Largazole Pharmacokinetics in Rats by LC-MS/MS


Abstract
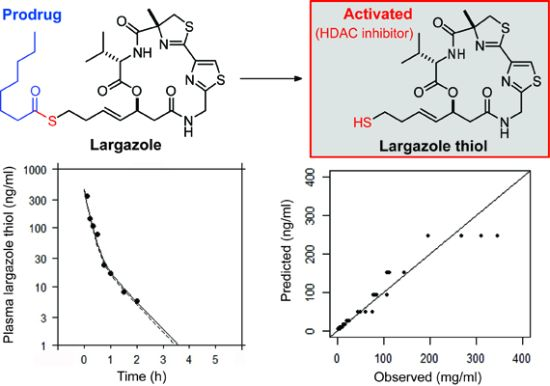
:
1. Introduction

2. Results
2.1. Assay Validation
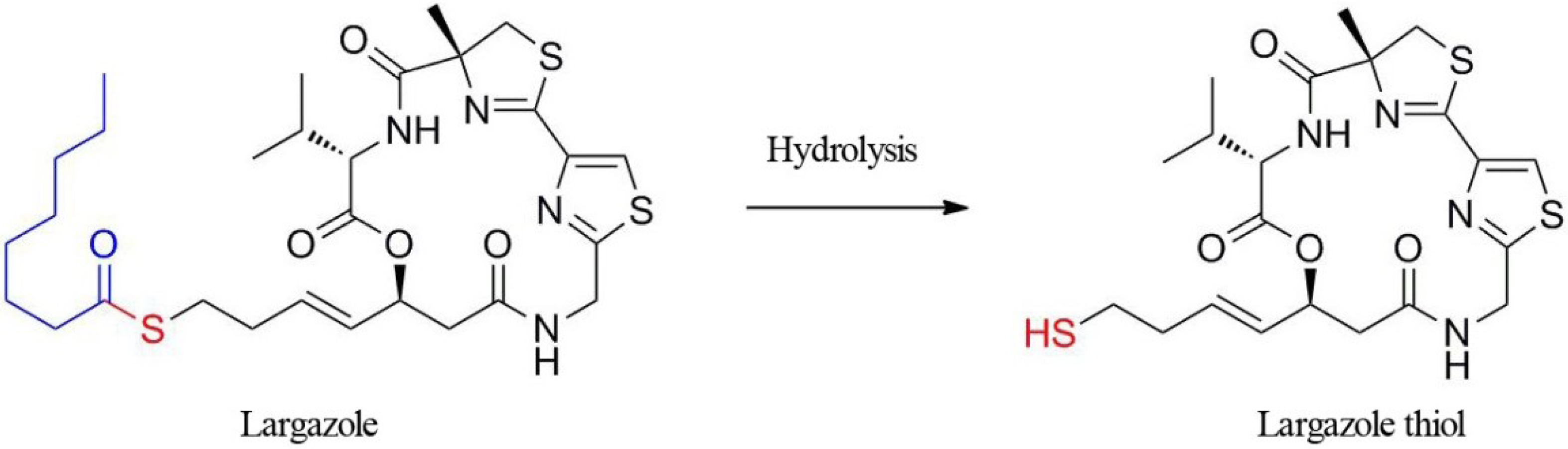
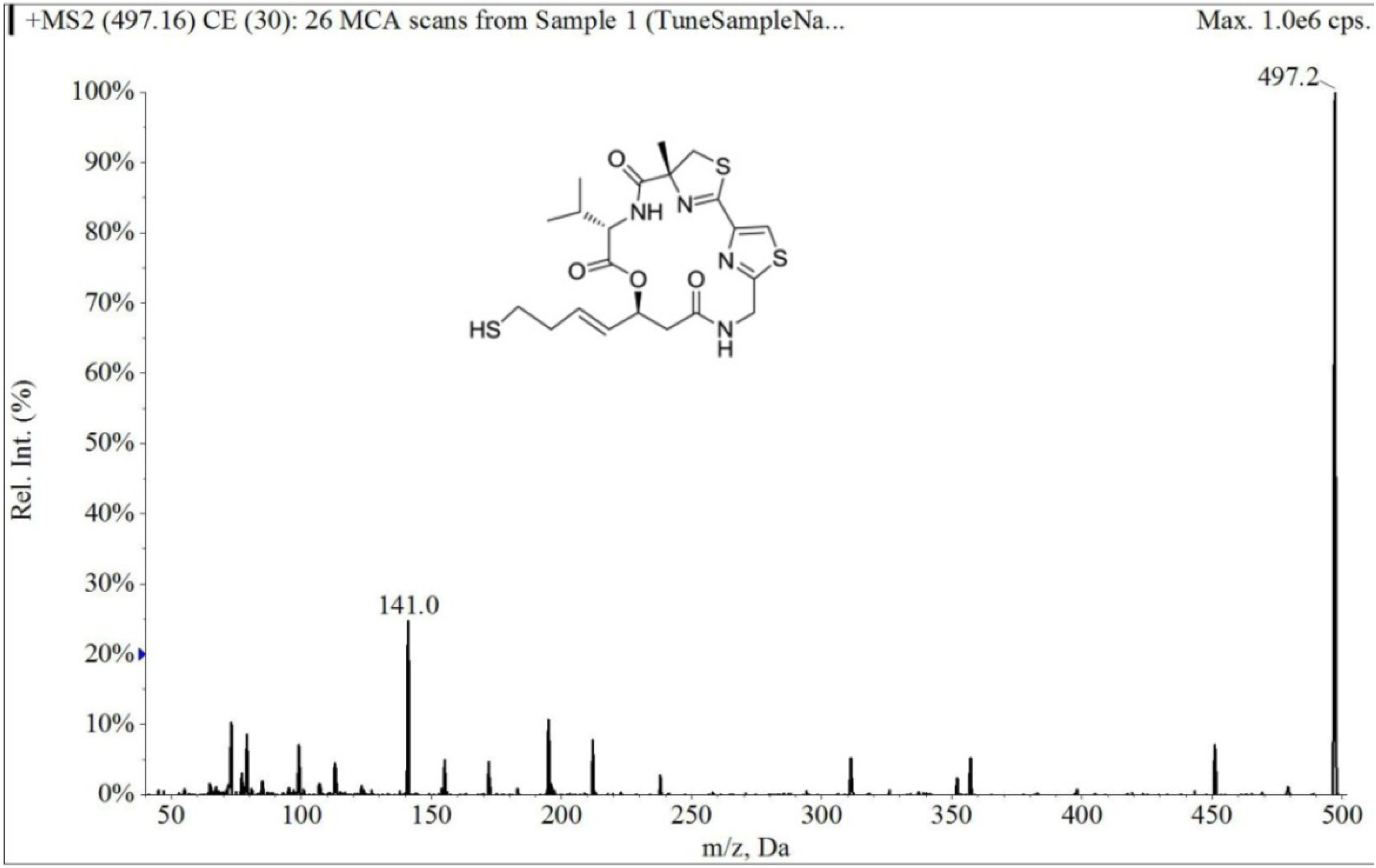
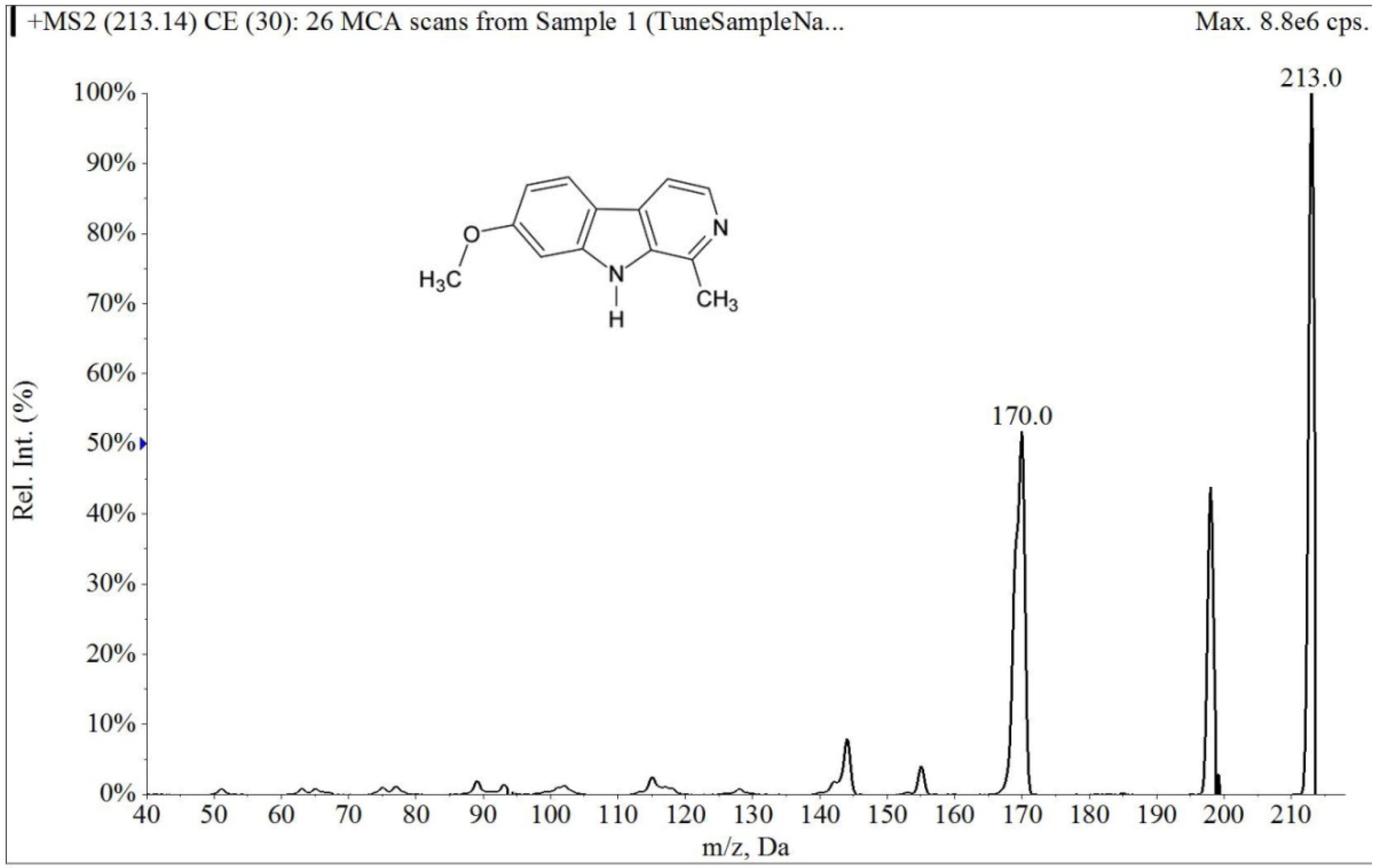
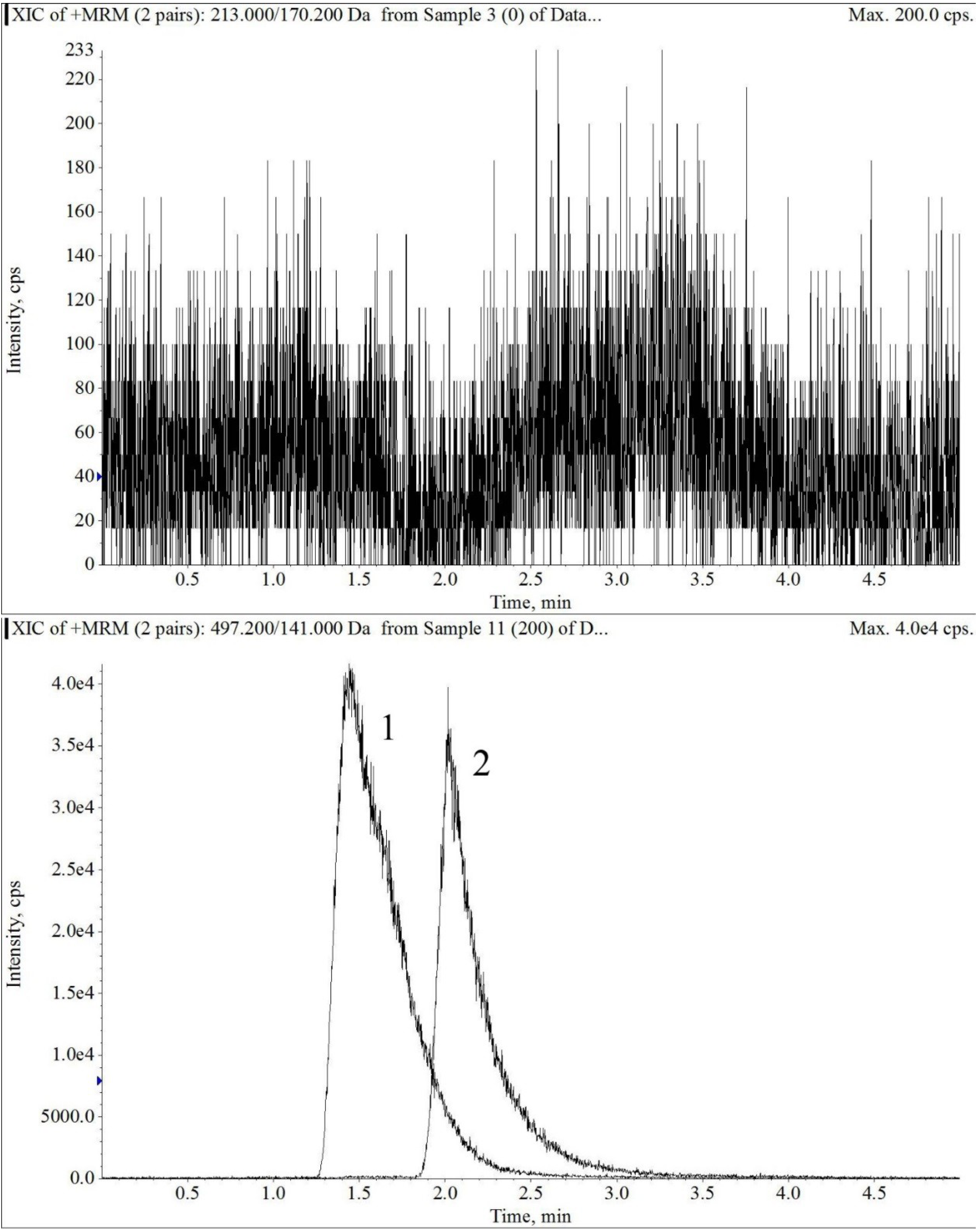
2.1.1. Specificity and Selectivity
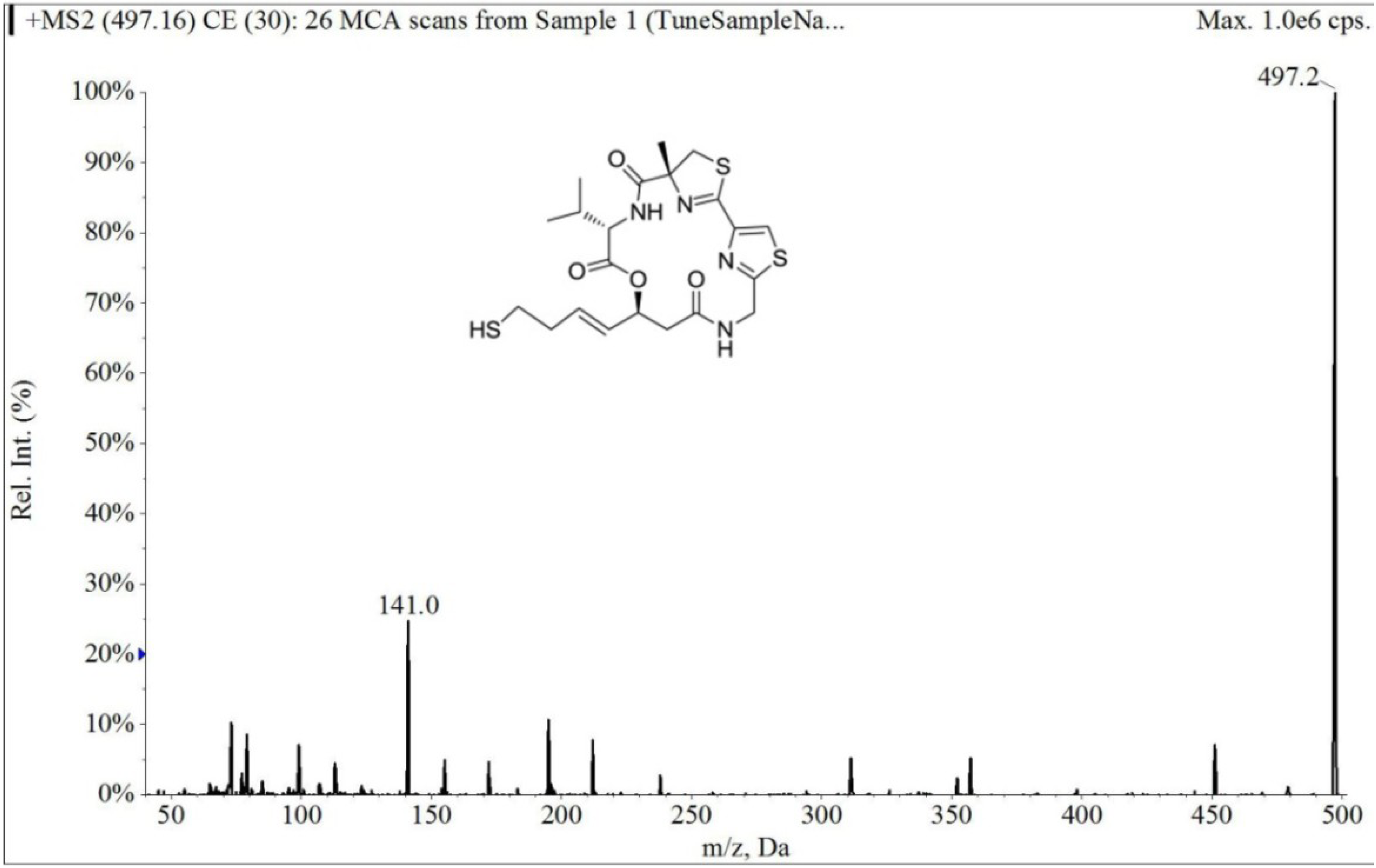


2.1.2. Linearity and Lower Limits of Quantification
2.1.3. Accuracy and Precision

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration (ng/mL) | Measured (mean ± S.D.) | Precision R.S.D. (%) | Accuracy Deviation (%) | |
|---|---|---|---|---|
| Day 1 | 37.5 | 39.38 ± 3.94 | 10.00 | 5.01 |
| 75 | 73.58 ± 6.14 | 8.35 | −1.89 | |
| 300 | 319.80 ± 26.74 | 8.36 | 6.60 | |
| Day 2 | 37.5 | 38.65 ± 2.05 | 5.12 | 3.01 |
| 75 | 71.38 ± 6.24 | 8.75 | −4.83 | |
| 300 | 333.00 ± 15.44 | 4.64 | 11.00 | |
| Day 3 | 37.5 | 35.78 ± 2.55 | 7.12 | 2.08 |
| 75 | 76.16 ± 9.57 | 12.56 | 1.55 | |
| 300 | 299.80 ± 11.61 | 3.87 | 0.067 |
| Nominal Concentration (ng/mL) | Measured (mean ± S.D.) | Precision R.S.D. (%) | Accuracy Deviation (%) |
|---|---|---|---|
| 37.5 | 37.85 ± 3.13 | 8.23 | 2.27 |
| 75 | 76.16 ± 7.19 | 9.77 | 1.55 |
| 300 | 317.53 ± 22.59 | 7.12 | 5.84 |
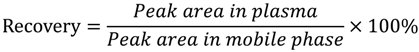
| Concentration (ng/mL) | Recovery (%) |
|---|---|
| 37.5 | 77.09 |
| 75 | 81.71 |
| 300 | 86.14 |
| Sample Condition | Nominal Concentration (ng/mL) | Measured (mean) | DEV (%) |
|---|---|---|---|
| 4 h at room temperature | 37.5 | 41.90 | 11.73 |
