOH (36%); (d) AcOH/H2SO4/Et3N (no yield given).
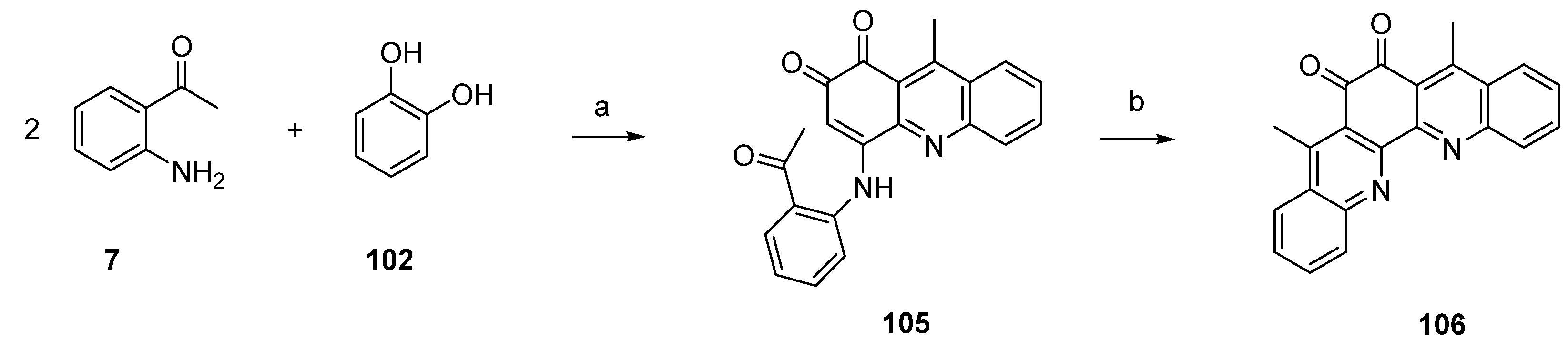
5. Styelsamine- and Cystodytin-Type Pyridoacridines
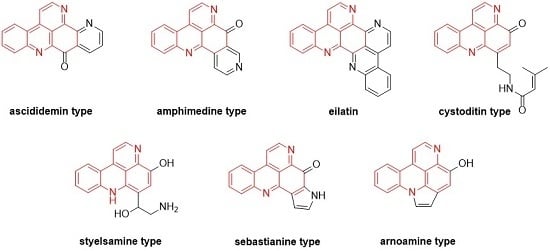
Styelsamine- and cystodytin-type pyridoacridines are teracyclic alkaloids bearing a functionalized side-chain at C-6. In contrast to cystodytin-type alkaloids, which possess the pyrido[4,3,2-mn]acridin-4-one skeleton, styelsamine alkaloids are characterized by the hydroxylated pyrido[4,3,2-mn]acridine core. Until today, four stylesamine-type (stylesamines A–D; 126–129) [39] and 11 cystodytin-type alkaloids (cystodytin A–K; 130–140) [40,41,42,43] have been isolated and characterized (Figure 5).
Figure 5.
Structures of the styelsamines A–D (126–129) and cystodytins A–K (130–140).
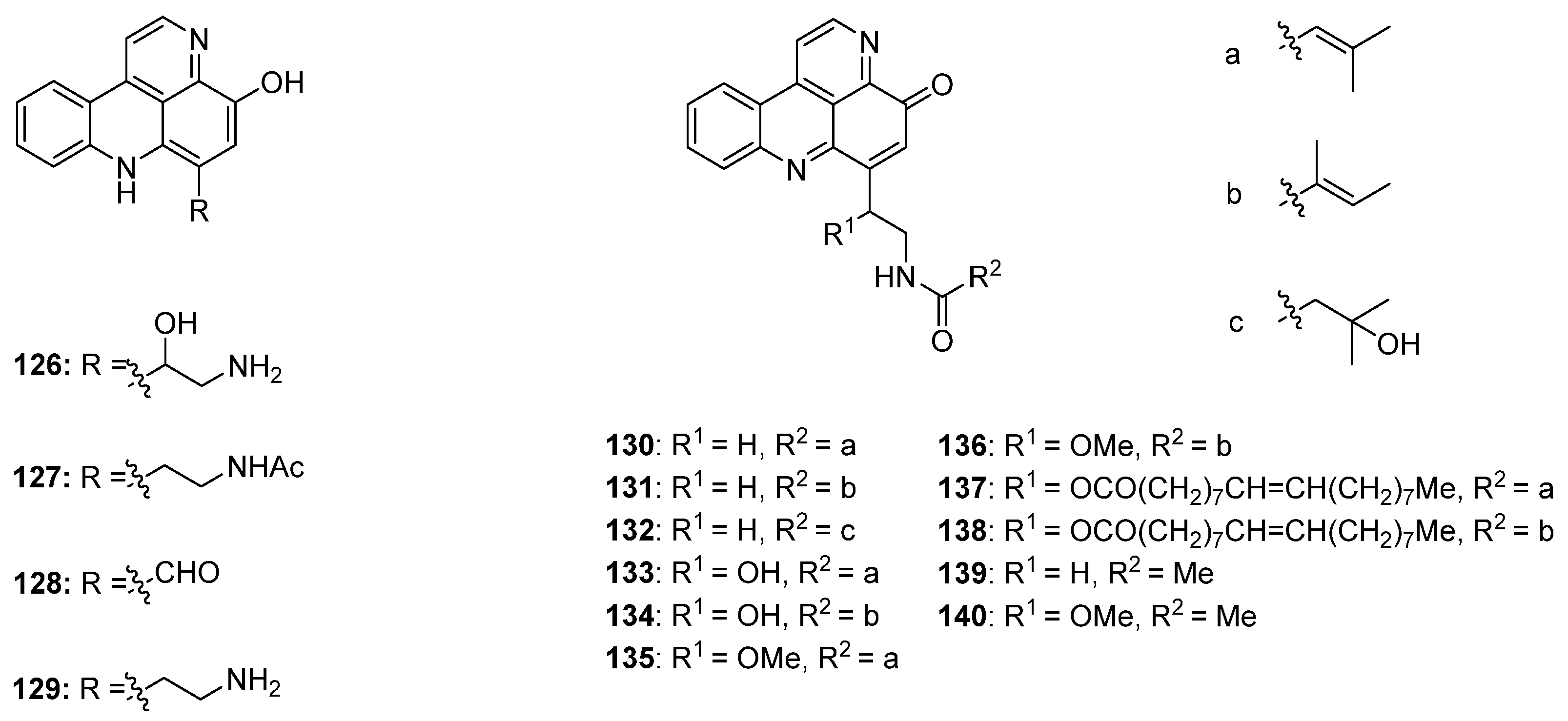
Based on Skyler and Heathcock’s biomimetic approach [44] to stylesamine B (127) and cystodytin J (139), Fong and Copp reported the synthesis of a series of side-chain substituted analogues of styelsamine and cystodytin alkaloids in 2013 [45]. The key step of this biomimetic strategy is a CeCl3-catalyzed oxidative coupling of kynuramine (141) and functionalized dopamine analogues 142–147 in the presence of silver oxide. While kynuramine (141) was prepared from a N-protected tryptamine via oxidative cleavage of the pyrrole ring, the dopamine analogues 142–147 were synthesized from 3,4-dimethoxyphenethylamine by N-acylation and subsequent O-demethylation (both syntheses are not shown in Scheme 23). In situ oxidation of the catechol moiety of the dopamines with silver oxide gives ortho-quinones, which undergo CeCl3-mediated nucleophilic addition of the aromatic amino group of kynuramine (141) (the more basic aliphatic side-chain amino group is prevented from this reaction by protonation) and re-oxidation to an aminoquinone. Subsequent treatment with 6 M hydrochloric acid resulted in cyclization to the acridines and, finally, closure of the fourth ring in an imine formation/tautomerization sequence related to a protocol that is known from Kashman’s biomimetic ascididemin synthesis [13]. The stylesamine-type pyridoacridines 127, 148–152 were isolated in yields of 6% to 20%. Subsequent oxidation of 127, 148–152 with one equivalent of silver oxide in bicarbonate-buffered methanol furnished cystodytin-type pyridoacridines 139, 153–157 in 13%–79% yields (Scheme 23) [45].
Scheme 23.
Synthesis of styelsamine (127, 148–152) and cystodytine analogues (139, 153–157): (a) CeCl3·7H2O, Ag2O, MeOH/AcOH (2:1), then 6 M HCl (6%–20%); (b) Ag2O (one equiv.), MeOH (13%–79%).
Scheme 23.
Synthesis of styelsamine (127, 148–152) and cystodytine analogues (139, 153–157): (a) CeCl3·7H2O, Ag2O, MeOH/AcOH (2:1), then 6 M HCl (6%–20%); (b) Ag2O (one equiv.), MeOH (13%–79%).

