1. Introduction
Along with the increase of average life expectancy, the prevalence of neurological/neurodegenerative diseases is rising, prompting the recent research focused on developing novel drugs targeting the central nervous system (CNS) [
1]. Inspired by the vastness and biodiversity richness of the marine environment, researchers have pursued the pharmacological potential of marine metabolites [
2].
Pharmacological studies with marine compounds affecting the CNS involve areas of neuropharmacology, such as those of stimulation of neurogenesis, modulation of receptors and voltage-dependent ion channels and enzymes inhibition [
3]. For instance, conotoxins peptides are currently being used as standard research tools in neuroscience, since they can interfere with receptors and channels, allowing a better understanding of how antagonist/agonist drugs bind to the binding sites [
4]. These researches have already culminated with Food and Drug Administration (FDA) approval of Ziconitide (Prialt
®), a synthetic equivalent of the ω-conotoxin MVIIA (isolated from
Conus magus L.), for pain and stroke treatment [
4,
5]. Moreover, several other marine compounds are being evaluated in preclinical trials, such as the α-conotoxin Vc1.1 (isolated from
Conus victoriae Reeve) and the χ-conotoxin MrIA/B (from
Conus marmoreus L.), for the treatment of neuropathic pain, and the anti-epileptic conantokin-G, isolated from
Conus geographus L. Currently undergoing a more advanced evaluation,
i.e., phase II trials, are ω-conotoxin CVID (from
Conus catus Hwass in Bruguière) for neuropathic pain treatment, and contulakin-G (from
C. geographus) for neuropathic and chronic inflammatory pain treatments [
5], as well as 3-(2,4-dimethoxybenzylidene)-anabaseine (DMXBA), the synthetic derivative produced from the alkaloid anabaseine (isolated from nemertines), to treat schizophrenia [
6] and Alzheimer’s disease (AD) [
7].
This review covers the studies performed with marine invertebrate drugs from the year 2000 until the present, focusing on their role in fighting neuroinflammation states and neurodegeneration. One hundred and eighty-four examples of marine drugs affecting neuronal growth and synaptic functions, neuroinflammation, CNS enzymes and CNS voltage and ligand-gated ion channels will be given. Towards the conclusion of this paper, the usefulness of marine skeletons in neural tissue engineering will be discussed. Recently, some review papers have been published focusing on some of the aspects considered in this review. The modulation of receptors, voltage-dependent channels and enzymes by conopeptides is, by far, the most extensively reviewed subject [
4,
8,
9,
10]. Sakai and Swanson [
11] presented a broad spectrum of marine drugs affecting those targets. Arias
et al. [
12] focused their attention on marine drugs affecting ion channels, and Al-Sabi
et al. [
13] reviewed data about marine toxins that target voltage-gated sodium channels. Kochanowska-Karamyan and Hamann [
14] covered the role of marine indole alkaloids as potential new antidepressant and anti-anxiety drug leads. Bharate
et al. [
15] and Skropeta
et al. [
16] gathered information concerning sponge drugs with protein kinase inhibitory activity. A broader spectrum of enzyme inhibited by marine drugs was covered by Nakao and Fusetani [
17]. Senthilkumar and Kim [
18] compiled information concerning marine invertebrate natural drugs for inflammatory and chronic diseases, including AD. Finally, information regarding preclinical and clinical candidates in the field of neurology was published by Martínez [
19], Twede
et al. [
10] and Bharate
et al. [
15].
2. The Nervous System
The nervous system is the network of specialized cells that conduct nerve impulses between parts of the body. The central nervous system (CNS) is responsible for driving and interpreting signals and for supplying excitatory stimuli to the peripheral nervous system (PNS); PNS nerves innervate muscle tissue, conducting sensory and excitatory stimuli to and from the spinal cord [
20].
Besides neurons, whose function is to propagate nerve impulses, CNS and PNS also contain another type of cells called glial cells or neuroglia. Neuroglia comprises four types of cells, namely, astrocytes, oligodendrocytes, microglia cells in the CNS and Schwann cells in the PNS. Astrocytes are a very heterogeneous population of cells and they can interfere in axon guidance, synaptic support, control of the blood–brain barrier (BBB) and blood flow [
21]. These are excitable cells like neurons, but they communicate by spontaneous or evoked cytosolic Ca
2+ variations, instead of membrane electrical signals [
22]. Oligodendrocytes and Schwann cells are responsible for the production of myelin [
21,
23]. Microglia cells are the immune cells of the CNS, contributing to CNS homeostasis during development, adulthood and ageing [
24]. They protect the brain from damage and infection, by engulfing dead cells and debris. They are also implicated in synaptic remodelling during the development of the nervous system and they are activated in many neurodegenerative diseases [
21,
23]. In the nervous system, glial cells are more abundant than neurons and have some capacity for cell division. Conversely, neurons have no capacity for mitotic division, but can regenerate portions under certain conditions [
20].
3. Regeneration of the CNS: Drawbacks and Challenges
Complete recovery from a CNS injury or neurological disorders has not yet been made possible [
25]. This is because an injury is a continuous process, with a primary damage triggering a cascade of deleterious events, such as blood–brain barrier disruption, excitotoxicity, inflammation, oedema, ischemia, increase of free radicals and altered cell signalling and gene expression [
26,
27]. Therefore, a massive death of neuronal and glial cells may occur along with the loss of both the 3D spatial organization and the connectivity of neuronal networks [
28].
Although neurite growth inhibitors are present in both CNS and PNS, the capacity for CNS nerves to regenerate is lower than that of peripheral nerves for several reasons. First, because astrocytes become “reactive astrocytes,” which produce glial scars that constitute a physical barrier to growth and up-regulate several extracellular-matrix-associated inhibitors of regeneration, such as chondroitin sulfate proteoglycans [
29]. Second, conversely to a PNS injury, in the case of a CNS injury, BBB and blood–spine barrier function as constrainers to the recruitment of macrophages from the blood circulation to remove myelin and axonal debris and resident microglia can only give a delayed and slow response [
24,
30,
31]. Moreover, in contrast to PNS, the up-regulation of regeneration-associated proteins (RAGs), which play a positive role in neurite outgrowth and axon regeneration, is relatively modest in the CNS after injury [
32,
33].
In order to counteract this low regenerating environment after a CNS injury, clinical trials have taken advantage of the recent progress in regenerative medicine, and new approaches for the treatment of CNS injuries have been explored, such as (i) cellular replacement with stem cells, (ii) delivery of brain-derived neurotrophic factor (BDNF), (iii) axon guidance with cell adhesion molecules and removal of growth inhibition molecules, (iv) manipulation of intracellular signalling with transcription factors, (v) bridging with a peripheral nerve bridge or foetal tissue or use of artificial substrates to guide axons across the scar, and (vi) modulation of the immune response [
25,
34]. Even though transplantation is a promising approach, therapeutic effects are currently limited due to the high level of donor cell death and lack of integration with the host brain tissue [
27]. Conversely, PNS injuries are usually treated surgically by reconnection of the damaged nerve ends (78%) or by using an autograft (15%) or conduit (4%) [
35,
36,
37]. Approximately 50% of surgical cases achieve normal to good function restoration [
35].
