Identification of Plakortide E from the Caribbean Sponge Plakortis halichondroides as a Trypanocidal Protease Inhibitor using Bioactivity-Guided Fractionation


Abstract
:1. Introduction
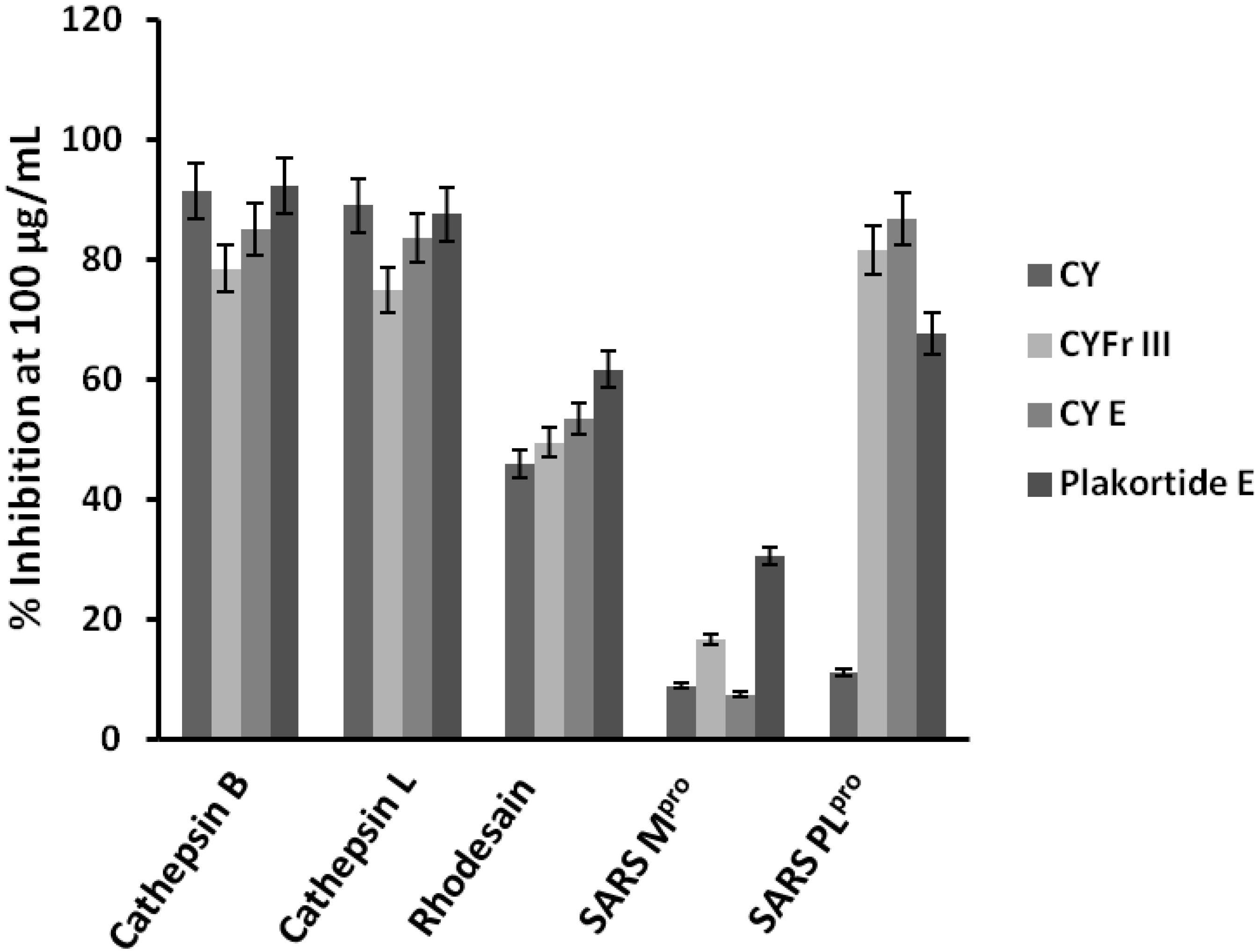
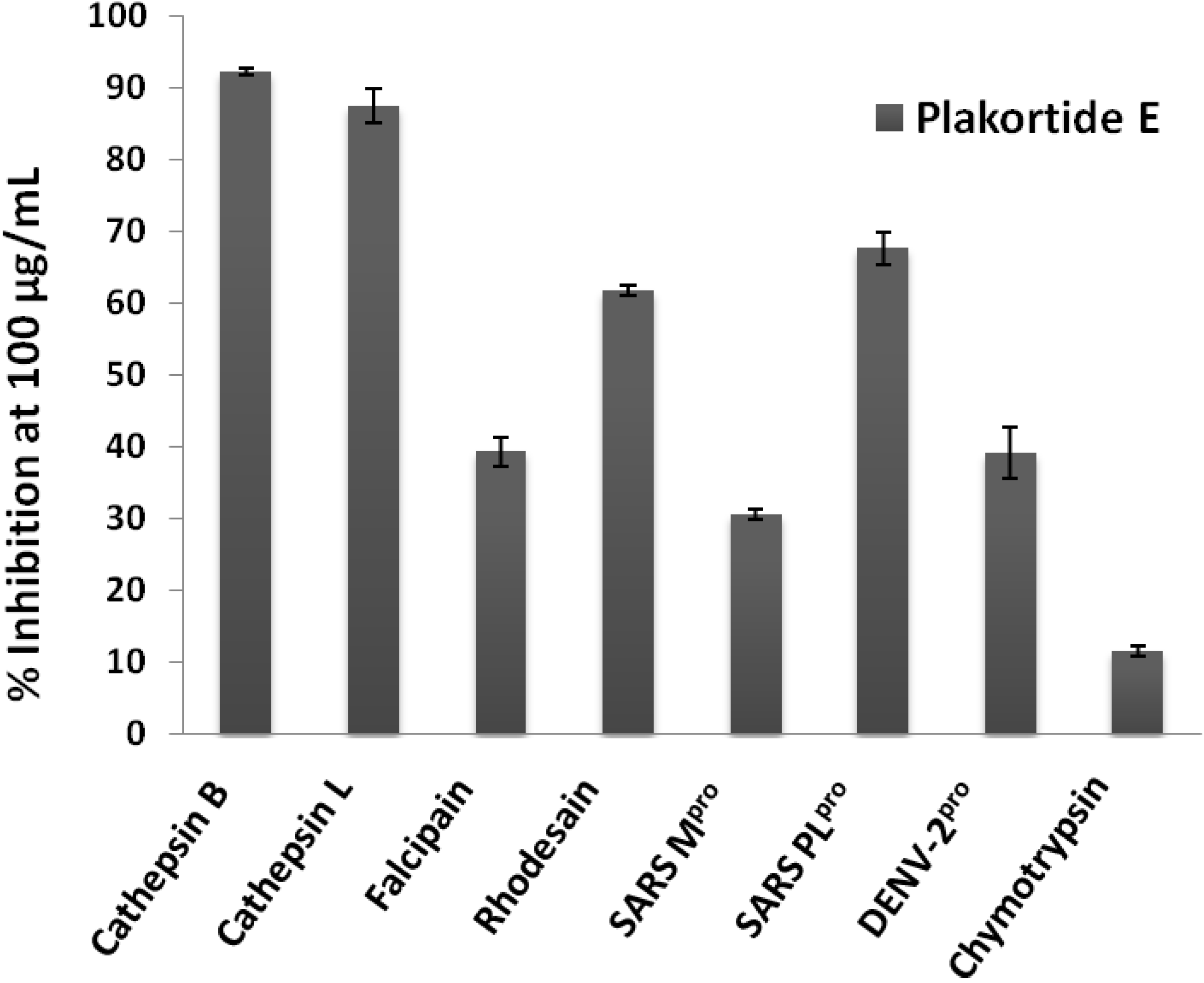
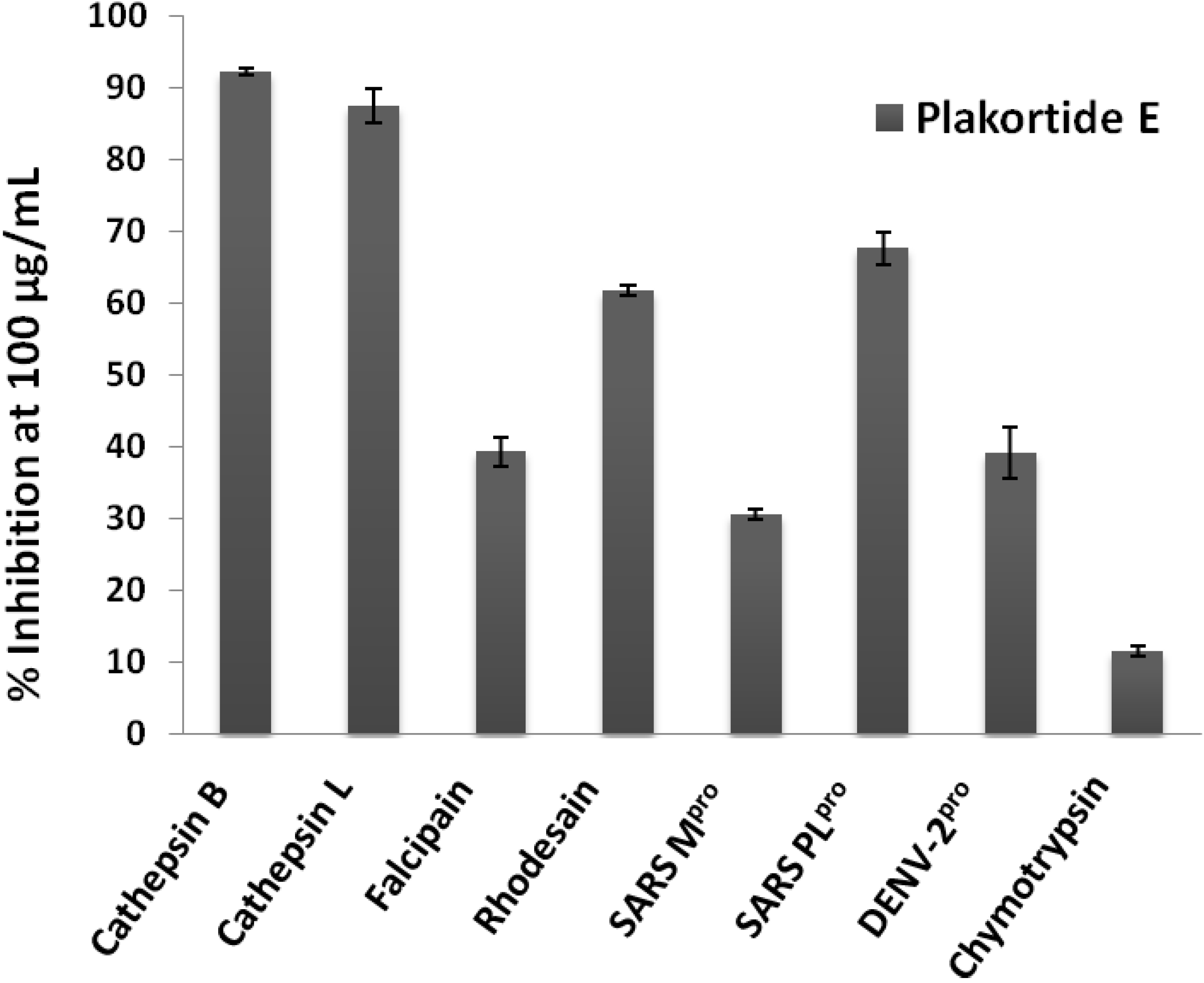
2. Results and Discussion
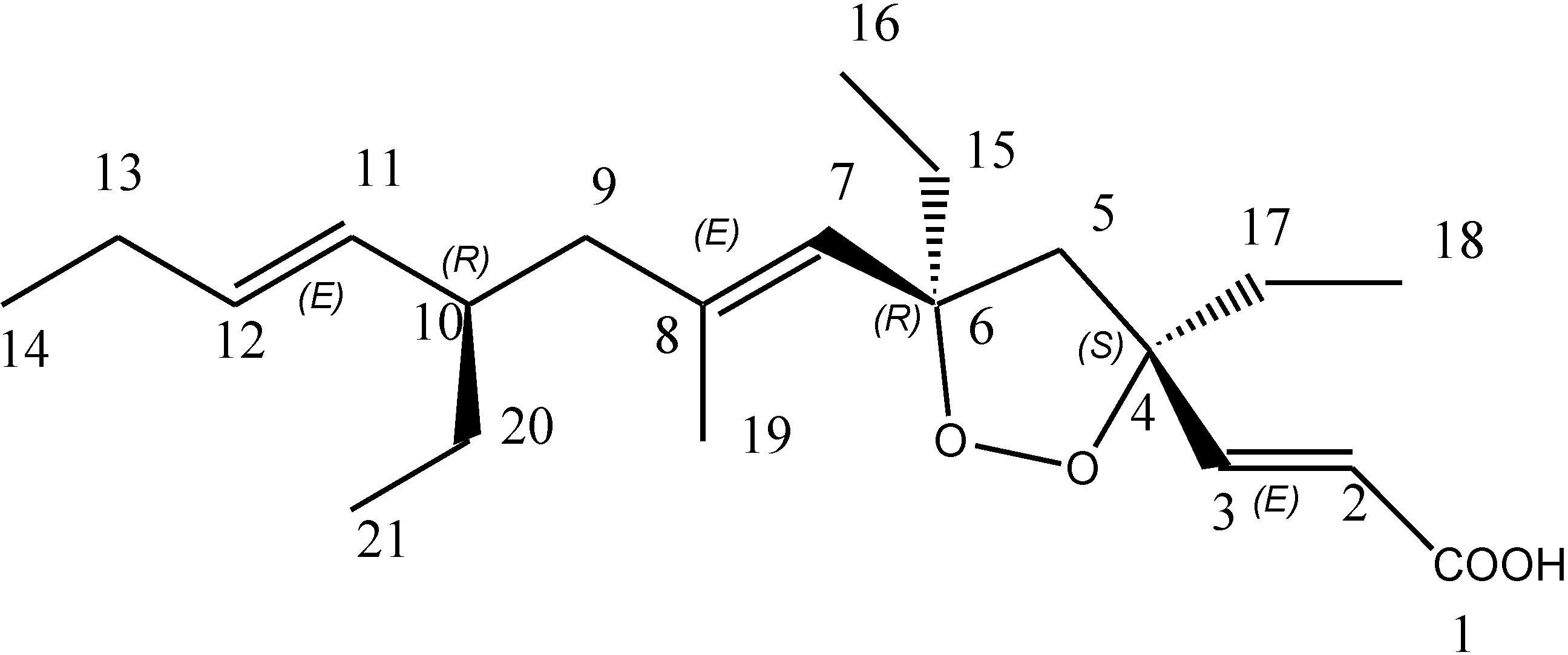
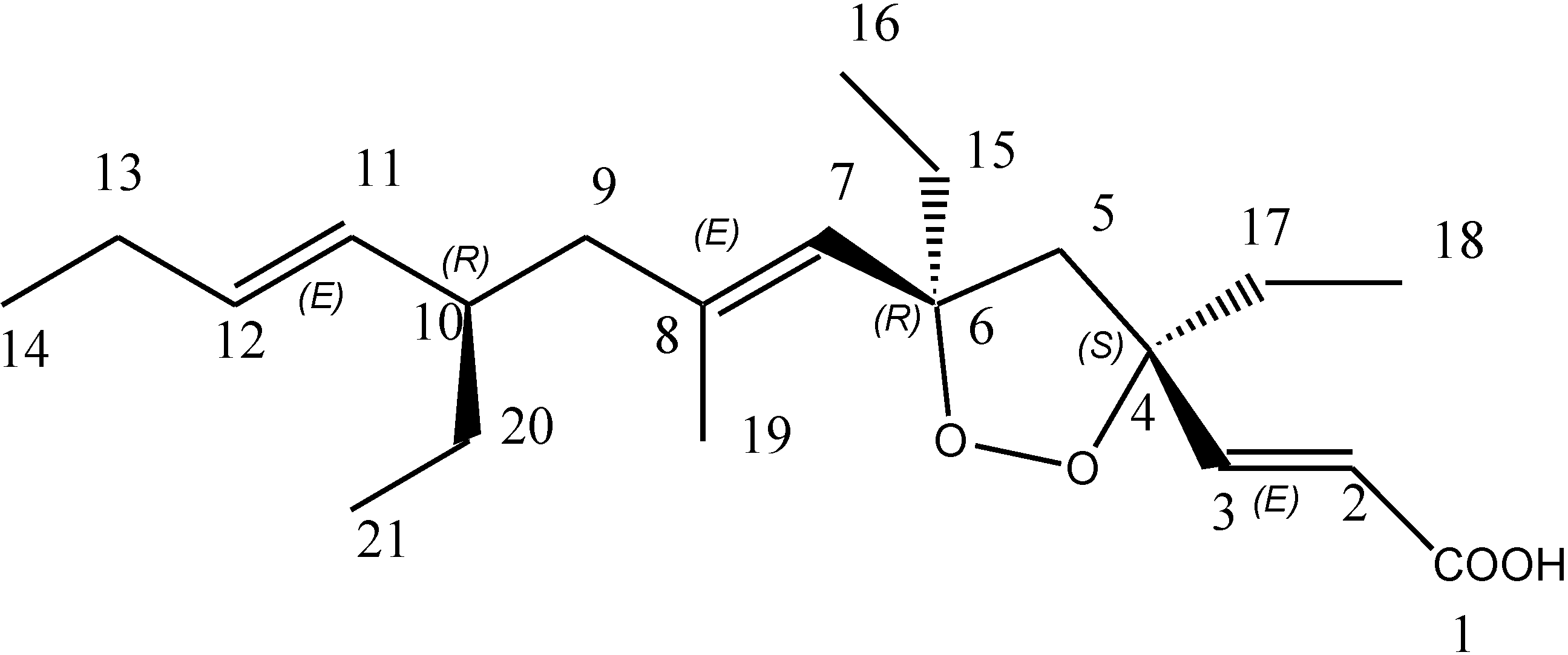
3. Experimental Section

{kind=link}
{kind=link}
{kind=link}
| Position | δC | Multiplicity | δH | Multiplicity | J (Hz) |
|---|---|---|---|---|---|
| 1 | 171.76 | C | |||
| 2 | 119.75 | CH | 6.09 | d | 15.8 |
| 3 | 152.31 | CH | 6.96 | d | 15.8 |
| 4 | 87.31 | C | |||
| 5 | 56.11 | CH2 | 2.55 | d | 12.0 |
| 2.45 | d | 12.0 | |||
| 6 | 89.39 | C | |||
| 7 | 126.71 | CH | 5.12 | s | |
| 8 | 136.82 | C | |||
| 9 | 46.67 | CH2 | 2.00 | m | |
| 1.85 | m | ||||
| 10 | 42.68 | CH | 1.99 | m | |
| 11 | 132.91 | CH | 5.05 | ddt | 15.2, 8.3 |
| 12 | 132.10 | CH | 5.34 | dt | 15.2, 6.3, 6.3 |
| 13 | 25.73 | CH2 | 1.96 | m | |
| 14 | 14.19 | CH3 | 0.94 | t | 7.4 |
| 15 | 32.36 | CH2 | 1.88 | m | |
| 1.64 | m | ||||
| 16 | 9.01 | CH3 | 0.89 | t | 7.4 |
| 17 | 30.88 | CH2 | 1.76 | m | |
| 18 | 8.95 | CH3 | 0.92 | t | 7.5 |
| 19 | 17.92 | CH3 | 1.62 | d | 1.2 |
| 20 | 27.79 | CH2 | 1.34 | m | |