In 2002, Skyler and Heathcock [3] reported that treatment of stylesamine B (127) with hydrochloric acid (4 M) in methanol afforded the alkylamino analogue of stylesamine D 158 in quantitative yield. Repeating this experiment, Copp isolated, besides the desired product 158, the unexpected O-methyl analogue 159. With this O-methyl analogue 159 in hand, the authors were able to prepare the N-acyl analogues 160–162. While acrylamide 160 and 2-phenylacetamide 161 were synthesized by treatment of 159 with appropriate carboxylic acids in DMF and PyBOP, 3-phenylpropanamide 162 was prepared from 159 by reaction with dihydrocinnamoyl chloride in THF (Scheme 24) [45].
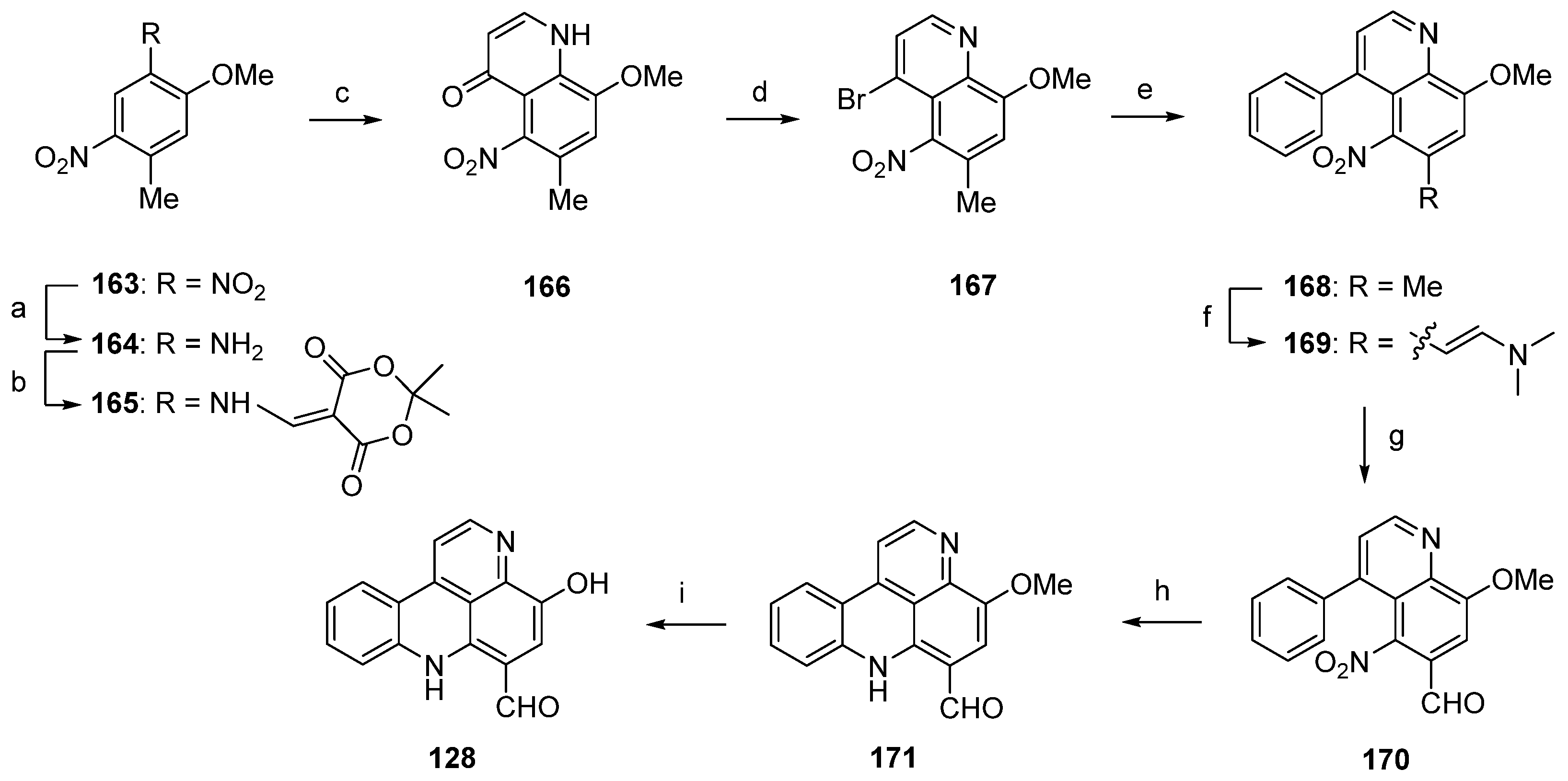
In 2003, Nakahara and Kubo reported the first total synthesis of styelsamine C (128) [46,47]. This nine-step approach with an overall yield of 16% started with selective transfer hydrogenation of 5-methoxy-2,4-dinitrotoluene (163) using cyclohexene and 10% palladium on carbon catalyst to give 2-methoxy-4-methyl-5-nitroaniline (164). The following treatment with Meldrum’s acid furnished enaminone 165, which was cyclized in refluxing diphenyl ether to afford 4-quinolone 166 (47% yield over three steps). Reaction of 166 with phosphoryl bromide gave 4-bromoquinoline 167 in 78% yield. The 4-phenylquinoline 168 was obtained in almost quantitative yield by the Suzuki cross-coupling reaction of 167 with phenylboronic acid under standard conditions. The acidic methyl group at C-6 of the nitroquinoline 168 was converted to an aldehyde in two steps. Treatment of 168 with N,N-dimethylformamide dimethyl acetal at 170 °C (in the full paper [47] the authors present different conditions, N,N-dimethylformamide dimethyl acetal at 145 °C, but the same yield) furnished enamine 169 in 91% yield. Subsequent oxidative cleavage with NaIO4 in THF/water afforded aldehyde 170 in 90% yield. The final cyclization step was achieved via an intramolecular nitrene insertion reaction by the heating of 170 with triethyl phosphite, yielding the tetracyclic compound 171 in 65% yield. Styelsamine C (128) was finally prepared by O-demethylation of 171 with boron tribromide in 86% yield (Scheme 25) [46,47].
Scheme 24.
Synthesis of analogues of styelsamine alkaloids: (a) 4 M HCl/MeOH (1:1) (158, 60% and 159, 45%); (b) for 160 and 161: corresponding carboxylic acid, DMF, CH2Cl2, Et3N, PyBOP (160, 88% and 161, 48%); for 162: dihydrocinnamoyl chloride, THF, Et3N (43%).
Scheme 24.
Synthesis of analogues of styelsamine alkaloids: (a) 4 M HCl/MeOH (1:1) (158, 60% and 159, 45%); (b) for 160 and 161: corresponding carboxylic acid, DMF, CH2Cl2, Et3N, PyBOP (160, 88% and 161, 48%); for 162: dihydrocinnamoyl chloride, THF, Et3N (43%).

Scheme 25.
Synthesis of styelsamine C (128): (a) 10% Pd/C, cyclohexene/EtOH (60%); (b) Meldrum’s acid, trimethyl orthoformate (94%); (c) diphenyl ether, reflux (83%); (d) POBr3 (78%); (e) phenylboronic acid, EtOH/toluene, K2CO3, Pd(PPh3)4 (94%); (f) N,N-dimethylformamide dimethyl acetal, 170 °C (91%); (g) NaIO4, THF/H2O (90%); (h) P(OEt)3 (65%); (i) BBr3 in CH2Cl2 (86% over two steps).
Scheme 25.
Synthesis of styelsamine C (128): (a) 10% Pd/C, cyclohexene/EtOH (60%); (b) Meldrum’s acid, trimethyl orthoformate (94%); (c) diphenyl ether, reflux (83%); (d) POBr3 (78%); (e) phenylboronic acid, EtOH/toluene, K2CO3, Pd(PPh3)4 (94%); (f) N,N-dimethylformamide dimethyl acetal, 170 °C (91%); (g) NaIO4, THF/H2O (90%); (h) P(OEt)3 (65%); (i) BBr3 in CH2Cl2 (86% over two steps).
6. Sebastianine A
Sebastianines A (172) and B (173) are members of a pyridoacridine subclass that possesses a pyrrole ring fused to ring A of the pyridoacridine ring system (Figure 6). In 2002, Torres et al. reported the isolation and structure elucidation of sebastianines A (172) and B (173) from the ascidian Cystodytes dellechiaijei [48].
Figure 6.
Structures of sebastianines A (172) and B (173).
