Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
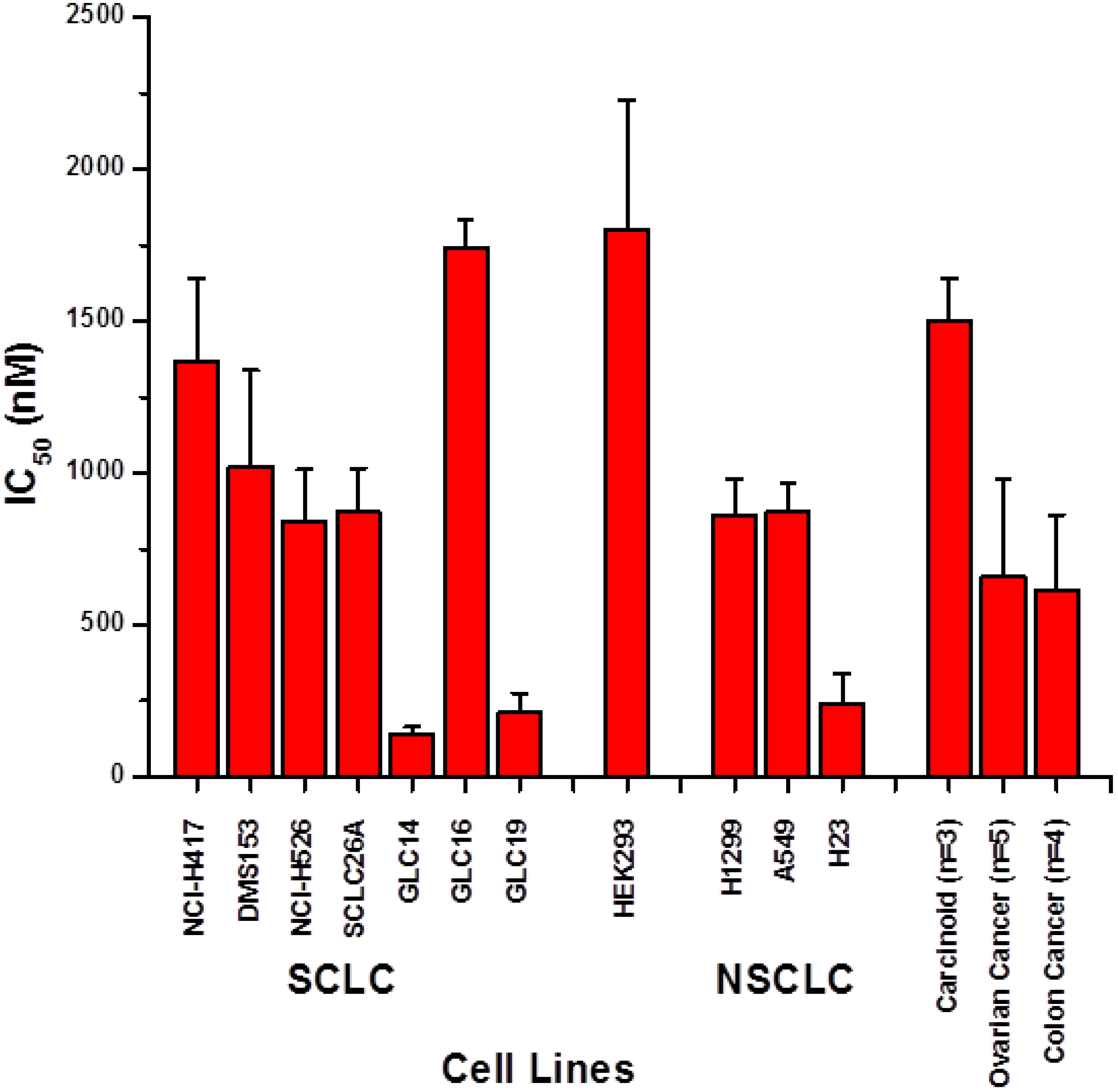
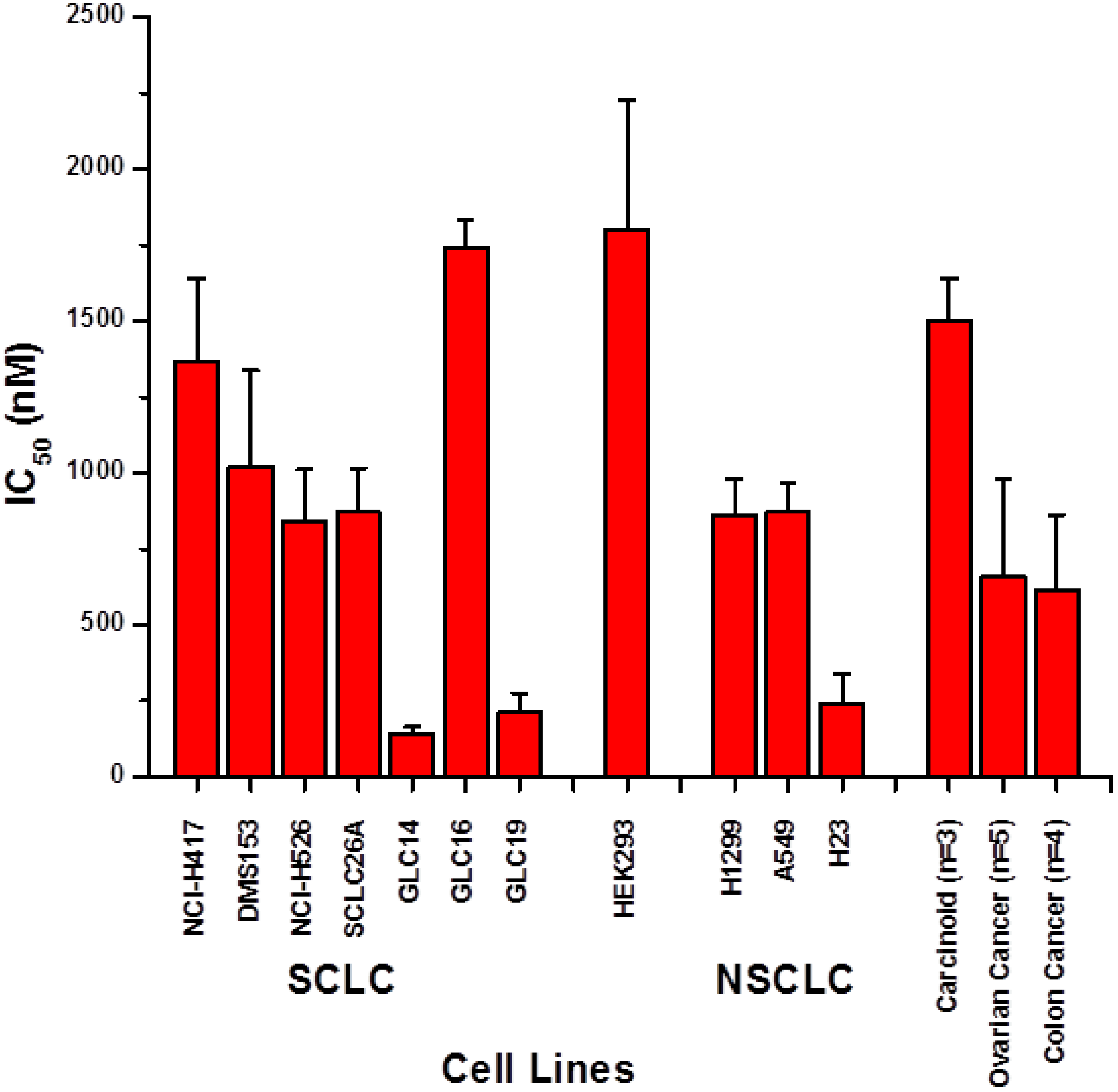
2.1. Screening of Cytotoxic Activity against Lung Cancer Cell Lines
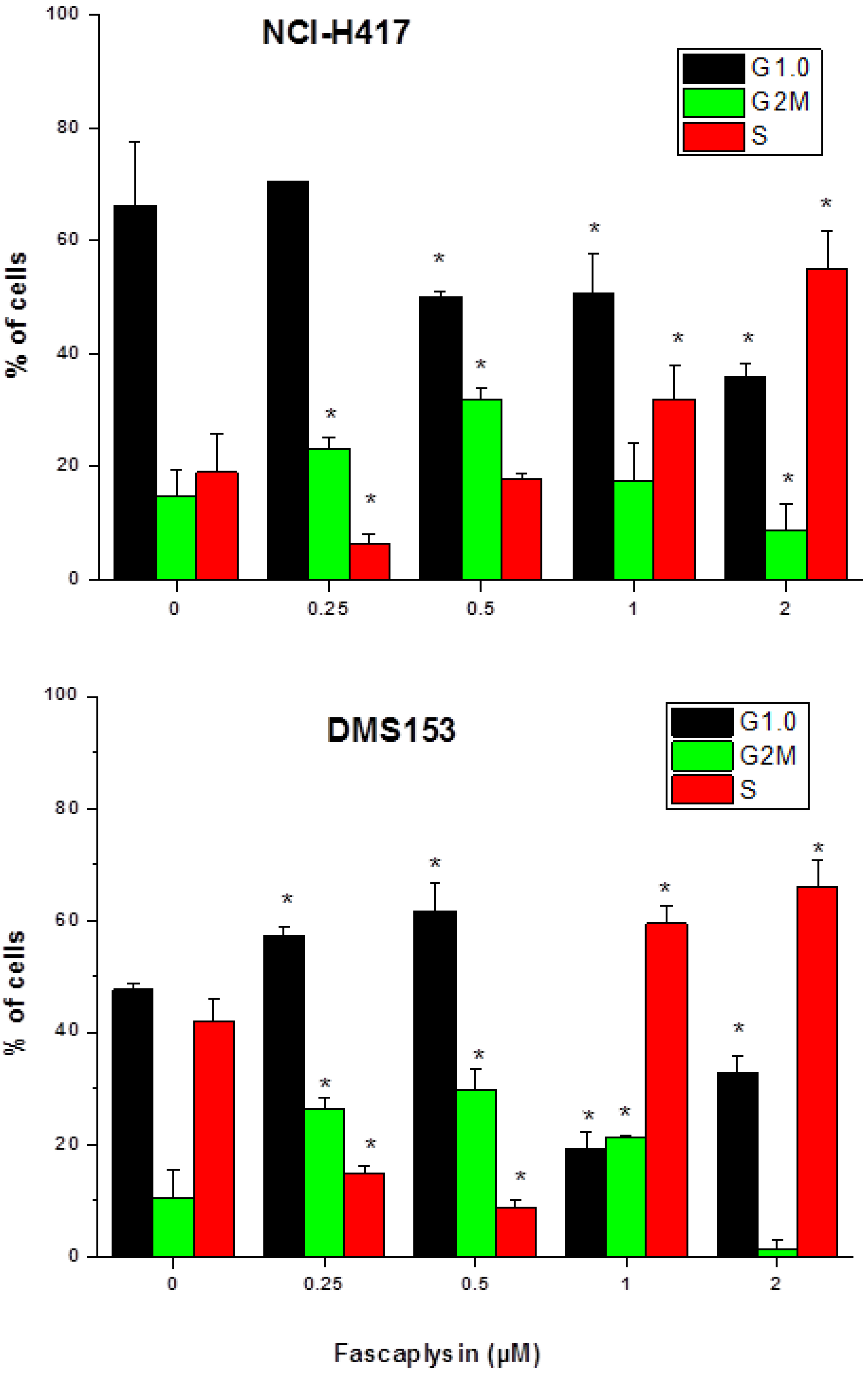
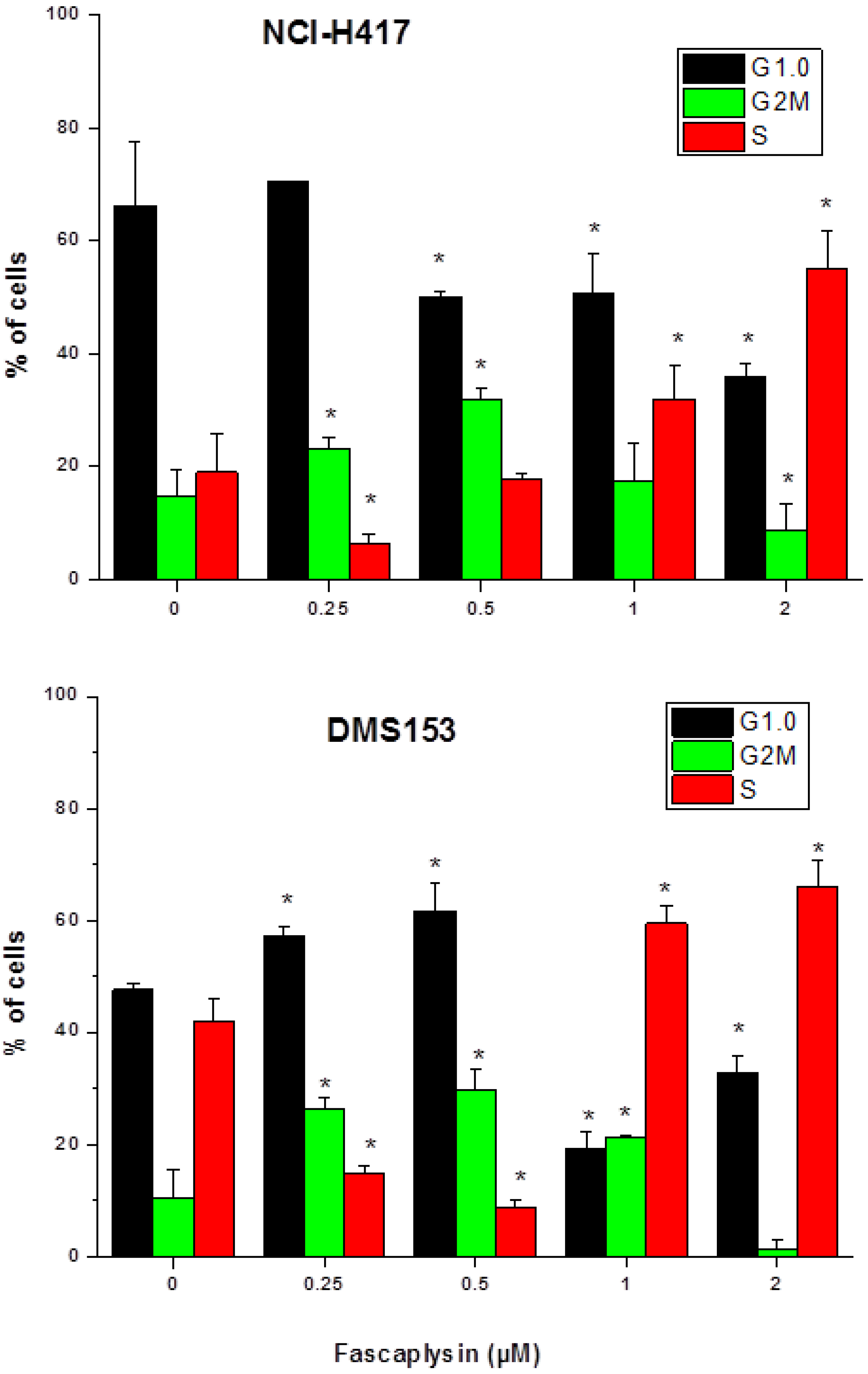
2.2. Effects of Fascaplysin on Cell Cycle Distribution of NCI-H417 and DMS153 Cells
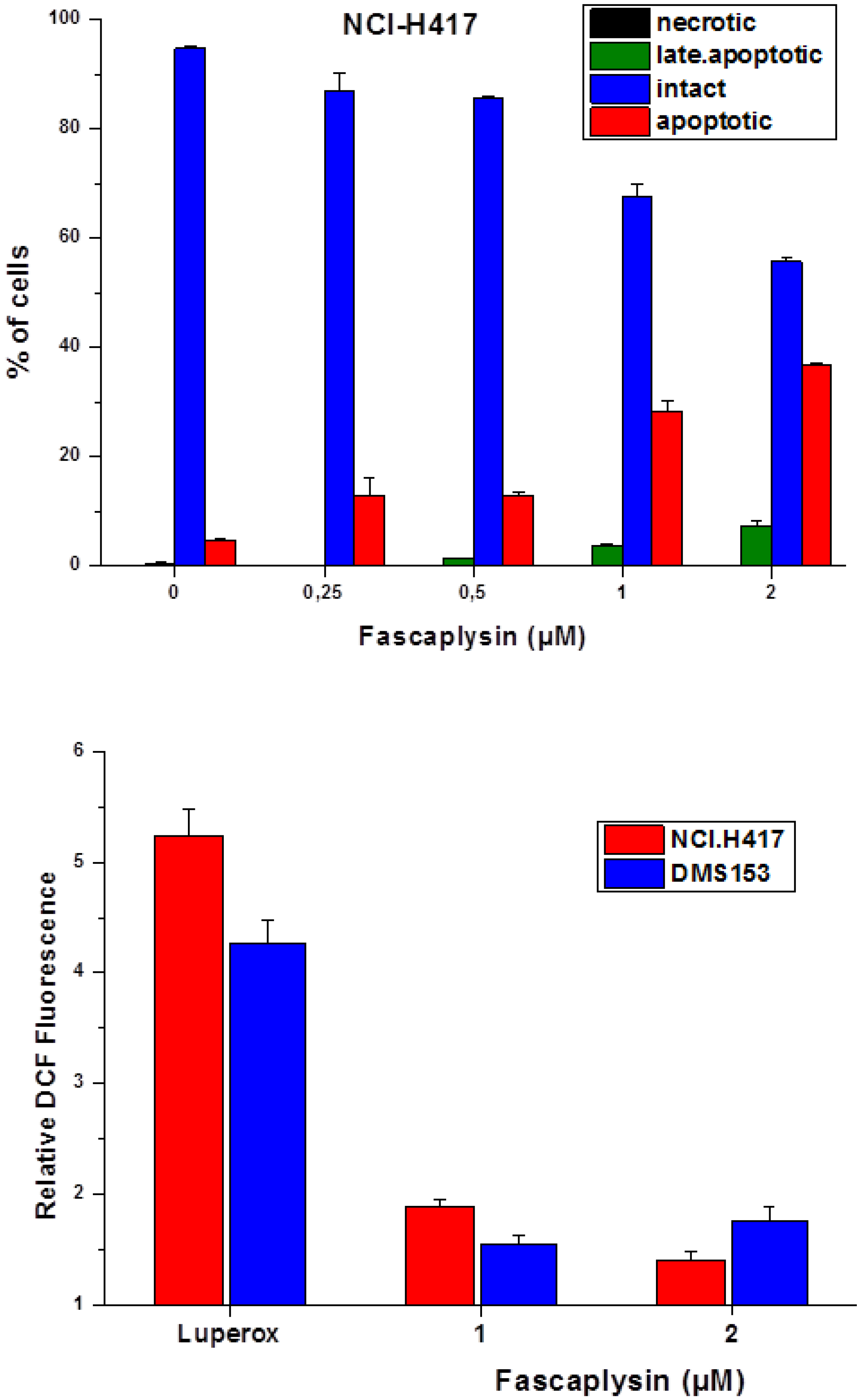
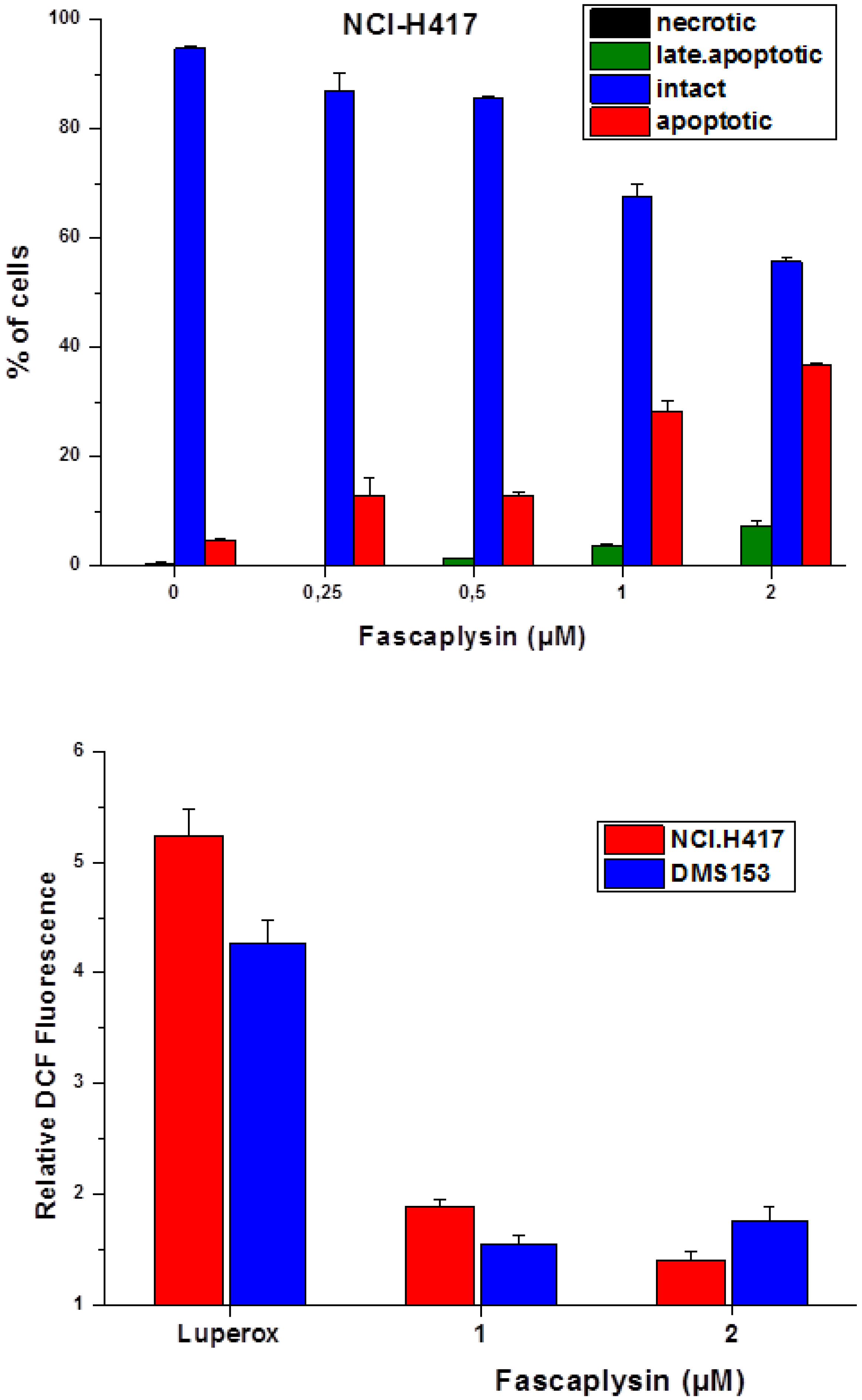
2.3. Fascaplysin-Induced Apoptosis and Generation of ROS in SCLC Lines
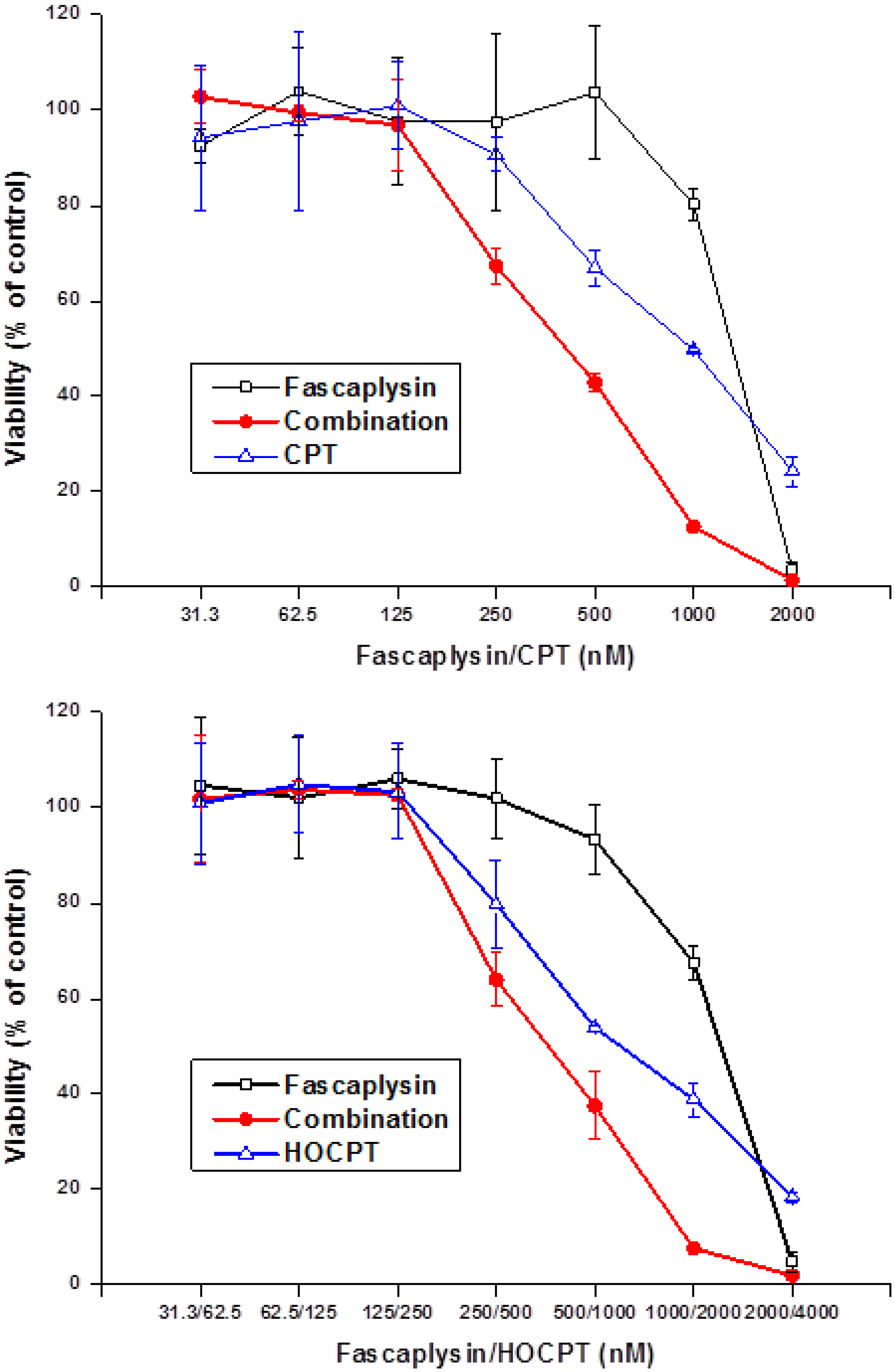
2.4. Synergistic Interaction of Fascaplysin and Camptothecins in NCI-H417 Cells
2.5. Effects of the BYK 204165 PARP Inhibitor on Fascaplysin-Induced Cell Death in SCLC Cell Lines
2.6. Discussion
3. Experimental Section
3.1. Reagents and Cell Lines
3.2. Chemosensitivity Assay
3.3. Measurement of Cell Cycle Distribution