| 300 | 320.33 | 6.78 | |
| Freeze/thaw cycle no. 1 | 37.5 | 39.70 | 5.87 |
| 300 | 335.67 | 11.89 | |
| Freeze/thaw cycle no. 2 | 37.5 | 41.13 | 9.68 |
| 300 | 298.33 | −0.56 | |
| Freeze/thaw cycle no. 3 | 37.5 | 40.40 | 7.73 |
| 300 | 315.67 | 5.22 |
2.1.4. Matrix Effect
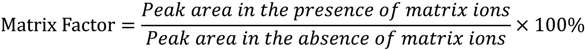
| Nominal Concentration (ng/mL) | Matrix Effect (%) |
|---|---|
| 37.5 | 74.21 |
| 75 | 72.17 |
| 300 | 72.30 |
2.1.5. Quality Control Samples of Rat Plasma
| Nominal Concentration (ng/mL) | Measured | DEV (%) |
|---|---|---|
| 37.5 | 36.18 | −3.52 |
| 75 | 76.14 | 1.52 |
| 300 | 323.40 | 7.8 |
2.2. Protein Binding
| Drug Concentration (µg/mL) | Human (mean ± S.D.) | Rat (mean ± S.D.) | |
|---|---|---|---|
| Protein binding (%) | 0.5 | 92.52 ± 0.083 | 79.27 ± 1.85 |
| 2 | 89.38 ± 0.94 | 74.39 ± 1.95 | |
| 5 | 88.48 ± 0.23 | 77.75 ± 0.86 |
2.3. Pharmacokinetics Study
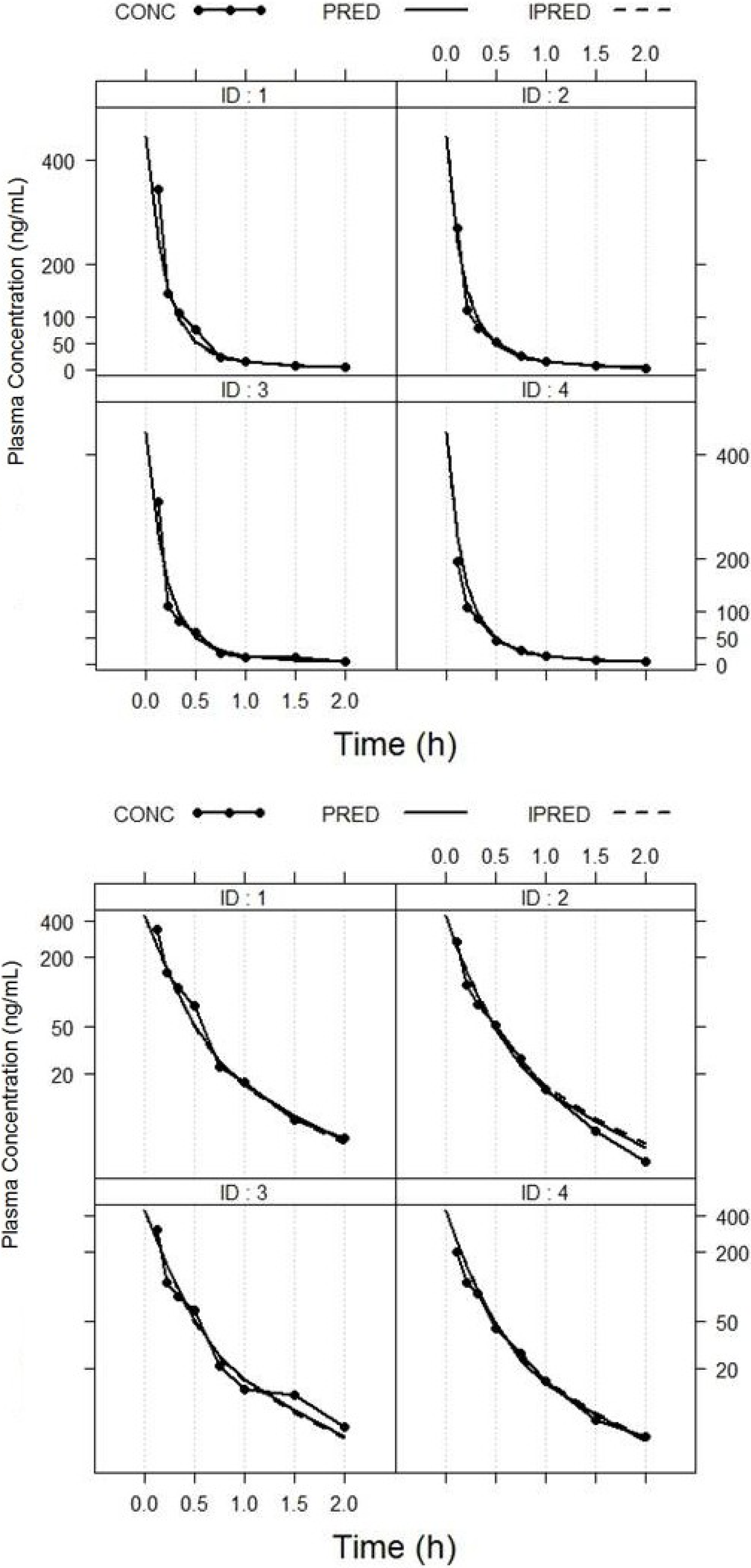
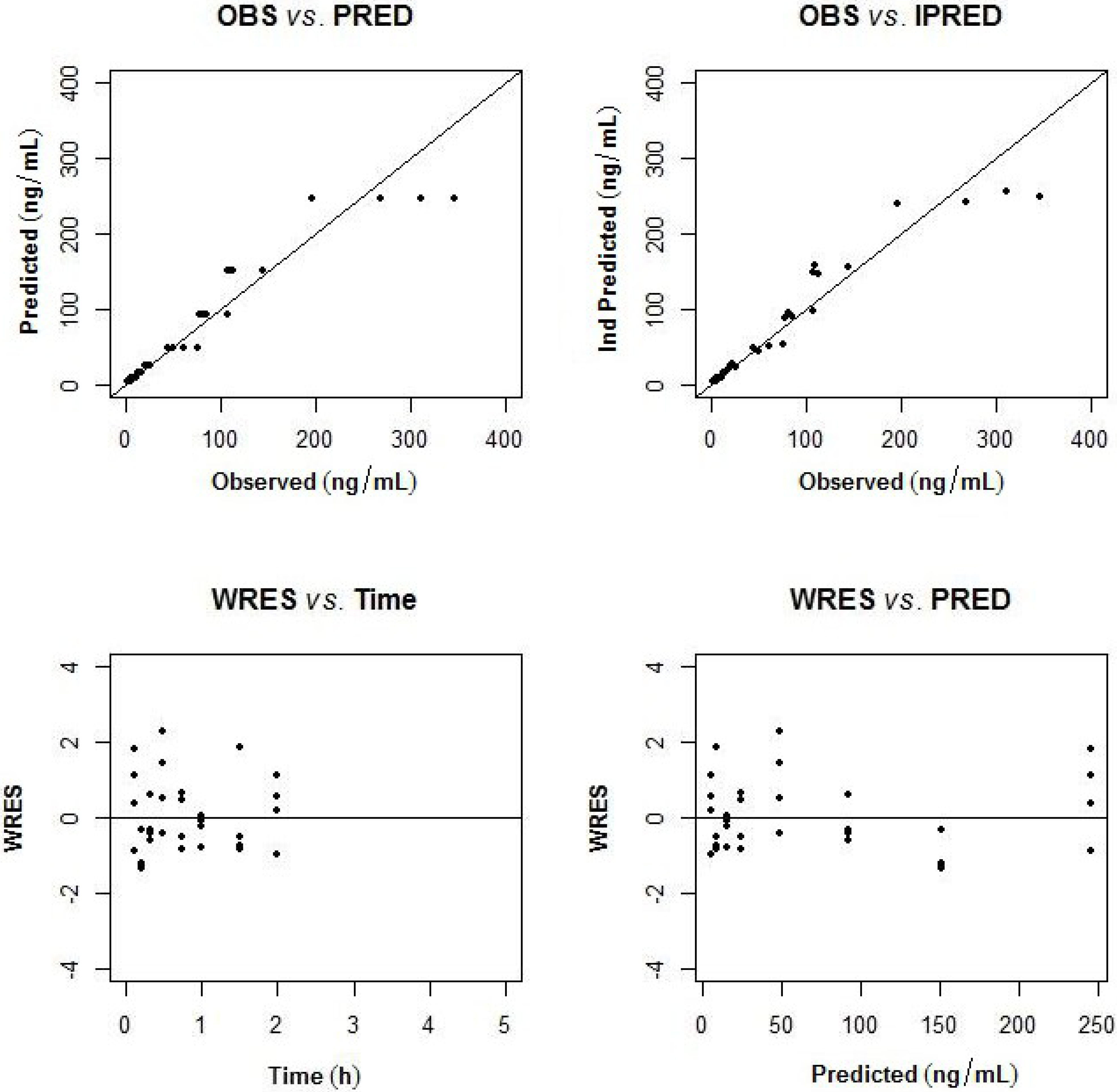
2.3.1. Population Pharmacokinetic Model
| Parameter | Mean | %SE |
|---|---|---|
| Structural model parameters | ||
| Clearance (CL) (L/h/kg) | 89.1 | 8.2 |
| Volume of central compartment (Vc) (L/kg) | 21.8 | 14.5 |
| Inter compartment clearance (Q) (L/h/kg) | 26.5 | 19.6 |
| Volume of peripheral compartment (Vp) (L/kg) | 17.4 | 14.4 |
| Interindividual variability | ||
| %CV of CL (ωCL) | 6.06 | 1.23 |
| %CV of Vc (ωV) | 31.6 | FIX |
| Residual variability | ||
| Proportional residual error | −0.208 | 11.9 |
| OFV | 168.8 |
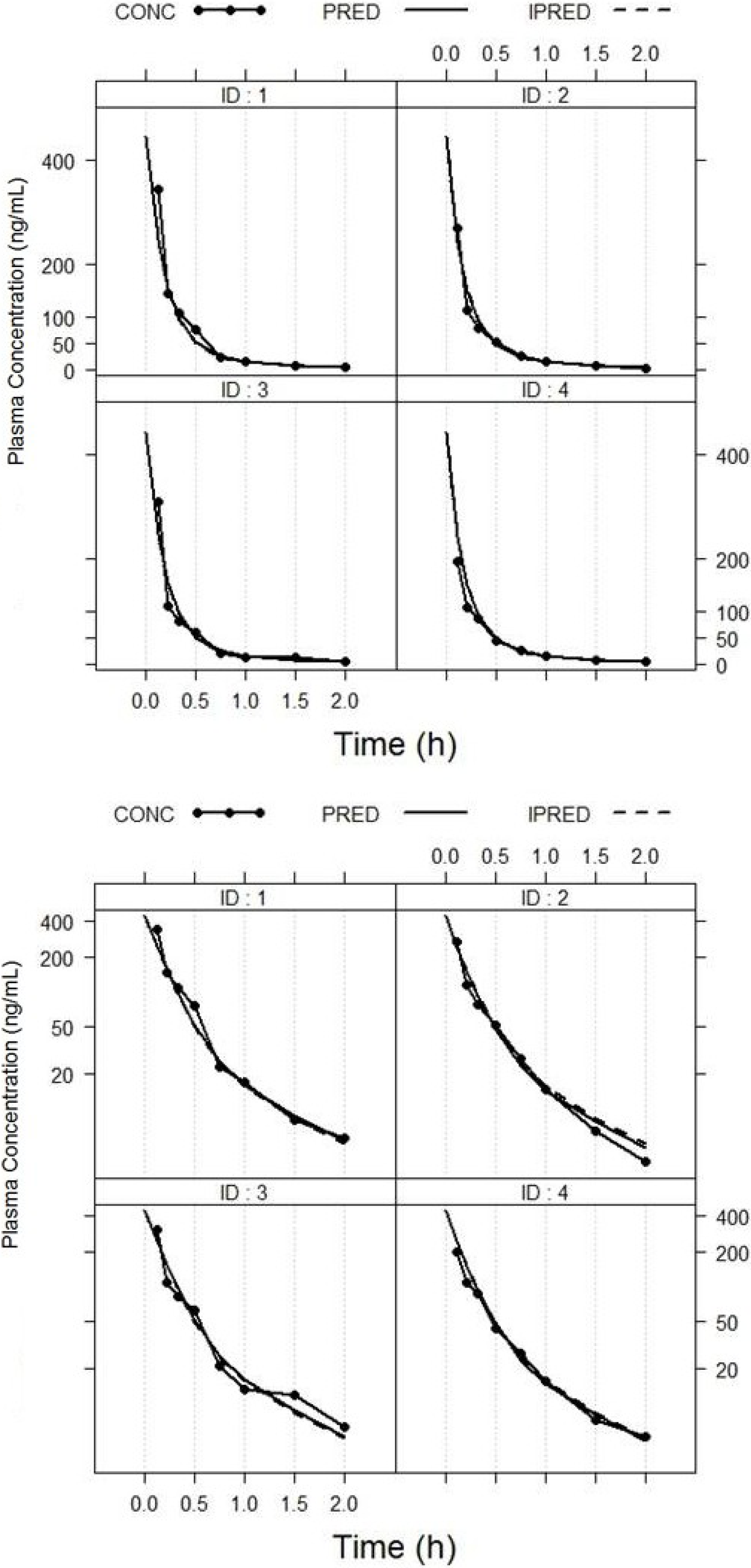