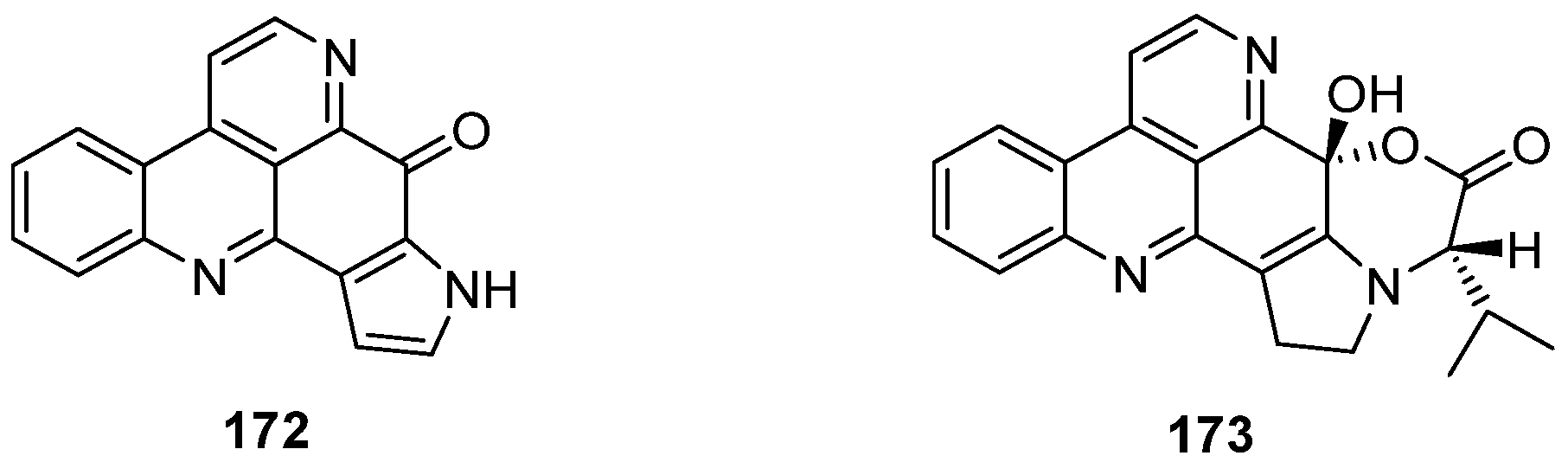
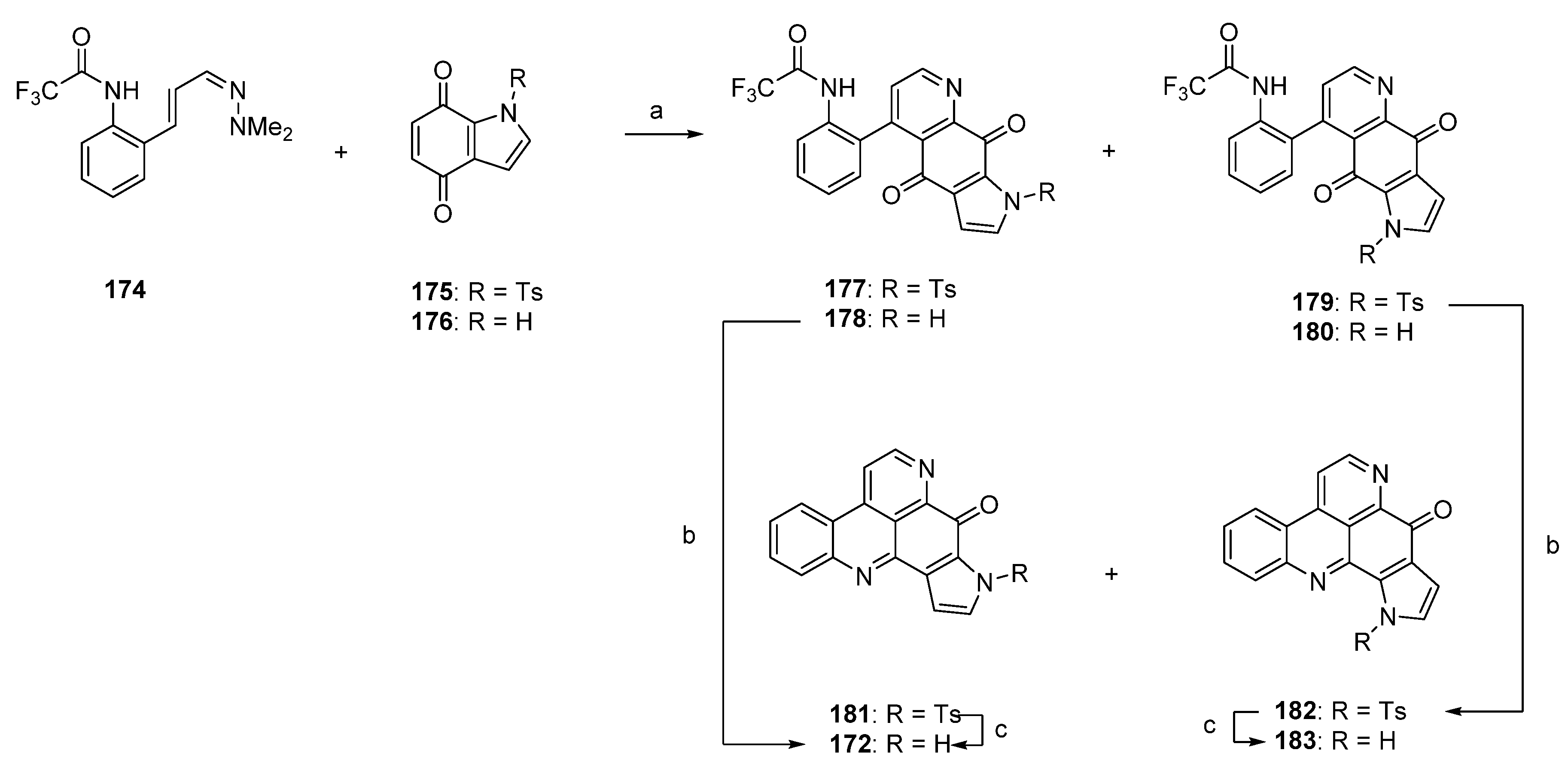
One year later, Delfourne published the first total synthesis of sebastianine A (172) [49]. This approach, which stands in close relationship to the above-mentioned synthesis of demethyldeoxyamphimedine (56) [12], again comprises a hetero-Diels-Alder cycloaddition as the key step. The required dienophiles, N-tosylindole-4,7-dione 175 as well as the corresponding unprotected indole-4,7-dione 176, were obtained via 4,7-dimethoxyindole in multistep procedures (five and six steps). Cycloadditions with cinnanaldehyde-derived 1-azadieene 174 and subsequent re-aromatization with MnO2 gave mixtures of regioisomers 177/179 (5:95) or 178/180 (60:40) in very poor yields (6%–8%). Structures of the isomers were assigned only by analogy to the outcome of related cycloadditions performed by another group. Subsequent cyclization of 178 under alkaline conditions afforded sebastianine A (172) in 85% yield, and its regioisomer 183 was obtained in high yield by prolonged treatment of N-tosyl intermediate 179 with NaOH under cyclization and subsequent N-detosylation. (Scheme 26) [49]. The more complex alkaloid sebastianine B (173) has not been synthesized until today.
Scheme 26.
Synthesis of sebastianine A (172) and its regioisomer 183: (a) Toluene, reflux, then MnO2 (no reaction conditions given) (R = Ts, 8%; R = H, 6%); (b) NaOH, CH2Cl2 (85%–92%); (c) NaOH, CH2Cl2 (95%–98%).
Scheme 26.
Synthesis of sebastianine A (172) and its regioisomer 183: (a) Toluene, reflux, then MnO2 (no reaction conditions given) (R = Ts, 8%; R = H, 6%); (b) NaOH, CH2Cl2 (85%–92%); (c) NaOH, CH2Cl2 (95%–98%).