4. Marine Drugs: Neuritogenic Activity, Neurotrophin-Mimic and Neurotrophin-Enhancer Agents
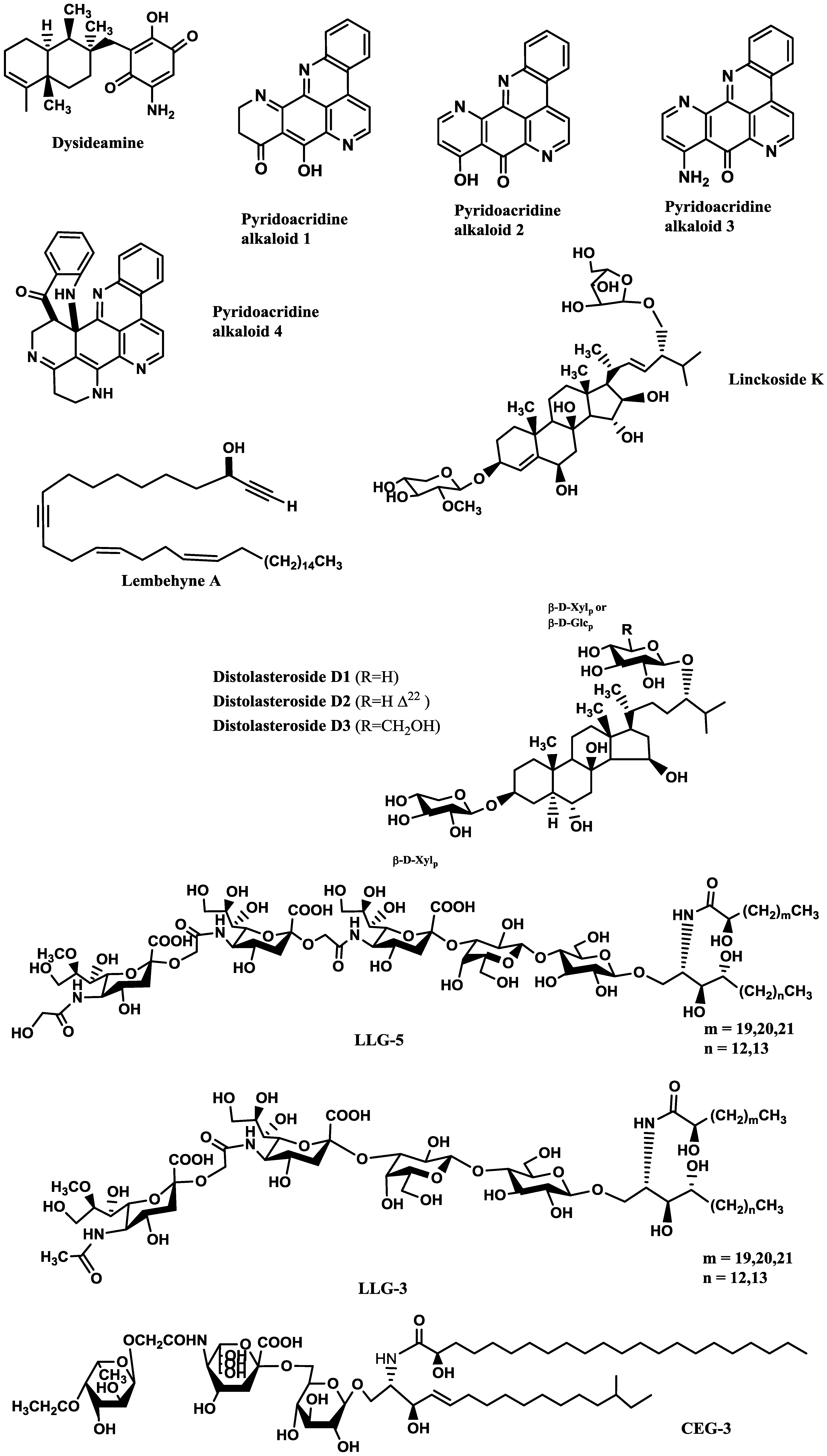
Compounds inducing neuronal growth are expected to become a new lead for medical treatment of CNS disorders, such as ischemic stroke and neurodegenerative diseases. Dysideamine (
Figure 1), a sesquiterpene aminoquinone from the marine sponge
Dysidea sp. 05C33, was shown to induce neurite outgrowth in mouse neuroblastoma Neuro 2A cells [
38]. More than 40% of the cells treated with 3 μM of this compound presented neurite outgrowth but, at 10 μM, slight cytotoxic effects were observed [
38]. Using the same cell system, as well as rat pheochromocytoma PC12 cells, Aoki
et al. [
39] studied the neuritogenic activity of four pyridoacridine alkaloids (
Figure 1) isolated from the marine sponge
Biemna fortis Topsent. None of these compounds were able to induce neurite outgrowth in rat pheochromocytoma PC12 cells. On the other hand, neurite outgrowth was induced in more than 50% of the Neuro 2A cells treated with compound 3 (0.01 μM), but at concentrations higher than 0.3 μM it was cytotoxic. Compounds 1 (labuanine A), 2 and 4 were less active. Taking into account the structure of these pyridoacridine alkaloids and the displayed activity, the authors suggested that the wide difference in neuritogenic activity between compounds 2 and 3 should be due to the presence of the amino group at C-9 in 3. Moreover, compound 3 provoked a four-fold increase of acetylcholinesterase (AChE) activity at 0.03 μM compared with the control, indicating that it induced both morphological and functional neuronal differentiation. Since neuronal differentiation closely relates to the cell cycle, the effect of the pyridoacridine alkaloids on the cell cycle of Neuro 2A cells was evaluated, revealing that, like topoisomerase II inhibitors, they arrested the cell cycle at G2/M phase. Thus, a possible mechanism suggested by the authors was that the induced neuronal differentiation could be related with inhibition of topoisomerase II.
In a similar study, lembehyne A (
Figure 1), a linear polyacetylene isolated from the sponge
Haliclona sp., induced neuritogenesis in both PC12 and Neuro 2A cell lines, at 2 and 0.1 µg/mL, respectively. Since treatment with an inhibitor of actin polymerization (cytochalasin B) or with an inhibitor of protein synthesis in eukaryotes (cycloheximide) inhibited the effect of lembehyne A, a mechanism dependent on actin polymerization and
de novo protein synthesis was suggested for this compound [
40,
41]. Aditionally, lembehyne A (1 and 3 µg/mL) arrested the cell cycle at the G1 phase, a response also known to be induced by nerve growth factor (NGF), and induced a two- and four-fold increase of AChE activity at 1 and 3 µg/mL, respectively [
41]. Later, the same research group investigated the structure–activity relationship among lembehynes A–C and five analogs using Neuro 2A cell system. They concluded that the features contributing to the activity were the carbon-chain length, since analogs with shorter carbon-chain were more active than lembehynes A–C, and that the presence of a hydroxyl group at C-3 was essential [
42].
Figure 1.
Potent marine drugs affecting neuronal growth and synaptic functions.
Figure 1.
Potent marine drugs affecting neuronal growth and synaptic functions.
NGF and BDNF are essential for neuronal differentiation, growth, survival, function maintenance and prevention of ageing in the CNS and PNS [
43,
44].
Although NGF and BDNF are expected to have therapeutic potential in the treatment of neuronal injuries, they do not cross the BBB due to their size. Therefore, low molecular weight compounds mimicking their activity should be interesting as promising therapeutic agents to treat traumatic or ischemic brain injuries and neurodegenerative diseases [
44]. In recent years, several low molecular weight substances from various natural sources have been shown to possess neurotrophic ability. Several marine drugs have proved to mimic and/or enhance NGF or BDNF activities.
Palyanova
et al. [
44] evaluated the neurotrophic potential of six sterols from
Asterina pectinifera Muller and Troschel (starfish) using C1300-NB cell line. C1300-NB, in contrast to PC12 cells, have the capacity to spontaneously differentiate; a residual differentiation of 14%–25% was thus observed. This differentiation was increased by distolasterosides D
1–D
3 (>5 nM;
Figure 1) more efficiently than by asterosaponin Р1 (>50 nM), (25
S)-5α-cholestane-3β,4β,6α,7α,8,15α,16β,26-octaol (>10 nM), and (25
S)-5α-cholestane-3β,6α,7α,8,15α,16β,26-heptaol (>50 nM). These compounds also synergistically enhanced NGF and BDNF activities.
Table 1 and
Table 2 report the neurotrophin mimic and neurotrophin-enhancement effects of several marine drugs in PC12 cells [
43,
45,
46,
47,
48,
49,
50].
Table 1.
Marine drugs with neurotrophin mimic activity in PC12 cell line.
Table 1.
Marine drugs with neurotrophin mimic activity in PC12 cell line.
| Compound/organism | Concentration tested (µM) | Neurites longer than soma diameter (%) |
|---|
| Linckoside A/blue starfish Linckia laevigata L. | 40 | 25.0 [47] |
| Linckoside B/blue starfish L. laevigata L. | 40 | 76.0 [47] |
| Linckoside F/blue starfish L. laevigata L. | 40 | 30.0 [43] |
| Linckoside G/blue starfish L. laevigata L. | 40 | <10.0 [43] |
| Linckoside H/blue starfish L. laevigata L. | 40 | <10.0 [43] |
| Linckoside I/blue starfish L. laevigata L. | 40 | 40.0 [43] |
| Linckoside J/blue starfish L. laevigata L. | 40 | <10.0 [43] |
| Linckoside K/blue starfish L. laevigata L. | 40 | 50.0 [43] |
| NGF | 10 * | 45.0 [47] |
Table 2.
Synergistic effect between NGF and marine drugs in PC12 cells.
Table 2.