3.4. Detection of Reactive Oxygen Species (ROS)
3.5. Statistical Analysis
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Segraves, N.L.; Lopez, S.; Johnson, T.A.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Structures and cytotoxicities of fascaplysin and related alkaloids from two marine phyla—Fascaplysinopsis sponges and Didemnum tunicates. Tetrahedron Lett. 2003, 44, 3471–3475. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar]
- Hormann, A.; Chaudhari, B.; Fretz, H. DNA binding properties of marine sponge pigment fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of cyclindependentkinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef]
- Soni, R.; O’Reilly, T.; Furet, P.; Muller, L.; Stephan, C.; Zumstein- Mecker, S.; Fretz, H.; Fabbro, D.; Chaudhuri, B. Selective in vivo and in vitro effects of a small molecule inhibitor of cyclindependent kinase-4. J. Natl. Cancer Inst. 2001, 93, 436–446. [Google Scholar]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. Fascaplysin as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS One 2012, 7, e42612. [Google Scholar]
- Goldberg, S.B.; Willers, H.; Heist, R.S. Multidisciplinary management of small cell lung cancer. Surg. Oncol. Clin. N. Am. 2013, 22, 329–343. [Google Scholar] [CrossRef]
- Lu, X.L.; Zheng, Y.L.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Anti-proliferation of human cervical cancer HeLa cell line by fascaplysin through apoptosis induction. Yao Xue Xue Bao 2009, 44, 980–986. [Google Scholar]
- Zheng, Y.L.; Lu, X.L.; Lin, J.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar]
- Lin, J.; Yan, X.J.; Chen, H.M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 59, 439–445. [Google Scholar] [CrossRef]