7. Arnoamine-Type Pyridoacridines
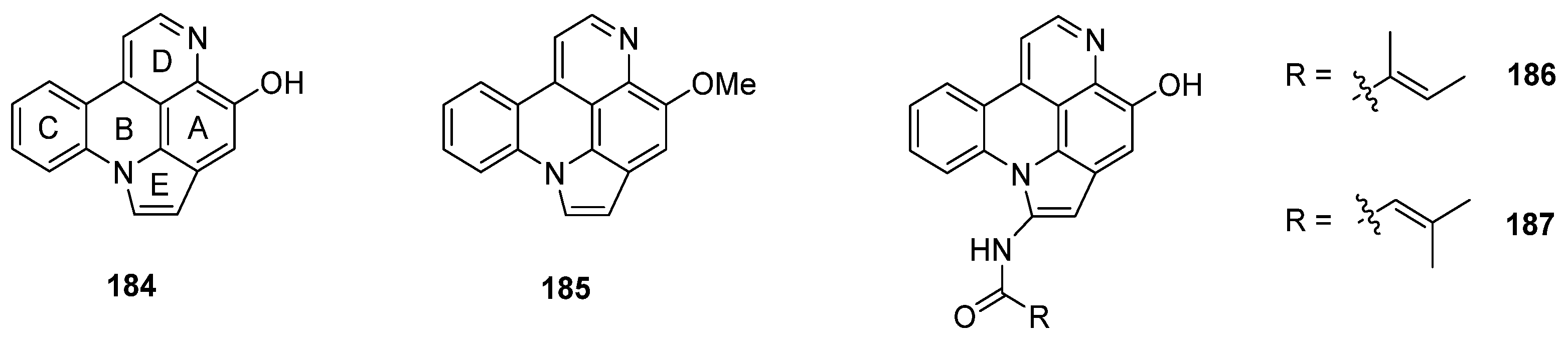
Arnoamines A–D (184–187) are a unique type of pyridoacridine alkaloids, since they bear a pyrrole ring fused to rings A and B of the pyrido[4,3,2-mn]acridine skeleton. Isolated in 1998 (arnoamines A and B) and 2013 (arnoamines C and D) from two different ascidians (Cystodytes species), these pentacycles are up to today the only known representatives of this pyridoacridine subclass (Figure 7) [50,51].
Figure 7.
Structures of arnoamines A (184), B (185), C (186), and D (187).
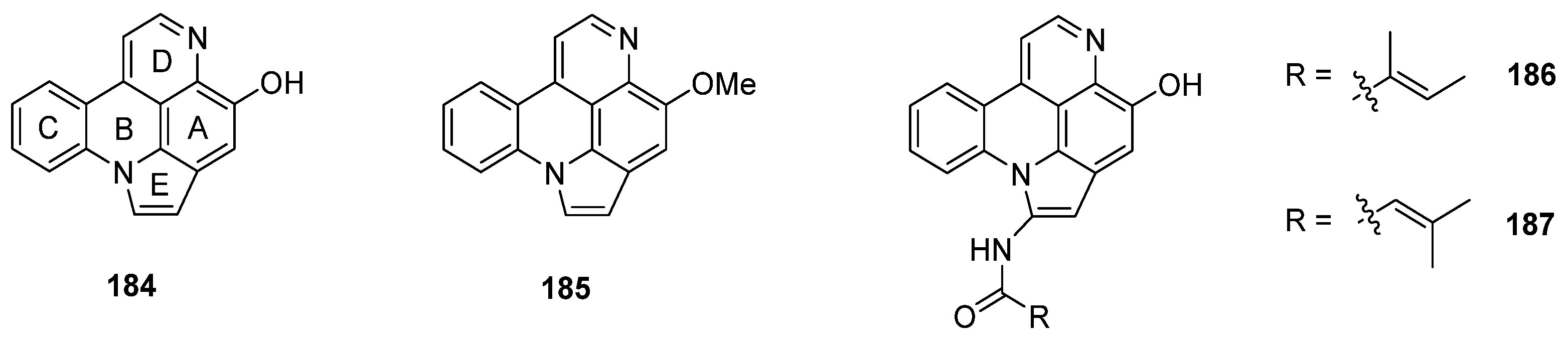
The first total syntheses of arnoamines A (184) and B (185) were reported in 2000 [52] and have already been described in Delfourne’s review [6]. The key steps of this approach were the thermolysis of an arylaminomethylene in Meldrum’s acid derivative for construction of the quinoline scaffold (rings A and D), a Suzuki cross-coupling reaction of a functionalized 4-bromoquinoline (188) for the introduction of the phenyl ring (C), annulation of the pyrrole ring via Fischer indole synthesis, followed by cyclization under intramolecular N-arylation of the pyrrole ring (E) [52].
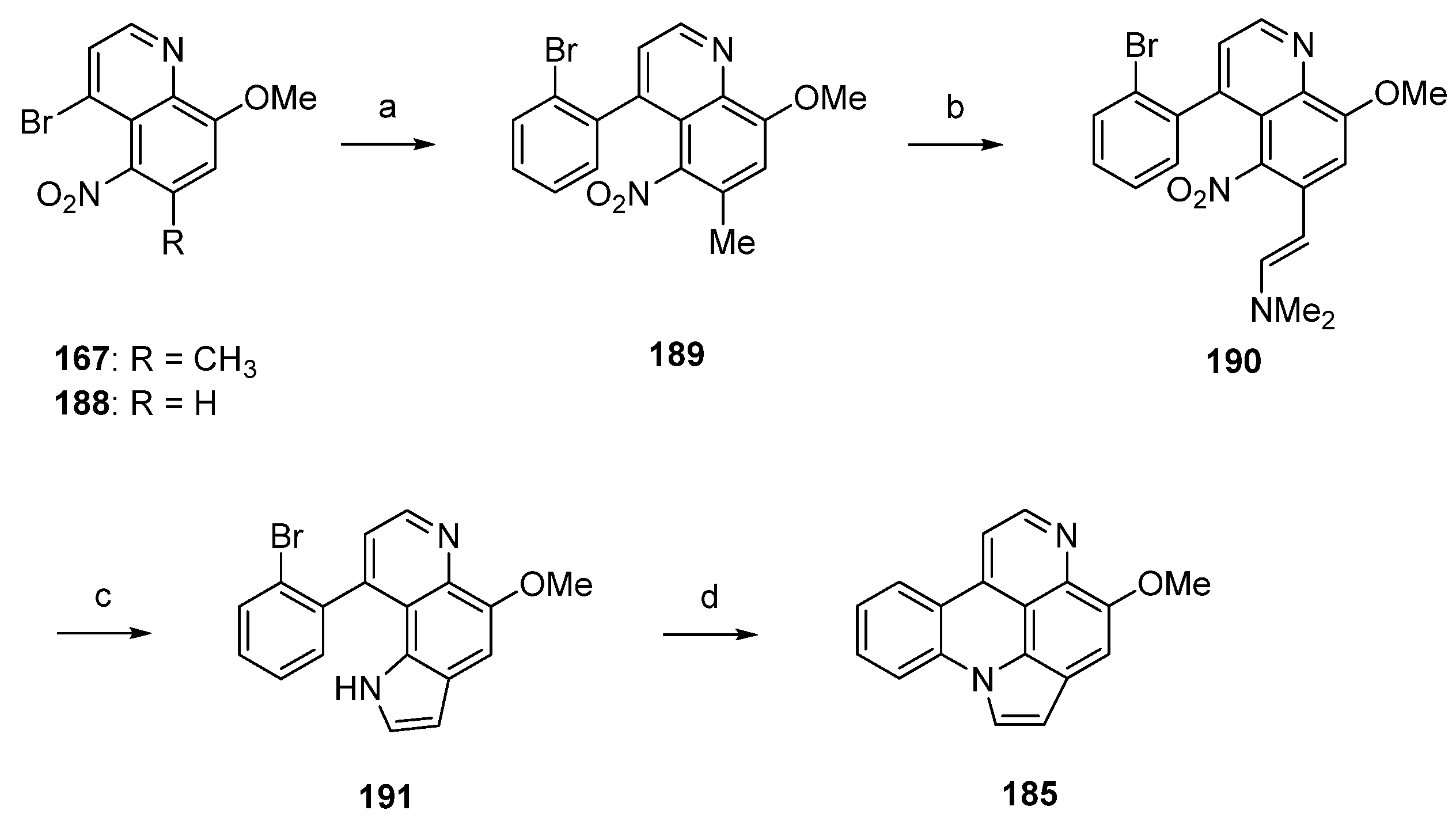
A related approach to arnoamine B (185) was reported by Nakahara et al. in 2007 [53]. Starting from readily available 5-nitroquinoline 167 (see Scheme 25), alkaloid 185 was prepared in four steps with an overall yield of 12%. In the first step, 167 was subjected to a palladium(0)-catalyzed Suzuki cross-coupling reaction to give 4-(2-bromophenyl)quinoline 189 in 85% yield. Annulation of the pyrrole ring was accomplished in low yield (17% over two steps) utilizing the Leimgruber-Batcho protocol by condensation of the CH-acidic 6-methyl group of 189 with N,N-dimethylformamide dimethyl acetal to give enamine 190 and subsequent reductive cyclization with zinc powder in 80% aqueous acetic acid. Despite the low yield, this protocol is more convenient than Delfourne’s protocol starting from the des-methyl compound 188 and utilizing a Fischer synthesis for construction of the pyrrole ring. Finally, cyclization to arnoamine B (185) was accomplished in 81% yield under intramolecular N-arylation of the pyrrole moiety of 191 under palladium(II) acetate catalysis (Scheme 27) [53].
Scheme 27.