Synergistic effect between NGF and marine drugs in PC12 cells.
| Compound/organism | [NGF] ng/mL | Effect of NGF alone (%) | [Drug] µM | Effect of NGF + marine drug (%) |
|---|
| Linckoside A/blue starfish Linckia laevigata L. | 2.5 | 5.0 | 40 | 62.0 [47] |
| Linckoside B/blue starfish L. laevigata L. | 2.5 | 5.0 | 40 | 87.0 [47] |
| Linckoside F/blue starfish L. laevigata L. | 1.5 | 6.0 | 40 | 90.0 [43] |
| Linckoside G/blue starfish L. laevigata L. | 1.5 | 6.0 | 40 | 40.0 [43] |
| Linckoside H/blue starfish L. laevigata L. | 1.5 | 6.0 | 40 | 46.0 [43] |
| Linckoside I/blue starfish L. laevigata L. | 1.5 | 6.0 | 40 | 95.0 [43] |
| Linckoside J/blue starfish L. laevigata L. | 1.5 | 6.0 | 40 | 46.0 [43] |
| Linckoside K/blue starfish L. laevigata L. | 1.5 | 6.0 | 40 | 98.0 [43] |
| LLG-5/blue starfish L. laevigata L. | 5.0 | 20.6 | 10 | 59.3 [46] |
| LLG-3/blue starfish L. laevigata L. | 5.0 | 20.6 | 10 | 63.1 [46] |
| Granulatoside A/blue starfish L. laevigata L. | 1.5 | <10.0 | 40 | 95.0 [45] |
| GP-3/starfish A. pectinifera Muller and Troschel | 5.0 | 20.6 | 10 | 38.2 [48] |
| CEG-6/sea cucumber Cucumaria echinata Von Marenzeller | 5.0 | 7.5 | 10 | 43.0 [49] |
| HLG-3/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 42.0 [49] |
| CEG-8/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 40.2 [49] |
| CEG-9/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 35.1 [49] |
| SJG-1/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 39.1 [50] |
| SJG-2/sea cucumber Stichopus japonicus Selenka | 5.0 | 20.6 | 10 | 64.8 [50] |
| CG-1/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 43.0 [50] |
| CEG-3/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 50.8 [50] |
| CEG-4/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 34.0 [50] |
| CEG-5/sea cucumber C. echinata Von Marenzeller | 5.0 | 7.5 | 10 | 35.7 [50] |
Some of the studies allowed establishing structure-activity relationships. Han
et al. [
43] tested six steroid glycosides (Linckosides F–K) from the blue starfish
Linckia laevigata L. and concluded that the carbon branch modified by a pentose at the side chain (present only in linckoside K;
Figure 1) and the 2′-
O-methyl ether group of xylose at C-3 (present in linckosides F and K) were the most important structures for the NGF-mimic activity. 2′-
O-Methyl ether group of xylose at C-3 plays a role for the significant NGF-enhancing activity. Another steroid glycoside, granulatoside A [
45], and two gangliosides, LLG-3 (
Figure 1) and LLG-5 (
Figure 1) [
46], isolated from the same blue starfish, were also very promising.
Kisa
et al. [
50] evaluated the NGF-mimic activity of five monosialo-gangliosides from the sea cucumber
Cucumaria echinata Von Marenzeller, SJG-1, CG-1, CEG-3, CEG-4 and CEG-5. The most active one was CEG-3 (
Figure 1), which possesses an acetyl group at the terminal fucose unit. Among the disialogangliosides (HLG-3 and CEG-6) and trisialogangliosides (CEG-8 and CEG-9) isolated from the same sea cucumber [
49], those displaying highest activity were CEG-6, HLG-3 and CEG-8, although lower than that of CEG-3. This was in accordance with the previous assumption made by the same authors, since CEG-6 and HLG-3 possess a terminal fucose without acetyl group and CEG-8 does not contain a terminal fucose. Despite their structural similarity, the different NGF-enhancement effect of linckosides A and B suggests that the sugar moiety at C-29 of the aglycon plays an important role for the activity of these steroid glycosides [
47].
5. Marine Drugs Affecting Enzymes Involved in Neurodegeneration
Neurodegenerative diseases, such as AD and Parkinson’s disease (PD), are characterized by the loss of particular neuronal populations and by intraneuronal and extracellular accumulation of fibrillary materials [
51]. AD is the most common form of dementia. It is an age-related neurodegenerative disorder characterized by extracellular deposition of plaques of aggregated β-amyloid protein (Aβ), intracellular deposition of neurofibrillary tangles that contain hyperphosphorylated tau (τ) protein, and a profound loss of basal forebrain cholinergic neurons that innervate the hippocampus and the neocortex [
52]. Current AD treatment consists of the administration of inhibitors of AChE and butyrylcholinesterase (BuChE) enzymes in order to counteract brain’s acetylcholine deficiency [
53]. However, other enzymes could be considered a target for future drug development, such as the proteases β-secretase (BACE1) and presenilin-dependent γ-secretase [
54,
55,
56] involved in the cleavage of amyloid-β precursor protein (APP) into Aβ fragments, and protein kinases that hyperphosphorylate τ protein making up paired helical filaments (PHFs) and straight filaments of neurofibrillary tangles (NFTs) in the brain [
57,
58,
59].
Protein kinases also display a pivotal role in other neurodegenerative disorders, such as in PD. Hyperphosphorylated α-synuclein, the major constituent of Lewy bodies, is one of the most important hallmarks of PD [
60,
61]. Several post-translational modifications to α-synuclein occur in PD, phosphorylation at serine (Ser)-129 residue being among them [
61,
62].
In the next sections, examples of marine compounds with inhibitory activity against cholinesterases (AChE and BuChE), BACE1 and protein kinases will be given.
5.1. Inhibition of Cholinesterases (ChEs) Activity
Beedessee
et al. [
53] evaluated the anticholinesterase effect of 134 extracts from 45 species of marine sponges and two of them showed strong AChE inhibition, namely the ethyl acetate extracts of
Pericharax heteroraphis Poléjaeff (90% inhibition at 0.1 mg/mL) and of
Amphimedon navalis Pulitzer-Finali (96% inhibition at 0.1 mg/mL). These extracts were rich in terpenoid compounds. Two other extracts obtained from the sponges
Latrunculia lendenfeldi Hentschel and
Latrunculia bocagei Ridley and Dendy displayed IC
50 = 1.3 and 9 ng/mL, respectively [
63].