- Wang, F.; Chen, H.; Yan, X.; Zhen, Y. Fascaplysin sensitizes cells to TRAIL-induced apoptosis through upregulating DR5 expression. Chin. J. Oceanol. Limnol. 2013, 31, 560–569. [Google Scholar] [CrossRef]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar] [CrossRef]
- Subramanian, B.; Nakeff, A.; Tenney, K.; Crews, P.; Gunatilaka, L.; Valeriote, F. A new paradigm for the development of anticancer agents from natural products. J. Exp. Ther. Oncol. 2006, 5, 195–204. [Google Scholar]
- Hamilton, G.; Olszewski, U.; Klameth, L.; Ulsperger, E.; Geissler, K. Synergistic anticancer activity of topotecan—cyclin-dependentkinase inhibitor combinations against drug-resistant small cell lung cancer (SCLC) cell lines. J. Cancer Ther. 2013, 4, 47–53. [Google Scholar]
- Berendsen, H.H.; de Leij, L.; de Vries, E.G.; Mesander, G.; Mulder, N.H.; de Jong, B.; Buys, C.H.; Postmus, E.P.; Poppema, S.; Sluiter, H.S.; et al. Characterization of three small cell lung cancer cell lines established from one patient during longitudinal follow-up. Cancer Res. 1988, 48, 6891–6899. [Google Scholar]
