Three New and Eleven Known Unusual C25 Steroids: Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy using Diethyl Sulphate

Abstract
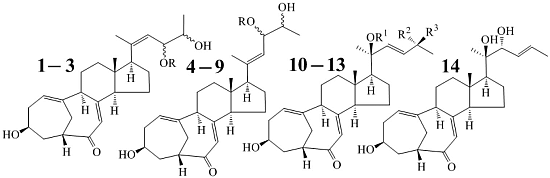
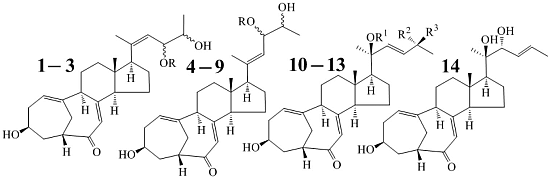
:
1. Introduction
2. Results and Discussions
2.1. Fermentation, Isolation of 1–14, and Identification of Known Steroids 4–14
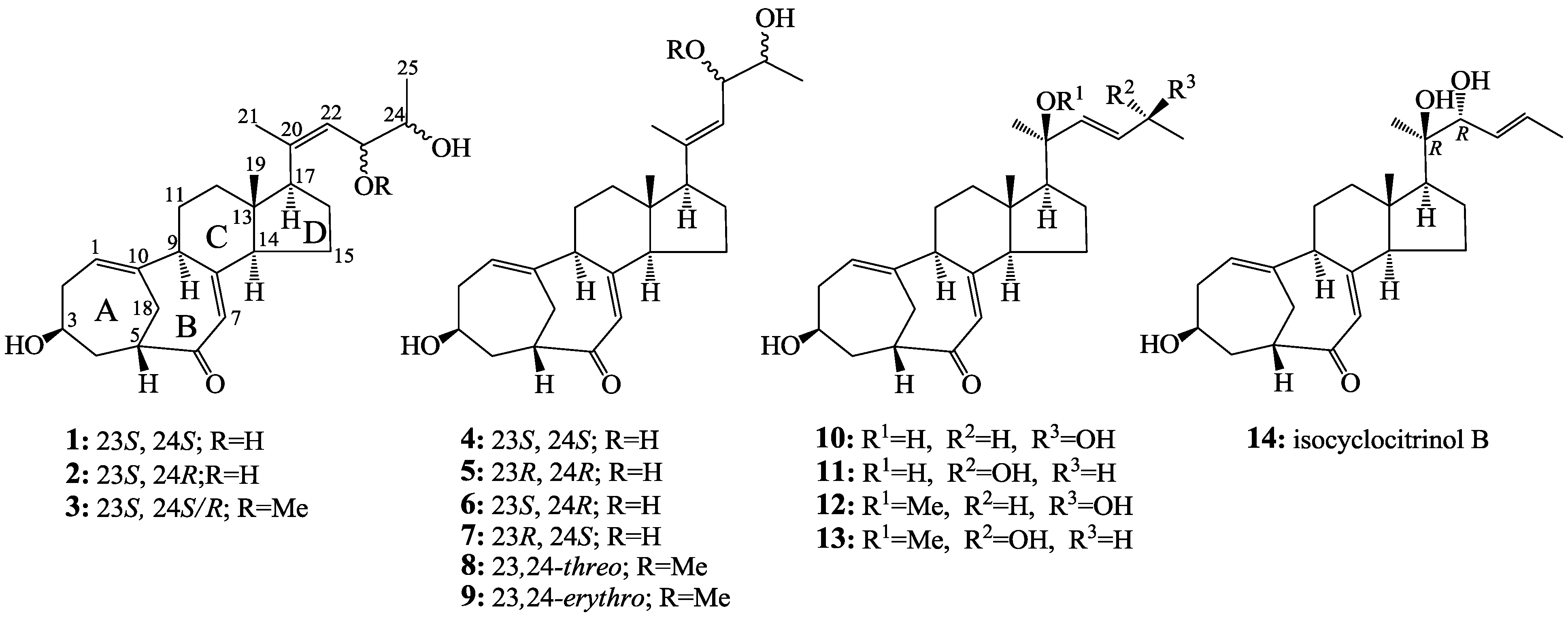
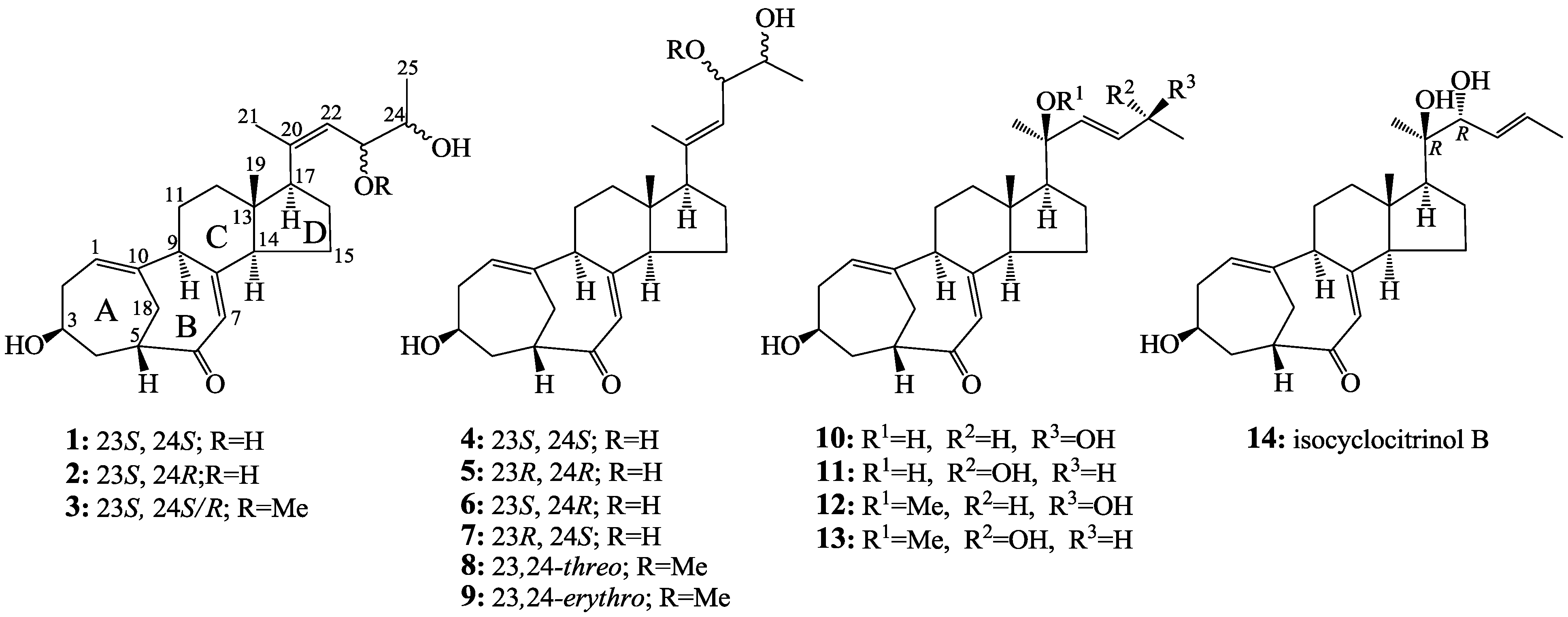
2.2. Structure Determination of New Compounds 1–3
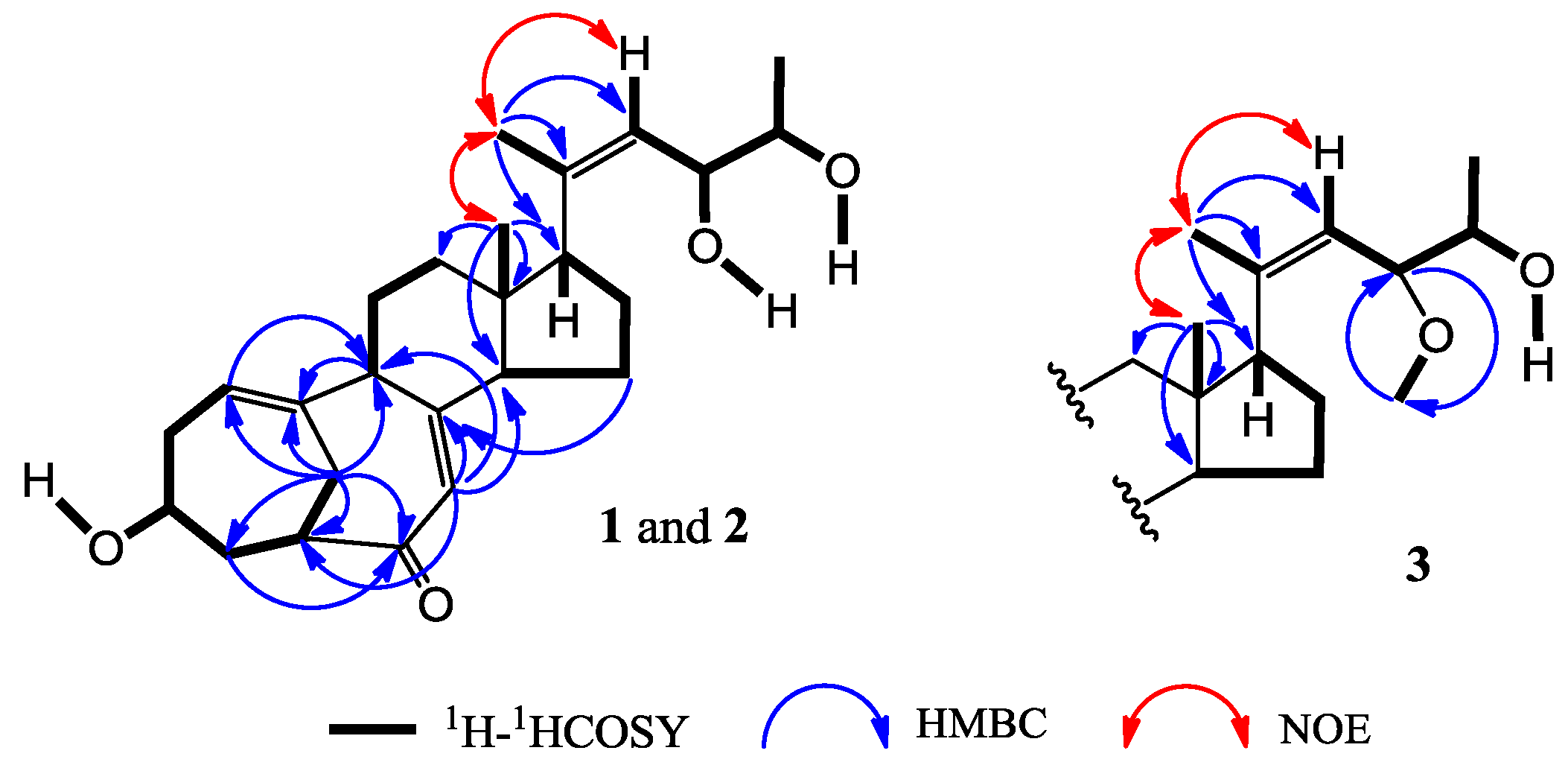
 +71.1 (c 0.23, MeOH) for 1 and m.p. 201–202 °C, +71.6 (c 0.30, MeOH) for 2, were assigned the molecular formula C25H36O4 by HRESIMS (measured 401.2689 [M + H]+ for 1 and 401.2684 [M + H]+ for 2; calculated for C25H37O4 [M + H]+ 401.2692). Their maximal UV absorptions around 243 nm and the IR absorptions around 1645 cm−1 revealed the presence of conjugated carbonyl groups in 1 and 2 [1]. Their IR spectra showed also the absorptions due to the OH and CH3/CH2 groups (see the UV and IR spectra in the Supplementary Information). Further, the positive ESIMS ion fragments detected at m/z 383 [M − H2O + H]+, 365 [M − 2H2O + H]+, and 347 [M − 3H2O + H]+ for both 1 and 2 suggested the presence of three hydroxyl groups in their structures [4]. In the 1H and 13C NMR spectra (the NMR spectra in the Supplementary Information), 1 and 2 showed 1H and 13C NMR signals that closely resembled the signals from the known steroids 4–7, except several proton and carbon signals from the structural parts around the side chains at C-17 in 1 and 2 differed slightly (Table 1 and Table 2). These NMR data indicated that the planar structures of 1 and 2 are the same as 4–7, but the stereo structures of the side chain moieties are slightly different. This was confirmed by detailed analyses of their DEPT, 1H-1H COSY, HMQC, and HMBC spectra (see Table S1 for 1 and Table S2 for 2 in the Supplementary Information) to establish their planar structures (Figure 2). The stereochemistry of 1 and 2 including their absolute configurations were determined as follows.
+71.1 (c 0.23, MeOH) for 1 and m.p. 201–202 °C, +71.6 (c 0.30, MeOH) for 2, were assigned the molecular formula C25H36O4 by HRESIMS (measured 401.2689 [M + H]+ for 1 and 401.2684 [M + H]+ for 2; calculated for C25H37O4 [M + H]+ 401.2692). Their maximal UV absorptions around 243 nm and the IR absorptions around 1645 cm−1 revealed the presence of conjugated carbonyl groups in 1 and 2 [1]. Their IR spectra showed also the absorptions due to the OH and CH3/CH2 groups (see the UV and IR spectra in the Supplementary Information). Further, the positive ESIMS ion fragments detected at m/z 383 [M − H2O + H]+, 365 [M − 2H2O + H]+, and 347 [M − 3H2O + H]+ for both 1 and 2 suggested the presence of three hydroxyl groups in their structures [4]. In the 1H and 13C NMR spectra (the NMR spectra in the Supplementary Information), 1 and 2 showed 1H and 13C NMR signals that closely resembled the signals from the known steroids 4–7, except several proton and carbon signals from the structural parts around the side chains at C-17 in 1 and 2 differed slightly (Table 1 and Table 2). These NMR data indicated that the planar structures of 1 and 2 are the same as 4–7, but the stereo structures of the side chain moieties are slightly different. This was confirmed by detailed analyses of their DEPT, 1H-1H COSY, HMQC, and HMBC spectra (see Table S1 for 1 and Table S2 for 2 in the Supplementary Information) to establish their planar structures (Figure 2). The stereochemistry of 1 and 2 including their absolute configurations were determined as follows.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 b | 2 b | 4 | 5 | 6 | 7 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|