Some examples of AChE inhibitors isolated from sponges, corals and molluscs are shown in
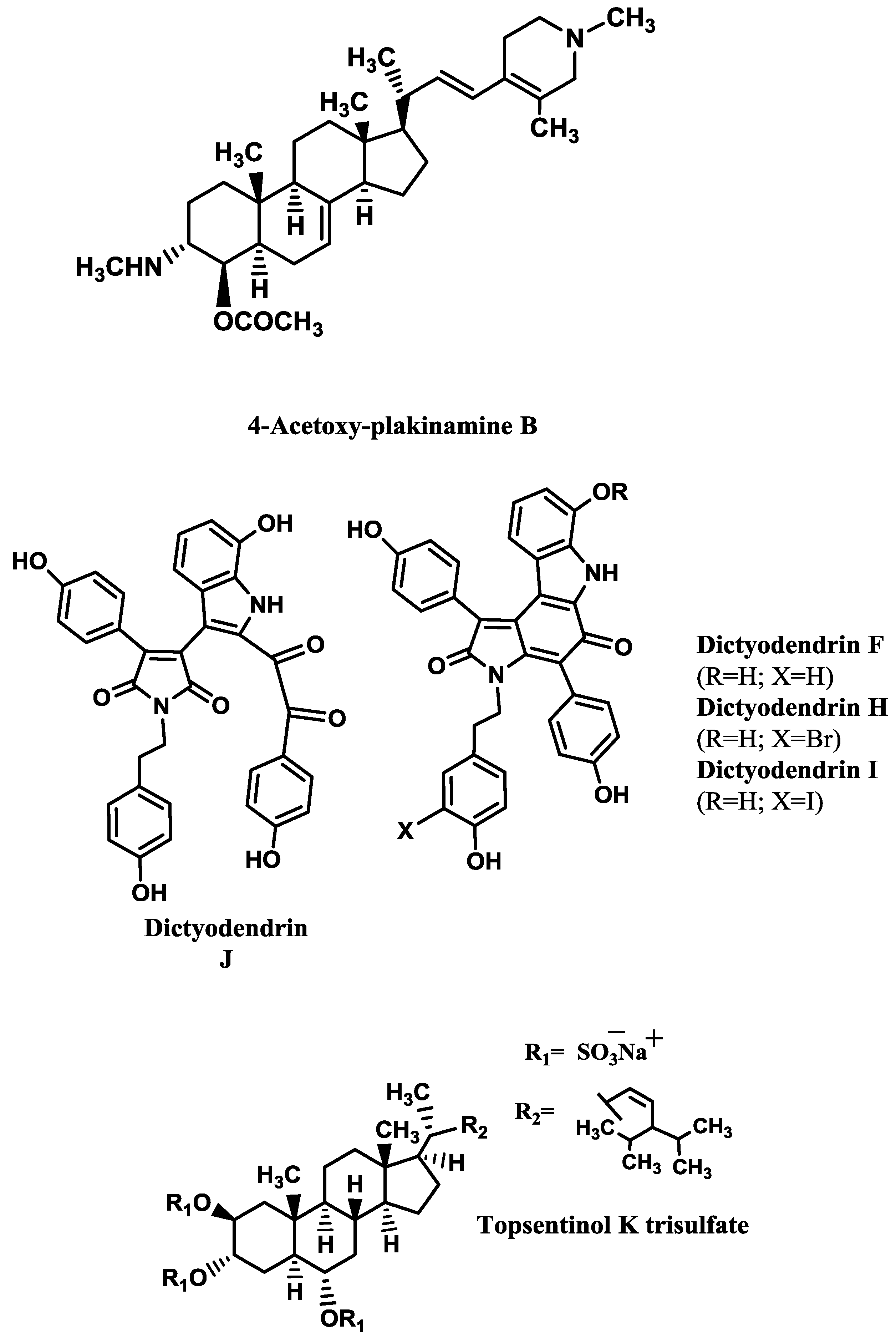
Table 3. The kinetics analysis of AChE inhibition promoted by the stigmastane-type steroidal alkaloid 4-acetoxy-plakinamine B (
Figure 2) suggested a mixed-competitive mode of inhibition [
64].
Table 3.
Marine drugs as AChE inhibitors.
Table 3.
Marine drugs as AChE inhibitors.
| Compound/Organism | IC50 (µM) |
|---|
| 4-acetoxy-plakinamine B/sponge Corticium sp. | 3.75 [64] |
| 2-Bromoamphimedine/sponge Petrosia n. sp. | 300 [65] |
| Petrosamine/sponge Petrosia n. sp. | 91 [65] |
| Cladidiol/soft coral Cladiella sp. | 67 [66] |
| Turbotoxin A/mollusc Turbo marmoratus L. | 28 [67] |
5.2. Inhibition of BACE1
Williams
et al. [
68] screened 130 pre-fractionated extracts from marine invertebrates and cyanobacteria against BACE1 activity, resulting in 7% of the extracts with outstanding inhibition (>90%) and 11% with activity between 70% and 89%. One group of submicromolar BACE1 inhibitors revealed by this study was the bastadins, a family of highly modified tetrapeptides occurring in some species of sponges, from which bastadin 9 is an example. Several metabolites isolated from sponges [
69,
70,
71,
72,
73,
74] showed BACE1 inhibitory activity (
Table 4). The most promising ones are dictyodendrins F and H–J (
Figure 2) [
74] and topsentinol K trisulfate (
Figure 2) [
70].
Dai
et al. [
69] tested several xestosaprols and concluded that the β-orientation of the C-3 alcohol (only present in xestosaprol H) was an important feature for the activity. Structure-activity relationships were also established for topsentinols. Topsentinol K trisulfate was the only active sterol isolated from the sponge
Topsentia sp., while topsentinols K and L were inactive. These results demonstrated that the presence of sulfate esters contribute to BACE1 activity [
70].
Table 4.
BACE1 inhibitors.
Table 4.
BACE1 inhibitors.
| Compound/organism | IC50 (μM) |
|---|
| Xestosaprol D/sponge Xestospongia sp. | 93.2 [72] |
| Xestosaprol F/sponge Xestospongia sp. | 135.0 [69] |
| Xestosaprol G/sponge Xestospongia sp. | 155.0 [69] |
| Xestosaprol H/sponge Xestospongia sp. | 82.0 [69] |
| Xestosaprol I/sponge Xestospongia sp. | 163.0 [69] |
| Xestosaprol J/sponge Xestospongia sp. | 90.0 [69] |
| Xestosaprol K/sponge Xestospongia sp. | 93.0 [69] |
| Xestosaprol L/sponge Xestospongia sp. | 98.0 [69] |
| Xestosaprol M/sponge Xestospongia sp. | 104.0 [69] |
| Dictyodendrin F/sponge Ianthella sp. | 1.5 [74] |
| Dictyodendrin H/sponge Ianthella sp. | 1.0 [74] |
| Dictyodendrin I/sponge Ianthella sp. | 2.0 [74] |
| Dictyodendrin J/sponge Ianthella sp. | 2.0 [74] |
| Dictazole A/sponge Smenospongia cerebriformis Duchassaing and Michelotti | 135.0 [71] |
| Topsentinol K trisulfate/sponge Topsentia sp. | 1.2 [70] |
| Lamellarin O/sponge Ianthella sp. | 40% (at 10 μM) [73] |
| Lamellarin O1/sponge Ianthella sp. | 60% (at 10 μM) [73] |
| Lamellarin O2/sponge Ianthella sp. | 40% (at 10 μM) [73] |
| Ianthellidone F/sponge Ianthella sp. | 40% (at 10 μM) [73] |
Figure 2.
AChE and BACE1 inhibitors isolated from marine invertebrates.
Figure 2.
AChE and BACE1 inhibitors isolated from marine invertebrates.
5.3. Inhibition of Protein Kinases
The human kinome codifies nearly 500 different protein kinases, which have serine/threonine (Ser/Thr) or tyrosine (Tyr) specificity. They catalyse phosphorylation pathways that regulate most of the biological processes, but abnormal phosphorylation is, normally, a cause or a consequence of disease [
61]. As stated above, inhibitors of these protein kinases can be useful to alleviate the symptoms of neurodegenerative disorders, such as AD and PD. In the next sections, a brief description on the involvement of protein kinases in neurodegeneration will be given, as well as some examples of marine protein kinases inhibitors and, when available, data about their inhibition mode.
5.3.1. Glycogen Synthase Kinase 3 (GSK-3)
GSK-3, also known as τ phosphorylating kinase I, is a multifunctional Ser/Thr kinase that is involved in glycogen metabolism, insulin signalling, cell proliferation, neuronal function, oncogenesis and embryonic development. There are two isoforms (α and β) with 98% homology and similar biological functions, but most of the research has been dedicated to the isoform β. GSK-3 is highly expressed in the brain and is associated with several CNS disorders, such as AD, bipolar disorder, Huntington’s disease and other neurodegenerative diseases [
75,
76].
GSK-3β phosphorylates transcription factors and cytoskeletal proteins, such as τ [
77]. There are, at least
in vitro, 40 different Ser and Thr residues in τ that can be phosphorylated by GSK-3 [
78,
79,
80,
81].
The human τ gene suffers extensive alternative splicing, giving rise to the expression of multiple spliced exons, exon 10 being one of them. The presence of exon 10 results in τ with four repeat microtubule-binding sequences (4R), while isoforms without exon 10 have only three (3R). Normally, the ratio of 3R and 4R tau transcripts is close to one. Although mutations in splicing regulatory elements are common in inherited tauopathies, in sporadic AD the ratio 4R/3R is also increased [
82]. In addition to hyperphosphorylate τ, GSK-3 can also induce τ splicing, because it phosphorylates the splicing factor SC35, an enhancer of splicing elements that regulate exon 10 splicing in τ [
79]. Hernández
et al. [
83] demonstrated that GSK-3 inhibition in cultured neurons affected τ splicing, resulting in increased τ mRNA containing exon 10.
Moreover, GSK-3β has been reported to play a role in the toxic effect mediated by Aβ since, in cultured cells, Aβ activates GSK-3, leading to the phosphorylation of SC35 [
79] and exposure of cortical and hippocampal primary neuronal cultures to Aβ induces activation of GSK-3β, τ hyperphosphorylation and cell death [
78]. Thus, inhibition of GSK-3 can contribute to the reduced formation of both Aβ plaques and neurofibrillary tangles [
84].