Synthesis of arnoamine B (185): (a) from 167: 2-bromophenylboronic acid, H2O/toluene, K2CO3, Pd(PPh3)4 (85%); (b) N,N-dimethylformamide dimethyl acetal, DMF (83%); (c) Zn, AcOH/H2O (21%); (d) Pd(OAc)2, P(CMe3)3, K2CO3, xylene (81%).
Scheme 27.
Synthesis of arnoamine B (185): (a) from 167: 2-bromophenylboronic acid, H2O/toluene, K2CO3, Pd(PPh3)4 (85%); (b) N,N-dimethylformamide dimethyl acetal, DMF (83%); (c) Zn, AcOH/H2O (21%); (d) Pd(OAc)2, P(CMe3)3, K2CO3, xylene (81%).
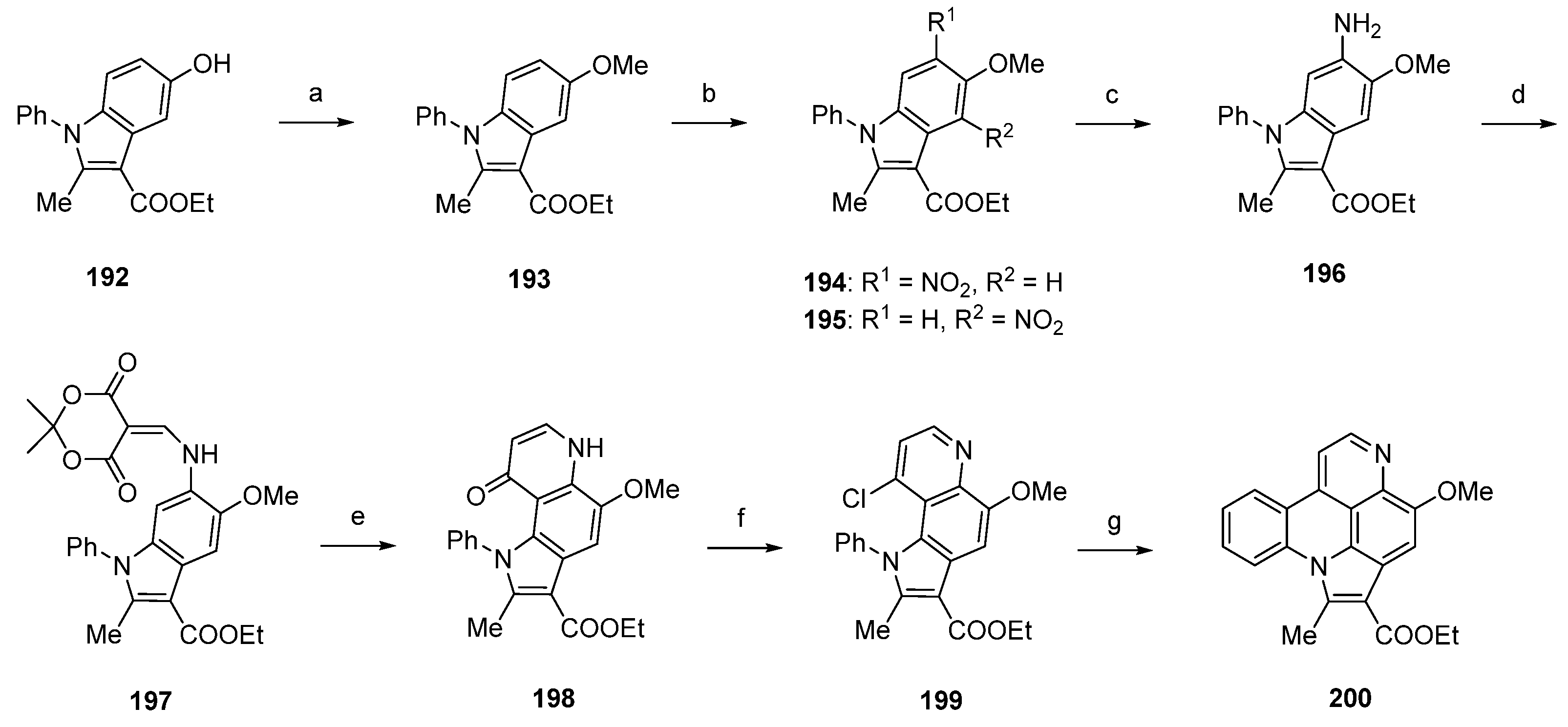
Using a different approach, starting from a N-phenylindole 192, Radchenko et al. worked out a simple and effective route to the pyrido[4,3,2-mn]pyrrolo[3,2,1-de]acridine core of arnoamine-type pyridoacridines [54]. Synthesis of the arnoamine B analogue 200 started with O-methylation of 192 with dimethyl sulfate and sodium hydroxide to afford methyl ether 193 in almost quantitative yield. The following nitration with nitric acid (75%) in acetic anhydride furnished a mixture of nitroindoles 194 and 195 in 63% and 30% yield. Raney nickel-catalyzed hydrogenation of 194 gave primary amine 196, which was converted to enamine 197 by treatment with Meldrum’s acid and trimethyl orthoformate in 95% yield. Subsequent thermal cyclization in refluxing diphenyl ether furnished pyrroloquinolone 198 in 80% yield. Arnoamine B analogue 200 was finally obtained by treating 198 with phosphoryl chloride (giving 199) and subsequent radical intramolecular cyclization using tributyltin hydride and azobis(isobutyronitrile) in 93% yield over two steps (Scheme 28) [54].
Scheme 28.
Synthesis of arnoamine B analogue 200: (a) Me2SO4, 2 M NaOH, H2O, dioxane (95%); (b) HNO3, Ac2O (194, 63% and 195, 30%); (c) from 194: H2, Raney nickel, isopropanol (no yield given); (d) Meldrum’s acid, trimethyl orthoformate (95%); (e) diphenyl ether, reflux (80%); (f) POCl3 (96%); (g) Bu3SnH, AIBN, benzene (97%).
Scheme 28.
Synthesis of arnoamine B analogue 200: (a) Me2SO4, 2 M NaOH, H2O, dioxane (95%); (b) HNO3, Ac2O (194, 63% and 195, 30%); (c) from 194: H2, Raney nickel, isopropanol (no yield given); (d) Meldrum’s acid, trimethyl orthoformate (95%); (e) diphenyl ether, reflux (80%); (f) POCl3 (96%); (g) Bu3SnH, AIBN, benzene (97%).
In close relationship to this approach, Nakahara et al. achieved the synthesis of arnoamine B (185) in five steps with an overall yield of 33% [55]. This total synthesis starts with a CuI-catalyzed N-arylation reaction of 5-methoxy-6-nitroindole (201) with iodobenzene in toluene to give indole 202 in 80% yield. Following catalytic hydrogenation over 10% palladium on carbon in methanol yielded 6-aminoindole 203. Annulation of the 4-pyridone ring was accomplished following the methods described above by treating 203 with Meldrum’s acid in trimethyl orthoformate (66% yield over two steps), followed by thermal cyclization of 204 upon heating in refluxing diphenyl ether. Subsequent treatment with phosphoryl chloride afforded chloro compound 205 in 70% yield over two steps. The final intramolecular cyclization step was performed using a large excess of tributyltin hydride and azobis(isobutyronitrile) in toluene to furnish the desired alkaloid 185 in 90% yield (Scheme 29) [55].
Arnoamines C (186) and D (187) have not been synthesized until today.
Scheme 29.
Synthesis of arnoamine B (185) by Kubo: (a) iodobenzene, CuI, (CH2NHMe)2, K3PO4, toluene (80%); (b) 10% Pd-C, methanol; (c) Meldrum’s acid, trimethyl orthoformate (66% over two steps); (d) Ph2O, reflux; then POCl3 (70% over two steps); (e) Bu3SnH (30 equiv.), AIBN (15 equiv.), toluene (90%).
Scheme 29.
Synthesis of arnoamine B (185) by Kubo: (a) iodobenzene, CuI, (CH2NHMe)2, K3PO4, toluene (80%); (b) 10% Pd-C, methanol; (c) Meldrum’s acid, trimethyl orthoformate (66% over two steps); (d) Ph2O, reflux; then POCl3 (70% over two steps); (e) Bu3SnH (30 equiv.), AIBN (15 equiv.), toluene (90%).