| 1 | 5.55 dd (8.0, 6.4) | 5.55 dd (8.2, 6.4) | 5.56 dd (8.2, 6.3) | 5.55 dd (8.1, 6.4) | 5.55 dd (8.1, 6.2) | 5.55 dd (8.1, 6.3) | 5.53 dd (8.4, 6.4) | 5.53 dd (8.3, 6.4) |
| 2α | 2.07 m | 2.07 m | 2.07 m | 2.07 m | 2.07 m | 2.08 m | 2.07 m | 2.07 m |
| β | 2.34 ddd (13.0, 11.4, 6.4) | 2.34 ddd (13.2, 11.2, 6.4) | 2.34 ddd (13.0, 11.5, 6.3) | 2.34 ddd (13.2, 11.2, 6.4) | 2.34 m | 2.34 ddd (12.7, 11.3, 6.3) | 2.33 ddd (13.1, 11.3, 6.3) | 2.33 ddd (13.0, 11.2, 6.3) |
| 3 | 3.12 m | 3.12 m | 3.12 m | 3.12 m | 3.12 m | 3.13 m | 3.11 m | 3.11 m |
| 4α | 2.63 br d (13.1) | 2.63 br d (12.8) | 2.63 br d (13.2) | 2.63 br d (13.1) | 2.63 br d (13.0) | 2.64 br d (13.1) | 2.62 br d (13.0) | 2.62 br d (12.9) |
| β | 1.53 m | 1.52 m | 1.53 m | 1.52 m | 1.52 m | 1.53 m | 1.51 m | 1.51 m |
| 5 | 2.69 m | 2.69 m | 2.68 m | 2.68 m | 2.68 m | 2.68 m | 2.67 m | 2.67 m |
| 7 | 5.43 s | 5.43 s | 5.42 s | 5.42 s | 5.42 s | 5.42 s | 5.37 s | 5.38 s |
| 9 | 2.86 dd (12.0, 5.5) | 2.86 dd (11.3, 5.6) | 2.85 dd (11.7, 5.4) | 2.84 dd (11.8, 5.6) | 2.85 dd (12.0, 5.4) | 2.85 dd (11.4, 5.7) | 2.79 dd (12.2, 5.4) | 2.79 dd (12.1, 5.5) |
| 11α | 1.54 m | 1.52 m | 1.54 m | 1.55 m | 1.54 m | 1.56 m | 1.50 m | 1.49 m |
| β | 1.75 m | 1.74 m | 1.80 m | 1.76 m | 1.80 m | 1.79 m | 1.75 m | 1.75 m |
| 12α | 1.41 td (12.7, 4.1) | 1.40 td (12.8, 4.3) | 1.50 m | 1.43 td (13.7, 4.6) | 1.47 m | 1.43 m | 1.42 m | 1.42 m |
| β | 1.63 m | 1.69 m | 1.74 m | 1.73 m | 1.74 m | 1.74 m | 2.12 m | 2.13 m |
| 14 | 2.26 br t (8.9) | 2.25 br t (8.6) | 2.26 br t (9.0) | 2.22 br t (8.8) | 2.25 br t (8.6) | 2.22 br t (9.0) | 2.10 m | 2.10 m |
| 15α | 1.62 m | 1.61 m | 1.56 m | 1.57 m | 1.56 m | 1.57 m | 1.38 m | 1.38 m |
| β | 1.62 m | 1.61 m | 1.52 m | 1.52 m | 1.49 m | 1.53 m | 1.46 m | 1.47 m |
| 16α | 1.66 m | 1.69 m | 1.83 m | 1.82 m | 1.83 m | 1.82 m | 1.56 m | 1.58 m |
| β | 1.84 m | 1.85 m | 1.65 m | 1.68 m | 1.67 m | 1.68 m | 1.66 m | 1.66 m |
| 17 | 2.89 br t (9.8) | 2.86 t (9.5) | 2.24 br t (9.5) | 2.27 br t (9.7) | 2.23 br t (9.1) | 2.27 br t (9.6) | 1.66 m | 1.66 m |
| 18α | 2.48 br s | 2.48 br s | 2.48 br s | 2.48 br s | 2.48 br s | 2.48 br s | 2.47 br s | 2.47 br s |
| β | 2.48 br s | 2.48 br s | 2.48 br s | 2.48 br s | 2.48 br s | 2.48 br s | 2.47 br s | 2.47 br s |
| 19 | 0.64 s | 0.63 s | 0.50 s | 0.53 s | 0.49 s | 0.53 s | 0.70 s | 0.70 s |
| 21 | 1.70 s | 1.70 d (1.1) | 1.68 s | 1.65 s | 1.65 s | 1.64 s | 1.20 s | 1.20 s |
| 22 | 5.33 d (9.2) | 5.33 dd (9.3, 1.1) | 5.17 d (8.7) | 5.15 d (8.8) | 5.22 d (8.2) | 5.22 d (8.4) | 5.62 dd (15.6, 0.9) | 5.64 dd (15.6, 1.1) |
| 23 | 3.99 ddd (9.2, 6.0, 4.6) | 4.11 ddd (9.3, 4.9, 4.1) | 3.96 ddd (8.7, 6.6, 4.3) | 3.95 ddd (8.8, 6.6, 4.3) | 4.09 ddd (8.2, 4.7, 4.0) | 4.06 ddd (8.4, 4.7, 3.9) | 5.49 dd (15.6, 5.9) | 5.49 dd (15.6, 5.3) |
| 24 | 3.41 m | 3.47 m | 3.41 m | 3.40 m | 3.49 m | 3.50 m | 4.09 m | 4.09 m |
| 25 | 1.00 d (6.3) | 1.01 d (6.3) | 0.95 d (6.3) | 0.96 d (6.3) | 0.96 d (6.3) | 0.98 d (6.3) | 1.08 d (6.4) | 1.08 d (6.4) |
| 3-OH | 4.62 d (4.3) | 4.62 d (4.3) | 4.59 d (4.4) | 4.63 d (4.3) | 4.62 d (4.3) | 4.60 d (4.2) | 4.61 d (4.3) | 4.61 d (4.3) |
| 20-OH | - | - | - | - | - | - | 4.23 s | 4.22 s |