Marine compounds [
76,
78,
80,
81,
85,
86,
87,
88,
89,
90] able to inhibit both isoforms of GSK-3 are shown in
Table 5 and
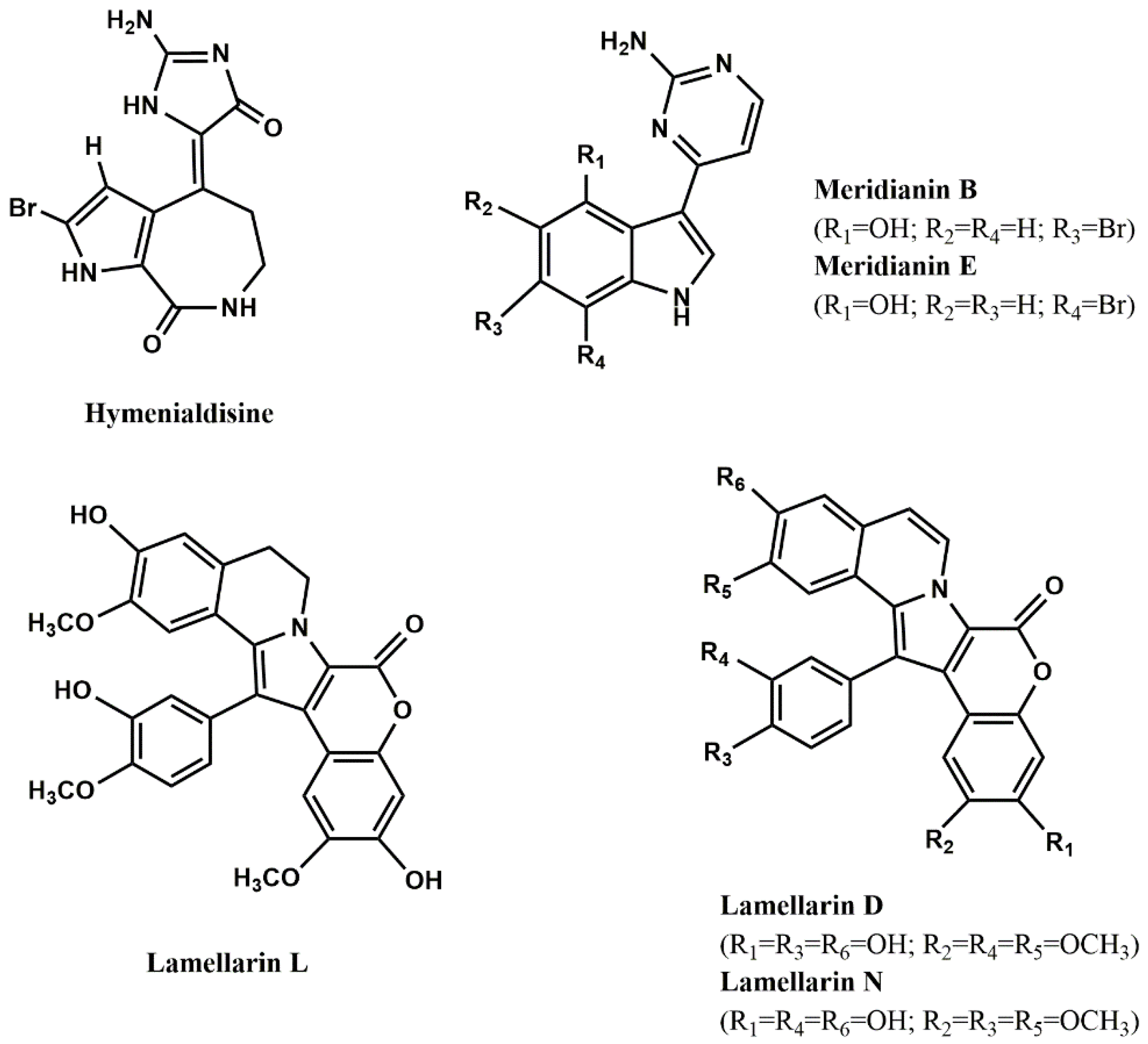
Figure 3. As it can be seen, hymenialdisine (
Figure 3), lamellarins (
Figure 3) and meridianins (
Figure 3) are the most active ones.
Few studies explored the mode of inhibition and the structural features contributing to high inhibitory activity of GSK-3 inhibitors. Concerning the first aspect, it is known that the alkaloid hymenialdisine and meridianins are competitive inhibitors at the ATP-binding site [
81,
91], while the alkaloid manzamine A [
80] and the furanoterpenoids tricantin [
89] and palinurin [
76] are non-ATP competitive. According to Eldar-Finkelman and Martinez [
91], ATP non-competitive GSK-3 inhibitors should be more selective than ATP-competitive ones, since they bind to unique regions within GSK-3, leading to a more subtle modulation of the kinase activity than by simply ATP entrance blockade.
Table 5.
GSK-3 inhibitors from marine organisms.
Table 5.
GSK-3 inhibitors from marine organisms.
| Compound/organism | Isoform | IC50 (μM) |
|---|
| Carteriosulfonic acid A/sponge Carteriospongia sp. | GSK-3β | 12.5 [88] |
| Carteriosulfonic acid B/sponge Carteriospongia sp. | GSK-3β | 6.8 [88] |
| Carteriosulfonic acid C/sponge Carteriospongia sp. | GSK-3β | 6.8 [88] |
| Hymenialdisine/sponge Axinella verrucosa Esper | GSK-3β | 10.0* [81] |
| Tricantin/sponge Ircinia sp. | GSK-3β | 7.5 [89] |
| Lamellarin α/ascidian Didemnum obscurum F. Monniot | GSK-3α/β | 1.4 [86] |
| Lamellarin D/prosobranch mollusc Lamellaria sp. | GSK-3α/β | 0.3 [86] |
| Lamellarin H/ascidian Didemnum chartaceum Sluiter | GSK-3α/β | 9.5 [86] |
| Lamellarin L/ascidian Didemnum sp. | GSK-3α/β | 40.0 * [86] |
| Lamellarin N/ascidian Didemnum sp. | GSK-3α/β | 5.0 * [86] |
| Leucettamine B/sponge Leucetta microraphis Haeckel | GSK-3α | 7.7 [85] |
| Leucettamine B/sponge L. microraphis Haeckel | GSK-3β | >10.0 [85] |
| Leucettamine B/sponge L. microraphis Haeckel | GSK-3α/β | 2.9 [85], 15.0 [90] |
| Manzamine A/sponge Acanthostrongylophora sp. | GSK-3β | 10.2 [80], 12.30 [78] |
| Meridianin A/ascidian Aplidium meridianum Sluiter | GSK-3β | 1.3 [87] |
| Meridianin B/ascidian A. meridianum Sluiter | GSK-3β | 0.5 [87] |
| Meridianin C/ascidian A. meridianum Sluiter | GSK-3β | 2.0 [87] |
| Meridianin D/ascidian A. meridianum Sluiter | GSK-3β | 2.5 [87] |
| Meridianin E/ascidian A. meridianum Sluiter | GSK-3β | 2.5 [87] |
| Meridianin F/ascidian A. meridianum Sluiter | GSK-3β | 2.0 [87] |
| Meridianin G/ascidian A. meridianum Sluiter | GSK-3β | 350.0 [87] |
| Palinurin/sponge Ircinia dendroides Schmidt | GSK-3β | 2.6 [76] |
| (Z)-5-(4-Hydroxybenzylidene)-hydantoin/sponge Hemimycale arabica Ilan, Gugel and van Soest | GSK-3β | 13.70 [78] |
Figure 3.
Most potent protein kinases inhibitors from marine organisms.
Figure 3.
Most potent protein kinases inhibitors from marine organisms.
Regarding the second issue, Hamann
et al. [
80] synthetized several manzamine A analogs to study the influence of several substituents on GSK-3 inhibition. They concluded that the entire molecule (carboline moiety and aliphatic heterocyclic system) contributed for the inhibitory activity. Concerning the carboline moiety, the substitution of nitrogen 9 by large groups, such as isobutyl, dodecyl or methylcarboxybutyl, produced non-active compounds, while shorter groups (methyl and ethyl) did not cause activity reduction. Changes in the aliphatic heterocyclic system also influence GSK-3 inhibition, because if conformational restriction is increased, compounds are more active.
Baunbæk
et al. [
86] evaluated the ability of several lamellarins (
Figure 3) and their analogs to inhibit not only GSK-3, but also other kinases (see next sections). Structure-activity studies led them to conclude that complex, but specific, interactions between lamellarins’ substituents and their kinase targets may exist, since different substituents influenced the inhibitory activity against different kinases.