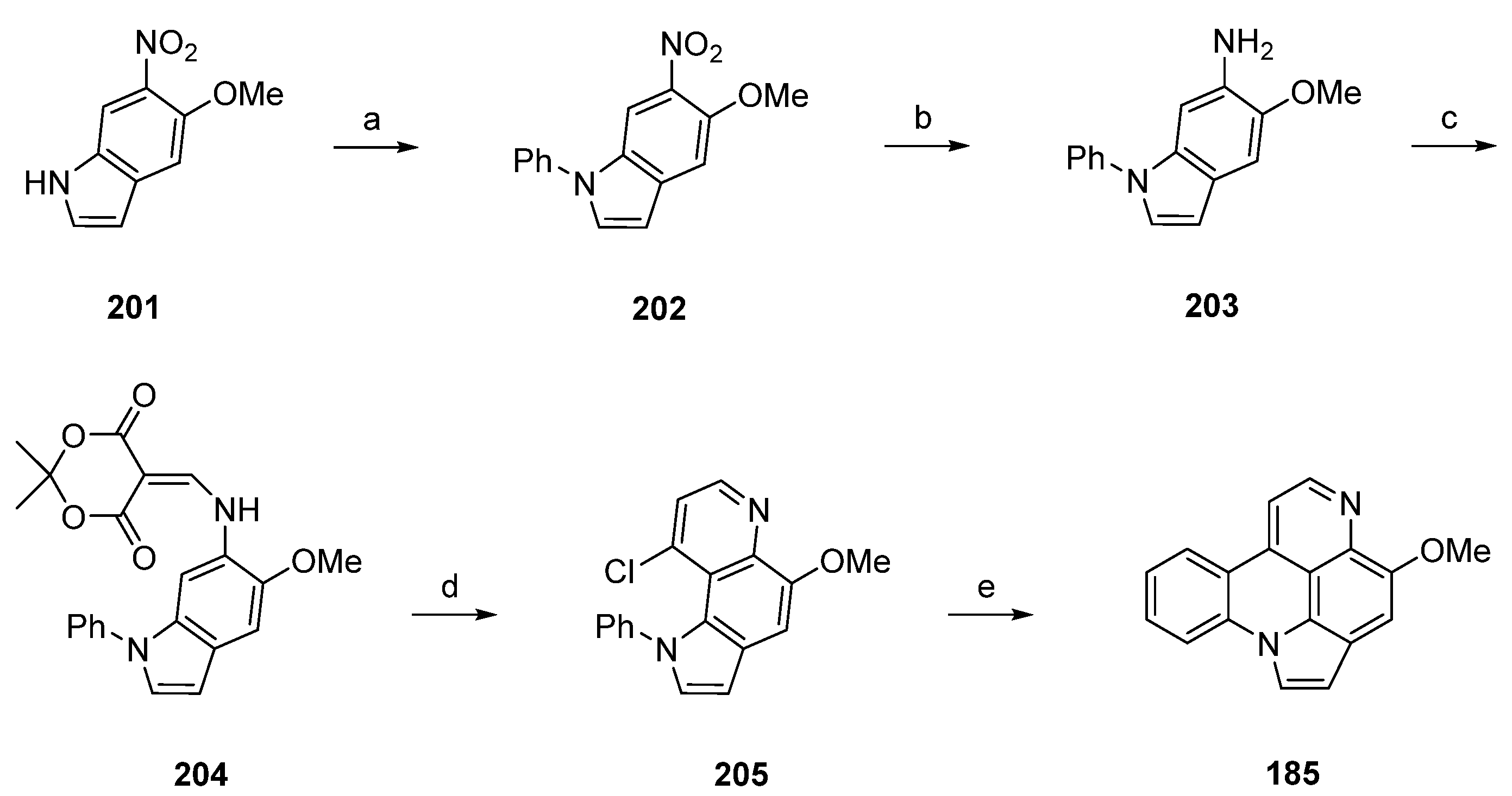
8. Subarine—A Formal Seco Analogue of Pyridoacridines
Subarine (212), a marine pyridyl benzo[c][2,7]naphthyridine alkaloid, is closely related to the pyridoacridine family, as it is formally a seco analogue of ascididemin-type alkaloids. This alkaloid was prepared following two different approaches by the Delfourne [56] and Bracher [57] groups.
Delfourne’s total synthesis of subarine (212) was accomplished in four steps with an overall yield of 70% starting from intermediate 1,10-phenanthrolin-4-ol (206), itself obtained in four steps from commercially available 8-aminoquinoline. Reaction of 206 with phosphoryl bromide and phosphorus tribromide gave the corresponding bromo derivative 207. Subsequent oxidative cleavage of 207 with potassium permanganate under alkaline conditions afforded the binicotinic-type dicarboxylic acid 208. The following conversion to diester 209 was accomplished by treatment of 208 with DCC and methanol. Palladium-catalyzed Stille cross-coupling reaction with N-(tert-butoxycarbonyl)-2-(trimethylstannyl)aniline afforded the expected phenyl-binicotinate 210, together with the already cyclized lactam 211. Subarine (212) was finally obtained by treating 211 with trifluoroacetic acid. In the case of 210, synthesis was accomplished through cleavage of the Boc protecting group and a subsequent intramolecular cyclization reaction (Scheme 30) [56].
Scheme 30.
Synthesis of subarine (212) by Delfourne: (a) POBr3, PBr3 (79%); (b) KMnO4, KOH, H2O; (c) DCC, MeOH (88% over two steps); (d) 2-(NHBoc)C6H4SnMe3, Pd(PPh3)4, 1,4-dioxane (210, 63% ; 211, 18%); (e) Et3N, CH2Cl2 (98% for both reactions).
Scheme 30.
Synthesis of subarine (212) by Delfourne: (a) POBr3, PBr3 (79%); (b) KMnO4, KOH, H2O; (c) DCC, MeOH (88% over two steps); (d) 2-(NHBoc)C6H4SnMe3, Pd(PPh3)4, 1,4-dioxane (210, 63% ; 211, 18%); (e) Et3N, CH2Cl2 (98% for both reactions).

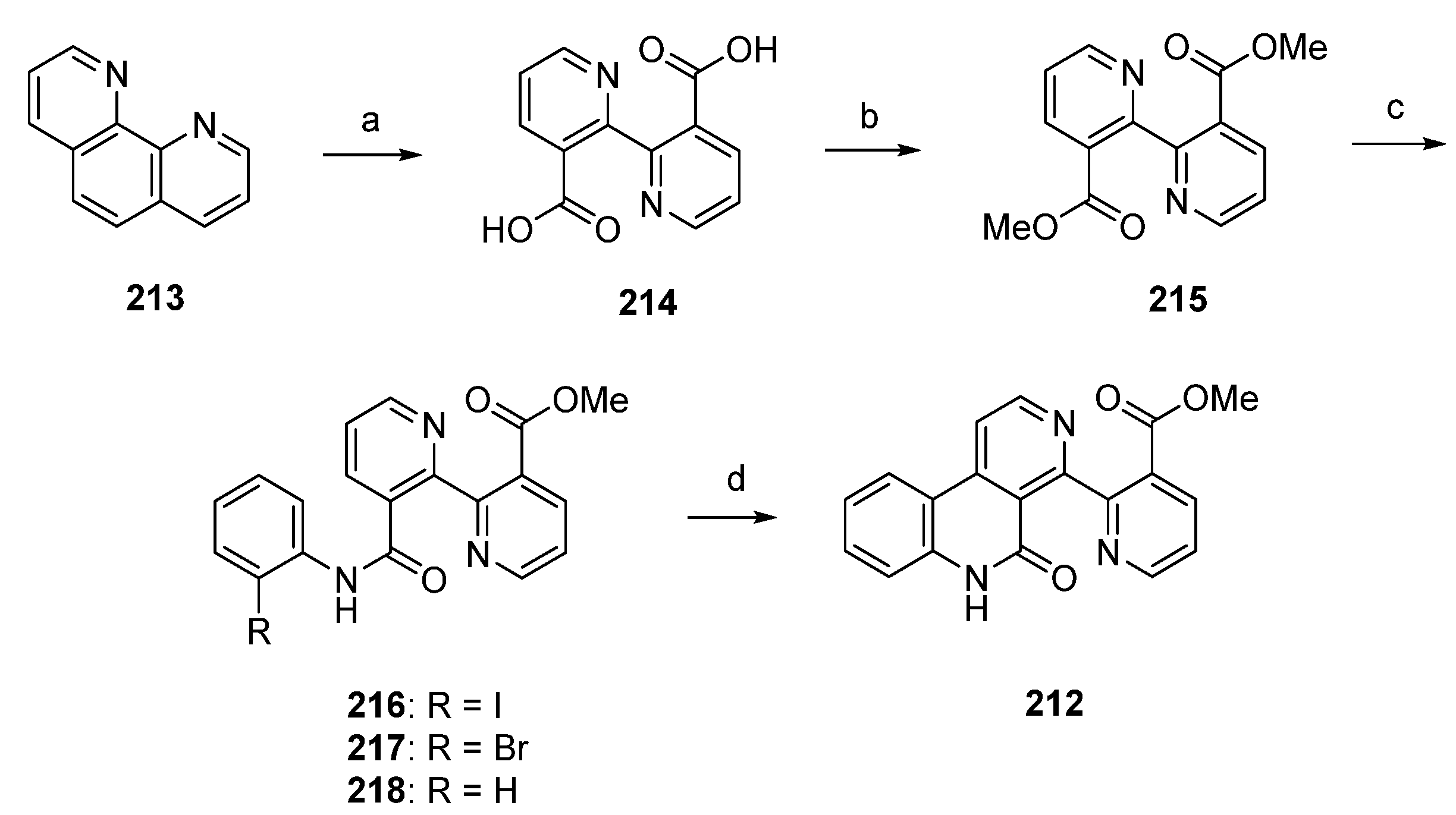
A significantly shorter synthetic route towards the alkaloid subarine (212) was reported by Lotter and Bracher in 2009 [57]. This four-step approach starts with the oxidation of 1,10-phenanthroline (213) using potassium permanganate. The so-obtained dicarboxylate 214 was esterificated by treatment with sulfuric acid in methanol to give the symmetric diester 215 in high yield. Amidation of 215 with 2-haloanilines under Weinreb conditions (trimethylaluminum, heptane) furnished the mono-2-haloanilides 216 and 217 in moderate yields, accompanied by the corresponding dianilides. The final cyclization step was accomplished by treating 2-iodoanilide 216 with tributyltin hydride and a catalytic amount of azobis(isobutyronitrile) in benzene to afford subarine (212) in very poor yield [57]. Significant amounts of deiodination product 218 were obtained in this reaction. Unfortunately, this approach gives only a very poor overall yield (Scheme 31).
