The Lipopolysaccharide Export Pathway in Escherichia coli: Structure, Organization and Regulated Assembly of the Lpt Machinery

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of OM Structure and Functions
2.1. OM Proteins and Lipoproteins
2.2. LPS Structure and Function
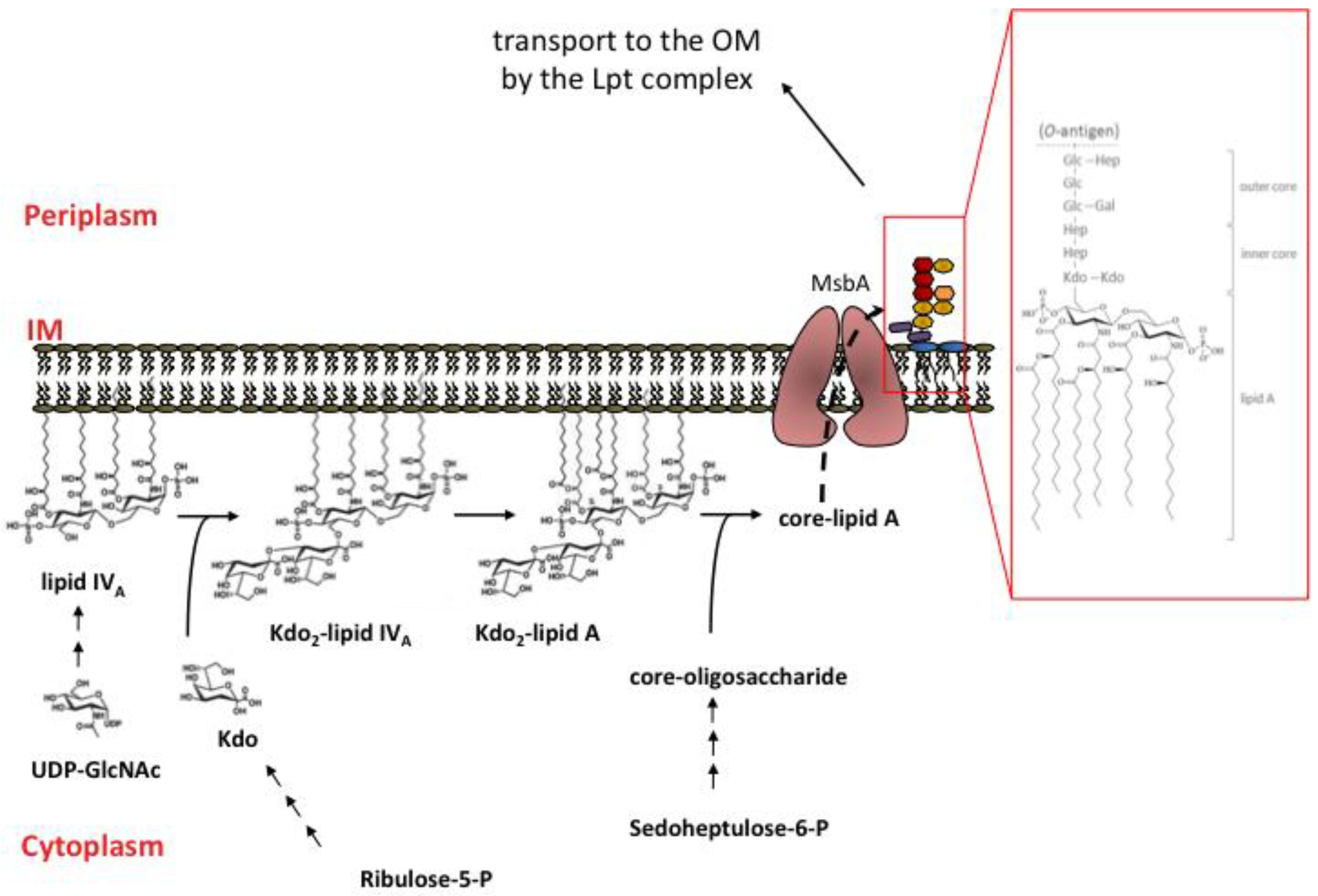
2.3. Overview of LPS Biosynthesis
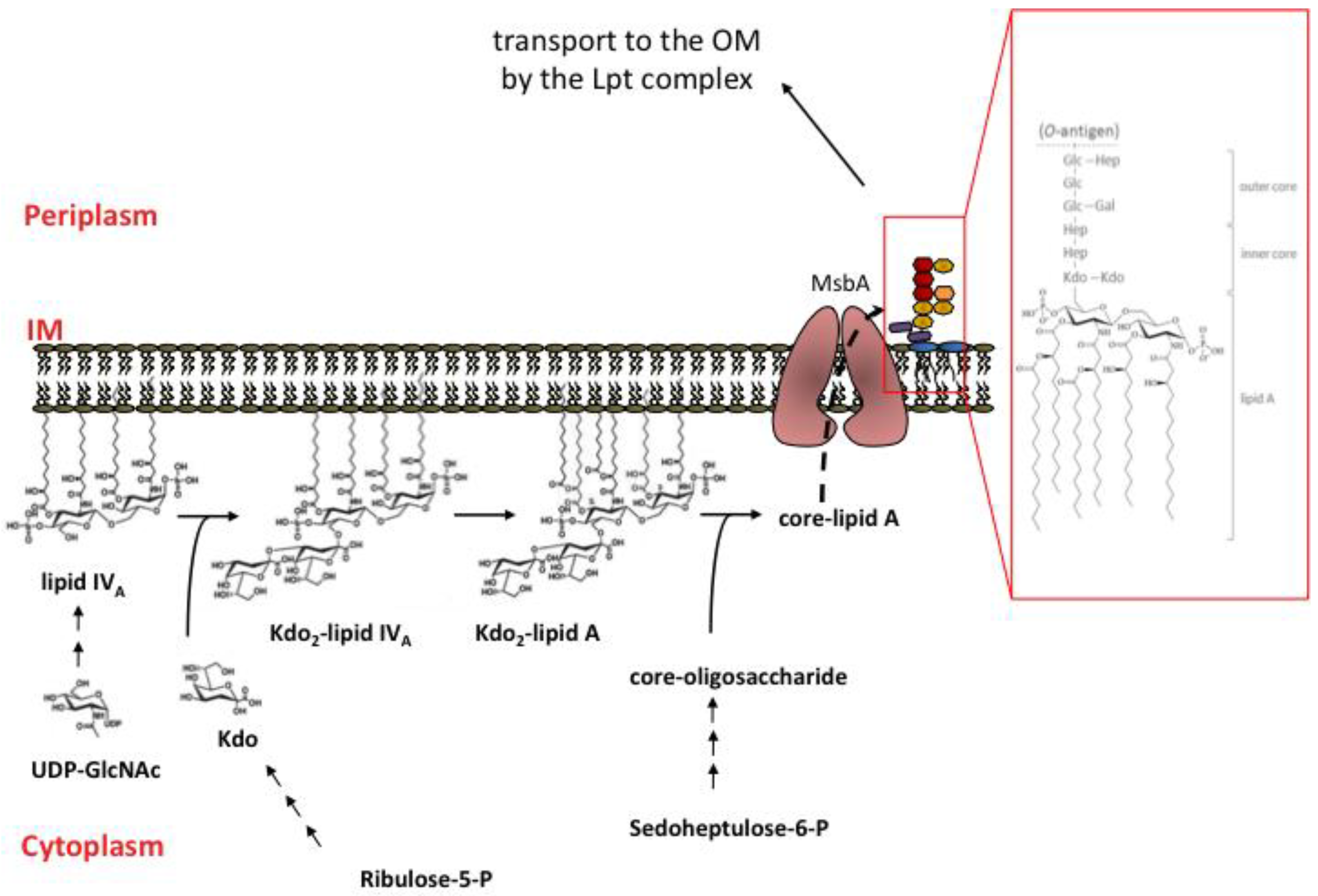
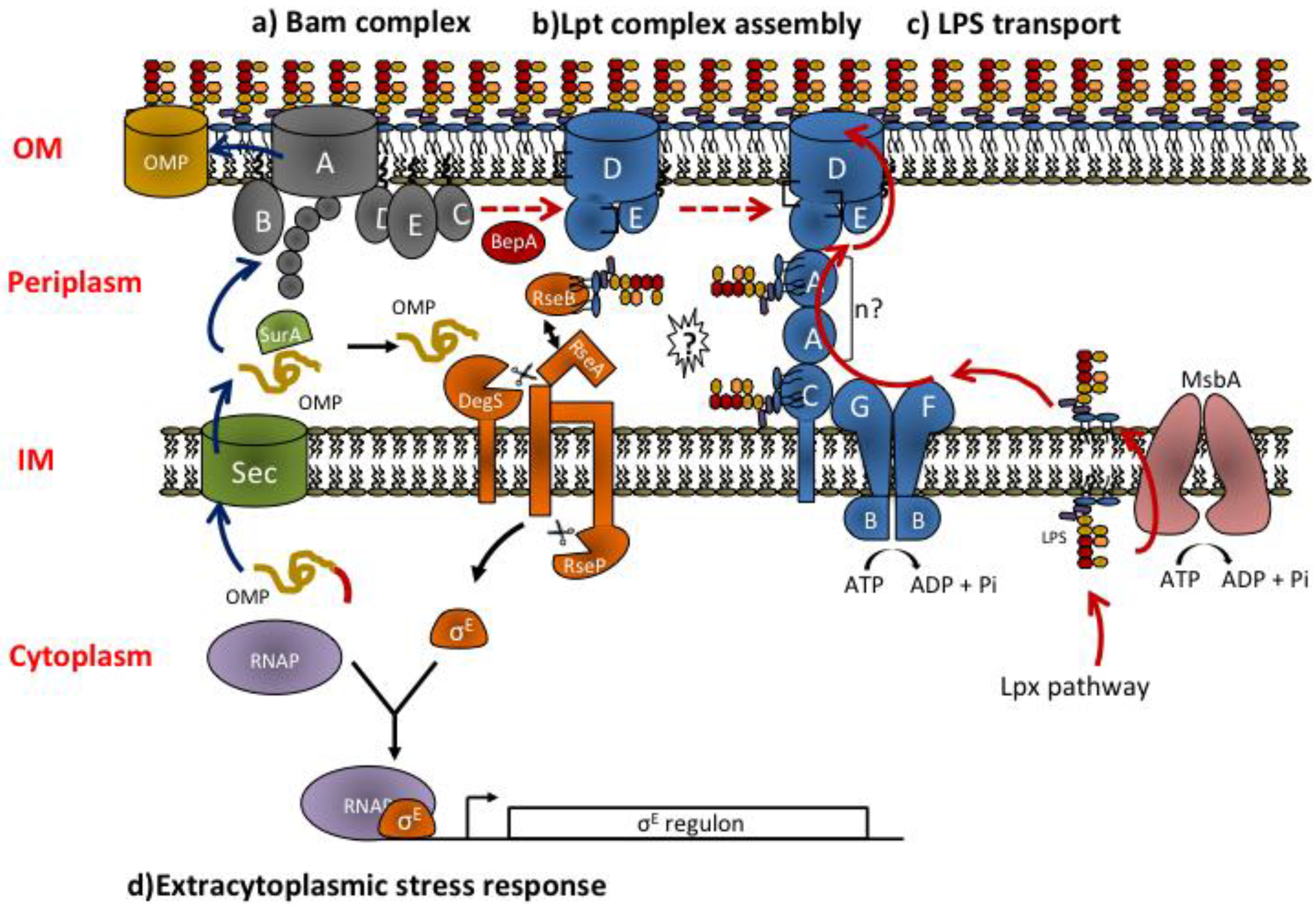
3. LPS transport to the Outer Membrane
3.1. The Lpt Machinery: Structure and Organization of the Components across IM and OM

3.2. Molecular Mechanism of Lpt Transport: Towards the Transenvelope Bridge Model
3.3. Regulation of Lpt Complex Assembly and LPS Transport Process
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Beveridge, T.J.; Davies, J.A. Cellular responses of Bacillus subtilis and Escherichia coli to the Gram stain. J. Bacteriol. 1983, 156, 846–858. [Google Scholar]
- Ellen, A.F.; Zolghadr, B.; Driessen, A.M.; Albers, S.V. Shaping the archaeal cell envelope. Archaea 2010, 2010, 608243. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2012, 2, a000414. [Google Scholar] [CrossRef]
- Gupta, R.S. What are archaebacteria: Life’s third domain or monoderm prokaryotes related to gram-positive bacteria? A new proposal for the classification of prokaryotic organisms. Mol. Microbiol. 1998, 29, 695–707. [Google Scholar] [CrossRef]
- Sutcliffe, I.C. A phylum level perspective on bacterial cell envelope architecture. Trends Microbiol. 2010, 18, 464–470. [Google Scholar] [CrossRef]
- Desvaux, M.; Hebraud, M.; Talon, R.; Henderson, I.R. Secretion and subcellular localizations of bacterial proteins: A semantic awareness issue. Trends Microbiol 2009, 17, 139–145. [Google Scholar] [CrossRef]
- Bos, M.P.; Robert, V.; Tommassen, J. Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 2007, 61, 191–214. [Google Scholar] [CrossRef]
- Gan, L.; Chen, S.; Jensen, G.J. Molecular organization of Gram-negative peptidoglycan. Proc. Natl. Acad. Sci. USA 2008, 105, 18953–18957. [Google Scholar] [CrossRef]
- Vollmer, W.; Seligman, S.J. Architecture of peptidoglycan: More data and more models. Trends Microbiol. 2010, 18, 59–66. [Google Scholar] [CrossRef]
- Vollmer, W.; Holtje, J.V. The architecture of the murein (peptidoglycan) in gram-negative bacteria: vertical scaffold or horizontal layer(s)? J. Bacteriol. 2004, 186, 5978–5987. [Google Scholar] [CrossRef]
- Oliver, D.B. Periplasm; ASM Press: Whashington, DC, USA, 1996. [Google Scholar]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Gunn, J.S. Mechanisms of bacterial resistance and response to bile. Microbes Infect. 2000, 2, 907–913. [Google Scholar] [CrossRef]