Other protein kinases function as activators for τ phosphorylations by GSK-3, such as casein kinase 1 (CK1) e 2 (CK2), dual specificity tyrosine phosphorylation-regulated kinase 1 A (DYRK1A), AMP-dependent protein kinase (PKA) and cyclin-dependent kinase-5 (CDK5) [
61,
79]. For instance, when CDK-5 phosphorylates τ at Ser-235 and Ser-404 residues, it promotes the subsequent τ phosphorylation by GSK-3 at Thr-231 and Ser-400, respectively. On the other hand, if PKA phosphorylates τ at Ser-214, it will activate τ phosphorylation by GSK-3 at Ser-210, Thr-205, Ser-199 and Ser-195 residues. However, some τ residues, such as Ser-396 and Ser-404, can be directly phosphorylated by GSK-3 without prior activity of other kinases [
77,
79].
5.3.2. DYRK1A
DIRK1A is located in chromosome 21 and codifies a protein kinase responsible for the phosphorylation of τ at Thr-212, Ser-202 and Ser-404 residues
in vitro and
in vivo. Studies indicate that overexpression of DYRK1A in the brains of Down’s syndrome patients may contribute to early onset of AD pathology through hyperphosphorylation of τ [
59].
Moreover, DYRK1A also phosphorylates other AD-related proteins,
in vitro and
in vivo. Phosphorylation of APP at Thr-668 residue leads to APP cleavage by BACE1 and γ-secretase and consequently to increased production of Aβ peptide [
92]. In a similar way, phosphorylation at Thr-354 residue of presenilin 1 (PS1), a key component of the γ-secretase complex, also induced an increased γ-secretase activity [
93]. Phosphorylation of septin-4 (SEPT-4) at Ser-68 and Ser-107 residues by DYRK1A may regulate specific protein–protein interactions, since septins are a family of filament-forming guanine nucleotide-binding proteins involved in cytokinesis, exocytosis and other cellular processes, such as synapse functions. It was shown that a complex formed by SEPT4, DYRK1A and α-synuclein may contribute to the development of α-synuclein-positive cytoplasmic aggregates characteristic of PD and, since SEPT4 has been found in neurofibrillary tangles, SEPT4/DIRK1A is also involved in the pathology of AD [
94,
95,
96]. Finally, DIRK1A also phosphorylates the regulator of calcineurin 1 (RCAN) at Ser-112 and Thr-192 residues, the latter enhancing τ phosphorylation [
97] and phosphorylating Munc18–1 at Thr-479 residue, stimulating its binding to Syntaxin 1 and X11α, two proteins involved in synaptic vesicle exocytosis and APP processing, respectively [
98]. Examples of marine compounds [
85,
86] that inhibit DYRK1A are shown in
Table 6 and
Figure 3.
Table 6.
DYRK1A inhibitors from marine organisms.
Table 6.
DYRK1A inhibitors from marine organisms.
| Compound/organism | IC50 (μM) |
|---|
| Lamellarin α/ascidian Didemnum obscurum F. Monniot | 5.0 [86] |
| Lamellarin D/prosobranch mollusc Lamellaria sp. | 0.5 [86] |
| Leucettamine B/sponge Leucetta microraphis Haeckel | 0.6–1.0 [85] |
| Lamellarin L/ascidian Didemnum sp. | 0.1 [86] |
| Lamellarin N/ascidian Didemnum sp. | 40.0 * [86] |
5.3.3. CK1 and CK2
In mammals, the CK1 family of protein kinases consist of monomeric enzymes assembled from seven isoforms (α, β, γ1, γ2, γ3, δ, and ε). They are responsible for the phosphorylation of cytoskeletal proteins, such as spectrin, troponin, myosin, ankyrin, τ and α-synuclein, but also of non-cytoskeletal proteins (SV40 T antigen, p53, and β-catenin). These phosphorylations modulate important physiological functions like vesicular trafficking, DNA repair, cell cycle kinetics and cell division [
99].
In AD patients’ brains, CK1α and CK1δ are co-localized with neurofibrillary lesions and granulovacuolar degeneration bodies. Furthermore, CK1α, CK1ε and CK1δ levels are increased in CA1 region of hippocampus, with a predominance of CK1δ. This CK1δ isoform phosphorylates τ at Ser-202, Thr-205, Ser-396 and Ser-404 residues and a combination of CK1δ and GSK-3 activities induce more than three-quarters of the Ser/Thr phosphorylations identified in τ-PHF, indicating that both protein kinases are involved in the pathogenesis of AD [
61]. Additionally, APP, BACE1 and γ-secretase contain multiple CK1 phosphorylation sites and CK1ε leads to an increase of Aβ peptide production. On the other hand, Aβ stimulates CK1 activity [
79,
100].
CK1 is also involved in PD pathology. It has been demonstrated that α-synuclein is phosphorylated at Ser-129 by CK1 [
61].
The CK2 holoenzyme forms a heterotetrameric complex with two catalytic (CK2α and CK2α′) and two regulatory (CK2β) subunits. Overexpression of CK2 leads to several pathological conditions, ranging from cardiovascular pathologies and cancer progression to infectious diseases and neurodegeneration. CK2 activity increases due to the presence of Aβ peptide and, thus, may accelerate τ phosphorylation. Besides CK2’s role in AD progression, CK2β subunits are present in Lewy bodies and phosphorylate α-synuclein at Ser-129 residue [
61].
Table 7 and
Figure 3 report some examples of marine compounds [
81,
86,
87] that display inhibitory activity against CK1 and CK2.
Hymenialdisine is a competitive inhibitor at the ATP-binding site [
81].
5.3.4. Cyclin-Dependent Kinase 5 (CDK5)
CDKs are a group of protein kinases that regulate cell-cycle control (CDK1–4, 6 and 7), thymocyte apoptosis (CDK2), neuronal functions (CDK5) and transcriptional control (CDK7–9). CDK5, initially known as brain proline-directed protein kinase or neuronal cdc2-like protein kinase, has been considered a major τ kinase that contributes to tauopathies. Interaction of CDK5 with either p35 or p39, two activator proteins, is necessary for its activation [
101]. CDK5/p35 is involved in several processes critical to CNS function during development and throughout maturity [
102]. CDK5/p35 is known to phosphorylate τ (at Ser-235, Ser-396 and Ser-404) and MAP-1B, Pak1 kinase and neurofilament subunits [
81] and its activity is promoted by Aβ peptide. Indeed, CDK5/p35 phosphorylates τ at Ser-396 and Ser-404 residues in response to Aβ25–35 [
103].
Table 7.
CK1 and CK2 inhibitors from marine organisms.
Table 7.
CK1 and CK2 inhibitors from marine organisms.
| Compound/organism | Enzyme | IC50 (μM) |
|---|
| Hymenialdisine/sponge Axinella verrucosa Esper | CK1 | 35.0 * [81] |
| Hymenialdisine/sponge A. verrucosa Esper | CK2 | 7.0 [81] |
| Lamellarin α/ascidian Didemnum obscurum F. Monniot | CK1 | 7.9 [86] |
| Lamellarin D/prosobranch mollusc Lamellaria sp. | CK1 | 13.0 [86] |
| Lamellarin K/ascidian Didemnum sp. | CK1 | 6.0 [86] |
| Lamellarin H/ascidian Didemnum chartaceum Sluiter | CK1 | 5.3 [86] |
| Meridianin B/ascidian Aplidium meridianum Sluiter | CK1 | 1.0 [87] |
| Meridianin C/ascidian A. meridianum Sluiter | CK1 | 30.0 [87] |
| Meridianin D/ascidian A. meridianum Sluiter | CK1 | 100.0 [87] |
| Meridianin E/ascidian A. meridianum Sluiter | CK1 | 0.4 [87] |
Aberrant CDK5 activity is induced by the conversion of p35 to p25 by calpain, a Ca
2+-dependent cysteine protease. CDK5/p25 plays a role in the pathogenesis of neurodegenerative diseases since it induces the formation of τ-PHF, τ aggregation and neuronal loss [
102,
104]. Other evidence from the involvement of Aβ peptide in τ hyperphosphorylation comes from the ability of Aβ to directly promote an increase of the levels of intracellular Ca
2+ ([Ca
2+]i) in neurons, this increment leading to calpain activation, which, in turn, cleaves p35 into p25 [
105].
