Helicusin E, Isochromophilone X and Isochromophilone XI: New Chloroazaphilones Produced by the Fungus Bartalinia robillardoides Strain LF550



Abstract
:1. Introduction
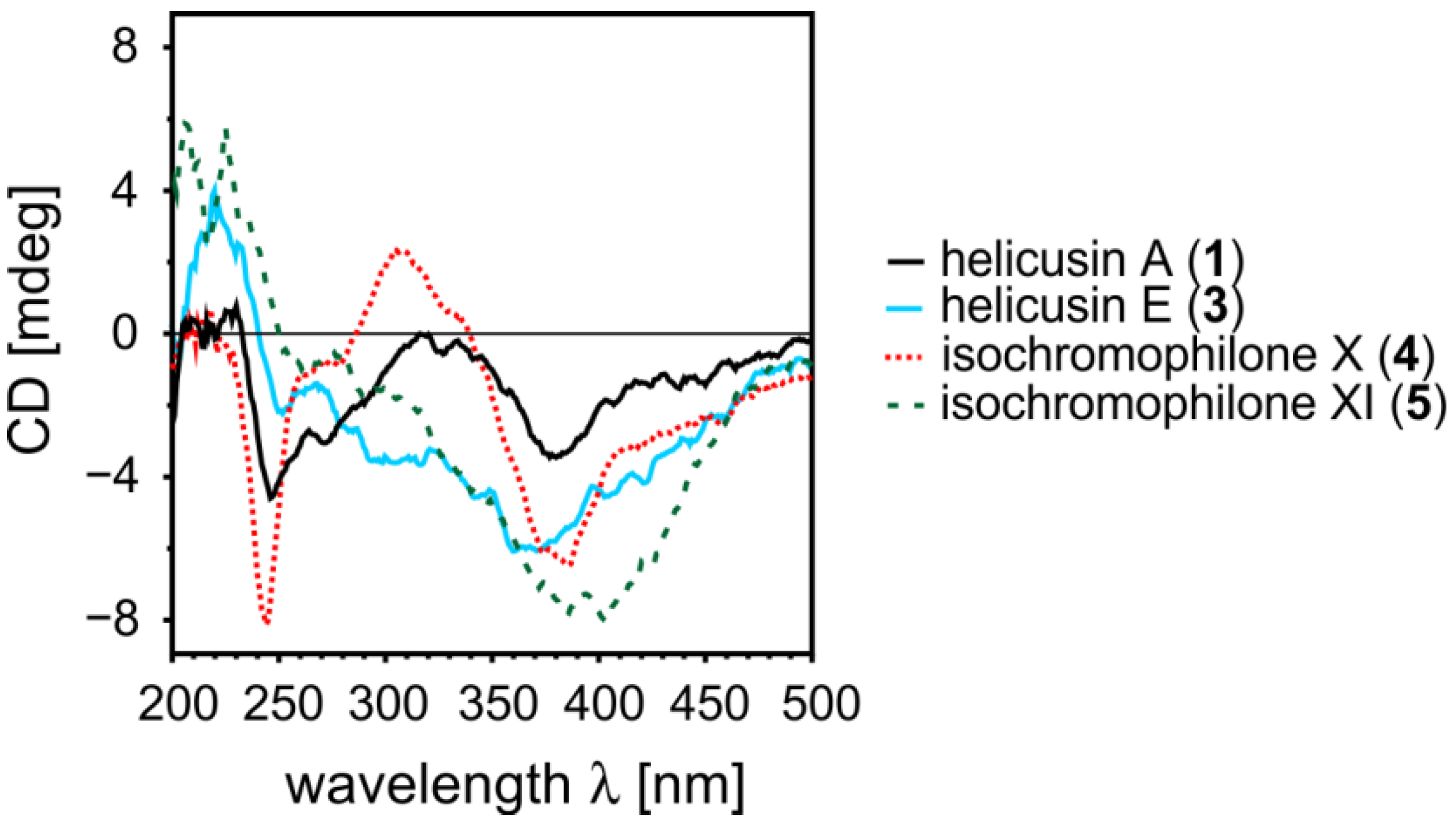
2. Results and Discussion
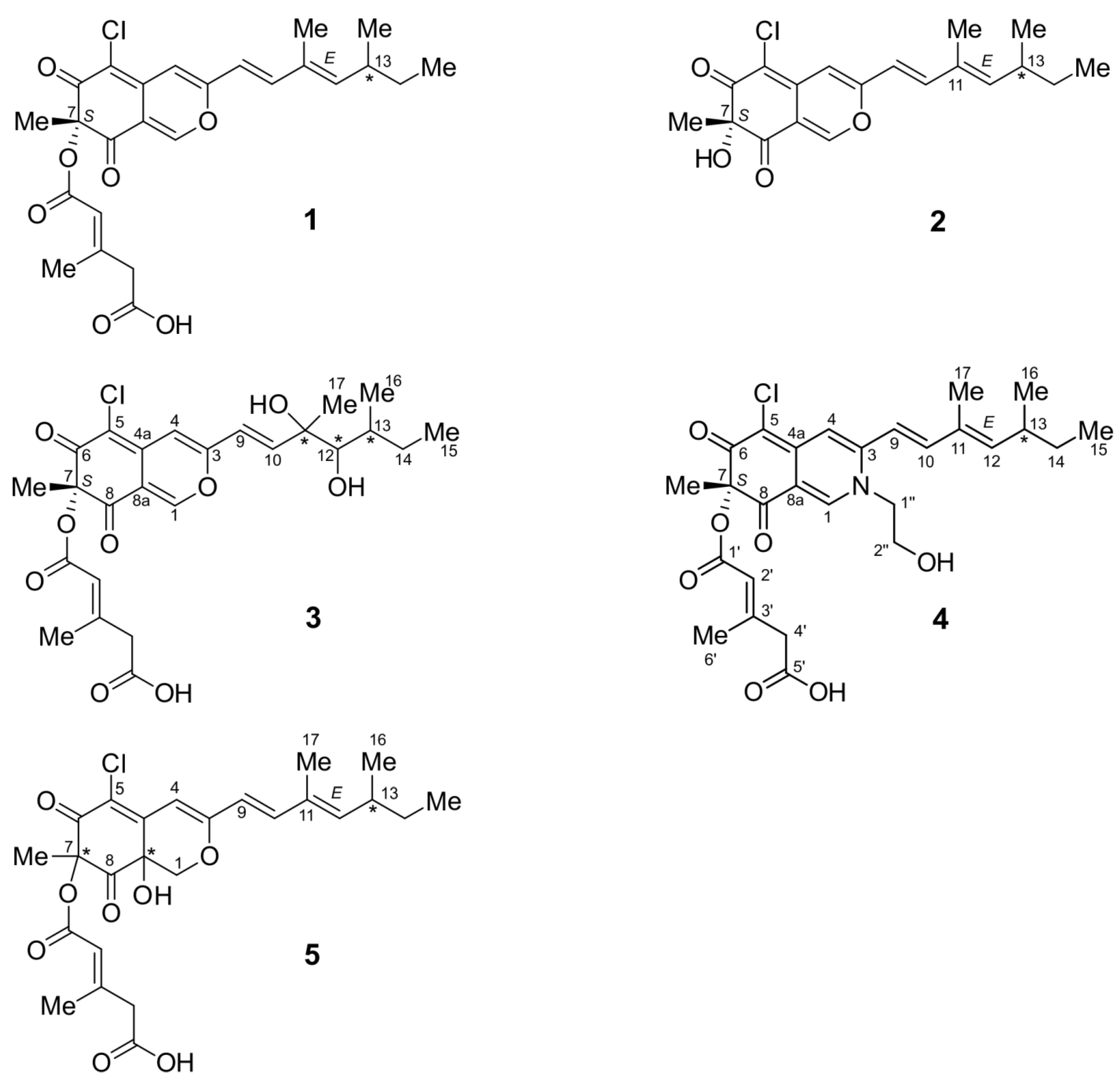
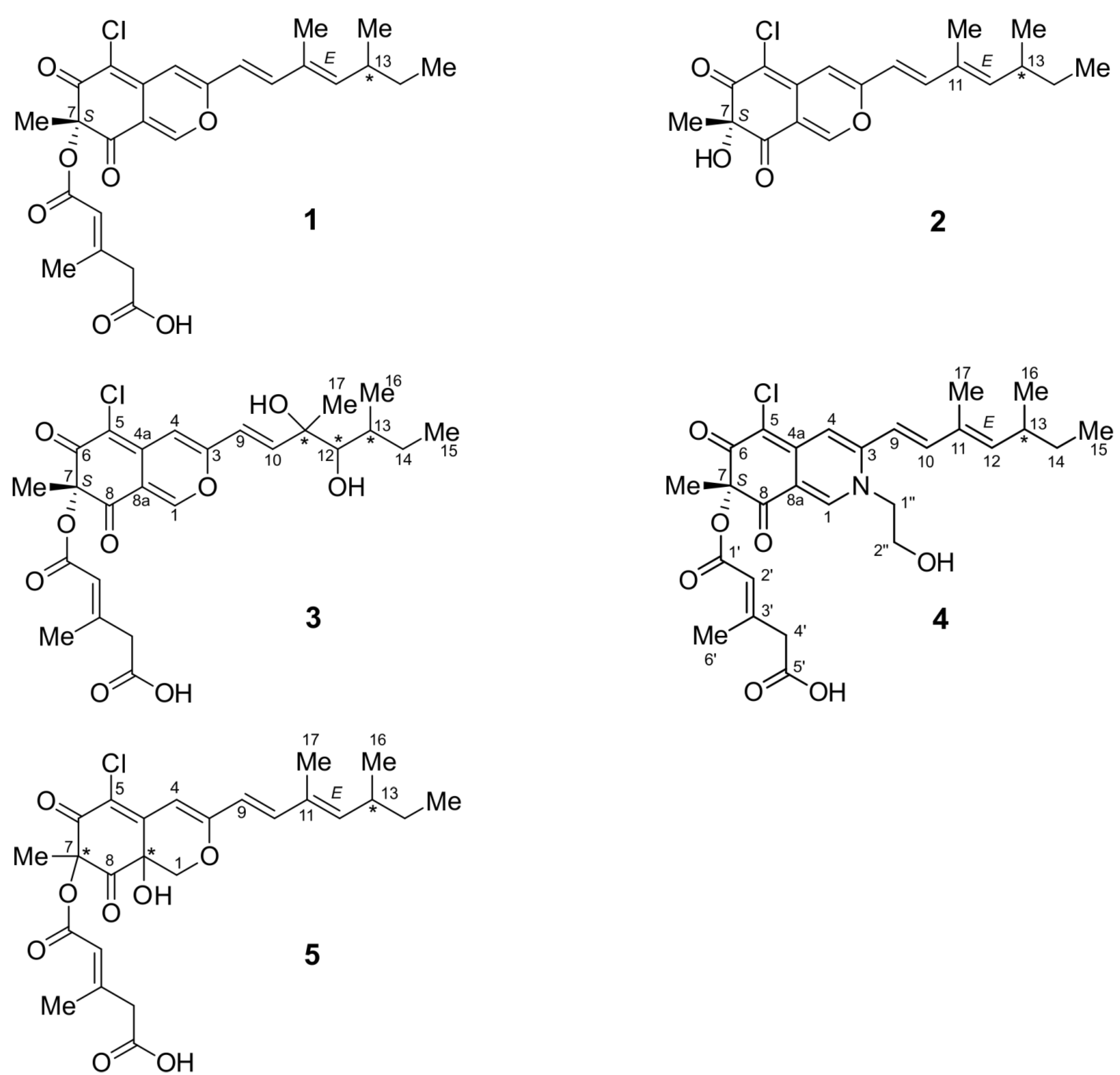
2.1. Structure Elucidation
2.1.1. Isochromophilone X (4)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, type | δH (J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 144.5, CH | 8.17, s | 3, 5, 4a, 8, 8a, 1″ | 1″ | |
| 3 | 151.9, C | ||||
| 4 | 112.6, CH | 7.20, s | 3, 5, 8, 8a, (9) | ||
| 4a | 148.3, C | ||||
| 5 | 116.4, C | ||||
| 6 | 185.7, C | ||||
| 7 | 85.7, C | ||||
| 8 | 195.2, C | ||||
| 8a | 101.4, C | ||||