- Giaccone, G.; Battey, J.; Gazdar, A.F.; Oie, H.; Draoui, M.; Moody, T.W. Neuromedin B is present in lung cancer cell lines. Cancer Res. 1992, 52, 2732s–2736s. [Google Scholar]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and expression of the c-myconcogene in human lung cancer cell lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef]
- Jiang, T.; Collins, B.J.; Jin, N.; Watkins, D.N.; Brock, M.V.; Matsui, W.; Nelkin, B.D.; Ball, D.W. Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res. 2009, 69, 845–854. [Google Scholar] [CrossRef]
- Pettengill, O.S.; Sorenson, G.D.; Wurster-Hill, D.H.; Curphey, T.J.; Noll, W.W.; Cate, C.C.; Maurer, L.H. Isolation and growth characteristics of continuous cell lines from small-cell carcinoma of the lung. Cancer 1980, 45, 906–918. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Otterson, G.A.; Kratzke, R.A.; Coxon, A.; Kim, Y.W.; Kaye, F.J. Absence of p16INK4 Protein Is Restricted to the Subset of Lung Cancer Lines that Retains Wildtype RB. Oncogene 1994, 9, 3375–3378. [Google Scholar]
- Wikman, H.; Kettunen, E. Regulation of the G1/S Phase of the Cell Cycle and Alterations in the RB Pathway in Human Lung Cancer. Expert Rev. Anticancer Ther. 2006, 6, 515–530. [Google Scholar] [CrossRef]
- Rygaard, K.; Sorenson, G.D.; Pettengill, O.S.; Cate, C.C.; Spang-Thomsen, M. Abnormalities in structure and expression of the retinoblastoma gene in small cell lung cancer cell lines and xenografts in nude mice. Cancer Res. 1990, 50, 5312–5317. [Google Scholar]
- Okamoto, A.; Hussain, S.P.; Hagiwara, K.; Spillare, E.A.; Rusin, M.R.; Demetrick, D.J.; Serrano, M.; Hannon, G.J.; Shiseki, M.; Zariwala, M.; et al. Mutations in the p16INK4/MTS1/CDKN2, p15INK4B/MTS2, and p18 Genes in Primary and Metastatic Lung Cancer. Cancer Res. 1995, 55, 1448–1451. [Google Scholar]