| 1.10 | m | ||||
| 21 | 11.72 | CH3 | 0.81 | t | 7.4 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Powers, J.C.; Asgian, J.L.; Ekici, Ö.D.; James, K.E. Irreversible inhibitors of serine, cysteine and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Morton, F.R. The MEROPS batch BLAST: A tool to detect peptidases and their non-peptidase homologues in a genome. Biochimie 2008, 90, 243–259. [Google Scholar] [CrossRef]
- Sata, N.; Abinsay, H.; Yoshida, W.Y.; Horgen, F.D.; Sitachitta, N.; Kelly, M.; Scheuer, P.J. Lehualides A–D, metabolites from a Hawaiian sponge of the genus Plakortis. J. Nat. Prod. 2005, 68, 1400–1403. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Bean, M.F.; Carte, B.K.; Westley, J.W.; Johnson, R.K.; Lahouratate, P. The plakortones, novel bicyclic lactones from the sponge Plakortis halichondroides: Activators of cardiac SR-Ca2+ pumping ATPase. Tetrahedron 1996, 52, 377–394. [Google Scholar] [CrossRef]
- Chen, Y.; McCarthy, P.J.; Harmody, D.K.; Schimoler-O’Rourke, R.; Chilson, K.; Selitrennikoff, C.; Pomponi, S.A.; Wright, A.E. New bioactive peroxides from marine sponges of the family plakiniidae. J. Nat. Prod. 2002, 65, 1509–1512. [Google Scholar] [CrossRef]