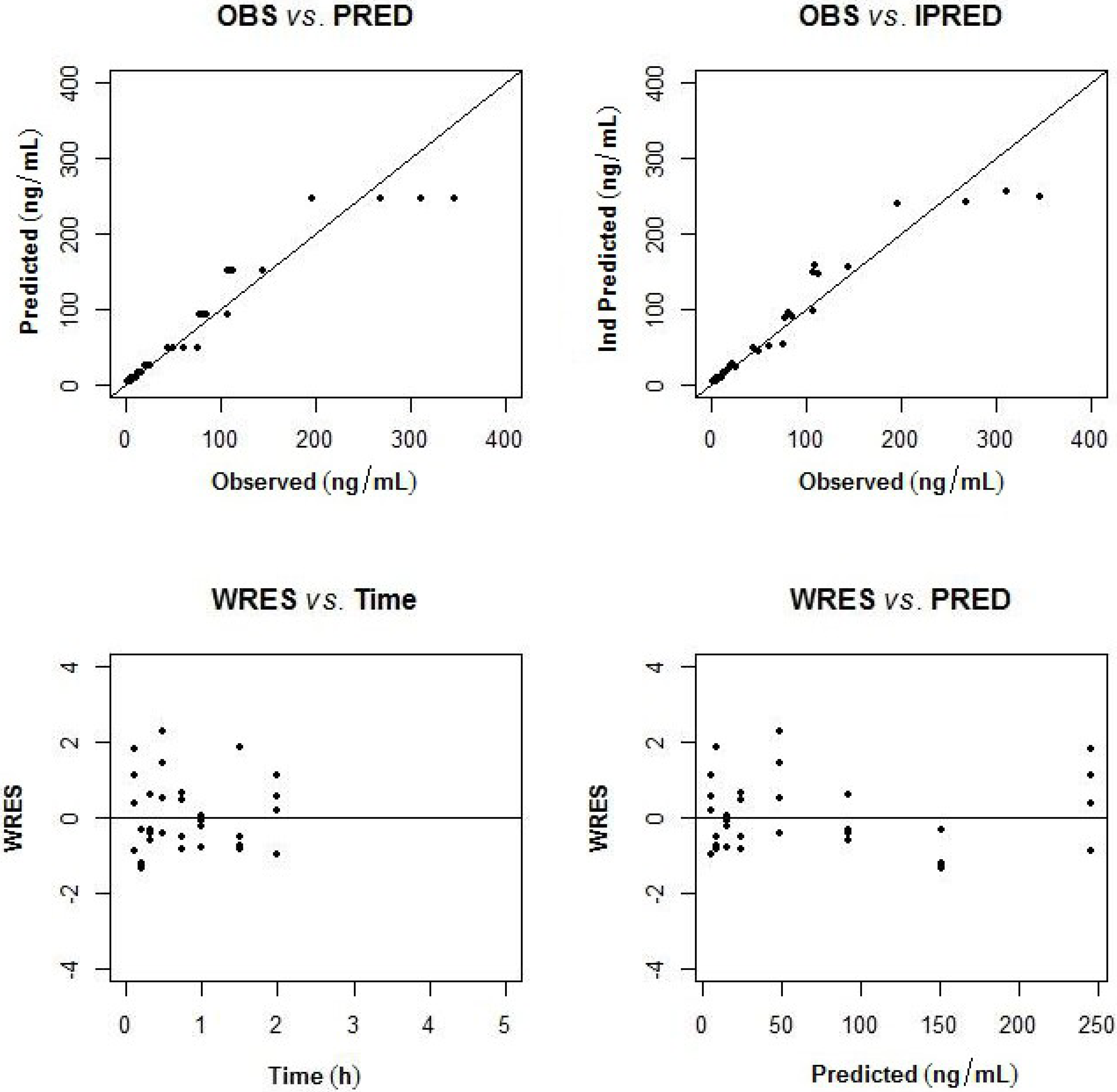
2.3.2. Non-Compartmental Analysis
| Parameters | Mean ± S.D. |
|---|---|
| Systemic clearance, CL_obs (L/h/kg) | 75.6 ± 17.9 |
| Volume based on terminal phase, Vzobs (L/kg) | 54.5 ± 12.1 |
| Volume at steady-state, Vssobs (L/kg) | 26.7 ± 10.9 |
| Extrapolated zero-time concentration, C0 (ng/L) | 804.5 ± 296.5 |
| Area under the curve to last measurable time, AUClast (h·µg/L) | 133.7 ± 29.1 |
| Area under the curve to infinity, AUCINFobs (h·µg/L) | 137.5 ± 29.6 |
| Mean residence time to infinity, MRTINFobs (h) | 0.34 ± 0.06 |
| Peak drug concentration, Cmax (ng/L) | 280.3 ± 63.6 |
| Last measurable drug concentration, Clast (ng/L) | 5.2 ± 1.2 |
| Area under the moment curve to last time point, AUMClast (h·h·µg/L) | 36.0 ± 4.0 |
| Area under the moment curve to infinity, AUMCINFobs (h·h·µg/L) | 46.4 ± 6.7 |
| Half-life, t1/2 (h) | 0.50 ± 0.07 |
3. Discussion
4. Experimental Section
4.1. Chemicals and Reagents
4.2. Instrumentation
LC-MS/MS System
4.3. Sample Extraction and Preparation
4.4. Assay Validation
4.5. Plasma Protein Binding
4.6. Pharmacokinetics Study in Rats
4.7. Data Analysis
5. Conclusions
Abbreviations
| API | Atmospheric pressure ionization |
| CE | Collision energy |
| Cps | Counts per second |
| DEV | Deviation |
| DMSO | Dimethyl sulfoxide |
| FDA | United States Food and Drug Administration |
| FK228 | Romidepsin |
| HDAC | Histone deacetylase |
| HCT116 | Human colon cancer |
| i.v. | Intravenous |
| LLOQ | Lower limit of quantification |
