The CHROMEVALOA Database: A Resource for the Evaluation of Okadaic Acid Contamination in the Marine Environment Based on the Chromatin-Associated Transcriptome of the Mussel Mytilus galloprovincialis


Abstract
:1. Introduction
2. Results and Discussion
2.1. Sequencing and Annotation of OA-Specific ESTs in M. galloprovincialis
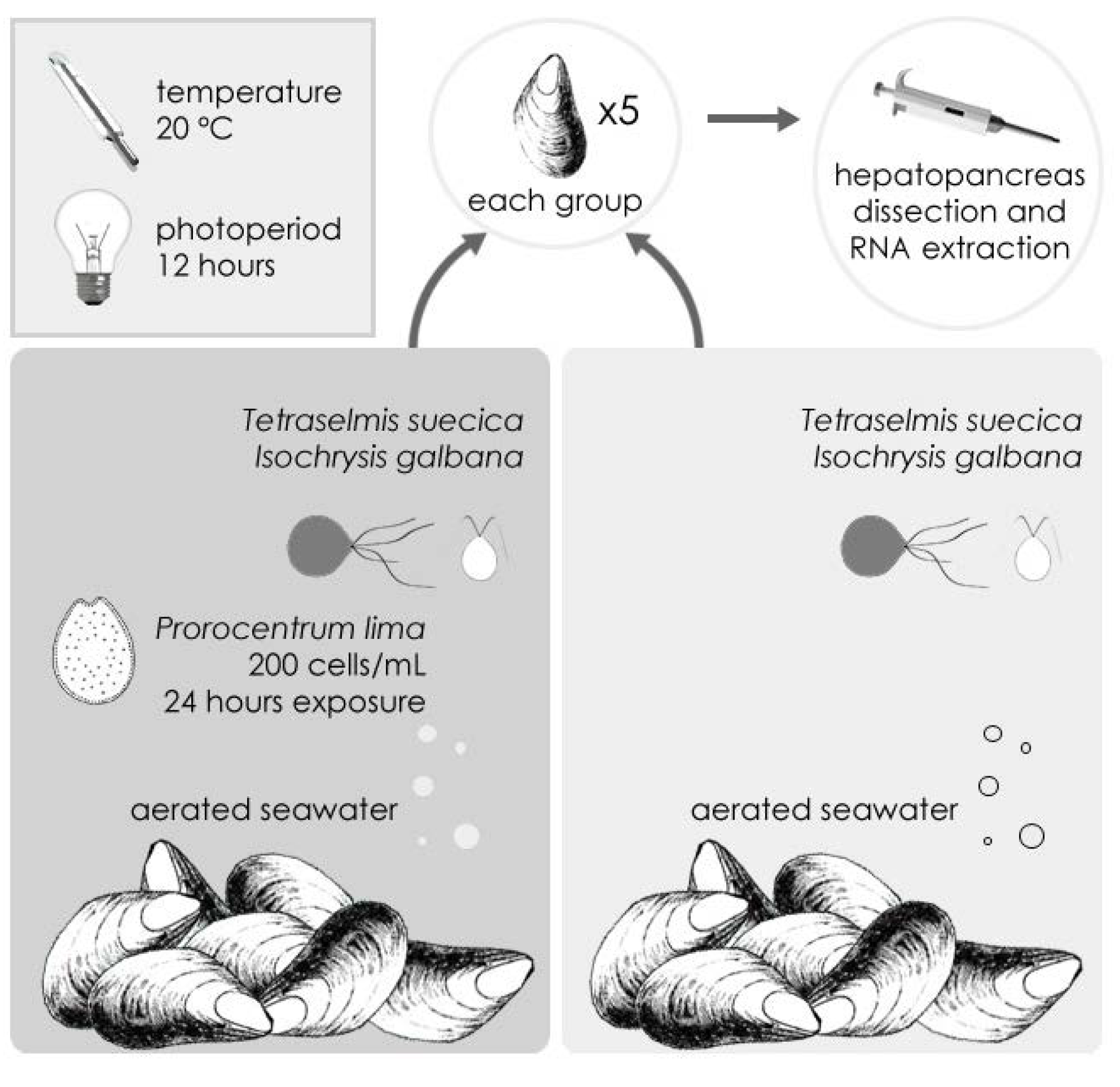

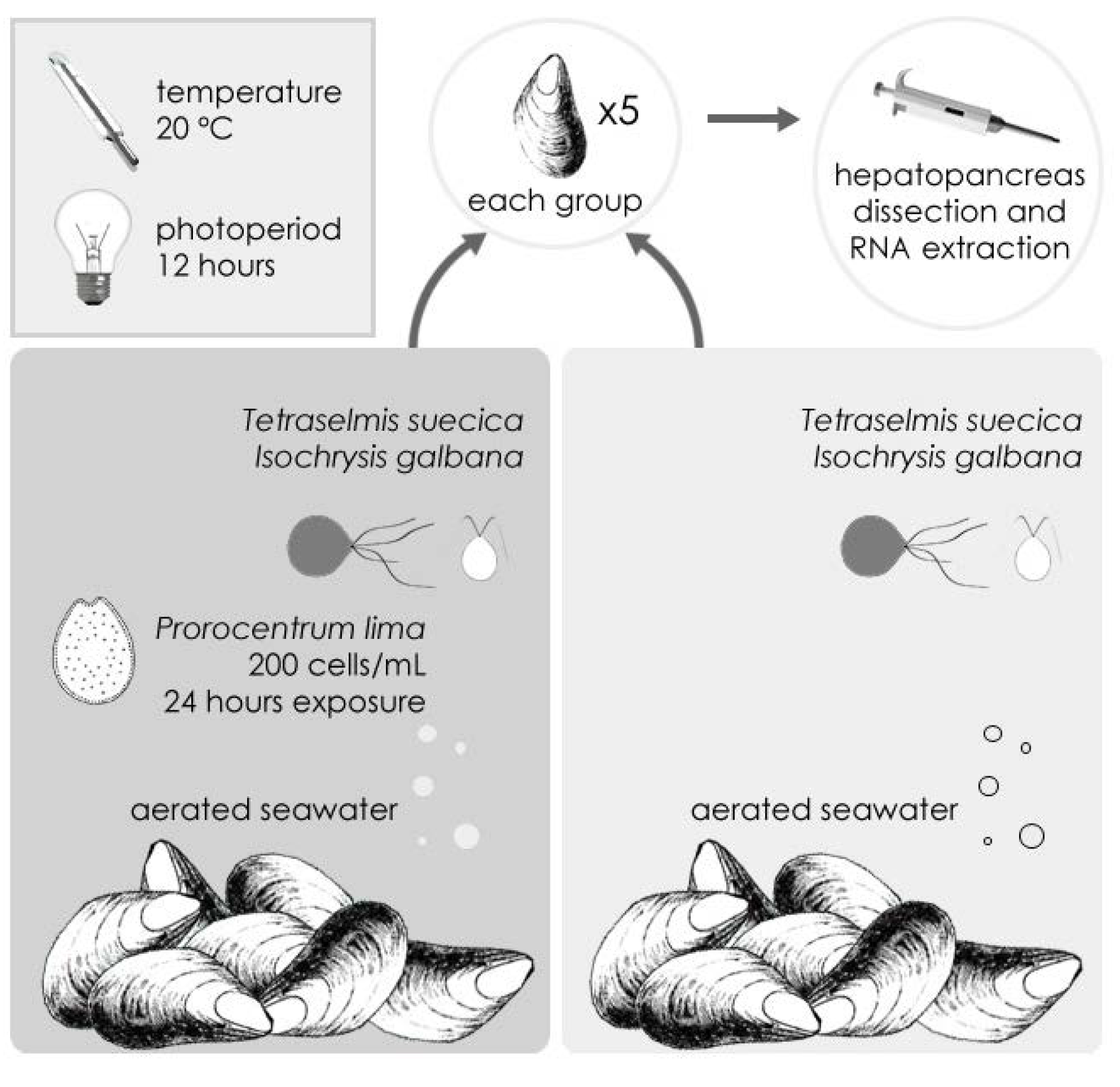{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental conditions | OA-content (ng/g) |
|---|---|
| Control | Below detection limit (~0) |
| OA-exposed | 18.27 |
| Library | Reads | Contigs | Annotated Sequences |
|---|---|---|---|
| NORM_MGC (control) | 493,440 | 16,395 | 7335 |
| NORM_MGT (OA-exposed) | 401,109 | 24,624 | 10,617 |
| Contigs | Unigenes | Differentially Expressed | |
| TOTAL | 41,019 | 2131 | 1254 |
| CHROMATIN-ASSOCIATED | 14,480 | 1124 | 90 |
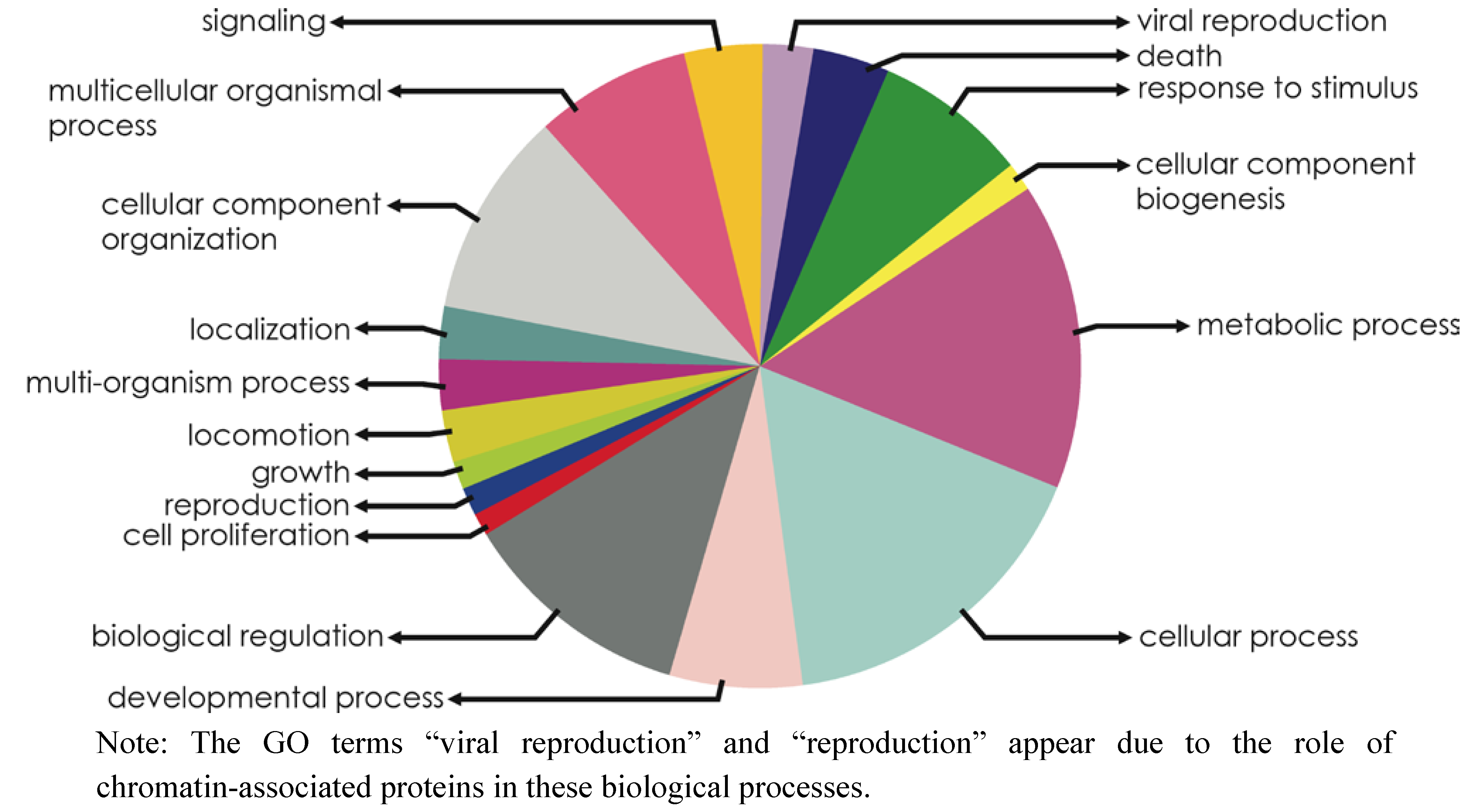
2.2. Novel Chromatin-Associated Transcripts in CHROMEVALOAdb

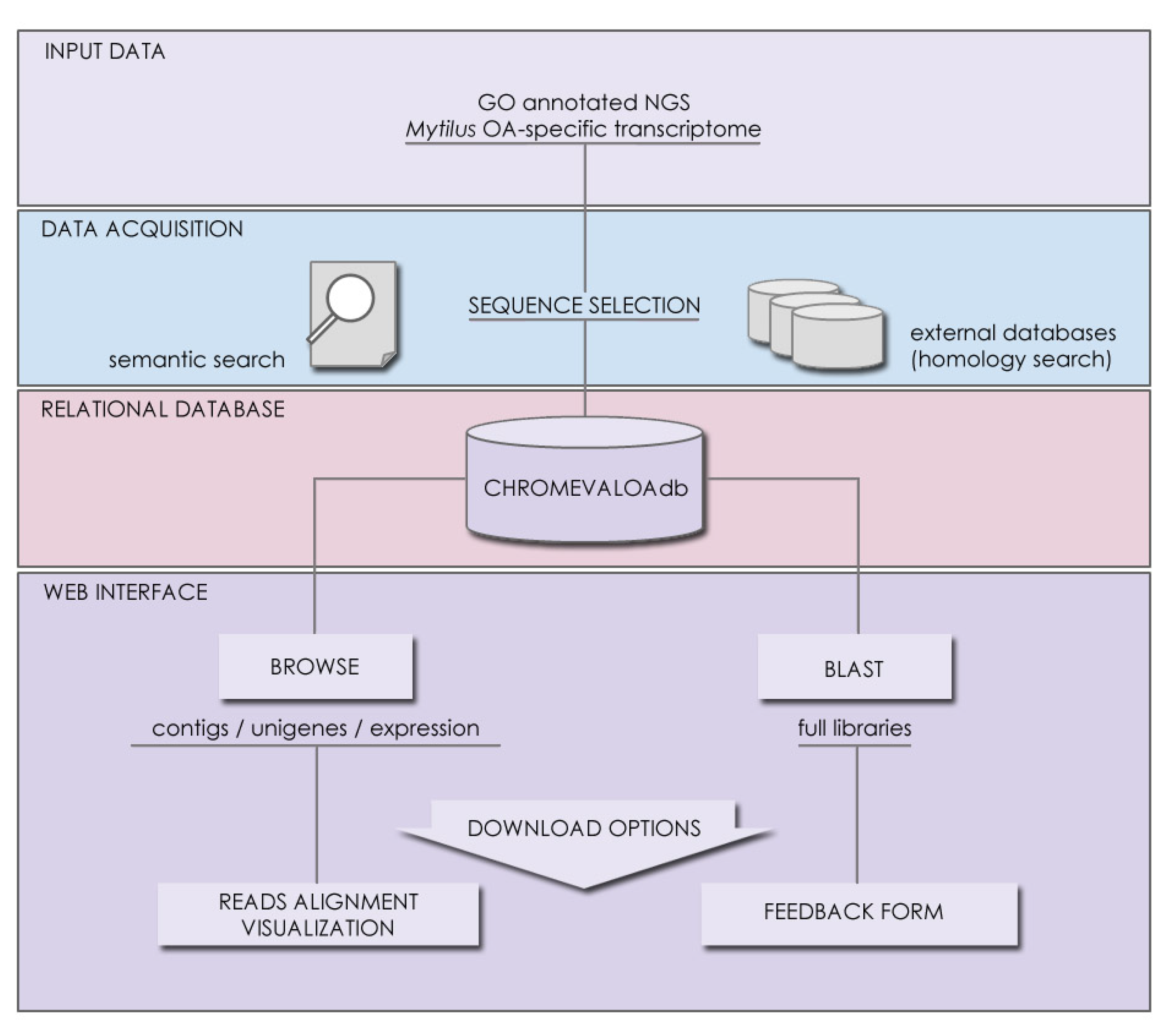
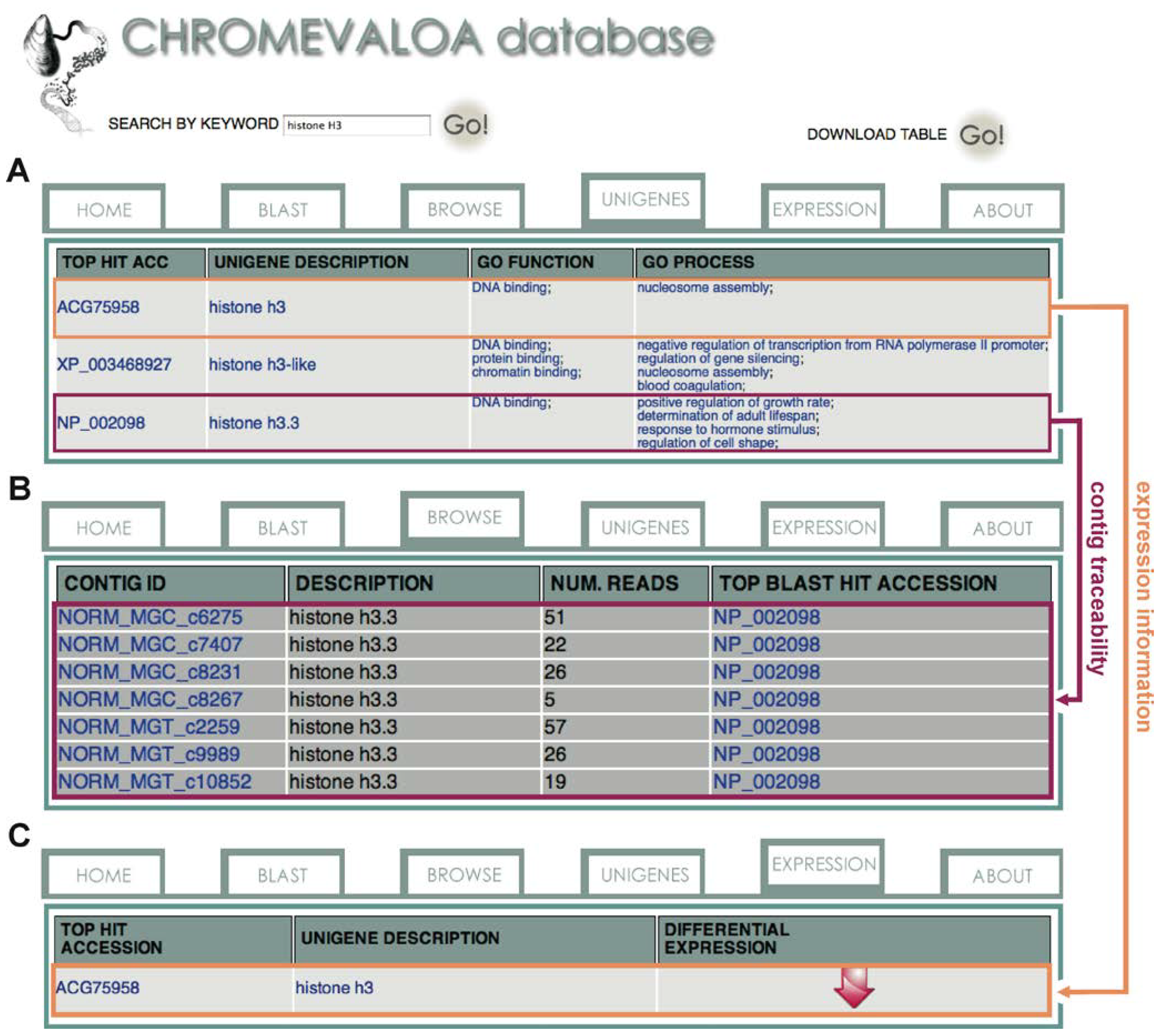