- Fattorusso, C.; Campiani, G.; Catalanotti, B.; Persico, M.; Basilico, N.; Parapini, S.; Taramelli, D.; Campagnuolo, C.; Fattorusso, E.; Romano, A.; et al. Endoperoxide derivatives from marine organisms: 1,2-Dioxanes of the plakortin family as novel antimalarial agents. J. Med. Chem. 2006, 49, 7088–7094. [Google Scholar] [CrossRef]
- Campagnuolo, C.; Fattorusso, E.; Romano, A.; Taglialatela-Scafati, O.; Basilico, N.; Parapini, S.; Taramelli, D. Antimalarial polyketide cycloperoxides from the marine sponge Plakortis simplex. Eur. J. Org. Chem. 2005, 23, 5077–5083. [Google Scholar]
- Hu, J.-F.; Gao, H.-F.; Kelly, M.; Hamann, M.T. Plakortides I–L, four new cyclic peroxides from an undescribed Jamaican sponge Plakortis sp. (Homosclerophorida, Plakinidae). Tetrahedron 2001, 57, 9379–9383. [Google Scholar] [CrossRef]
- Fattorusso, E.; Parapini, S.; Campagnuolo, C.; Basilico, N.; Taglialatela-Scafati, O.; Taramelli, D. Activity against Plasmodium falciparum of cycloperoxide compounds obtained from the sponge Plakortis simplex. J. Antimicrob. Chemother. 2002, 50, 883–888. [Google Scholar] [CrossRef]
- Palermo, C.; Joyce, J.A. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharm. Sci. 2008, 29, 22–28. [Google Scholar]
- Lankelma, J.M.; Voorend, D.M.; Barwari, T.; Koetsveld, J.; Van der Spek, A.H.; de Porto, A.P.N.A.; Van Rooijen, G.; Van Noorden, C.J.F. Cathepsin L target in cancer treatment? Life Sci. 2010, 86, 225–233. [Google Scholar] [CrossRef]