Scheme 31.
Synthesis of subarine (212) by Lotter and Bracher: (a) KMnO4, KOH, H2O (b) H2SO4, MeOH (73% over two steps); (c) 2-iodoaniline/2-bromoaniline, CH2Cl2, Me3Al, heptane (216, 40%; 217, 35%); (d) Bu3SnH, AIBN, benzene (7% from 216).
Scheme 31.
Synthesis of subarine (212) by Lotter and Bracher: (a) KMnO4, KOH, H2O (b) H2SO4, MeOH (73% over two steps); (c) 2-iodoaniline/2-bromoaniline, CH2Cl2, Me3Al, heptane (216, 40%; 217, 35%); (d) Bu3SnH, AIBN, benzene (7% from 216).
Author Contributions
The authors participated equally in the review concept.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marshall, K.M.; Barrows, L.R. Biological activities of pyridoacridines. Nat. Prod. Rep. 2004, 21, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Agarwal, S.K.; Gunasekera, S.P.; Schmidt, P.G.; Shoolery, J.N. Amphimedine, new aromatic alkaloid from pacific sponge, Amphimedon sp. Carbon connectivity determination from natural abundance carbon-13-carbon-13 coupling constants. J. Am. Chem. Soc. 1983, 105, 4835–4836. [Google Scholar] [CrossRef]
- Skyler, D.; Heathcock, C.H. The pyridoacridine family tree: A useful scheme for designing synthesis and predicting undiscovered natural products. J. Nat. Prod. 2002, 65, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F. Marine pyridoacridine alkaloids: Structure, synthesis, and biological chemistry. Chem. Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef]
- Sharma, V.; Sharma, P.C.; Kumar, V. A mini review on pyridoacridines: Prospective lead compounds in medicinal chemistry. J. Adv. Res. 2014, 6, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Bastide, J. Marine pyridoacridine alkaloids and synthetic analogues as antitumor agents. Med. Res. Rev. 2002, 23, 234–252. [Google Scholar] [CrossRef] [PubMed]
- Bracher, F. Total synthesis of the pentacyclic alkaloid ascididemin. Heterocycles 1989, 29, 2093–2095. [Google Scholar] [CrossRef]
- Koller, A.; Rudi, A.; Garcia, G.M.; Kashman, Y. Synthesis and electrophilic substitution of pyrido[2,3,4-kl]-acridines. Molecules 2001, 6, 300–322. [Google Scholar] [CrossRef]
- Cuerva, J.M.; Cárdenas, D.J.; Echavarren, A.M. Intramolecular Michael-type addition of azadienes to 1,4-naphthoquinones instead of aza-Diels-Alder cycloaddition: A synthesis of ascididemin. J. Chem. Soc. Perkin Trans. 1 2002, 11, 1360–1365. [Google Scholar] [CrossRef]
- Lindsay, B.S.; Pearce, A.N.; Copp, B.R. Efficient and convenient pyridine ring-E formation of the cytotoxic marine alkaloid ascididemin and related analogues. Synth. Commun. 1997, 27, 2587–2592. [Google Scholar] [CrossRef]
- Peterson, J.R.; Zjawiony, J.K.; Liu, S.; Hufford, C.D.; Clark, A.M.; Rogers, R.D. Copyrine alkaloids: Synthesis, spectroscopic characterization, and antimycotic/antimycobacterial activity of A- and B-ring-functionalized sampangines. J. Med. Chem. 1992, 35, 4069–4077. [Google Scholar] [CrossRef] [PubMed]
- Brahic, C.; Darro, F.; Belloir, M.; Bastide, J.; Kiss, R.; Delfourne, E. Synthesis and cytotoxic evaluation of analogues of the marine pyridoacridine amphimedine. Bioorg. Med. Chem. 2002, 10, 2845–2853. [Google Scholar] [CrossRef]
- Gellerman, G.; Rudi, A.; Kashman, Y. Biomimetic synthesis of ascididemin and derivatives. Synthesis 1994, 3, 239–241. [Google Scholar] [CrossRef]
- Cuerva, J.M.; Cárdenas, D.J.; Echavarren, A.M. New synthesis of pyridoacridines based on an intramolecular aza-Diels-Alder reaction followed by an unprecedented rearrangement. Chem. Commun. 1999, 17, 1721–1722. [Google Scholar] [CrossRef]
- Yin, H.; Shan, N.; Wang, S.; Yao, Z.-J. Total synthesis of ascididemin-type alkaloids using alkyne building blocks. J. Org. Chem. 2014, 79, 9748–9753. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, B.S.; Barrows, L.R.; Copp, B.R. Structural requirements for biological activity of the marine alkaloid ascididemin. Bioorg. Med. Chem. 1995, 5, 739–742. [Google Scholar] [CrossRef]
- Copp, B.R.; Kayser, O.; Brun, R.; Kiderlen, A.F. Antiparasitic activity of marine pyridoacridone alkaloids related to the ascididemins. Planta Med. 2003, 69, 527–531. [Google Scholar] [PubMed]
- Appleton, D.R.; Pearce, A.N.; Copp, B.R. Anti-tuberculosis natural products: Synthesis and biological evaluation of pyridoacridine alkaloids related to ascididemin. Tetrahedron 2010, 66, 4977–4986. [Google Scholar] [CrossRef]
- Debnath, B.; Gayen, S.; Bhattacharya, S.; Samanta, S.; Jha, T. QSAR study on some pyridoacridine ascididemin analogues as anti-tumor agents. Bioorg. Med. Chem. 2003, 11, 5493–5499. [Google Scholar] [CrossRef] [PubMed]
- Petersen, I.N.; Crestey, F.; Kristensen, J.L. Total synthesis of ascididemin via anionic cascade ring closure. Chem. Commun. 2012, 48, 9092–9094. [Google Scholar] [CrossRef] [PubMed]
- Petersen, I.N.; Kristensen, J.L. Synthesis of pyridoacridines through anionic cascade ring closure. Synthesis 2014, 46, 1469–1474. [Google Scholar]
- Raeder, S.; Bracher, F. A novel approach to the pyridoacridine ring system: Synthesis of the topoisomerase inhibitor 13-deazaascididemin. Arch. Pharm. Chem. Life Sci. 2012, 345, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Bracher, F. First synthesis of the benzo[f]pyrido[2′,3′:3,4]pyrrolo[2,1-a][2,7]naphthyridine ring system. J. Heterocyclic Chem. 1993, 30, 157–159. [Google Scholar] [CrossRef]
- Plodek, A.; Raeder, S.; Bracher, F. A novel approach to ring A analogues of the marine pyridoacridine alkaloid ascididemin. Tetrahedron 2013, 69, 9857–9864. [Google Scholar] [CrossRef]
- Plodek, A.; König, M.; Bracher, F. Synthesis of the azaoxoaporphine alkaloid sampangine and ascididemin-type pyridoacridines through TMPMgCl·LiCl-mediated ring closure. Eur. J. Org. Chem. 2015, 6, 1302–1308. [Google Scholar] [CrossRef]
- Melzer, B.; Plodek, A.; Bracher, F. Total synthesis of the marine pyridoacridine alkaloid demethyldeoxyamphimedine. J. Org. Chem. 2014, 79, 7239–7242. [Google Scholar] [CrossRef] [PubMed]
- Bry, D.; Banaigs, B.; Long, C.; Bontemps, N. New pyridoacridine alkaloids from the purple morph of the ascidian Cystodytes dellechiajei. Tetrahedron Lett. 2011, 52, 3041–3044. [Google Scholar] [CrossRef]