| MCA | Mass cumulative acquisition |
| MDA-MB-231 | Human breast adenocarcinoma 231 |
| MS2 | Tandem mass spectrometry |
| NIH | National Institutes of Health |
| NM-TRAN | NONMEM translator |
| NONMEM | Non-linear mixed effects modeling |
| OFV | Object function value |
| PB | Protein binding |
| PBS | Phosphate buffered saline |
| PEG400 | Polyethylene glycol 400 |
| PREDPP | Prediction for observation population pharmacokinetics |
| QC | Quality control |
| Rel. Int. | Relative intensity |
| R.S.D. | Relative standard deviation |
| SAHA | Suberoylanilide hydroxamic acid |
| SE | Stand error |
| TRANS4 | Translator4 |
| Vdss | Volume of distribution at steady state |
| XIC | Extracted-ion chromatogram |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. The Total Synthesis and biological mode of action of largazole: A potent class I histone deacetylase (HDAC) inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar]
- Hong, J.; Luesch, H. Largazole: from discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Luesch, H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef]
- Nasveschuk, C.G.; Ungermannova, D.; Liu, X.; Phillips, A.J. A concise total synthesis of largazole, solution structure, and some preliminary structure activity relationships. Org. Lett. 2008, 10, 3595–3598. [Google Scholar] [CrossRef]
- Ren, Q.; Dai, L.; Zhang, H.; Tan, W.; Xu, Z.; Ye, T. Total synthesis of largazole. Synlett 2008, 2008, 2379–2383. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, L.-P.; Jiao, X.-Z.; Liu, X.-Y.; Wu, Q.; Xie, P. Concise total synthesis of largazole. J. Asian Nat. Prod. Res. 2010, 12, 940–949. [Google Scholar] [CrossRef]