- Luirink, J.; Yu, Z.; Wagner, S.; de Gier, J.W. Biogenesis of inner membrane proteins in Escherichia coli. Biochim. Biophys. Acta 2012, 1817, 965–976. [Google Scholar]
- Rigel, N.W.; Silhavy, T.J. Making a β-barrel: Assembly of outer membrane proteins in Gram-negative bacteria. Curr. Opin. Microbiol. 2012, 15, 189–193. [Google Scholar] [CrossRef]
- Sankaran, K.; Wu, H.C. Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. J. Biol. Chem. 1994, 269, 19701–19706. [Google Scholar]
- Braun, V. Covalent lipoprotein from the outer membrane of Escherichia coli. Biochim. Biophys. Acta 1975, 415, 335–377. [Google Scholar] [CrossRef]
- Du Plessis, D.J.; Nouwen, N.; Driessen, A.J. The Sec translocase. Biochim. Biophys. Acta 2011, 1808, 851–865. [Google Scholar] [CrossRef]
- Tokuda, H. Biogenesis of outer membranes in Gram-negative bacteria. Biosci. Biotechnol. Biochem. 2009, 73, 465–473. [Google Scholar] [CrossRef]
- Okuda, S.; Tokuda, H. Lipoprotein sorting in bacteria. Annu. Rev. Microbiol. 2011, 65, 239–259. [Google Scholar] [CrossRef]
- Sklar, J.G.; Wu, T.; Kahne, D.; Silhavy, T.J. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 2007, 21, 2473–2484. [Google Scholar] [CrossRef]
- Ricci, D.P.; Silhavy, T.J. The Bam machine: A molecular cooper. Biochim. Biophys. Acta 2012, 1818, 1067–1084. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Holst, O. The structures of core regions from enterobacterial lipopolysaccharides—An update. FEMS Microbiol. Lett. 2007, 271, 3–11. [Google Scholar] [CrossRef]
- Steeghs, L.; den Hartog, R.; den Boer, A.; Zomer, B.; Roholl, P.; van der Ley, P. Meningitis bacterium is viable without endotoxin. Nature 1998, 392, 449–450. [Google Scholar] [CrossRef]
- Meredith, T.C.; Aggarwal, P.; Mamat, U.; Lindner, B.; Woodard, R.W. Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem. Biol. 2006, 1, 33–42. [Google Scholar] [CrossRef]
- Rahim, R.; Burrows, L.L.; Monteiro, M.A.; Perry, M.B.; Lam, J.S. Involvement of the rml locus in core oligosaccharide and O polysaccharide assembly in Pseudomonas aeruginosa. Microbiology 2000, 146, 2803–2814. [Google Scholar]
- Walsh, A.G.; Matewish, M.J.; Burrows, L.L.; Monteiro, M.A.; Perry, M.B.; Lam, J.S. Lipopolysaccharide core phosphates are required for viability and intrinsic drug resistance in Pseudomonas aeruginosa. Mol. Microbiol. 2000, 35, 718–727. [Google Scholar] [CrossRef]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 2005, 3, 36–46. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Miyake, K. Roles for accessory molecules in microbial recognition by Toll-like receptors. J. Endotoxin Res. 2006, 12, 195–204. [Google Scholar] [CrossRef]