Table 8.
CDK5 inhibitors from marine organisms.
Table 8.
CDK5 inhibitors from marine organisms.
| Compound/organism | Enzyme | IC50 (μM) |
|---|
| Lamellarin α/ascidian Didemnum obscurum F. Monniot | CDK5/p25 | >10.0 [86] |
| Lamellarin D/prosobranch mollusc Lamellaria sp. | CDK5/p25 | 0.6 [86] |
| Lamellarin L/ascidian Didemnum sp. | CDK5/p25 | 0.1 [86] |
| Lamellarin N/ascidian Didemnum sp. | CDK5/p25 | 25.0 * [86] |
| Fascaplysin/sponge Fascaplysinopsis sp. | CDK5/p35 | 20.0 [106] |
| Manzamine A/sponge Acanthostrongylophora sp. | CDK5/p35 | 1.5 [80] |
| Meridianin A/ascidian Aplidium meridianum Sluiter | CDK5/p25 | 3.0 [87] |
| Meridianin B/ascidian A. meridianum Sluiter | CDK5/p25 | 1.0 [87] |
| Meridianin C/ascidian A. meridianum Sluiter | CDK5/p25 | 6.0 [87] |
| Meridianin D/ascidian A. meridianum Sluiter | CDK5/p25 | 5.5 [87] |
| Meridianin E/ascidian A. meridianum Sluiter | CDK5/p25 | 0.2 [87] |
| Meridianin F/ascidian A. meridianum Sluiter | CDK5/p25 | 20.0 [87] |
| Meridianin G/ascidian A. meridianum Sluiter | CDK5/p25 | 140.0 [87] |
5.3.5. PKA Inhibitors
PKA is the first element of cAMP signal transduction cascade, one of the several second messenger-dependent pathways that generate intracellular responses to extracellular signals. PKA mediates most of cAMP actions by phosphorylation [
107].
Phosphorylation of τ at Ser-214 residue by PKA affects the interaction between τ and microtubules by reducing the tau’s affinity for them. This phenomenon also occurs with the phosphorylation caused by GSK-3β and CDK5 [
108].
Table 9.
PKA inhibitors from marine organisms.
Table 9.
PKA inhibitors from marine organisms.
| Compound/organism | IC50 (μM) |
|---|
| Meridianin A/ascidian Aplidium meridianum Sluiter | 11.0 [87] |
| Meridianin B/ascidian A. meridianum Sluiter | 0.2 [87] |
| Meridianin C/ascidian A. meridianum Sluiter | 0.7 [87] |
| Meridianin D/ascidian A. meridianum Sluiter | 1.0 [87] |
| Meridianin E/ascidian A. meridianum Sluiter | 90.0 * [87] |
| Meridianin F/ascidian A. meridianum Sluiter | 3.2 [87] |
| Meridianin G/ascidian A. meridianum Sluiter | 120.0 [87] |
8. Anti-Neuroinflammatory Activity of Marine Drugs
Neuroinflammation is a complex process involved in the pathology of several CNS diseases, such as AD, PD, multiple sclerosis and ischemic stroke, and involves activated microglia [
183,
184]. Activated microglial cells activate inflammatory mediators, such as proteolytic enzymes [
185], ROS and reactive nitrogen species [
183,
184,
185,
186], eicosanoids [
186,
187], pro-inflammatory cytokines [
185,
186,
188] and chemokines [
185,
186,
189], which can promote nociceptive transmission by causing activation of dorsal horn neurons. Many studies have indicated that inhibition of microglial activation attenuates the development of neuropathy [
183].
Two COX isozymes, COX-1 and COX-2, catalyse the rate-limiting steps of eicosanoids (prostaglandin (PG) and thromboxane) synthesis, by converting arachidonic acid into PGG2 and PGH2 and then into PGE2, PGF2α, PGD2, PGI2 and tromboxanes (TXB2) [
187]. Prostaglandins are critically involved in peripheral and spinal nociceptive sensitization. In general, COX1 is considered to be constitutive, while COX2 is considered as inducible, especially under inflammatory conditions. In the brain, COX2 is constitutively expressed only by specific neuronal populations, particularly in the hippocampus, being necessary for synaptic plasticity and memory acquisition. Inhibition of COX-2, but not of COX-1, by selective inhibitors attenuates hyperalgesia in neuropathic rats [
190]. Moreover, although nitric oxide (•NO) acts as cellular messenger and modulates neurotransmition, its overproduction has been associated with neuropathological disorders, such as stroke, AD and PD [
191]. Therefore, COX 1 and COX 2, as well as the enzyme neuronal nitric oxide synthase (nNOS), responsible for the synthesis of •NO, represent important therapeutic targets for the development of novel anti-neuroinflammatory drugs.
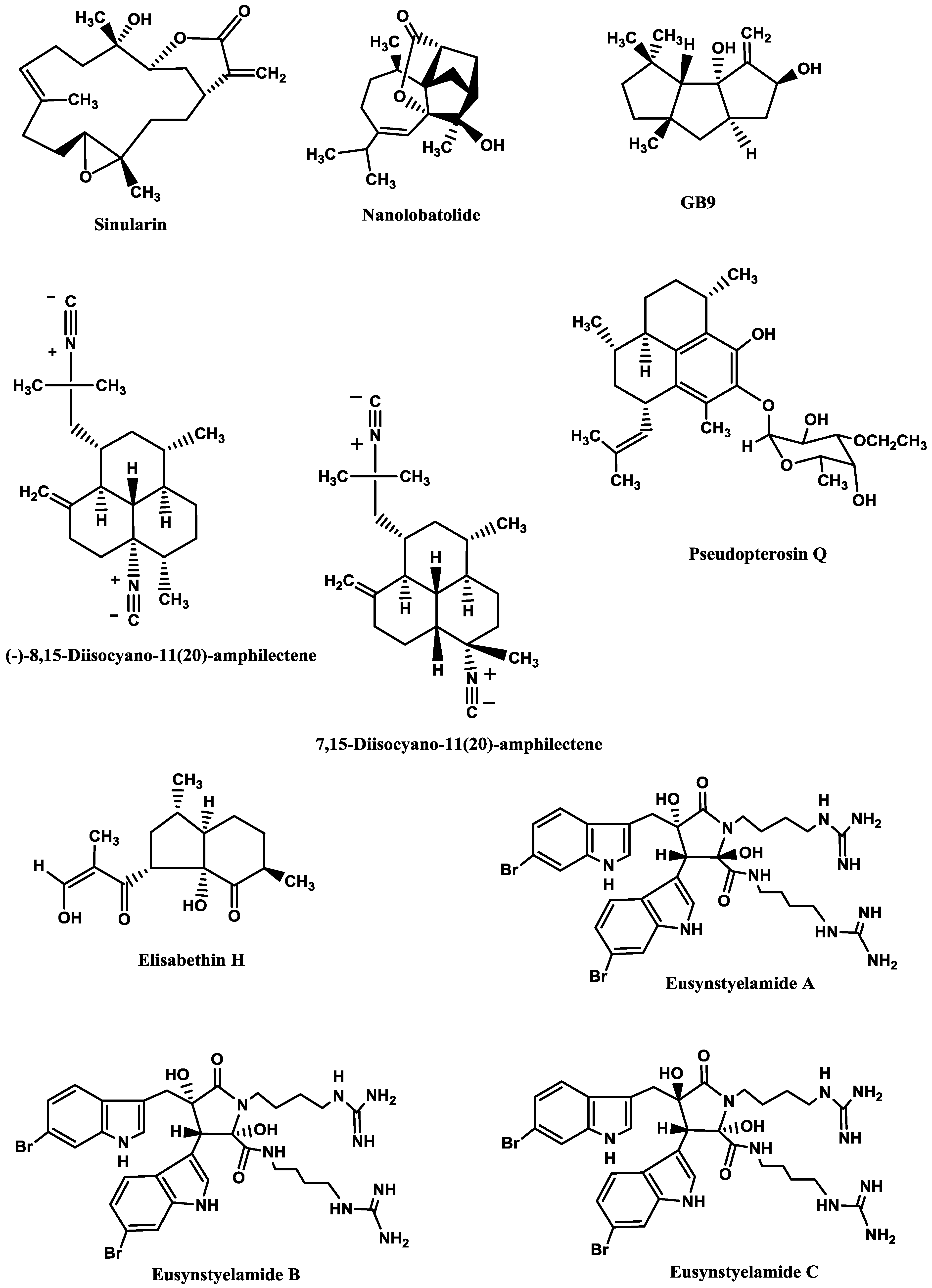
Sinularin (
Figure 6), a cembranolide diterpene isolated from the soft coral
Sinularia querciformis Pratt, displayed
in vitro anti-inflammatory activity by significantly inhibiting up-regulation of pro-inflammatory proteins (inducible NOS (iNOS) and COX-2) in LPS-stimulated murine macrophage RAW 264.7 cells. Sinularin (0.1–20 μM) dose-dependently reduced the levels of iNOS and increased those of TGF-β, while COX-2 levels were only reduced at 10 and 20 μM.