- Seiser, T.; Kamena, F.; Cramer, N. Synthesis and biological activity of largazole and derivatives. Angew. Chem. Int. Ed. Engl. 2008, 47, 6483–6485. [Google Scholar] [CrossRef]
- Zeng, X.; Yin, B.; Hu, Z.; Liao, C.; Liu, J.; Li, S.; Li, Z.; Nicklaus, M.C.; Zhou, G.; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef]
- Wang, B.; Forsyth, C. Total synthesis of largazole—devolution of a novel synthetic strategy. Synthesis 2009, 2009, 2873–2880. [Google Scholar] [CrossRef]
- Benelkebir, H.; Marie, S.; Hayden, A.L.; Lyle, J.; Loadman, P.M.; Crabb, S.J.; Packham, G. Ganesan, a Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg. Med. Chem. 2011, 19, 3650–3658. [Google Scholar] [CrossRef]
- Numajiri, Y.; Takahashi, T.; Takagi, M.; Shin-ya, K.; Doi, T. Total synthesis of largazole and its biological evaluation. Synlett 2008, 2008, 2483–2486. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kulkarni, S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org. Lett. 2008, 10, 3907–3909. [Google Scholar] [CrossRef]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar]
- Salvador, L.; Luesch, H. HDAC Inhibitors and Other Histone Modifying Natural Products as Emerging Anticancer Agents. In Natural Products and Cancer Drug Discovery; Koehn, F.E., Ed.; Springer: New York, NY, USA, 2013; pp. 59–95. [Google Scholar]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.