2.3. Availability, Management and Application of Data Stored in CHROMEVALOAdb
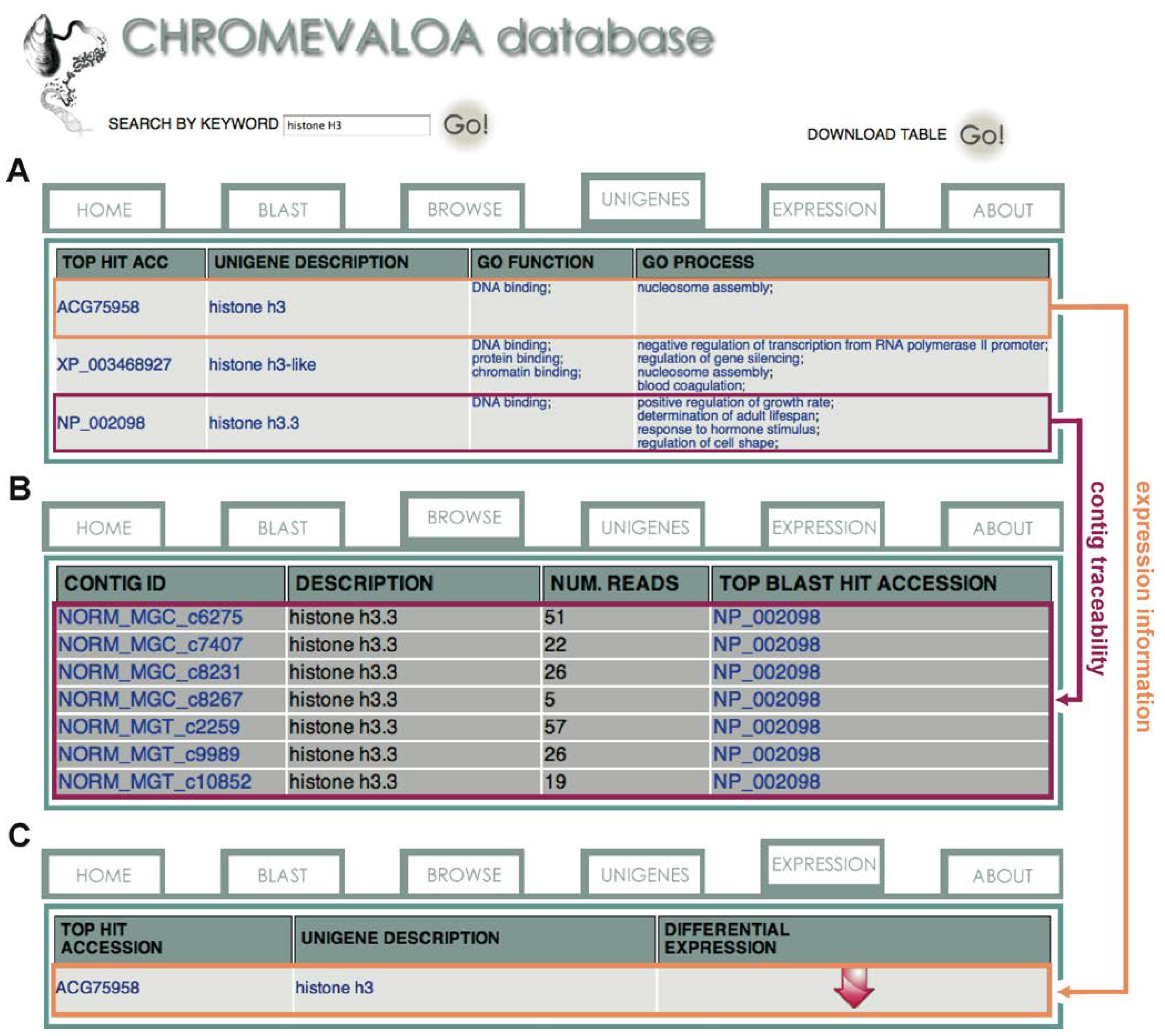
3. Experimental Section
3.1. Synthesis of ESTs Libraries and Transcriptome Assembly
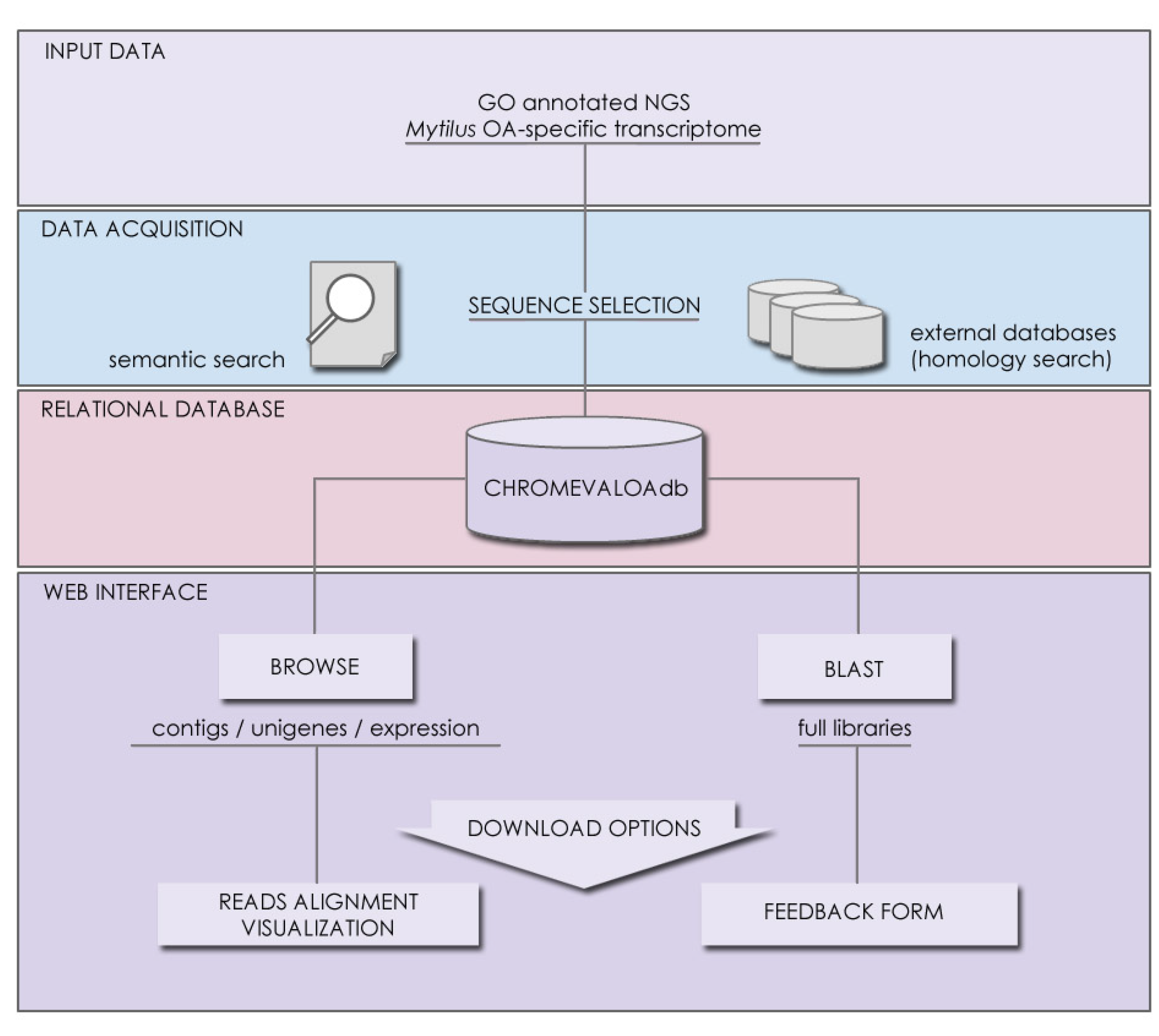
3.2. Database Contents, Accessibility and Tool Implementation
3.3. Gene Annotation and Expression Analysis
4. Conclusions
Acknowledgments
- Samples Availability: Available from the authors.
References
- Cardozo, K.H.; Guaratini, T.; Barros, M.P.; Falcao, V.R.; Tonon, A.P.; Lopes, N.P.; Campos, S.; Torres, M.A.; Souza, A.O.; Colepicolo, P.; et al. Metabolites from algae with economical impact. Comp. Biochem. Physiol. C 2007, 146, 60–78. [Google Scholar]
- Aune, T.; Yndestad, M. Diarrhetic Shellfish Poisoning. In Algal Toxins in Seafood and Drinking Water; Falconer, I.R., Ed.; Academic Press: London, UK, 1993; pp. 87–104. [Google Scholar]
- James, K.J.; Carey, B.; O’Halloran, J.; van Pelt, F.; Skrabakova, Z. Shellfish toxicity: Human health implications of marine algal toxins. Epidemiol. Infect. 2010, 138, 927–940. [Google Scholar] [CrossRef]