| 23-OH | 4.30 d (4.6) | 4.28 d (4.9) | 4.46 d (4.3) | 4.48 d (4.3) | 4.40 d (4.7) | 4.35 d (4.7) | - | - |
| 24-OH | 4.31 d (4.7) | 4.31 d (4.9) | 4.33 d (4.1) | 4.39 d (4.1) | 4.29 d (5.0) | 4.27 d (4.6) | 4.57 d (4.4) | 4.57 d (4.6) |
| Position | 1 b | 2 b | 3 b,c | 4 | 5 | 6 | 7 | 8 c | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 122.1 | 122.1 | 122.1 | 122.1 | 122.1 | 122.1 | 122.1 | 122.1 | 122.1 | 122.0 | 121.9 | 122.0 | 122.0 | 122.1 |
| 2 | 36.0 | 36.0 | 36.0 | 35.9 | 36.0 | 36.0 | 36.0 | 35.9 | 36.0 | 36.0 | 35.9 | 35.8 | 35.7 | 35.8 |
| 3 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 | 64.6 | 64.6 | 64.7 |
| 4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.4 | 41.3 | 41.8 | 41.7 | 41.8 |
| 5 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.1 | 48.7 | 48.7 | 48.7 |
| 6 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 204.1 | 205.5 | 205.5 | 205.4 |
| 7 | 124.5 | 124.5 | 124.5 | 124.3 | 124.3 | 124.2 | 124.3 | 124.3 | 124.3 | 124.6 | 124.56 | 125.1 | 125.1 | 125.3 |
| 8 | 157.0 | 157.1 | 157.0 | 156.9 | 157.0 | 157.0 | 157.0 | 156.9 | 156.93/156.90 | 157.1 | 157.1 | 158.0 | 158.0 | 157.4 |
| 9 | 53.2 | 53.3 | 53.3 | 53.1 | 53.3 | 53.2 | 53.3 | 53.2 | 53.2 | 53.2 | 53.2 | 54.2 | 54.2 | 54.1 |
| 10 | 145.5 | 145.5 | 145.5 | 145.5 | 145.5 | 145.6 | 145.6 | 145.5 | 145.5 | 145.7 | 145.7 | 146.1 | 146.0 | 146.0 |
| 11 | 27.2 | 27.3 | 27.3 | 27.5 | 27.4 | 27.5 | 27.4 | 27.4 | 27.5 | 27.5 | 27.5 | 27.8 | 27.8 | 27.8 |
| 12 | 36.7 | 36.6 | 36.4 | 37.3 | 36.9 | 37.2 | 36.9 | 37.07/37.13 | 37.08/37.13 | 38.9 | 38.9 | 39.5 | 39.5 | 39.4 |
| 13 | 47.8 | 47.8 | 47.73/47.66 | 46.6 | 46.9 | 46.6 | 46.9 | 46.76/46.84 | 46.74/46.76 | 45.9 | 45.9 | 46.5 | 46.4 | 46.5 |
| 14 | 54.2 | 54.3 | 54.2 | 54.4 | 54.3 | 54.5 | 54.3 | 54.3 | 54.4 | 55.3 | 55.2 | 56.1 | 56.0 | 55.9 |
| 15 | 22.6 | 22.6 | 22.7 | 22.2 | 22.4 | 22.3 | 22.4 | 22.3 | 22.34/22.38 | 22.1 | 22.1 | 22.7 | 22.6 | 22.9 |
| 16 | 24.1 | 24.0 | 24.1/24.0 | 23.7 | 23.7 | 23.8 | 23.7 | 23.8/23.7 | 23.82/23.80 | 22.3 | 22.3 | 22.6 | 22.6 | 21.4 |
| 17 | 50.9 | 51.0 | 51.2 | 58.6 | 58.9 | 58.6 | 58.9 | 58.9 | 58.87/58.92 | 60.1 | 60.1 | 60.3 | 60.4 | 55.4 |
| 18 | 27.2 | 27.2 | 27.3 | 27.2 | 27.2 | 27.2 | 27.2 | 27.2 | 27.2 | 27.2 | 27.1 | 27.7 | 27.7 | 27.7 |
| 19 | 14.8 | 14.7 | 14.7 | 13.3 | 13.5 | 13.3 | 13.4 | 13.53/13.49 | 13.48/13.44 | 14.3 | 14.3 | 14.9 | 15.0 | 14.3 |
| 20 | 136.7 | 136.6 | 140.1/140.0 | 135.8 | 135.8 | 134.9 | 135.0 | 139.5 | 138.78/138.67 | 73.3 | 73.3 | 79.5 | 79.7 | 76.9 |
| 21 | 22.2 | 22.3 | 22.2/22.0 | 18.9 | 17.3 | 18.8 | 17.3 | 18.2/18.5 | 18.04/18.27 | 29.0 | 28.9 | 21.8 | 21.8 | 20.8 |
| 22 | 130.7 | 130.8 | 126.9 | 127.1 | 128.2 | 127.3 | 128.2 | 124.3/124.1 | 124.4/124.3 | 136.2 | 136.0 | 134.5 | 134.5 | 77.6 |
| 23 | 70.4 | 70.3 | 79.9/79.7 | 72.2 | 72.0 | 71.6 | 71.5 | 81.86/81.94 | 81.3/81.4 | 130.8 | 130.8 | 134.2 | 134.4 | 129.4 |
| 24 | 70.1 | 69.8 | 68.6 | 70.3 | 70.3 | 69.8 | 69.8 | 68.7 | 68.32/68.38 | 66.7 | 66.3 | 68.8 | 68.8 | 130.4 |
| 25 | 19.8 | 18.5 | 18.8/19.9 | 19.1 | 18.9 | 18.3 | 18.5 | 18.9 | 18.9/18.8 | 24.1 | 24.0 | 23.8 | 23.7 | 18.2 |
| 23-OCH3 | - | - | 55.0/54.9 | - | - | - | - | 55.44/55.39 | 55.5 | - | - | 49.8 | 49.9 | - |

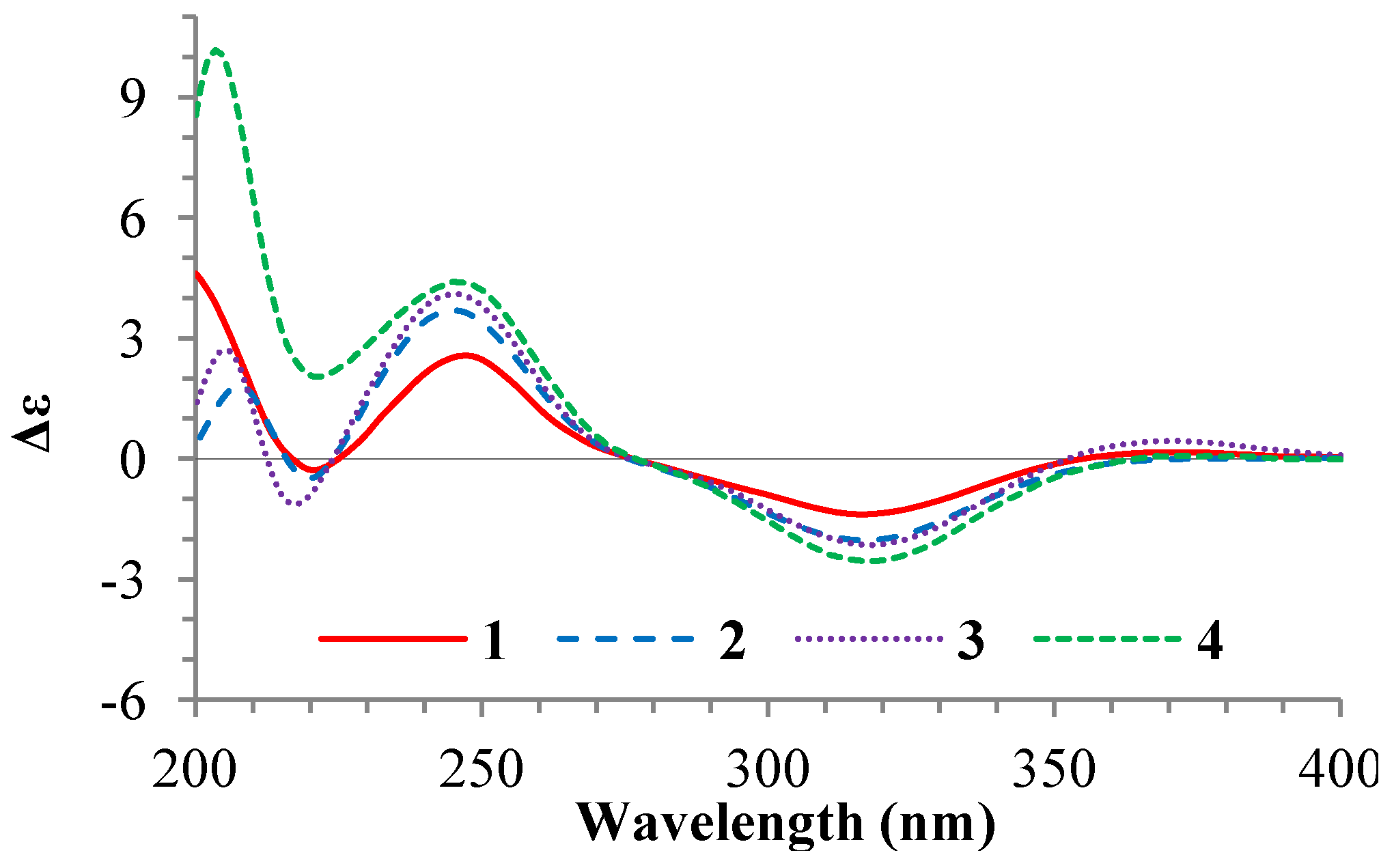
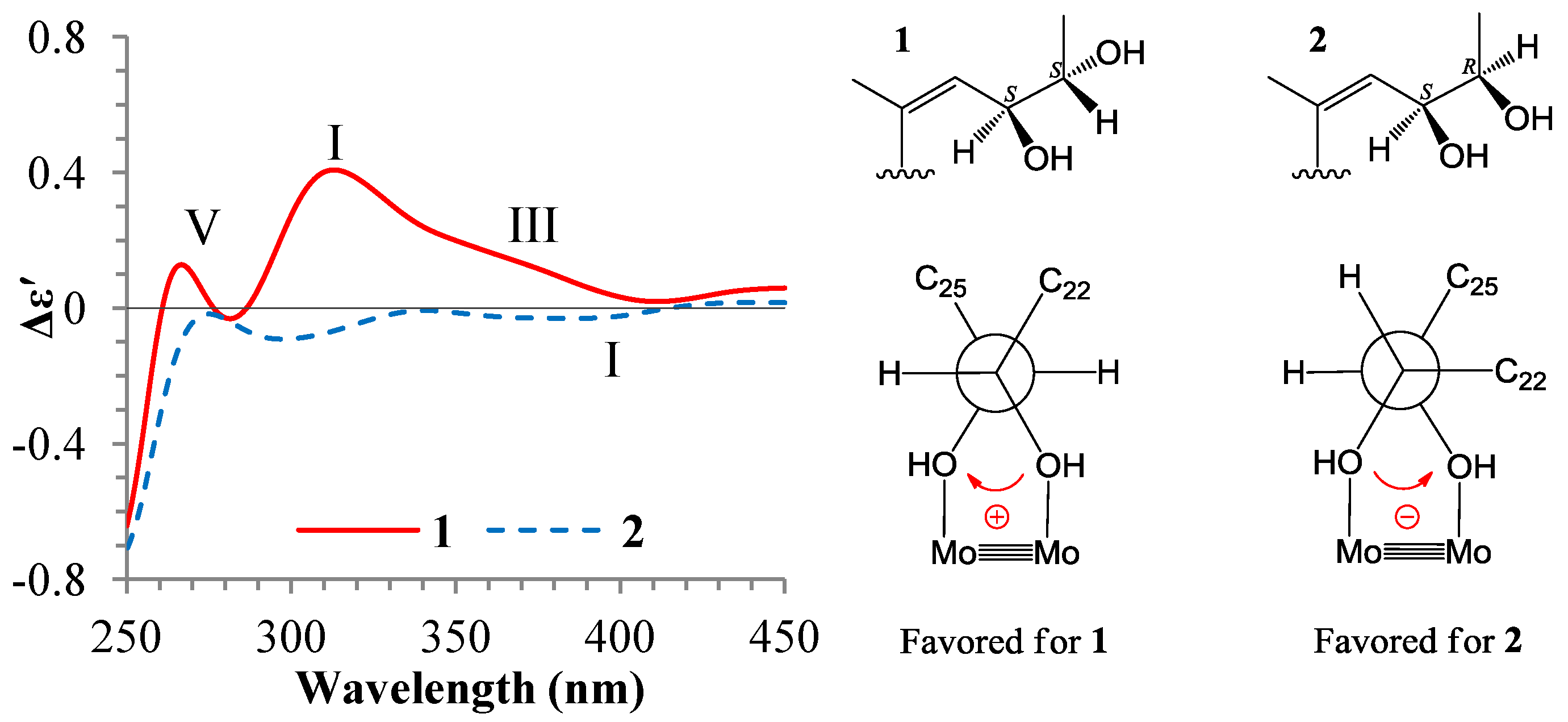