- Kerr, I.D.; Wu, P.; Marion-Tsukamaki, R.; Mackey, Z.B.; Brinen, L.S. Crystal structures of TbCatB and rhodesain, potential chemotherapeutic targets and major cysteine proteases of Trypanosoma brucei. PLoS Negl. Trop. Dis. 2010, 4. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Han, Y.-S.; Chang, G.-G.; Juo, C.-G.; Lee, H.-J.; Yeh, S.-H.; Hsu, J.T.-A.; Chen, X. Papain-like protease 2 (PLP2) from Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV): Expression, purification, characterization, and inhibition. Biochemistry 2005, 44, 10349–10359. [Google Scholar] [CrossRef]
- Rosenthal, P. Falcipains and other cysteine proteases of malaria parasites. In Cysteine Proteases of Pathogenic Organisms; Robinson, M., Dalton, J., Eds.; Springer: San Francisco, California, USA, 2011; Volume 712, pp. 30–48. [Google Scholar]
- Bessaud, M.; Pastorino, B.A.M.; Peyrefitte, C.N.; Rolland, D.; Grandadam, M.; Tolou, H.J. Functional characterization of the NS2B/NS3 protease complex from seven viruses belonging to different groups inside the genus Flavivirus. Virus Res. 2006, 120, 79–90. [Google Scholar]