- Valvano, M.A. Export of O-specific lipopolysaccharide. Front. Biosci. 2003, 8, s452–s471. [Google Scholar] [CrossRef]
- Samuel, G.; Reeves, P. Biosynthesis of O-antigens: Genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 2003, 338, 2503–2519. [Google Scholar] [CrossRef]
- Jackman, J.E.; Raetz, C.R.; Fierke, C.A. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry 1999, 38, 1902–1911. [Google Scholar]
- Raetz, C.R.; Dowhan, W. Biosynthesis and function of phospholipids in Escherichia coli. J. Biol. Chem. 1990, 265, 12351–1238. [Google Scholar]
- Mohan, S.; Kelly, T.M.; Eveland, S.S.; Raetz, C.R.; Anderson, M.S. An Escherichia coli gene (FabZ) encoding (3R)-hydroxymyristoyl acyl carrier protein dehydrase. Relation to fabA and suppression of mutations in lipid A biosynthesis. J. Biol. Chem. 1994, 269, 32896–32903. [Google Scholar]
- Clementz, T.; Bednarski, J.J.; Raetz, C.R. Function of the htrB high temperature requirement gene of Escherchia coli in the acylation of lipid A: HtrB catalyzed incorporation of laurate. J. Biol. Chem. 1996, 271, 12095–12102. [Google Scholar] [CrossRef]
- Clementz, T.; Zhou, Z.; Raetz, C.R. Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. Acylation by MsbB follows laurate incorporation by HtrB. J. Biol. Chem. 1997, 272, 10353–10360. [Google Scholar] [CrossRef]
- Polissi, A.; Georgopoulos, C. Mutational analysis and properties of the msbA gene of Escherichia coli, coding for an essential ABC family transporter. Mol. Microbiol. 1996, 20, 1221–1233. [Google Scholar] [CrossRef]
- Zhou, Z.; White, K.A.; Polissi, A.; Georgopoulos, C.; Raetz, C.R. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 1998, 273, 12466–12475. [Google Scholar]
- Perez, J.M.; McGarry, M.A.; Marolda, C.L.; Valvano, M.A. Functional analysis of the large periplasmic loop of the Escherichia coli K-12 WaaL O-antigen ligase. Mol. Microbiol. 2008, 70, 1424–1440. [Google Scholar] [CrossRef]
- Reeves, P.R.; Hobbs, M.; Valvano, M.A.; Skurnik, M.; Whitfield, C.; Coplin, D.; Kido, N.; Klena, J.; Maskell, D.; Raetz, C.R.; et al. Bacterial polysaccharide synthesis and gene nomenclature. Trends Microbiol. 1996, 4, 495–503. [Google Scholar] [CrossRef]
- Rubires, X.; Saigi, F.; Pique, N.; Climent, N.; Merino, S.; Alberti, S.; Tomas, J.M.; Regue, M. A gene (wbbL) from Serratia marcescens N28b (O4) complements the rfb-50 mutation of Escherichia coli K-12 derivatives. J. Bacteriol. 1997, 179, 7581–7586. [Google Scholar]
- Trent, M.S.; Stead, C.M.; Tran, A.X.; Hankins, J.V. Diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 2006, 12, 205–223. [Google Scholar] [CrossRef]