- Vale, P. Profiles of fatty acids and 7-O-acyl okadaic acid esters in bivalves: Can bacteria be involved in acyl esterification of okadaic acid? Comp. Biochem. Physiol. C 2010, 151, 18–24. [Google Scholar]
- Yasumoto, T.; Oshima, Y.; Sugawara, W.; Fukuyo, Y.; Oguri, H.; Igarashi, T.; Fujita, N. Identification of Dinophysis fortii as the causative organism of diarrhetic shellfish poisoning. Bull. Jpn. Soc. Sci. Fish. 1980, 46, 1405–1411. [Google Scholar]
- Leira, F.; Alvarez, C.; Vieites, J.M.; Vieytes, M.R.; Botana, L.M. Study of cytoskeletal changes induced by okadaic acid in BE(2)-M17 cells by means of a quantitative fluorimetric microplate assay. Toxicol. In Vitro 2001, 15, 277–282. [Google Scholar]
- Suganuma, M.; Fujiki, H.; Suguri, H.; Yoshizawa, S.; Hirota, M.; Nakayasu, M.; Ojika, M.; Wakamatsu, K.; Yamada, K.; Sugimura, T. Okadaic acid: An additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc. Natl. Acad. Sci. USA 1988, 85, 1768–1771. [Google Scholar]