| 9 | 117.3, CH | 6.58, d (15.5) | 10 | 4, 10, 11, 12 | 17, 1″ |
| 10 | 146.4, CH | 7.11, d (15.5) | 9 | 3, 9, 11, 12, 17 | 12 |
| 11 | 133.9, C | ||||
| 12 | 148.7, CH | 5.78, d (9.6) | 13, (17) | 10, 13, 14, 17, 16 | 10, 14b, 16 |
| 13 | 36.2, CH | 2.54, m | 12, 14, 16 | 11, 12, 14, 16, 15 | 14a, 16, 17 |
| 14a | 31.2, CH2 | 1.47, m | 13, 15 | 12, 13, 15, 16 | 13 |
| 14b | 1.36, m | 13, 15 | 12, 13, 15, 16 | 12 | |
| 15 | 12.4, CH3 | 0.90, t (7.4) | 14 | 13, 14 | |
| 16 | 20.6, CH3 | 1.04, d (6.7) | 13 | 12, 13, 14 | 12, 13 |
| 17 | 12.7, CH3 | 1.92, d (1.0) | (12) | 10, 11, 12, (14), (16) | 9, 13 |
| 18 | 23.7, CH3 | 1.52, s | 6, 7, 8 | ||
| 1′ | 166.3, C | ||||
| 2′ | 118.7, CH | 5.97, d (1.1) | (6′) | 1′, 3′, 4′, 6′ | 4′ |
| 3′ | 156.1, C | ||||