- Raetz, C.R.; Guan, Z.; Ingram, B.O.; Six, D.A.; Song, F.; Wang, X.; Zhao, J. Discovery of new biosynthetic pathways: the lipid A story. J. Lipid Res. 2009, 50, S103–S108. [Google Scholar]
- Ogura, T.; Inoue, K.; Tatsuta, T.; Suzaki, T.; Karata, K.; Young, K.; Su, L.H.; Fierke, C.A.; Jackman, J.E.; Raetz, C.R.; et al. Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 1999, 31, 833–844. [Google Scholar] [CrossRef]
- Sorensen, P.G.; Lutkenhaus, J.; Young, K.; Eveland, S.S.; Anderson, M.S.; Raetz, C.R. Regulation of UDP-3-O-[R-3-hydroxymyristoyl]-N-acetylglucosamine deacetylase in Escherichia coli. The second enzymatic step of lipid a biosynthesis. J. Biol. Chem. 1996, 271, 25898–25905. [Google Scholar]
- Katz, C.; Ron, E.Z. Dual role of FtsH in regulating lipopolysaccharide biosynthesis in Escherichia coli. J. Bacteriol. 2008, 190, 7117–7122. [Google Scholar] [CrossRef]
- Schakermann, M.; Langklotz, S.; Narberhaus, F. FtsH-mediated coordination of lipopolysaccharide biosynthesis in Escherichia coli correlates with the growth rate and the alarmone (p)ppGpp. J. Bacteriol. 2013, 195, 1912–1919. [Google Scholar] [CrossRef]
- Mahalakshmi, S.; Sunayana, M.R.; Saisree, L.; Reddy, M. yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol. Microbiol. 2014, 91, 145–157. [Google Scholar] [CrossRef]
- Langklotz, S.; Schakermann, M.; Narberhaus, F. Control of lipopolysaccharide biosynthesis by FtsH-mediated proteolysis of LpxC is conserved in enterobacteria but not in all gram-negative bacteria. J. Bacteriol. 2011, 193, 1090–1097. [Google Scholar] [CrossRef]
- Sperandeo, P.; Pozzi, C.; Deho, G.; Polissi, A. Non-essential KDO biosynthesis and new essential cell envelope biogenesis genes in the Escherichia coli yrbG-yhbG locus. Res. Microbiol. 2006, 157, 547–558. [Google Scholar] [CrossRef]
- Braun, M.; Silhavy, T.J. Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 2002, 45, 1289–1302. [Google Scholar] [CrossRef]
- Wu, T.; McCandlish, A.C.; Gronenberg, L.S.; Chng, S.S.; Silhavy, T.J.; Kahne, D. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2006, 103, 11754–11759. [Google Scholar]
- Ruiz, N.; Gronenberg, L.S.; Kahne, D.; Silhavy, T.J. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 5537–4552. [Google Scholar] [CrossRef]
- Sperandeo, P.; Deho, G.; Polissi, A. The lipopolysaccharide transport system of Gram-negative bacteria. Biochim. Biophys. Acta 2009, 1791, 594–602. [Google Scholar]
- Ruiz, N.; Kahne, D.; Silhavy, T.J. Transport of lipopolysaccharide across the cell envelope: The long road of discovery. Nat. Rev. Microbiol. 2009, 7, 677–683. [Google Scholar] [CrossRef]
- Sperandeo, P.; Cescutti, R.; Villa, R.; Di Benedetto, C.; Candia, D.; Deho, G.; Polissi, A. Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 2007, 189, 244–253. [Google Scholar] [CrossRef]