 +77.4 (c 0.38, MeOH), was assigned the molecular formula C26H38O4 by HRESIMS (measured 415.2850 [M + H]+, calculated for C26H39O4 [M + H]+ 415.2848). The maximal UV absorption at 243.8 nm (log ε 4.10) in MeOH and the strong IR absorption at 1651 cm−1 indicated the presence of a conjugated carbonyl group in 3 [1]. The IR spectrum also showed the absorptions due to OH (3374 cm−1) and CH3/CH2 (2946, 2910, 2878 and 1378 cm−1) groups. The 1H and 13C NMR spectra of 3 in DMSO-d6 gave 1H and 13C signals (Table 2 and Table 3) resembled those of 1 and 2 (Table 1 and Table 2), except additional 1H and 13C signals from a methoxy group were detected along with disappearance of a hydroxyl signal in the 1H NMR spectrum. This indicated that 3 was a methoxylated derivative of 1 and 2. The MS ion fragments detected at m/z 383 [M − CH3OH + H]+ in the positive ESIMS and at m/z 381 [M − CH3OH − H]− in the negative ESIMS also supported the presence of a methoxy group in 3. In addition, several 1H and 13C NMR signals of 3 appeared as pairs in an approximate 4:1 ratio, and significant changes were mainly observed on the signals from side chain moieties in 1–3 (Table 1, Table 2 and Table 3). These observations revealed that the methoxy group in 3 should be in the side chain and 3 was a mixture. Detailed analysis of the 1H and 13C NMR spectra with the aide of DEPT, 1D difference NOE, 1H-1H COSY, HMQC, HMBC and NOESY techniques (Table S3 in the Supplementary Information) established the structure of 3 as shown in Figure 1. The methoxy group in 3 was located at C-23 by the HMBCs shown in Figure 2. The NOEs between H3-21 and H-22 demonstrated the Z-configuration of the 20,22-double bond in 3. The relative stereochemistry of the ring system in 3 was determined to be the same as 1 and 2 on the basis of the NOEs between pairs of the protons, H3-21/H3-19, H3-19/Hα-18 and H-9/H-14, and the almost identical 1H and 13C signals of the A/B ring moieties in 1–3 (Table 1, Table 2 and Table 3). The absolute configuration of the A–D ring system in 3 was established to be the same as 1–2 and 4 by the closely resembled CD spectra of 1–4 as shown in Figure 3. The couplings of H-23 and H-24 in 3, 5.9 Hz for major isomer and 4.0 Hz for minor isomer, indicated that the major isomer in 3 was the 23,24-erythro and the minor isomer was the 23,24-thero [1,21]. This was supported also by the downfield resonances of H-23 and H-24 in the major erythro isomer than in the minor thero isomer (see Table 3) [1]. Further, the chemical shifts of C-21 and C-23 in the major and minor isomers were consistent (Table 2) and indicated the same configuration at C-23 [1]. Thus, 3 was the 4:1 mixture of 23-methoxylated 2 and 1.
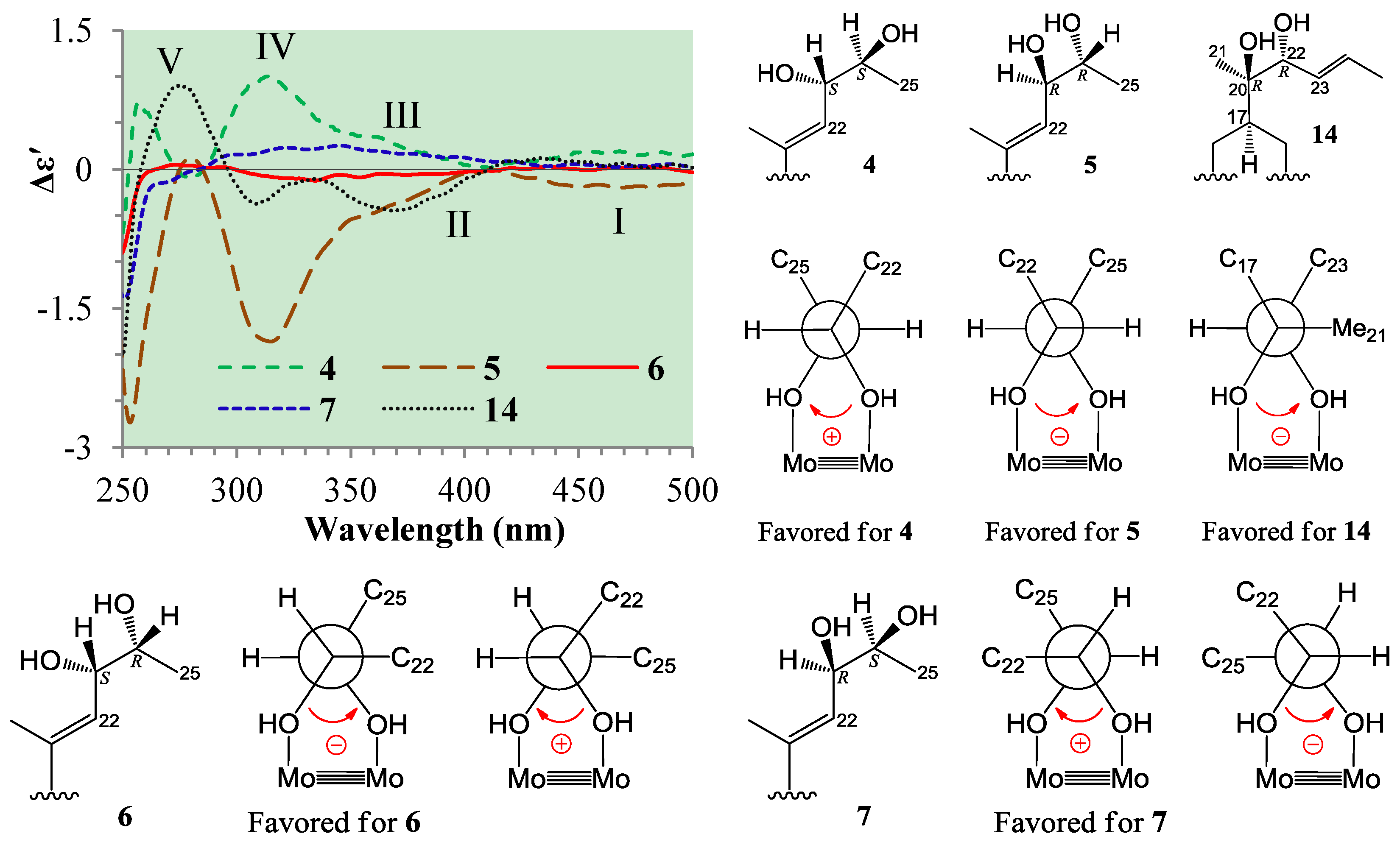
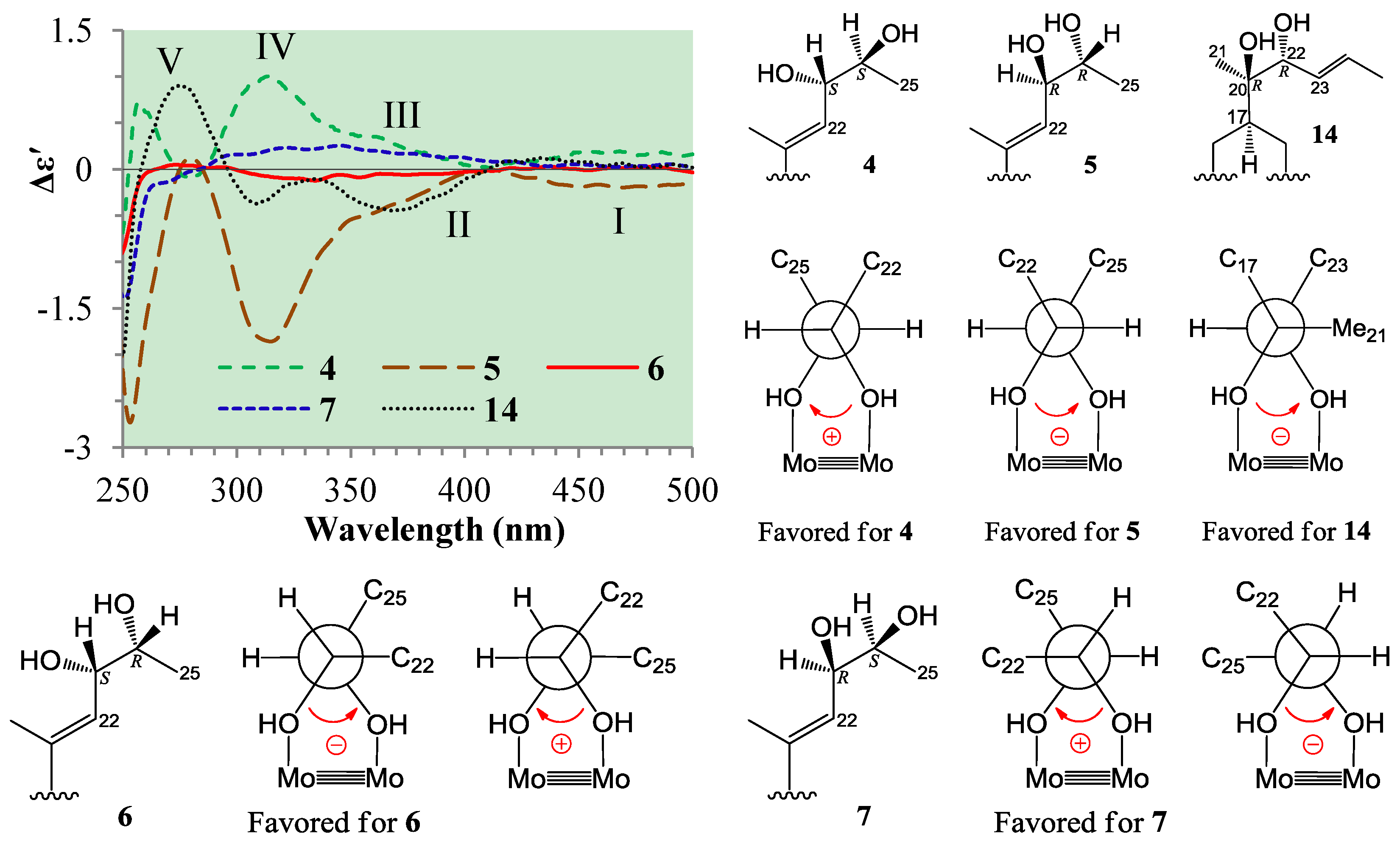
+77.4 (c 0.38, MeOH), was assigned the molecular formula C26H38O4 by HRESIMS (measured 415.2850 [M + H]+, calculated for C26H39O4 [M + H]+ 415.2848). The maximal UV absorption at 243.8 nm (log ε 4.10) in MeOH and the strong IR absorption at 1651 cm−1 indicated the presence of a conjugated carbonyl group in 3 [1]. The IR spectrum also showed the absorptions due to OH (3374 cm−1) and CH3/CH2 (2946, 2910, 2878 and 1378 cm−1) groups. The 1H and 13C NMR spectra of 3 in DMSO-d6 gave 1H and 13C signals (Table 2 and Table 3) resembled those of 1 and 2 (Table 1 and Table 2), except additional 1H and 13C signals from a methoxy group were detected along with disappearance of a hydroxyl signal in the 1H NMR spectrum. This indicated that 3 was a methoxylated derivative of 1 and 2. The MS ion fragments detected at m/z 383 [M − CH3OH + H]+ in the positive ESIMS and at m/z 381 [M − CH3OH − H]− in the