| 4′ | 46.6, CH2 | 3.21, s | 2′, 5′, 6′ | 2′, 6′ | |
| 5′ | 173.4, C | ||||
| 6′ | 19.3, CH3 | 2.15, d (1.2) | (2′) | 1′, 2′, 3′, 4′, 5′ | 4′ |
| 1″ | 57.4, CH2 | 4.24, d (4.4) | 2″ | 1, 3, 2″ | 1, 9 |
| 2″ | 61.1, CH2 | 3.85, t (5.2) | 1″ | 1″ |
2.1.2. Isochromophilone XI (5)
2.1.3. Helicusin E (3)
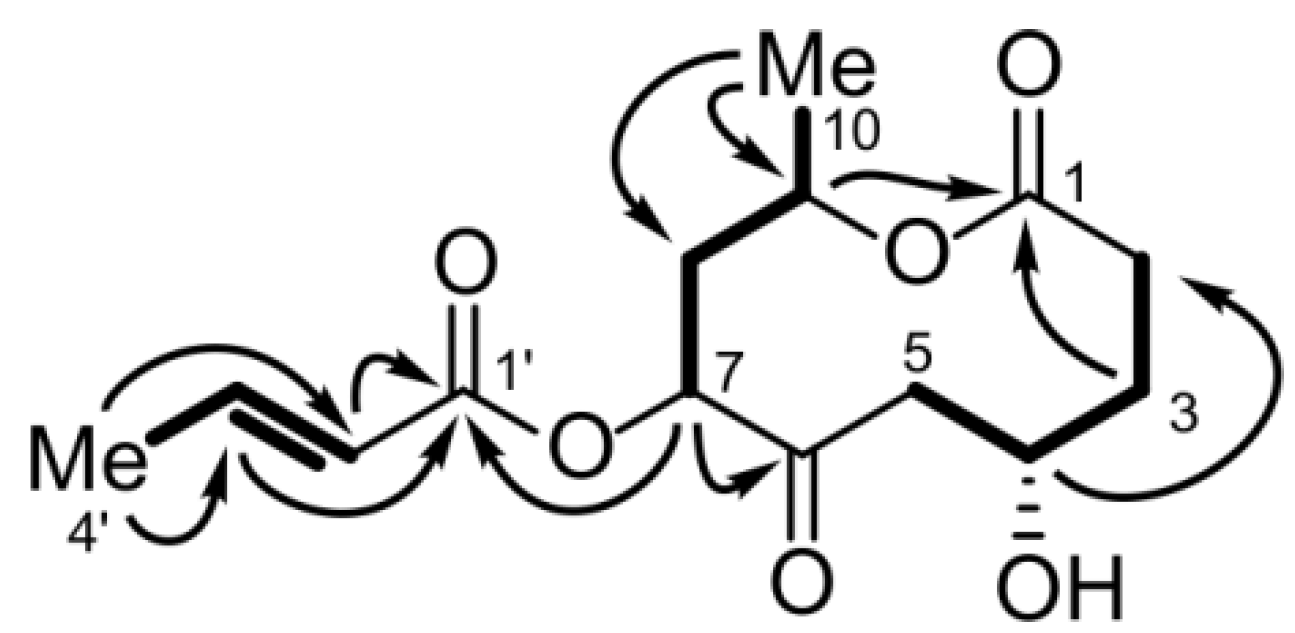
2.1.4. Bartanolide (6)
| Position | δC, type | δH (J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 173.5, C | ||||
| 2a | 28.5, CH2 | 2.58, ddd (18.2, 6.3, 2.3) | 3 | 1, 3, 4 | 2b, 3b, 3a, 5a |
| 2b | 2.38, ddd (18.2, 12.8, 1.8) | 2a, 3b, 3a | |||
| 3a | 27.8, CH2 | 2.20, dddd (15.5, 12.8, 2.3, 2.3) | 2, 4 | 1, 2, 5 | 2a, 2b, 3b, 4 |
| 3b | 1.68, dddd (15.5, 6.3, 4.5, 1.8) | 3a, 4 | |||
| 4 | 66.2, CH | 4.38, dddd (11.5, 4.5, 4.5, 2.3) | 3, 5 | 2, 5 | 3a, 3b, 5a, 5b |