- Narita, S.; Tokuda, H. Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides. FEBS Lett. 2009, 583, 2160–2164. [Google Scholar] [CrossRef]
- Sperandeo, P.; Lau, F.K.; Carpentieri, A.; de Castro, C.; Molinaro, A.; Deho, G.; Silhavy, T.J.; Polissi, A. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 2008, 190, 4460–4469. [Google Scholar] [CrossRef]
- Saurin, W.; Hofnung, M.; Dassa, E. Getting in or out: Early segregation between importers and exporters in the evolution of ATP-binding cassette (ABC) transporters. J. Mol. Evol. 1999, 48, 22–41. [Google Scholar] [CrossRef]
- Chng, S.S.; Ruiz, N.; Chimalakonda, G.; Silhavy, T.J.; Kahne, D. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc. Natl. Acad. Sci. USA 2010, 107, 5363–5368. [Google Scholar] [CrossRef]
- Freinkman, E.; Okuda, S.; Ruiz, N.; Kahne, D. Regulated Assembly of the Transenvelope Protein Complex Required for Lipopolysaccharide Export. Biochemistry 2012, 51, 4800–4806. [Google Scholar] [CrossRef]
- Chng, S.S.; Gronenberg, L.S.; Kahne, D. Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 2010, 49, 4565–4567. [Google Scholar] [CrossRef]
- Suits, M.D.; Sperandeo, P.; Deho, G.; Polissi, A.; Jia, Z. Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. J. Mol. Biol. 2008, 380, 476–488. [Google Scholar] [CrossRef]
- Tran, A.X.; Trent, M.S.; Whitfield, C. The LptA protein of Escherichia coli is a periplasmic lipid A-binding protein involved in the lipopolysaccharide export pathway. J. Biol. Chem. 2008, 283, 20342–20349. [Google Scholar] [CrossRef]
- Okuda, S.; Freinkman, E.; Kahne, D. Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 2012, 338, 1214–1217. [Google Scholar] [CrossRef]
- Tran, A.X.; Dong, C.; Whitfield, C. Structure and functional analysis of LptC, a conserved membrane protein involved in the lipopolysaccharide export pathway in Escherichia coli. J. Biol. Chem. 2010, 285, 33529–33539. [Google Scholar] [CrossRef]
- Santambrogio, C.; Sperandeo, P.; Villa, R.; Sobott, F.; Polissi, A.; Grandori, R. LptA assembles into rod-like oligomers involving disorder-to-order transitions. J. Am. Soc. Mass Spectrom. 2013, 24, 1593–1602. [Google Scholar]
- Tefsen, B.; Geurtsen, J.; Beckers, F.; Tommassen, J.; de Cock, H. Lipopolysaccharide transport to the bacterial outer membrane in spheroplasts. J. Biol. Chem. 2005, 280, 4504–4509. [Google Scholar]
- Symmons, M.F.; Bokma, E.; Koronakis, E.; Hughes, C.; Koronakis, V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. USA 2009, 106, 7173–7178. [Google Scholar] [CrossRef]