- Valdiglesias, V.; Laffon, B.; Pasaro, E.; Mendez, J. Evaluation of okadaic acid-induced genotoxicity in human cells using the micronucleus test and gammaH2AX analysis. J. Toxicol. Environ. Health A 2012, 74, 980–992. [Google Scholar] [CrossRef]
- Florez-Barros, F.; Prado-Alvarez, M.; Mendez, J.; Fernandez-Tajes, J. Evaluation of genotoxicity in gills and hemolymph of clam Ruditapes decussatus fed with the toxic dinoflagellate Prorocentrum lima. J. Toxicol. Environ. Health A 2011, 74, 971–979. [Google Scholar] [CrossRef]
- Van Dolah, F.M.; Ramsdell, J.S. Okadaic acid inhibits a protein phosphatase activity involved in formation of the mitotic spindle of GH4 rat pituitary cells. J. Cell. Physiol. 1992, 151, 190–198. [Google Scholar] [CrossRef]
- Wells, P.G.; Depledge, M.H.; Butler, J.N.; Manock, J.J.; Knap, A.H. Rapid toxicity assessment and biomonitoring of marine contaminants—Exploiting the potential of rapid biomarker assays and microscale toxicity tests. Mar. Pollut. Bull. 2001, 42, 799–804. [Google Scholar]