negative ESIMS also supported the presence of a methoxy group in 3. In addition, several 1H and 13C NMR signals of 3 appeared as pairs in an approximate 4:1 ratio, and significant changes were mainly observed on the signals from side chain moieties in 1–3 (Table 1, Table 2 and Table 3). These observations revealed that the methoxy group in 3 should be in the side chain and 3 was a mixture. Detailed analysis of the 1H and 13C NMR spectra with the aide of DEPT, 1D difference NOE, 1H-1H COSY, HMQC, HMBC and NOESY techniques (Table S3 in the Supplementary Information) established the structure of 3 as shown in Figure 1. The methoxy group in 3 was located at C-23 by the HMBCs shown in Figure 2. The NOEs between H3-21 and H-22 demonstrated the Z-configuration of the 20,22-double bond in 3. The relative stereochemistry of the ring system in 3 was determined to be the same as 1 and 2 on the basis of the NOEs between pairs of the protons, H3-21/H3-19, H3-19/Hα-18 and H-9/H-14, and the almost identical 1H and 13C signals of the A/B ring moieties in 1–3 (Table 1, Table 2 and Table 3). The absolute configuration of the A–D ring system in 3 was established to be the same as 1–2 and 4 by the closely resembled CD spectra of 1–4 as shown in Figure 3. The couplings of H-23 and H-24 in 3, 5.9 Hz for major isomer and 4.0 Hz for minor isomer, indicated that the major isomer in 3 was the 23,24-erythro and the minor isomer was the 23,24-thero [1,21]. This was supported also by the downfield resonances of H-23 and H-24 in the major erythro isomer than in the minor thero isomer (see Table 3) [1]. Further, the chemical shifts of C-21 and C-23 in the major and minor isomers were consistent (Table 2) and indicated the same configuration at C-23 [1]. Thus, 3 was the 4:1 mixture of 23-methoxylated 2 and 1.2.3. Absolute Configuration Assignment of vic-Diols in 4–7 and 14 by ICD Analysis
| Position | 3 b | 8 | 9 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|
| 1 | 5.55 dd (8.1, 6.4) | 5.56 dd (8.3, 6.3) | 5.55 dd (8.1, 6.5) | 5.55 dd (8.2, 6.2) | 5.54 m | 5.56 dd (8.4, 6.3) |
| 2α | 2.07 m | 2.07 m | 2.07 m | 2.23 m | 2.22 m | 2.24 ddt (13.4, 8.4, 2.2) |
| β | 2.34 ddd (13.3, 11.4, 6.4) | 2.34 m | 2.34 m | 2.47 ddd (13.4, 11.3, 6.2) | 2.46 ddd (13.1, 11.4, 6.0) | 2.48 ddd (13.4, 11.3, 6.3) |
| 3 | 3.11 m | 3.12 m | 3.12 m | 3.47 m | 3.46 m | 3.50 m |
| 4α | 2.63 br d (13.0) | 2.63 br d (13.2) | 2.63 br d (13.1) | 2.87 br d (12.8) | 2.87 br d (12.8) | 2.89 br d (13.0) |
| β | 1.52 m | 1.53 m | 1.52 m | 1.68 m | 1.67 m | 1.66 m |
| 5 | 2.69 m | 2.68 m | 2.68 m | 2.72 m | 2.71 m | 2.74 m |
| 7 | 5.43 s | 5.43 s | 5.43 s | 5.55 br s | 5.54 br s | 5.58 br s |
| 9 | 2.84 dd (11.2, 6.0) | 2.85 dd (11.5, 5.5) | 2.85 dd (11.5, 5.4) | 2.74 dd (12.5, 5.8) | 2.73 dd (12.6, 5.8) | 2.76 dd (12.0, 5.8) |
| 11α | 1.50 m | 1.54 m | 1.54 m | 1.56 m | 1.55 m | 1.59 m |
| β | 1.77 m | 1.79 m | 1.78 m | 1.65 m | 1.64 m | 1.86 m |
| 12α | 1.41 td (12.4, 4.0) | 1.49 m | 1.49 m | 1.43 td (13.0, 4.8) | 1.42 m | 1.47 td (13.1, 4.4) |
| β | 1.68 m | 1.75 m | 1.75 m | 2.18 m | 2.17 m | 2.16 m |
| 14 | 2.29 br t (7.9) | 2.26 br t (9.2) | 2.25 br t (9.2) | 2.06 ddd (12.0, 6.4, 1.5) | 2.05 ddd (12.4, 6.6, 1.4) | 2.08 ddd (12.0, 6.1, 1.6) |
| 15α | 1.62 m | 1.57 m | 1.57 m | 1.46 m | 1.46 m | 1.62 m |
| β | 1.62 m | 1.53 m | 1.52 m | 1.58 m | 1.57 m | 1.54 m |
| 16α | 1.71 m | 1.84 m | 1.83 m | 1.74 m | 1.71 m | 1.91 m |
| β | 1.90 m | 1.70 m | 1.70 m | 1.78 m | 1.77 m | 1.67 m |