- Breuning, A.; Degel, B.; Schulz, F.; Buchold, C.; Stempka, M.; Machon, U.; Heppner, S.; Gelhaus, C.; Leippe, M.; Leyh, M.; et al. Michael acceptor based antiplasmodial and antitrypanosomal cysteine protease inhibitors with unusual amino acids. J. Med. Chem. 2010, 53, 1951–1963. [Google Scholar] [CrossRef]
- Copeland, R.A. A Guide for Medicinal Chemists and Pharmacologists. In Evaluation of Enzyme Inhibitors in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Yang, P.-Y.; Wang, M.; He, C.Y.; Yao, S.Q. Proteomic profiling and potential cellular target identification of K11777, a clinical cysteine protease inhibitor, in Trypanosoma brucei. Chem. Commun. 2012, 48, 835–837. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Vicik, R.; Schultheis, M.; Schirmeister, T.; Moll, H. Aziridine-2,3-dicarboxylates, peptidomimetic cysteine protease inhibitors with antileishmanial activity. Antimicrob. Agents Chemother. 2006, 50, 2439–2447. [Google Scholar] [CrossRef]
- Sun, X.Y.; Tian, X.Y.; Li, Z.W.; Peng, X.S.; Wong, H.N. Total synthesis of plakortide E and biomimetic synthesis of plakortone B. Chemistry 2011, 17, 5874–5880. [Google Scholar] [CrossRef]
- Vicik, R.; Busemann, M.; Gelhaus, C.; Stiefl, N.; Scheiber, J.; Schmitz, W.; Schulz, F.; Mladenovic, M.; Engels, B.; Leippe, M.; et al. Aziridide-based inhibitors of cathepsin L: synthesis, inhibition activity, and docking studies. ChemMedChem 2006, 1, 1126–1141. [Google Scholar] [CrossRef]