- Sassolas, A.; Hayat, A.; Catanante, G.; Marty, J.-L. Detection of the marine toxin okadaic acid: Assessing seafood safety. Talanta 2012, in press. [Google Scholar]
- Marcaillou-Le Baut, C.; Amzil, Z.; Vernoux, J.P.; Pouchus, Y.F.; Bohec, M.; Simon, J.F. Studies on the detection of okadaic acid in mussels: Preliminary comparison of bioassays. Nat. Toxins 1994, 2, 312–317. [Google Scholar] [CrossRef]
- Ledreux, A.; Serandour, A.L.; Morin, B.; Derick, S.; Lanceleur, R.; Hamlaoui, S.; Furger, C.; Bire, R.; Krys, S.; Fessard, V.; et al. Collaborative study for the detection of toxic compounds in shellfish extracts using cell-based assays. Part II: Application to shellfish extracts spiked with lipophilic marine toxins. Anal. Bioanal. Chem. 2012, 403, 1995–2007. [Google Scholar]
- Manfrin, C.; Dreos, R.; Battistella, S.; Beran, A.; Gerdol, M.; Varotto, L.; Lanfranchi, G.; Venier, P.; Pallavicini, A. Mediterranean mussel gene expression profile induced by okadaic acid exposure. Environ. Sci. Technol. 2010, 44, 8276–8283. [Google Scholar] [CrossRef]