-I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; Tsuneyoshi, M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; Nam, S.W. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Law, M.E.; Corsino, P.E.; Jahn, S.C.; Davis, B.J.; Chen, S.; Patel, B.; Pham, K.; Lu, J.; Sheppard, B.; Nørgaard, P.; et al. Glucocorticoids and histone deacetylase inhibitors cooperate to block the invasiveness of basal-like breast cancer cells through novel mechanisms. Oncogene 2013, 32, 1316–1329. [Google Scholar] [CrossRef]
- Lee, S.-U.; Kwak, H.B.; Pi, S.-H.; You, H.-K.; Byeon, S.R.; Ying, Y.; Luesch, H.; Hong, J.; Kim, S.H. In vitro and in vivo osteogenic activity of largazole. ACS Med. Chem. Lett. 2011, 2, 248–251. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Wang, J.; Lam, W.; Kwong, S.; Li, F.; Friedman, S.L.; Zhou, S.; Ren, Q.; Xu, Z.; et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-β and vascular endothelial growth factor signaling. Liver Int. 2013, 33, 504–515. [Google Scholar] [CrossRef]
- FDA Pharmacology Toxicology Review and Evaluation of Romidepsin. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022393s000_PharmR_P2.pdf (accessed on 20 December 2013).
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; Horinouchi, S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Kantharaj, E.; Jayaraman, R. Histone Deacetylase Inhibitors as Therapeutic Agents for Cancer Therapy: Drug Metabolism and Pharmacokinetic Properties. In Drug Development—A Case Study Based Insight into Modern Strategies; Rundfeldt, C., Ed.; InTech: Rijeka, Croatia, 2011; pp. 101–111. [Google Scholar]