- Kubori, T.; Matsushima, Y.; Nakamura, D.; Uralil, J.; Lara-Tejero, M.; Sukhan, A.; Galan, J.E.; Aizawa, S.I. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 1998, 280, 602–605. [Google Scholar] [CrossRef]
- Sperandeo, P.; Villa, R.; Martorana, A.M.; Samalikova, M.; Grandori, R.; Deho, G.; Polissi, A. New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA–LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex. J. Bacteriol. 2011, 193, 1042–1053. [Google Scholar] [CrossRef]
- Bayer, M.E. Areas of adhesion between wall and membrane of Escherichia coli. J. Gen. Microbiol. 1968, 53, 395–404. [Google Scholar] [CrossRef]
- Bayer, M.E. Zones of membrane adhesion in the cryofixed envelope of Escherichia coli. J. Struct. Biol. 1991, 107, 268–280. [Google Scholar] [CrossRef]
- Muhlradt, P.F.; Menzel, J.; Golecki, J.R.; Speth, V. Outer membrane of Salmonella. Sites of export of newly synthesised lipopolysaccharide on the bacterial surface. Eur. J. Biochem. 1973, 35, 471–481. [Google Scholar] [CrossRef]
- Ishidate, K.; Creeger, E.S.; Zrike, J.; Deb, S.; Glauner, B.; MacAlister, T.J.; Rothfield, L.I. Isolation of differentiated membrane domains from Escherichia coli and Salmonella typhimurium, including a fraction containing attachment sites between the inner and outer membranes and the murein skeleton of the cell envelope. J. Biol. Chem. 1986, 261, 428–443. [Google Scholar]
- Chin, J.W.; Schultz, P.G. In vivo photocrosslinking with unnatural amino Acid mutagenesis. Chembiochem 2002, 3, 1135–1137. [Google Scholar] [CrossRef]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef]
- Freinkman, E.; Chng, S.S.; Kahne, D. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. USA 2011, 108, 24862–2491. [Google Scholar]
- Villa, R.; Martorana, A.M.; Okuda, S.; Gourlay, L.J.; Nardini, M.; Sperandeo, P.; Deho, G.; Bolognesi, M.; Kahne, D.; Polissi, A. The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J. Bacteriol. 2013, 195, 1100–1108. [Google Scholar] [CrossRef]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2008, 36, D281–D288. [Google Scholar] [CrossRef]
- Ried, G.; Hindennach, I.; Henning, U. Role of lipopolysaccharide in assembly of Escherichia coli outer membrane proteins OmpA, OmpC, and OmpF. J. Bacteriol. 1990, 172, 6048–6053. [Google Scholar]
- Sen, K.; Nikaido, H. In vitro trimerization of OmpF porin secreted by spheroplasts of Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 743–747. [Google Scholar] [CrossRef]
- Sen, K.; Nikaido, H. Lipopolysaccharide structure required for in vitro trimerization of Escherichia coli OmpF porin. J. Bacteriol. 1991, 173, 926–928. [Google Scholar]
- Schindler, H.; Rosenbusch, J.P. Matrix protein in planar membranes: clusters of channels in a native environment and their functional reassembly. Proc. Natl. Acad. Sci. USA 1981, 78, 2302–2306. [Google Scholar] [CrossRef]