| 5a | 42.8, CH2 | 2.82, dd (18.3, 11.5) | 4 | 3, 4, 6 | 2a, 4, 5b, 9 |
| 5b | 2.28, dd (18.3, 4.5) | 5a, 4 | |||
| 6 | 209.4, C | ||||
| 7 | 77.0, CH | 5.02, dd (6.0, 1.8) | 8 | 6, 8, 9, 1′ | 8a, 8b |
| 8a | 40.5, CH2 | 2.35, ddd (14.6, 12.5, 1.8) | 7, 9 | 6 | 7, 8b, 9, 10 |
| 8b | 2.18, ddd (14.6, 6.0, 2.4) | 7, 9 | |||
| 9 | 67.9, CH | 5.28, ddq (12.5, 6.5, 2.4) | 8, 10 | 1, 7, 8, 10 | 3′, 5a, 8a, 8b, 10 |
| 10 | 20.1, CH3 | 1.24, d (6.5) | 9 | 8, 9 | 8a, 8b, 9 |
| 1′ | 167.0, C | ||||
| 2′ | 122.7, CH | 6.00, dq (15.6, 1.8) | 3′, 4′ | 1′, 4′ | 3′, 4′, 5a |
| 3′ | 148.5, CH | 7.15, dq (15.6, 7.0) | 2′, 4′ | 1′, 2′, 4′ | 5a, 9, 2′, 4′ |
| 4′ | 18.3, CH3 | 1.96, dd (1.8, 7.0) | 2′, 3′ | 2′, 3′ | 2′, 3′ |
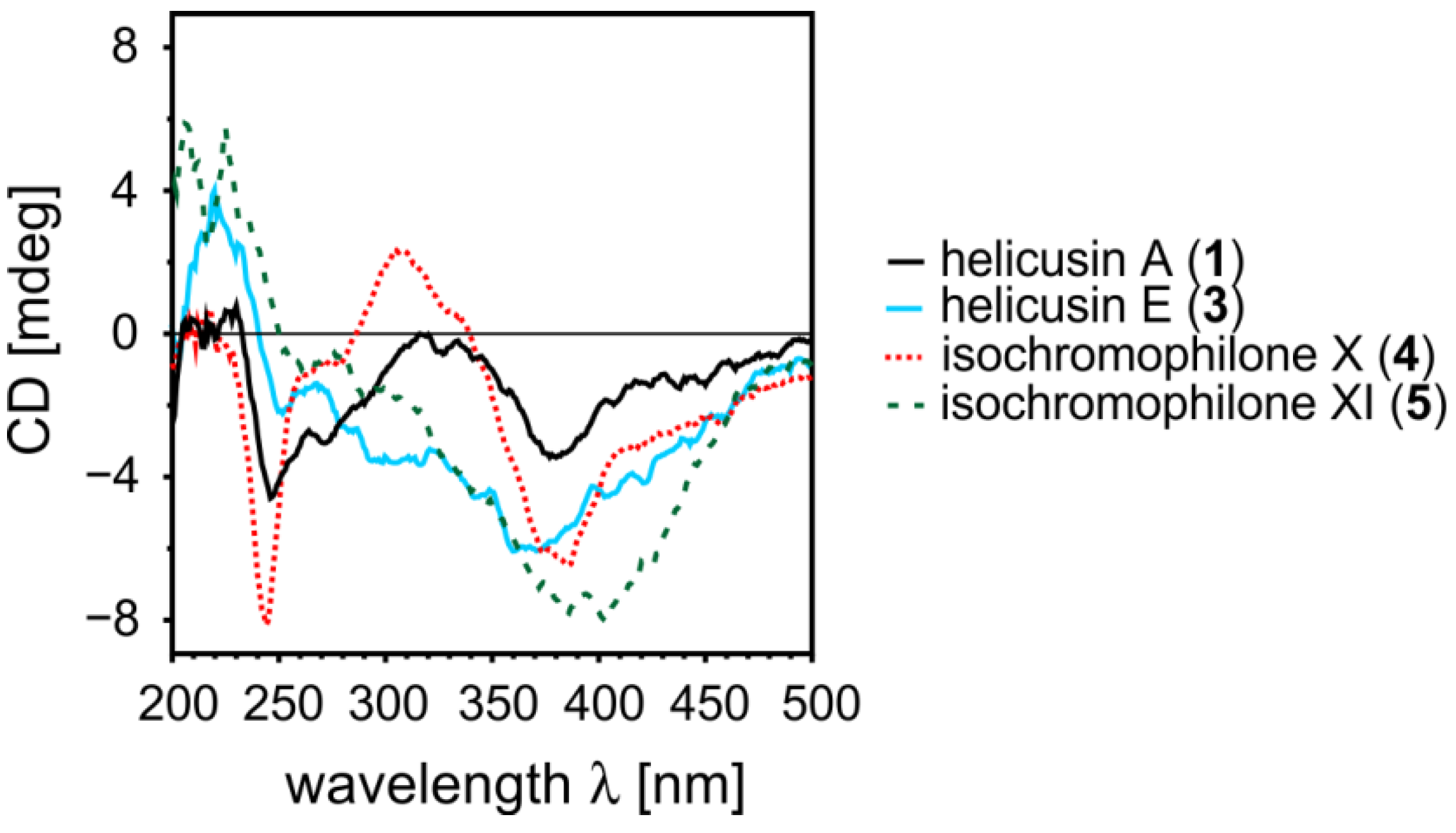