- Combes, F.P.; Retout, S.; Frey, N.; Mentré, F. Prediction of shrinkage of individual parameters using the bayesian information matrix in non-linear mixed effect models with evaluation in pharmacokinetics. Pharm. Res. 2013, 30, 2355–2367. [Google Scholar] [CrossRef]
- Chan, K.K.; Bakhtiar, R.; Jiang, C. Depsipeptide (FR901228, NSC-630176) pharmacokinetics in the rat by LC/MS/MS. Invest. New Drugs 1997, 15, 195–206. [Google Scholar] [CrossRef]
- Masuoka, Y.; Shindoh, N.; Inamura, N. Histone deacetylase inhibitors from microorganisms: The Astellas experience. Fortschr. Arzneimittelforsch. 2008, 66, 335, 337–359. [Google Scholar]
- Migdalof, B.H.; Antonaccio, M.J.; McKinstry, D.N.; Singhvi, S.M.; Lan, S.J.; Egli, P.; Kripalani, K.J. Captopril: Pharmacology, metabolism and disposition. Drug Metab. Rev. 1984, 15, 841–869. [Google Scholar] [CrossRef]
- Kubo, M.; Mizooku, Y.; Hirao, Y.; Osumi, T. Development and validation of an LC-MS/MS method for the quantitative determination of aripiprazole and its main metabolite, OPC-14857, in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 822, 294–299. [Google Scholar] [CrossRef]
- Koseki, N.; Kawashita, H.; Hara, H.; Niina, M.; Tanaka, M.; Kawai, R.; Nagae, Y.; Masuda, N. Development and validation of a method for quantitative determination of valsartan in human plasma by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2007, 43, 1769–1774. [Google Scholar] [CrossRef]