| 17 | 2.84 m (covered by H-9 signals) | 2.33 m | 2.33 m | 1.80 m | 1.79 m | 1.68 m |
| 18α | 2.48 br s | 2.48 br s | 2.48 br s | 2.54 br s | 2.53 m | 2.54 d (13.6) |
| β | 2.48 br s | 2.48 br s | 2.48 br s | 2.55 m | 2.54 m | 2.59 dd (13.6, 6.0) |
| 19 | 0.65 s/0.67 s | 0.55 s/0.53 s | 0.54 s/0.53 s | 0.73 s | 0.71 s | 0.86 s |
| 21 | 1.77 s | 1.71 s | 1.68 s | 1.28 s | 1.27 s | 1.25 s |
| 22 | 5.17 dd (10.2, 1.0)/5.14 dd (10.2, 1.0) | 5.02 d (9.4) | 5.12 d (8.9) | 5.66 d (16.0) | 5.67 d (16.0) | 3.90 dd (8.3, 0.6) |
| 23 | 3.84 dd (10.0, 4.0)/3.74 dd (10.0, 5.9) | 3.71 dd (9.4, 6.7) | 3.77 dd (8.9, 4.0)/3.79 dd (8.9, 3.8) | 5.55 dd (16.0, 6.0) | 5.54 dd (16.0, 6.1) | 5.41 ddq (15.3, 8.2, 1.6) |
| 24 | 3.56 m/3.50 m | 3.53 m | 3.60 m | 4.34 m | 4.33 m | 5.77 dqd (15.3, 6.5, 0.6) |
| 25 | 1.03 d (6.3)/1.01 d (6.3) | 0.95 d (6.4)/0.94 d (6.3) | 0.99 d (6.3)/0.97d (6.1) | 1.28 d (6.3) | 1.27 d (6.3) | 1.73 dd (6.5, 1.6) |
| 3-OH | 4.63 d (4.3) | 4.62 d (4.2) | 4.63 d (3.8) | Not detected | Not detected | Not detected |
| 20-OH | - | - | - | - | - | Not detected |
| 24-OH | 4.47 d (4.9) /4.44 d (4.7) | 4.45 d (3.9)/4.44 d (3.8) | 4.44 d (3.6) | Not detected | Not detected | Not detected |
| 20-OCH3 | - | - | - | 3.11 s | 3.12 d (0.7) | - |
| 23-OCH3 | 3.11 s | 3.147 s/3.152 s | 3.15 s/3.16 s | - | - | - |
2.4. Inhibitory Effects of 1–14 on Several Human Cancer Cell Lines
2.5. Discussions
3. Experimental Section
3.1. General Experimental
3.2. MTT Assay
3.3. Fermentation and EtOAc Extract Preparation
3.3.1. Initial Fungal Strain and its Mutant the 1–14 Producing Strain
3.3.2. Preparation of Spore Suspensions
3.3.3. Fermentation and Extraction
3.4. Isolation of Compounds 1–14
3.5. Physicochemical and Spectroscopic Data for 1–14
+71.1 (c 0.23, MeOH). Positive ESIMS m/z: 347 [M − 3H2O + H]+, 365 [M − 2H2O + H]+, 383 [M − H2O + H]+, 401 [M + H]+, 423 [M + Na]+, 439 [M + K]+, 823 [2M + Na]+; negative ESI-MS m/z: 363 [M − 2H2O − H]−, 381 [M − H2O − H]−, 399 [M − H]−, 435 [M + Cl]−, 445 [M + HCOO]−, 799 [2M − H]−, 835 [2M + Cl]−. Positive HRESIMS m/z: measured 401.2689 [M + H]+, calculated for C25H37O4 [M + H]+ 401.2692; measured 423.2508 [M + Na]+, calculated for C25H36O4Na [M + Na]+ 423.2511. UV λmax nm (log ε) in MeOH: 203.7 (3.97), 243.8 (3.95). IR νmax cm−1 (Diamond ATR crystal): 3517, 3295, 2966, 2938, 1645, 1460, 1433, 1368, 1294, 1128, 1078, 1012, 915, 899, 869, 827, 764. CD Δε (nm) in MeOH: +4.61 (200.0), 0 (217.0), −0.28 (221), 0 (225.0), +2.57 (247.0), 0 (276.5), −1.38 (316.5), 0 (355.0), +0.16 (372.0), 0 (398.0). 1H and 13C NMR data: see Table 1 and Table 2, respectively. +71.6 (c 0.30, MeOH). Positive ESIMS m/z: 347 [M − 3H2O + H]+, 365 [M − 2H2O + H]+, 383 [M − H2O + H]+, 401 [M + H]+, 423 [M + Na]+, 439 [M + K]+; negative ESI-MS m/z: 363 [M − 2H2O − H]−, 381 [M − H2O − H]−, 399 [M − H]−, 435 [M + Cl]−. Positive HRESIMS m/z: measured 401.2684 [M + H]+, calculated for C25H37O4 [M + H]+ 401.2692; measured 423.2500 [M + Na]+, calculated for C25H36O4Na [M + Na]+ 423.2511. UV λmax nm (log ε) in MeOH: 205.4 (4.11), 243.0 (4.10). IR νmax cm−1 (Diamond ATR crystal): 3353, 2956, 2874, 1641, 1460, 1380, 1295, 1176, 1153, 1125, 1078, 1025, 1008, 915, 897, 867, 822, 763. CD Δε (nm) in MeOH: +0.31 (200.0), 0 (216.0), −0.49 (220.0), 0 (224.0), +3.69 (245.0), 0 (275.5), −2.03 (317.0), 0 (374.0). 