- Vicik, R.; Hoerr, V.; Glaser, M.; Schultheis, M.; Hansell, E.; McKerrow, J.H.; Holzgrabe, U.; Caffrey, C.R.; Ponte-Sucre, A.; Moll, H.; Stich, A.; et al. Aziridine-2,3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg. Med. Chem. Lett. 2006, 16, 2753–2757. [Google Scholar] [CrossRef]
- Ehmke, V.; Heindl, C.; Rottmann, M.; Freymond, C.; Schweizer, W.B.; Brun, R.; Stich, A.; Schirmeister, T.; Diederich, F. Potent and selective inhibition of cysteine proteases from Plasmodium falciparum and Trypanosoma brucei. ChemMedChem 2011, 6, 273–278. [Google Scholar] [CrossRef]
- Ettari, R.; Zappala, M.; Micale, N.; Giofre, S.; Schirmeister, T.; Grasso, S. Peptidomimetics containing a vinyl ketone warhead as falcipain-2 inhibitor. Eur. J. Med. Chem. 2011, 46, 2058–2065. [Google Scholar] [CrossRef]
- Käppler, U.; Stiefl, N.; Schiller, M.; Vicik, R.; Breuning, A.; Schmitz, W.; Rupprecht, D.; Schmuck, C.; Baumann, K.; Ziebuhr, J.; et al. A new lead for non-peptidic active-site-directed inhibitors of the SARS coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem. 2005, 48, 6832–6842. [Google Scholar] [CrossRef]
- Steuer, C.; Heinonen, K.H.; Kattner, L.; Klein, C.D. Optimization of assay conditions for dengue virus protease: Effect of various polyols and nonionic detergents. J. Biomol. Screen. 2009, 14, 1102–1108. [Google Scholar] [CrossRef]