- LaBarbera, D.V.; Bugni, T.S.; Ireland, C.M. The total synthesis of neoamphimedine. J. Org. Chem. 2007, 72, 8501–8505. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Abraham, A.D.; Zhou, Q.; Ali, H.; O’Brien, J.V.; Hamill, B.D.; Arcaroli, J.J.; Messersmith, W.A.; LaBarbera, D.V. An improved high yield total synthesis and cytotoxicity study of the marine alkaloid neoamphimedine: An ATP-competitive inhibitor of topoisomerase IIα and potent anticancer agent. Mar. Drugs 2014, 12, 4833–4850. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Mukai, Y.; Kubo, A. Synthesis of neoamphimedine. Heterocycles 2012, 85, 933–940. [Google Scholar] [CrossRef]
- Rudi, A.; Benayahu, Y.; Goldberg, I.; Kashman, Y. Eilatin, a novel alkaloid from the marine tunicate Eudistoma sp. Tetrahedron Lett. 1988, 29, 6655–6656. [Google Scholar] [CrossRef]
- Nakahara, S.; Tanaka, Y.; Kubo, A. Total synthesis of eilatin. Heterocycles 1993, 36, 1139–1144. [Google Scholar]
- Kubo, A.; Nakahara, S. Synthesis of amphimedine, a new fused aromatic alkaloid from a pacific sponge, Amphimedon sp. Heterocycles 1988, 27, 2095–2098. [Google Scholar] [CrossRef]
- Gellerman, G.; Babad, M.; Kashman, Y. A two step biomimetic total synthesis of eilatin. Tetrahedron Lett. 1993, 34, 1827–1830. [Google Scholar] [CrossRef]
- Gellerman, G.; Rudi, A.; Kashman, Y. Biomimetic synthesis of pyrido[2,3,4-kl]acridines. Tetrahedron Lett. 1993, 34, 1823–1826. [Google Scholar] [CrossRef]
- Glazer, E.C.; Tor, Y. RuII complexes of “large-surface” ligands. Angew. Chem. Int. Ed. 2002, 41, 4022–4026. [Google Scholar] [CrossRef]
- Plodek, A.; Bracher, F. A divergent approach to the total synthesis of the marine pyridoacridine alkaloid eilatin and its synthetic isomer isoeilatin. Tetrahedron Lett. 2015, 56, 4194–4197. [Google Scholar] [CrossRef]
- Gellerman, G.; Rudi, A.; Kashman, Y. The biomimetic synthesis of marine alkaloid related pyrido- and pyrrolo[2,3,4-kl]acridines. Tetrahedron 1994, 50, 12959–12972. [Google Scholar] [CrossRef]
- Copp, B.R.; Jompa, J.; Tahir, A.; Ireland, C.M. Styelsamines A–D: New tetracyclic pyridoacridine alkaloids from the indonesian ascidian Eusynstyela latericius. J. Org. Chem. 1998, 63, 8024–8026. [Google Scholar] [CrossRef]
- Kobayashi, J.; Cheng, J.-F.; Wälchli, M.R.; Nakamura, H.; Hirata, Y.; Sasaki, T.; Ohizumi, Y. Cystodytins A, B, and C, novel tetracyclic aromatic alkaloids with potent antineoplastic activity from the Okinawan tunicate Cystodytes dellechiajei. J. Org. Chem. 1988, 53, 1800–1804. [Google Scholar] [CrossRef]
- Kobayashi, J.; Tsuda, M.; Tanabe, A.; Ishibashi, M.; Cheng, J.-F.; Yamamura, S.; Sasaki, T. Cystodytins D–I, new cytotoxic tetracyclic aromatic alkaloids from the Okinawan marine tunicate Cystodytes dellechiajei. J. Nat. Prod. 1991, 54, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.A.; Eldredge, G.S.; Barrows, L.R.; Ireland, C.M. Inhibition of topoisomerase II catalytic activity by pyridoacridine alkaloids from a Cystodytes sp. ascidian: A mechanism for the apparent intercalator-induced inhibition of topoisomerase II. J. Med. Chem. 1994, 37, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Appleton, D.R.; Pearce, A.N.; Lambert, G.; Babcock, R.C.; Copp, B.R. Isodiplamine, cystodytin K and lissoclinidine: Novel bioactive alkaloids from the New Zealand ascidian Lissoclinum notti. Tetrahedron 2002, 58, 9779–9783. [Google Scholar] [CrossRef]
- Skyler, D.; Heathcock, C.H. A simple biomimetic synthesis of styelsamine B. Org. Lett. 2001, 3, 4323–4324. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.K.H.; Copp, B.R. Synthesis, DNA binding and antitumor evaluation of styelsamine and cystodytin analogues. Mar. Drugs 2013, 11, 274–299. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Kubo, A. Total synthesis of styelsamine C, a cytotoxic fused tetracyclic aromatic alkaloid. Heterocycles 2003, 60, 2017–2018. [Google Scholar] [CrossRef]
- Nakahara, S.; Kubo, A. Total synthesis of styelsamine C, and formal synthesis of norsegoline. Heterocycles 2005, 65, 1925–1929. [Google Scholar] [CrossRef]
- Torres, Y.R.; Bugni, T.S.; Berlinck, R.G.S.; Ireland, C.M.; Magalhães, A.; Ferreira, A.G.; Moreira da Rocha, R. Sebastianines A and B, novel biologically active pyridoacridine alkaloids from the Brazilian ascidian Cystodytes dellechiajei. J. Org. Chem. 2002, 67, 5429–5432. [Google Scholar] [CrossRef] [PubMed]
- Legentil, L.; Bastide, J.; Delfourne, E. Total synthesis of the marine pyridoacridine alkaloid sebastianine A. Tetrahedron Lett. 2003, 44, 2473–2475. [Google Scholar] [CrossRef]
- Plubrukarn, A.; Davidson, B.S. Arnoamines A and B, new cytotoxic pentacyclic pyridoacridine alkaloids from the ascidian Cystodytes sp. J. Org. Chem. 1998, 63, 1657–1659. [Google Scholar] [CrossRef]
- Bontemps, N.; Gattacceca, F.; Long, C.; Thomas, O.P.; Banaigs, B. Additional cytotoxic pyridoacridine alkaloids from the ascidian Cystodytes violatinctus and biogenetic considerations. J. Nat. Prod. 2013, 76, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Roubin, C.; Bastide, J. The first synthesis of the pentacyclic pyridoacridine marine alkaloids: Arnoamines A and B. J. Org. Chem. 2000, 65, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Kubo, A.; Mikami, Y.; Mitani, H. Synthesis of arnoamine B and related compounds. Heterocycles 2007, 71, 1801–1806. [Google Scholar] [CrossRef]
- Radchenko, O.S.; Balaneva, N.N.; Denisenko, V.A.; Novikov, V.L. A simple and effective approach to the synthesis of pyrido[4,3,2-mn]pyrrolo[3,2,1-de]acridine skeleton of arnoamines A and B, pentacyclic marine alkaloids from the ascidian Cystodytes sp. Tetrahedron Lett. 2006, 47, 7819–7822. [Google Scholar] [CrossRef]
- Nakahara, S.; Sadachi, T.; Kubo, A. Synthesis of arnoamine B. Heterocycles 2010, 81, 145–148. [Google Scholar] [CrossRef]
- Bijeire, L.; Legentil, L.; Bastide, J.; Darro, F.; Rochart, C.; Delfourne, E. A total synthesis of subarine, a marine alkaloid related to the pyridoacridine family. Eur. J. Org. Chem. 2004, 9, 1891–1893. [Google Scholar] [CrossRef]
- Lotter, M.; Bracher, F. Short total synthesis of the marine alkaloid subarine. Sci. Pharm. 2009, 77, 1–7. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Plodek, A.; Bracher, F. New Perspectives in the Chemistry of Marine Pyridoacridine Alkaloids. Mar. Drugs 2016, 14, 26. https://doi.org/10.3390/md14020026
AMA Style
Plodek A, Bracher F. New Perspectives in the Chemistry of Marine Pyridoacridine Alkaloids. Marine Drugs. 2016; 14(2):26. https://doi.org/10.3390/md14020026
Chicago/Turabian StylePlodek, Alois, and Franz Bracher. 2016. "New Perspectives in the Chemistry of Marine Pyridoacridine Alkaloids" Marine Drugs 14, no. 2: 26. https://doi.org/10.3390/md14020026
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.