- Dinant, C.; Houtsmuller, A.B.; Vermeulen, W. Chromatin structure and DNA damage repair. Epigenetics Chromatin 2008, 1, 9. [Google Scholar] [CrossRef]
- Dickey, J.S.; Redon, C.E.; Nakamura, A.J.; Baird, B.J.; Sedelnikova, O.A.; Bonner, W. H2AX: Functional roles and potential applications. Chromosoma 2009, 118, 683–695. [Google Scholar] [CrossRef]
- Watters, G.P.; Smart, D.J.; Harvey, J.S.; Austin, C.A. H2AX phosphorylation as a genotoxicity endpoint. Mutat. Res. 2009, 679, 50–58. [Google Scholar] [CrossRef]
- Albino, A.P.; Jorgensen, E.D.; Rainey, P.; Gillman, G.; Clark, T.J.; Zhao, H.; Traganos, F.; Darzynkiewicz, Z. Gama-H2AX: A potential DNA damage response biomarker for assessing toxicological risk of tobacco products. Mutat. Res. 2009, 678, 43–52. [Google Scholar] [CrossRef]
- Gonzalez-Romero, R.; Rivera-Casas, C.; Fernandez-Tajes, J.; Ausio, J.; Méndez, J.; Eirín-López, J.M. Chromatin specialization in bivalve molluscs: A leap forward for the evaluation of okadaic acid genotoxicity in the marine environment. Comp. Biochem. Physiol. C 2012, 155, 175–181. [Google Scholar]
- Eirín-López, J.M.; Lewis, J.D.; Howe, L.; Ausió, J. Common phylogenetic origin of protamine-like (PL) proteins and histone H1: Evidence from bivalve PL genes. Mol. Biol. Evol. 2006, 23, 1304–1317. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; Ruiz, M.F.; González-Tizón, A.M.; Martínez, A.; Sánchez, L.; Méndez, J. Molecular evolutionary characterization of the mussel Mytilus histone multigene family: First record of a tandemly repeated unit of five histone genes containing an H1 subtype with “orphon” features. J. Mol. Evol. 2004, 58, 131–144. [Google Scholar] [CrossRef]
- González-Romero, R.; Rivera-Casas, C.; Frehlick, L.J.; Méndez, J.; Ausió, J.; Eirín-López, J.M. Histone H2A (H2A.X and H2A.Z) variants in molluscs: Molecular characterization and potential implications for chromatin dynamics. PLoS One 2012, 7, e30006. [Google Scholar]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar]
- CHROMEVALOAdb. Available online: http://chromevaloa.udc.es (accessed on 1 September 2012).