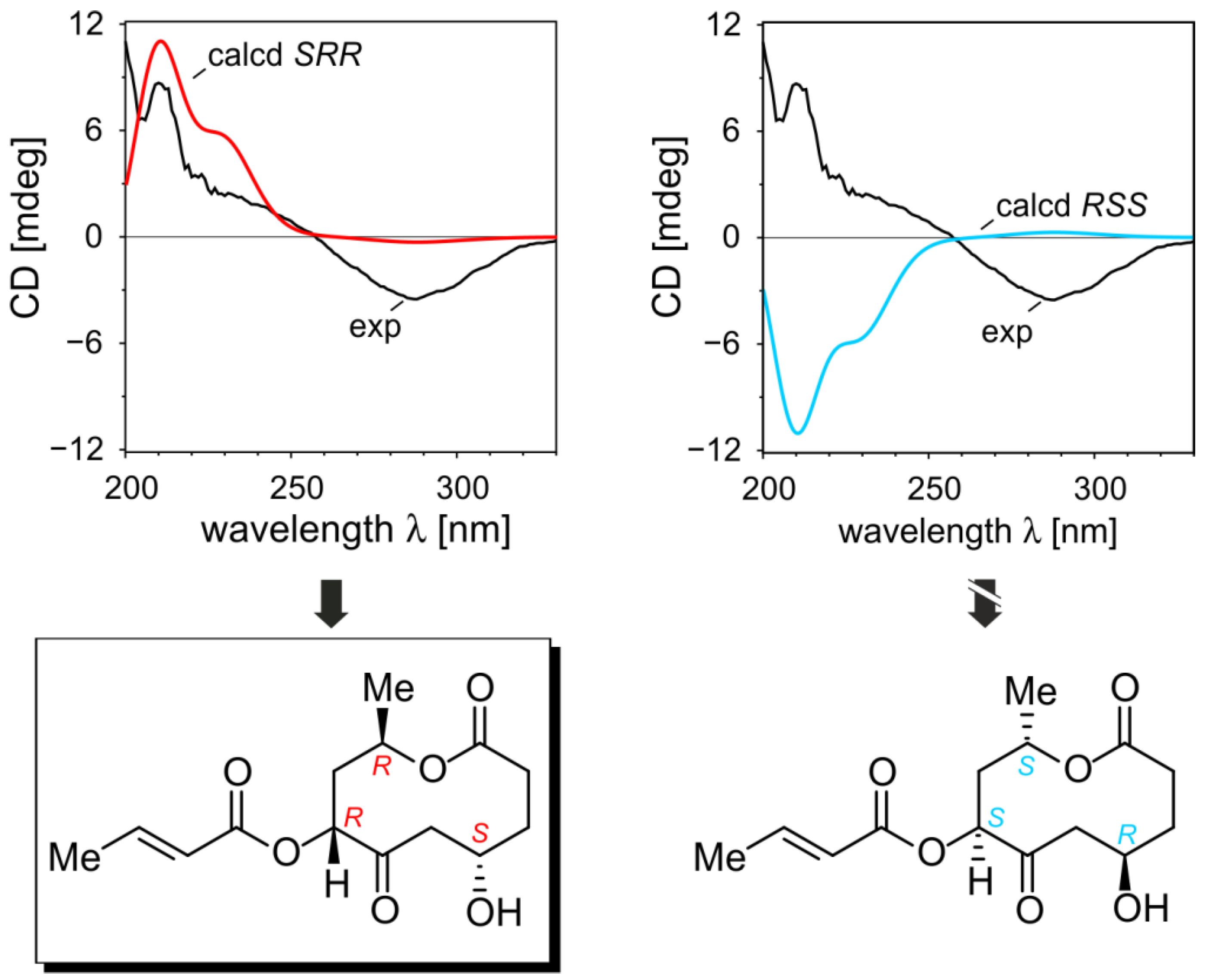
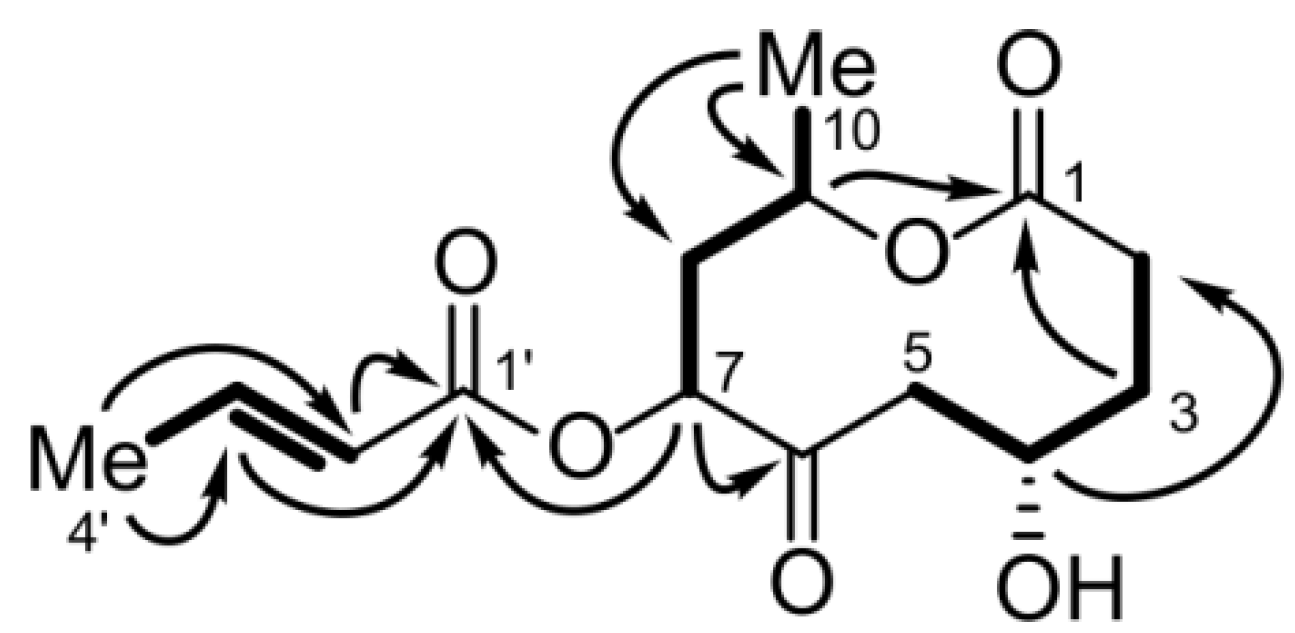
2.1.5. Elucidation of the Absolute Configurations of 1–6
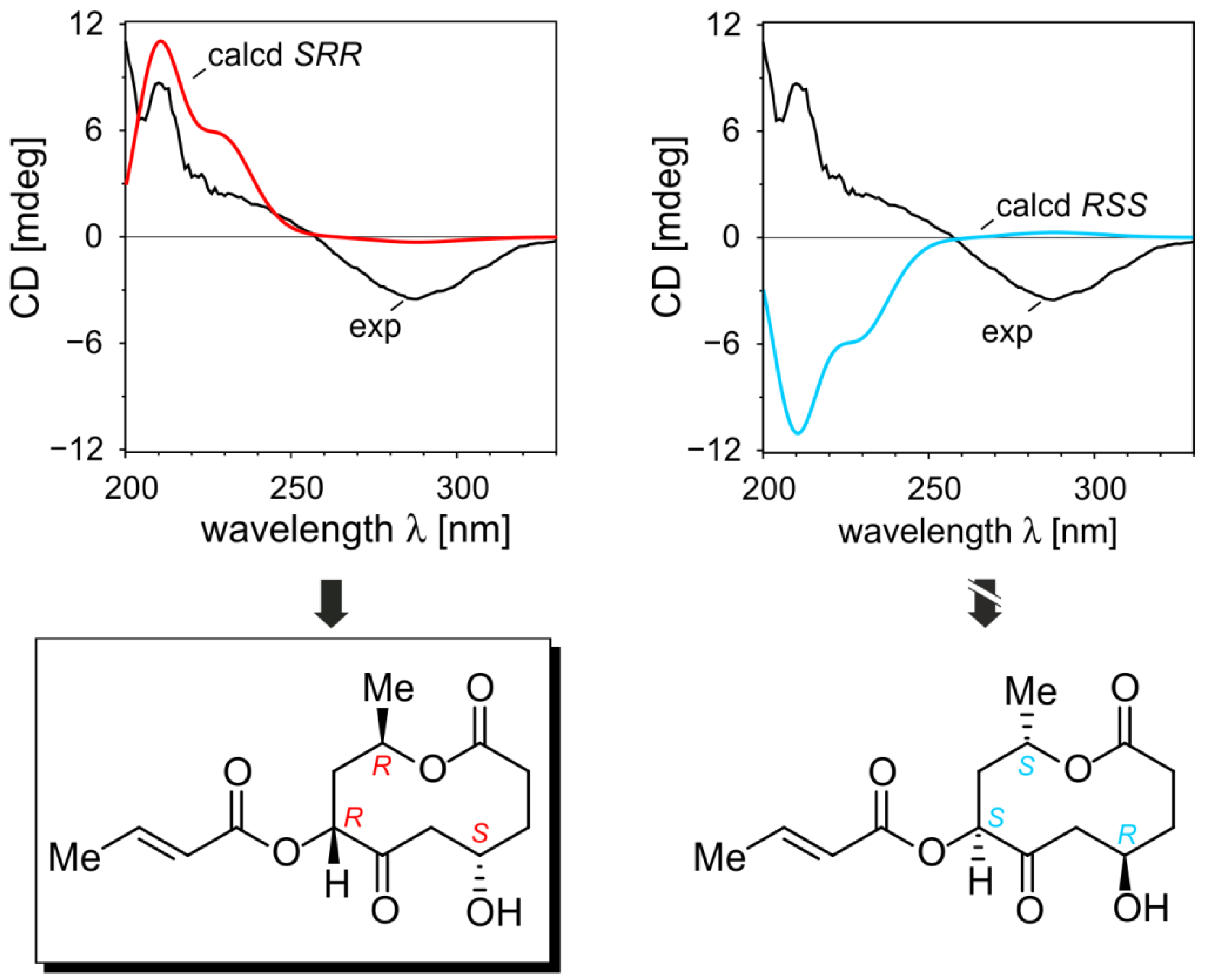
2.2. Inhibitory Activities of the Compounds
| Antibacterial | Antifungal | Enzyme Assays | |||||
|---|---|---|---|---|---|---|---|
| Bacillus subtilis | Staphylococcus lentus | Candida albicans | Septoria tritici | Trichophyton rubrum | Phosphodiesterase 4 | Acetylcholinesterase | |
| helicusin A (1) | >100 μM | >100 μM | 24.4 μM | 7.6 μM (±2.20) | 7.23 μM (±1.20) | >10 μM | 2.10 μM (±0.36) |
| deacetylsclerotiorin (2) | 38.8 μM | 43.6 μM | 24.0 μM | 7.45 μM (±2.05) | 2.83 μM (±0.59) | 2.79 μM (±0.05) | >50 μM |
| helicusin E (3) | >100 μM | >100 μM | >200 μM | >100 μM | >100 μM | >10 μM | >50 μM |