In vivo, subcutaneous administration of sinularin (80 mg/kg, intraplantar) to rats had analgesic effects and inhibited carrageenan-induced spinal neuroinflammation, up-regulation of microglial and astrocyte activation and up-regulation of iNOS in the dorsal horn of the lumbar spinal cord. Furthermore, treatment with sinularin (80 mg/kg) clearly inhibited carrageenan-induced leukocyte infiltration and up-regulated TGF-β1, demonstrating its analgesic effect [
192]. Nanolobatolide (
Figure 6), a C
18 terpene from the soft coral
Sinularia nanolobata Verseveldt, at 10 µM, also reduced the accumulation of iNOS in microglial cells stimulated with INFγ to 45.5% [
193].
Δ9(12)-Capnellene-8β,10α-diol (
Figure 6; GB9), a sesquiterpene isolated from the soft coral
Capnella imbricata Quoy and Gaimard, was able to down-regulate the expression of pro-inflammatory iNOS (IC
50 = 17.1 µM) and COX-2 (IC
50 = 6.21 µM) in INFγ-stimulated mouse microglial cells (BV2). Moreover, GB9 revealed an analgesic effect
in vivo. GB9 (10 mg/kg, intraperitoneal) significantly inhibited chronic constriction injury (CCI)-induced thermal hyperalgesia behaviour in rats, as well as inhibited CCI-induced elevation of microglial and neuronal COX-2 in the spinal cord [
183].
Diterpene isocyanides isolated from marine sponge
Hymeniacidon sp. (7-isocyano-11(20)-15(16)-amphilectadiene, (−)-8,15-diisocyano-11(20)-amphilectene (
Figure 6), 7,15-diisocyano-11(20)-amphilectene (
Figure 6), 8-isocyano-11(20)-ene-15-amphilectaformamide and monamphilectine A) were screened for anti-neuroinflammatory activity in LPS-activated rat brain microglia. They inhibited TXB2 generation (IC
50 = 0.20–4.69 µM), (−)-8,15-diisocyano-11(20)-amphilectene (IC
50 = 0.23 µM) and 7,15-diisocyano-11(20)-amphilectene (IC
50 = 0.20 µM) being the most active ones. However, all demonstrated minimal effect on O
2•− release (IC
50 > 10 µM) [
184]. Using the same system, Rodriguéz
et al. [
194] and Shi
et al. [
195] tested the anti-inflammatory activity of diterpenoid compounds isolated from the gorgonian
Pseudopterogorgia elisabethae Bayer. The most promising ones were pseudopterosin Q (
Figure 6) (IC
50 = 4.7 µM against TXB2 and IC
50 = 11.2 µM against O
2•− [
194] and elisabethin H (
Figure 6; IC
50 = 7.0 μM against O
2•−) [
195]. However, due to its cytotoxicity (LDH release > 50% at 3.4 µM), the inhibition of TXB2 by pseudopterosin Q could result, at least in part, from a toxic rather than a pharmacological effect [
194].
Figure 6.
Anti-neuroinflammatory marine compounds.
Figure 6.
Anti-neuroinflammatory marine compounds.
Finally, the brominated tryptophan-derived eusynstyelamides A, B, and C (
Figure 6), isolated from the ascidian
Eusynstyela latericius Sluiter, exhibited inhibitory activity against nNOS in rat cerebella, with IC
50 values of 41.7, 4.3 and 5.8 μM, respectively [
191].
9. Marine Skeletons as Scaffolds for Neural Tissue Engineering
When large tissue volume is lost due to an injury, tissue implantation is advantageous over cell therapy because it enables controlled organization of neurons into intricate networks before implantation [
28]. Thus, the aim of bioengineering is to deliver cells and signalling factors to a target tissue in combination with a non-cellular scaffolding material, which is an immobilization matrix that facilitates tissue ingrowth and regeneration [
196]. This three-dimensional (3D) cell cultures mimic the cytoarchitecture of
in situ tissue to a higher degree than cells grown on non-physiological hard surfaces (2D) and, therefore, 3D cultures have been shown to result in longer neurite outgrowth, higher levels of survival and distinct patterns of differentiation as compared to 2D monolayers [
197]. An ideal 3D scaffold must not only facilitate the adherence, spread and outgrowth of neurons and neuronal process, but also possess a large number of pores to allow cell expansion and diffusion transport of nutrients and waste molecules to and from the cells. Naturally derived scaffolds (e.g., gels of collagen and chitosan, polysaccharide fibres and aragonite) and synthetic polymers, such as methyl cellulose, poly(α-hydroxyacids), poly(glycolic acid), poly(L-lactic acid) and poly(lactic-co-glycolic acid) are being tested [
198,
199,
200,
201].
One of the most efficient scaffolds for neural development is the biodegradable and biocompatible aragonite, a needle-like crystalline form of calcium carbonate (CaCO
3) present in the exoskeleton of foraminiferans, sponges, corals, hydrozoans, molluscs (gastropods, bivalves, cephalopods), worms, arthropods (ostracods, barnacles). In invertebrates, this skeleton provides mechanical support for the soft tissues and act as a storage system withdrawing ions during times of special physiological demand [
196,
202,
203]. This scaffold presents several advantages over other templates, like hydrogels, not only because its pores are much larger than those of the hydrogels (160 mm
vs. few microns), allowing many more cells to accumulate, but also because it can release Ca
2+ to the medium, promoting cell adhesion, cell–cell contact and survival. Moreover, it also provides higher mechanical strength than hydrogels and the absence of a gel covering the cells may facilitate the explant–tissue interactions [
202].
Shanny
et al. [
198] grew rat hippocampal primary neurons on aragonite skeleton of the coral
Porites lutea Link and observed that the neurons usually grew on a sheet of glial cells and acquired the morphology of hippocampal pyramidal and granule neurons. Moreover, dendrites were branched and long, sometimes extending more than 100 µm away from the cell body, and axons were thinner than dendrites and grew up to hundreds of µm in length in all directions, covering the entire surface of the aragonite support. Synaptic connections were active in these neurons, since the presynaptic sites expressed the synaptic vesicle protein 2 (SV2) and post-synaptic spines contained the glutamate receptors GluR1. Peretz
et al. [
202] proved that
P. lutea aragonite matrix not only was a good support to cell growth
in vitro, but also
in vivo. They implanted the scaffolds in cortical regions of postnatal rat brains and observed that the implants did not cause any severe inflammation or rejection response and did not have significant influence on animal survival or behaviour. The implants were invaded by neural tissue and, besides supporting the survival of neurons in the cortex, they induced their invasion into the injured area.
Using a different marine scaffold, Baranes
et al. [
28] grew co-cultures of primary neurons and glia from rat hippocampi on aragonite matrices from the hydrozoan
Millepora dichotoma Forsskål. Conversely to the
P. lutea matrix, this scaffold supported ganglion-like cell spheres, rather than multi-layer cells, which included both astrocytes and mature neurons with active synaptic processes. The spheres were interconnected through fibres of neuronal and astrocytic processes and most of the cells had only cell–cell and no cell–matrix interactions. This cell organization resembles more the
in vivo situation, where neurons do not exhibit substrate contact. Moreover, it has also several advantages for neural tissue engineering, because most cells in the spheres are in contact with other cells, instead of with the matrix surface, and it is easier to detach cells from the scaffold since they are connected to the surface through a neck [
28].
M. dichotoma-derived aragonite matrix was also used to study the Ca
2+ uptake by neuronal and glial cells [
199]. The authors found that hippocampal cells growing on
45Ca
2+ or calcein-labelled aragonite took up aragonite-derived Ca
2+ and enhanced this uptake when extracellular Ca
2+ ions were chelated by EGTA. These ions activate Ca
2+-dependent adhesion molecules, like cadherins, which play important roles in cell migration, cell rearrangement and maintenance of tissue integrity.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}