- Zandi, R.; Selivanova, G.; Christensen, C.L. PRIMA-1Met/APR-246 Induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef]
- Zhang, F.; Lau, S.S.; Monks, T.J. The cytoprotective effect of N-acetyl-l-cysteine against ROS-induced cytotoxicity is independent of its ability to enhance glutathione synthesis. Toxicol. Sci. 2011, 120, 87–97. [Google Scholar] [CrossRef]
- Caputo, F.; Vegliante, R.; Ghibelli, L. Redox modulation of the DNA damage response. Biochem. Pharmacol. 2012, 84, 1292–1306. [Google Scholar] [CrossRef]
- Eltze, T.; Boer, R.; Wagner, T.; Weinbrenner, S.; McDonald, M.C.; Thiemermann, C.; Bürkle, A.; Klein, T. Imidazoquinolinone, imidazopyridine, and isoquinolindione derivatives as novel and potent inhibitors of the poly(ADP-ribose) polymerase (PARP): A comparison with standard PARP inhibitors. Mol. Pharmacol. 2008, 74, 1587–1598. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Horton, J.K.; Wilson, S.H. Strategic combination of DNA-damaging agent and PARP inhibitor results in enhanced cytotoxicity. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 2009, 218, 193–202. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs 2014, 12, 1377-1389. https://doi.org/10.3390/md12031377
Hamilton G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Marine Drugs. 2014; 12(3):1377-1389. https://doi.org/10.3390/md12031377
Chicago/Turabian StyleHamilton, Gerhard. 2014. "Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines" Marine Drugs 12, no. 3: 1377-1389. https://doi.org/10.3390/md12031377
APA StyleHamilton, G. (2014). Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Marine Drugs, 12(3), 1377-1389. https://doi.org/10.3390/md12031377