1H and 13C NMR data: see Table 1 and Table 2, respectively. +77.4 (c 0.38, MeOH). Positive ESIMS m/z: 347 [M − CH3OH − 2H2O + H]+, 365 [M − CH3OH − H2O + H]+, 383 [M − CH3OH + H]+, 415 [M + H]+, 437 [M + Na]+, 453 [M + K]+, 851 [2M + Na]+, 867 [2M + K]+; negative ESI-MS m/z: 363 [M − CH3OH − H2O − H]−, 381 [M − CH3OH − H]−, 413 [M − H]−, 449 [M + Cl]−. Positive HRESIMS m/z: measured 415.2850 [M + H]+, calculated for C26H39O4 [M + H]+ 415.2848; measured 437.2671 [M + Na]+, calculated for C26H38O4Na [M + Na]+ 437.2668. UV λmax nm (log ε) in MeOH: 205.4 (4.14), 243.8 (4.10). IR νmax cm−1 (Diamond ATR crystal): 3374, 2946, 2910, 2878, 1651, 1587, 1459, 1378, 1297, 1153, 1026, 1007, 916, 869, 820, 761. CD Δε (nm) in MeOH: +1.28 (200.0), +2.69 (205.0), 0 (212.5), −1.13 (217.5), 0 (225.0), +4.10 (246.0), 0 (276.0), −2.15 (318.0), 0 (352.5), +0.45 (371.5), +0.08 (400.0). 1H and 13C NMR data: see Table 2 and Table 3, respectively. +126.1 (c 0.38, MeOH). Positive ESIMS m/z: 365 [M − 2H2O + H]+, 401 [M + H]+, 423 [M + Na]+, 823 [2M + Na]+; negative ESI-MS m/z: 381 [M − H2O − H]−, 399 [M − H]−, 445 [M + HCOO]−, 799 [2M − H]−. CD Δε (nm) in MeOH: +8.45 (200.0), +10.17 (203.5), +2.05 (221.5), +4.40 (245.0), 0 (277), −2.55 (318.0), 0 (364.5), 0.08 (372.0), 0 (390.0), −0.02 (400.0). 1H and 13C NMR data: see Table 1 and Table 2, respectively.3.6. ICD Measurements for 1, 2, 4–7 and 14 Using Dimolybdenum Tetracetate
3.7. HPLC-PDAD-UV and HPLC-ESI-MS Analyses
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Du, L.; Zhu, T.J.; Fang, Y.; Gu, Q.; Zhu, W. Unusual C25 steroid isomers with bicyclo[4.4.1]A/B rings from a volcano ash-derived fungus Penicillium citrinum. J. Nat. Prod. 2008, 71, 1343–1351. [Google Scholar] [CrossRef]
- Kozlovsky, A.G.; Zhelifonova, V.P.; Ozerskaya, S.M.; Vinokurova, N.G.; Adanin, V.M.; Gräfe, U. Cyclocitrinol, a new fungal metabolite from Penicillium citrinum. Pharmazie 2000, 55, 470–471. [Google Scholar]
- Amagata, T.; Amagata, A.; Tenney, K.; Valeriote, F.A.; Lobkovsky, E.; Clardy, J.; Crews, P. Unusual C25 steroids produced by a sponge-derived Penicillium citrinum. Org. Lett. 2003, 5, 4393–4396. [Google Scholar] [CrossRef]
- Marinhoa, A.M.R.; Rodrigues-Filho, E.; Ferreira, A.G.; Santos, L.S. C25 steroid epimers produced by Penicillium janthinellum, a fungus isolated from fruits Melia azedarach. J. Braz. Chem. Soc. 2005, 16, 1342–1346. [Google Scholar] [CrossRef]
- Schneider, P.; Misiek, M.; Hoffmeister, D. In vivo and in vitro production options for fungal secondary metabolites. Mol. Pharm. 2008, 5, 234–242. [Google Scholar] [CrossRef]
- Saleem, M.; Ali, M.S.; Hussain, S.; Jabbar, A.; Ashraf, M.; Lee, Y.S. Marine natural products of fungal origin. Nat. Prod. Rep. 2007, 24, 1142–1152. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar]
- Brakhage, A.A.; Schroeckh, V. Fungal secondary metabolites—Strategies to activate silent gene clusters. Fungal Genet. Biol. 2011, 48, 15–22. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar]
- Henrikson, J.C.; Hoover, A.R.; Joyner, P.M.; Cichewicz, R.H. A chemical epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from Aspergillus niger. Org. Biomol. Chem. 2009, 7, 435–438. [Google Scholar] [CrossRef]
- Takahashi, J.A.; Teles, A.P.C.; Bracarense, A.A.P.; Gomes, D.C. Classical and epigenetic approaches to metabolite diversification in filamentous fungi. Phytochem. Rev. 2013, 12, 773–789. [Google Scholar] [CrossRef]
- Ochi, K. From microbial differentiation to ribosome engineering. Biosci. Biotechnol. Biochem. 2007, 71, 1373–1386. [Google Scholar] [CrossRef]