- Ruiz, N.; Chng, S.S.; Hiniker, A.; Kahne, D.; Silhavy, T.J. Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc. Natl. Acad. Sci. USA 2010, 107, 12245–12250. [Google Scholar] [CrossRef]
- Driessen, A.J.; Nouwen, N. Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 2008, 77, 643–667. [Google Scholar] [CrossRef]
- Vertommen, D.; Ruiz, N.; Leverrier, P.; Silhavy, T.J.; Collet, J.F. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 2009, 9, 2432–2443. [Google Scholar] [CrossRef]
- Rizzitello, A.E.; Harper, J.R.; Silhavy, T.J. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 2001, 183, 6794–6800. [Google Scholar] [CrossRef]
- Schwalm, J.; Mahoney, T.F.; Soltes, G.R.; Silhavy, T.J. Role for Skp in LptD assembly in Escherichia coli. J. Bacteriol. 2013, 195, 3734–3742. [Google Scholar] [CrossRef]
- Harms, N.; Koningstein, G.; Dontje, W.; Muller, M.; Oudega, B.; Luirink, J.; de Cock, H. The early interaction of the outer membrane protein phoe with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J. Biol. Chem. 2001, 276, 18804–18811. [Google Scholar]
- Chimalakonda, G.; Ruiz, N.; Chng, S.S.; Garner, R.A.; Kahne, D.; Silhavy, T.J. Lipoprotein LptE is required for the assembly of LptD by the β-barrel assembly machine in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2011, 108, 2492–2497. [Google Scholar] [CrossRef]
- Kadokura, H.; Tian, H.; Zander, T.; Bardwell, J.C.; Beckwith, J. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 2004, 303, 534–537. [Google Scholar] [CrossRef]
- Chng, S.S.; Xue, M.; Garner, R.A.; Kadokura, H.; Boyd, D.; Beckwith, J.; Kahne, D. Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 2012, 337, 1665–1668. [Google Scholar] [CrossRef]
- Narita, S.; Masui, C.; Suzuki, T.; Dohmae, N.; Akiyama, Y. Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, E3612–E3621. [Google Scholar] [CrossRef]
- Grabowicz, M.; Yeh, J.; Silhavy, T.J. Dominant negative lptE mutation that supports a role for LptE as a plug in the LptD barrel. J. Bacteriol. 2013, 195, 1327–1334. [Google Scholar] [CrossRef]
- Lima, S.; Guo, M.S.; Chaba, R.; Gross, C.A.; Sauer, R.T. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science 2013, 340, 837–841. [Google Scholar] [CrossRef]
- Ades, S.E. Regulation by destruction: Design of the sigmaE envelope stress response. Curr. Opin. Microbiol. 2008, 11, 535–540. [Google Scholar] [CrossRef]
- Martorana, A.M.; Sperandeo, P.; Polissi, A.; Deho, G. Complex transcriptional organization regulates an Escherichia coli locus implicated in lipopolysaccharide biogenesis. Res. Microbiol. 2011, 162, 470–482. [Google Scholar] [CrossRef]