- Baltz, T.; Baltz, D.; Giroud, C.; Crocket, J. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 1985, 4, 1273–1277. [Google Scholar]
- Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
- Merschjohann, K.; Sporer, F.; Steverding, D.; Wink, M. In vitro effect of alkaloids on bloodstream forms of Trypanosoma brucei and T. congolense. Planta Med. 2001, 67, 623–627. [Google Scholar] [CrossRef]
- Huber, W.; Koella, J.C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–261. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Gogal, R.M.; Walsh, J.E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H] thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Degel, B.; Staib, P.; Rohrer, S.; Scheiber, J.; Martina, E.; Büchold, C.; Baumann, K.; Morschhäuser, J.; Schirmeister, T. Cis-configured aziridines are new pseudo-irreversible dual-mode inhibitors of Candida albicans secreted aspartic protease 2. ChemMedChem 2008, 3, 302–315. [Google Scholar] [CrossRef]
- Büchold, C.; Hemberger, Y.; Heindl, C.; Welker, A.; Degel, B.; Pfeuffer, T.; Staib, P.; Schneider, S.; Rosenthal, P.J.; Gut, J.; et al. New cis-configured aziridine-2-carboxylates as aspartic acid protease inhibitors. ChemMedChem 2011, 6, 141–152. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oli, S.; Abdelmohsen, U.R.; Hentschel, U.; Schirmeister, T. Identification of Plakortide E from the Caribbean Sponge Plakortis halichondroides as a Trypanocidal Protease Inhibitor using Bioactivity-Guided Fractionation. Mar. Drugs 2014, 12, 2614-2622. https://doi.org/10.3390/md12052614
Oli S, Abdelmohsen UR, Hentschel U, Schirmeister T. Identification of Plakortide E from the Caribbean Sponge Plakortis halichondroides as a Trypanocidal Protease Inhibitor using Bioactivity-Guided Fractionation. Marine Drugs. 2014; 12(5):2614-2622. https://doi.org/10.3390/md12052614
Chicago/Turabian StyleOli, Swarna, Usama Ramadan Abdelmohsen, Ute Hentschel, and Tanja Schirmeister. 2014. "Identification of Plakortide E from the Caribbean Sponge Plakortis halichondroides as a Trypanocidal Protease Inhibitor using Bioactivity-Guided Fractionation" Marine Drugs 12, no. 5: 2614-2622. https://doi.org/10.3390/md12052614
APA StyleOli, S., Abdelmohsen, U. R., Hentschel, U., & Schirmeister, T. (2014). Identification of Plakortide E from the Caribbean Sponge Plakortis halichondroides as a Trypanocidal Protease Inhibitor using Bioactivity-Guided Fractionation. Marine Drugs, 12(5), 2614-2622. https://doi.org/10.3390/md12052614