- Viswanathan, C.T.; Bansal, S.; Booth, B.; DeStefano, A.J.; Rose, M.J.; Sailstad, J.; Shah, V.P.; Skelly, J.P.; Swann, P.G.; Weiner, R. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm. Res. 2007, 24, 1962–1973. [Google Scholar] [CrossRef]
- De Moraes, N.V.; Lauretti, G.R.; Napolitano, M.N.; Santos, N.R.; Godoy, A.L.; Lanchote, V.L. Enantioselective analysis of unbound tramadol, O-desmethyltramadol and N-desmethyltramadol in plasma by ultrafiltration and LC-MS/MS: application to clinical pharmacokinetics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 880, 140–147. [Google Scholar] [CrossRef]
- Pillai, G.C.; Mentré, F.; Steimer, J.-L. Non-linear mixed effects modeling—from methodology and software development to driving implementation in drug development science. J. Pharmacokinet. Pharmacodyn. 2005, 32, 161–183. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, M.; Salvador, L.A.; Sy, S.K.B.; Tang, Y.; Singh, R.S.P.; Chen, Q.-Y.; Liu, Y.; Hong, J.; Derendorf, H.; Luesch, H. Largazole Pharmacokinetics in Rats by LC-MS/MS. Mar. Drugs 2014, 12, 1623-1640. https://doi.org/10.3390/md12031623
Yu M, Salvador LA, Sy SKB, Tang Y, Singh RSP, Chen Q-Y, Liu Y, Hong J, Derendorf H, Luesch H. Largazole Pharmacokinetics in Rats by LC-MS/MS. Marine Drugs. 2014; 12(3):1623-1640. https://doi.org/10.3390/md12031623
Chicago/Turabian StyleYu, Mingming, Lilibeth A. Salvador, Sherwin K. B. Sy, Yufei Tang, Ravi S. P. Singh, Qi-Yin Chen, Yanxia Liu, Jiyong Hong, Hartmut Derendorf, and Hendrik Luesch. 2014. "Largazole Pharmacokinetics in Rats by LC-MS/MS" Marine Drugs 12, no. 3: 1623-1640. https://doi.org/10.3390/md12031623