- Dartigalongue, C.; Missiakas, D.; Raina, S. Characterization of the Escherichia coli sigma E regulon. J. Biol. Chem. 2001, 276, 20866–20875. [Google Scholar] [CrossRef]
- Cezairliyan, B.O.; Sauer, R.T. Inhibition of regulated proteolysis by RseB. Proc. Natl. Acad. Sci. USA 2007, 104, 3771–3776. [Google Scholar] [CrossRef]
- Kim, D.Y.; Jin, K.S.; Kwon, E.; Ree, M.; Kim, K.K. Crystal structure of RseB and a model of its binding mode to RseA. Proc. Natl. Acad. Sci. USA 2007, 104, 8779–8784. [Google Scholar]
- Walsh, N.P.; Alba, B.M.; Bose, B.; Gross, C.A.; Sauer, R.T. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 2003, 113, 61–71. [Google Scholar] [CrossRef]
- Chaba, R.; Alba, B.M.; Guo, M.S.; Sohn, J.; Ahuja, N.; Sauer, R.T.; Gross, C.A. Signal integration by DegS and RseB governs the σE-mediated envelope stress response in Escherichia coli. Proc. Natl. Acad. Sci. USA 2011, 108, 2106–2111. [Google Scholar]
- Liechti, G.; Goldberg, J.B. Outer membrane biogenesis in Escherichia coli, Neisseria meningitidis, and Helicobacter pylori: paradigm deviations in H. pylori. Front. Cell. Infect. Microbiol. 2012, 2, 29. [Google Scholar] [CrossRef]
- Zhang, G.; Meredith, T.C.; Kahne, D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [Google Scholar] [CrossRef]
- Kobayashi, N.; Nishino, K.; Yamaguchi, A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J. Bacteriol. 2001, 183, 5639–5644. [Google Scholar] [CrossRef]
- Lu, S.; Zgurskaya, H.I. MacA, a periplasmic membrane fusion protein of the macrolide transporter MacAB-TolC, binds lipopolysaccharide core specifically and with high affinity. J. Bacteriol. 2013, 195, 4865–4872. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Polissi, A.; Sperandeo, P. The Lipopolysaccharide Export Pathway in Escherichia coli: Structure, Organization and Regulated Assembly of the Lpt Machinery. Mar. Drugs 2014, 12, 1023-1042. https://doi.org/10.3390/md12021023
Polissi A, Sperandeo P. The Lipopolysaccharide Export Pathway in Escherichia coli: Structure, Organization and Regulated Assembly of the Lpt Machinery. Marine Drugs. 2014; 12(2):1023-1042. https://doi.org/10.3390/md12021023
Chicago/Turabian StylePolissi, Alessandra, and Paola Sperandeo. 2014. "The Lipopolysaccharide Export Pathway in Escherichia coli: Structure, Organization and Regulated Assembly of the Lpt Machinery" Marine Drugs 12, no. 2: 1023-1042. https://doi.org/10.3390/md12021023
APA StylePolissi, A., & Sperandeo, P. (2014). The Lipopolysaccharide Export Pathway in Escherichia coli: Structure, Organization and Regulated Assembly of the Lpt Machinery. Marine Drugs, 12(2), 1023-1042. https://doi.org/10.3390/md12021023