- Hosaka, T.; Ohnishi-Kameyama, M.; Muramatsu, H.; Murakami, K.; Tsurumi, Y.; Kodani, S.; Yoshida, M.; Fujie, A.; Ochi, K. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat. Biotechnol. 2009, 27, 462–464. [Google Scholar]
- Chai, Y.-J.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Tian, C.-K.; Hua, W. Activation of the dormant secondary metabolite production by introducing gentamicin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 559–582. [Google Scholar] [CrossRef]
- Wu, C.J.; Cui, C.B.; Tian, C.K.; Li, C.W. Antitumor metabolites produced by two Penicillium purpurogenum G59 mutants. J. Int. Pharm. Res. 2010, 37, 122–126. [Google Scholar]
- Fang, S.-M.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Zhang, Z.-J.; Li, L.; Huang, X.-J.; Ye, W.-C. Purpurogemutantin and purpurogemutantidin, new drimenyl cyclohexenone derivatives produced by a mutant obtained by diethyl sulfate mutagenesis of a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 1266–1287. [Google Scholar] [CrossRef]
- Fang, S.-M.; Wu, C.-J.; Li, C.-W.; Cui, C.-B. A practical strategy to discover new antitumor compounds by activating silent metabolite production in fungi by diethyl sulphate mutagenesis. Mar. Drugs 2014, in press. [Google Scholar]
- Wu, C.-J.; Li, C.-W.; Cui, C.-B. Seven new and two known lipopeptides as well as five known polyketides: The activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, in press. [Google Scholar]
- Tian, C.K.; Cui, C.B.; Han, X.X. Isolation of fungal strains in unusual environment and screening for their antitumor activity. J. Int. Pharm. Res. 2008, 35, 401–405. [Google Scholar]
- Jarvis, B.B.; Wang, S.; Ammon, H.L. Trichoverroid stereoisomers. J. Nat. Prod. 1996, 59, 254–261. [Google Scholar] [CrossRef]
- Bari, L.D.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef]
- Frelek, J.; Ruśkowska, P.; Suszczyńska, A.; Szewczyk, K.; Osuch, A.; Jarosz, S.; Jagodziński, J. Configurational assignment of sugar erythro-1,2-diols from their electronic circular dichroism spectra with dimolybdenum tetraacetate. Tetrahedron Asymmetry 2008, 19, 1709–1713. [Google Scholar] [CrossRef]
- Górecki, M.; Jabłońska, E.; Kruszewska, A.; Suszczyńska, A.; Urbańczyk-Lipkowska, Z.; Gerards, M.; Morzycki, J.W.; Szczepek, W.J.; Frelek, J. Practical method for the absolute configuration assignment of tert/tert 1,2-diols using their complexes with Mo2(OAc)4. J. Org. Chem. 2007, 72, 2906–2916. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xia, M.-W.; Cui, C.-B.; Li, C.-W.; Wu, C.-J. Three New and Eleven Known Unusual C25 Steroids: Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy using Diethyl Sulphate. Mar. Drugs 2014, 12, 1545-1568. https://doi.org/10.3390/md12031545
Xia M-W, Cui C-B, Li C-W, Wu C-J. Three New and Eleven Known Unusual C25 Steroids: Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy using Diethyl Sulphate. Marine Drugs. 2014; 12(3):1545-1568. https://doi.org/10.3390/md12031545
Chicago/Turabian StyleXia, Ming-Wen, Cheng-Bin Cui, Chang-Wei Li, and Chang-Jing Wu. 2014. "Three New and Eleven Known Unusual C25 Steroids: Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy using Diethyl Sulphate" Marine Drugs 12, no. 3: 1545-1568. https://doi.org/10.3390/md12031545
APA StyleXia, M.-W., Cui, C.-B., Li, C.-W., & Wu, C.-J. (2014). Three New and Eleven Known Unusual C25 Steroids: Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy using Diethyl Sulphate. Marine Drugs, 12(3), 1545-1568. https://doi.org/10.3390/md12031545