- Pinto-Silva, C.R.; Creppy, E.E.; Matias, W.G. Micronucleus test in mussels Perna perna fed with the toxic dinoflagellate Prorocentrum lima. Arch. Toxicol. 2005, 79, 422–426. [Google Scholar]
- Venier, P.; de Pitta, C.; Bernante, F.; Varotto, L.; de Nardi, B.; Bovo, G.; Roch, P.; Novoa, B.; Figueras, A.; Pallavicini, A.; et al. MytiBase: A knowledgebase of mussel (M. galloprovincialis) transcribed sequences. BMC Genomics 2009, 10, 72. [Google Scholar]
- Svensson, S. Effects, dynamics and management of okadaic acid in blue mussels, Mytilus edulis. Ph.D Thesis, Göteborg University, Strömstad, Sweden, 2003. [Google Scholar]
- Niu, B.; Fu, L.; Sun, S.; Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinforma. 2010, 11, 187. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- SeqtrimNext. Available online: http://www.scbi.uma.es/seqtrimnext (accessed on 5 June 2012).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, pp. 10–12. Available online: http://journal.embnet.org/index.php/embnetjournal/article/view/200 (accessed on 21 May 2012).
- Chevreux, B.; Wetter, T.; Suhai, S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. In Proceedings of the German Conference on Bioinformatics (GCB); Computer Science and Biology: Hannover, Germany, 1999; pp. 45–56. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position specific gap penalties and weight matrix choice. Nucl. Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Clamp, M.; Cuff, J.; Searle, S.M.; Barton, G.J. The Jalview Java alignment editor. Bioinformatics 2004, 20, 426–427. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucl. Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Myhre, S.; Tveit, H.; Mollestad, T.; Laegreid, A. Additional gene ontology structure for improved biological reasoning. Bioinformatics 2006, 22, 2020–2027. [Google Scholar]
- Lara, A.J.; Perez-Trabado, G.; Villalobos, D.P.; Diaz-Moreno, S.; Canton, F.R.; Claros, M.G. A Web Tool to Discover Full-Length Sequences: Full-Lengther. In Innovations in Hibrid Intelligent Systems; Corchado, E., Corchado, J.M., Abraham, A., Eds.; Springer: Berlin, Germany, 2007. [Google Scholar]
- Marino-Ramirez, L.; Levine, K.M.; Morales, M.; Zhang, S.; Moreland, R.T.; Baxevanis, A.D.; Landsman, D. The Histone Database: An integrated resource for histones and histone fold-containing proteins. Database (Oxford) 2011, 2011, bar048. [Google Scholar]
- Gendler, K.; Paulsen, T.; Napoli, C. ChromDB: The chromatin database. Nucl. Acids Res. 2008, 36, D298–D302. [Google Scholar] [CrossRef]
- Shipra, A.; Chetan, K.; Rao, M.R. CREMOFAC-A database of chromatin remodeling factors. Bioinformatics 2006, 22, 2940–2944. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suárez-Ulloa, V.; Fernández-Tajes, J.; Aguiar-Pulido, V.; Rivera-Casas, C.; González-Romero, R.; Ausio, J.; Méndez, J.; Dorado, J.; Eirín-López, J.M. The CHROMEVALOA Database: A Resource for the Evaluation of Okadaic Acid Contamination in the Marine Environment Based on the Chromatin-Associated Transcriptome of the Mussel Mytilus galloprovincialis. Mar. Drugs 2013, 11, 830-841. https://doi.org/10.3390/md11030830
Suárez-Ulloa V, Fernández-Tajes J, Aguiar-Pulido V, Rivera-Casas C, González-Romero R, Ausio J, Méndez J, Dorado J, Eirín-López JM. The CHROMEVALOA Database: A Resource for the Evaluation of Okadaic Acid Contamination in the Marine Environment Based on the Chromatin-Associated Transcriptome of the Mussel Mytilus galloprovincialis. Marine Drugs. 2013; 11(3):830-841. https://doi.org/10.3390/md11030830
Chicago/Turabian StyleSuárez-Ulloa, Victoria, Juan Fernández-Tajes, Vanessa Aguiar-Pulido, Ciro Rivera-Casas, Rodrigo González-Romero, Juan Ausio, Josefina Méndez, Julián Dorado, and José M. Eirín-López. 2013. "The CHROMEVALOA Database: A Resource for the Evaluation of Okadaic Acid Contamination in the Marine Environment Based on the Chromatin-Associated Transcriptome of the Mussel Mytilus galloprovincialis" Marine Drugs 11, no. 3: 830-841. https://doi.org/10.3390/md11030830