| isochromophilone X (4) | >100 μM | >100 μM | >100 μM | >100 μM | >80 μM | 11.7 μM (±0.80) | not determined |
| isochromophilone XI (5) | 55.6 μM | 78.4 μM | >100 μM | >100 μM | 41.5 μM | 8.30 μM (±1.14) | >50 μM |
| IC50 values of the positive controls | chloram-phenicol 1.45 μM (±0.13) | chloram-phenicol 2.13 μM (±0.11) | nystatin 5.80 μM (±2.80) | boscalid 0.53 μM | Clotrimazole 0.2 μM | rolipam 0.75 μM (±0.05) | huperzine <0.1 μM |
3. Experimental Section
3.1. General
3.2. Cultivation, Extraction and Substance Characterization
3.3. Bioassays
3.4. Computational Details
4. Conclusions
Acknowledgments
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Gangadevi, V.; Muthumary, J. Taxol, an anticancer drug produced by an endophytic fungus Bartalinia robillardoides Tassi, isolated from a medicinal plant, Aegle marmelos Correa ex Roxb. World J. Microb. Biot. 2008, 24, 717–724. [Google Scholar] [CrossRef]
- Chruma, J.J.; Moon, S.-J.; Sanford, W.E., Jr. Azaphilone α-bromoacetates (AzBs): Fluorescent linchpin reagents for the inter- and intramolecular cross-linkage of primary amines to thiols. Lett. Org. Chem. 2009, 6, 367–371. [Google Scholar] [CrossRef]
- Wei, W.G.; Yao, Z.J. Synthesis studies toward chloroazaphilone and vinylogous gamma-pyridones: Two common natural product core structures. J. Org. Chem. 2005, 70, 4585–4590. [Google Scholar] [CrossRef]
- Osmanova, N.; Schultze, W.; Ayoub, N. Azaphilones: A class of fungal metabolites with diverse biological activities. Phytochem. Rev. 2010, 9, 315–342. [Google Scholar] [CrossRef]
- Wiese, J.; Ohlendorf, B.; Blümel, M.; Schmaljohann, R.; Imhoff, J.F. Phylogenetic identification of fungi isolated from the marine sponge Tethya aurantium and identification of their secondary metabolites. Mar. Drugs 2011, 9, 561–585. [Google Scholar] [CrossRef]
- Yoshida, E.; Fujimoto, H.; Baba, M.; Yamazaki, M. Four new chlorinated azaphilones, helicusins A–D, closely related to 7-epi-sclerotiorin, from an ascomycetous fungus, Talaromyces helicus. Chem. Pharm. Bull. (Tokyo) 1995, 43, 1307–1310. [Google Scholar] [CrossRef]
- Sun, X.L.; Takayanagi, H.; Matsuzaki, K.; Tanaka, H.; Furuhata, K.; Omura, S. Synthesis and inhibitory activities of isochromophilone analogues against gp120-CD4 binding. J. Antibiot. (Tokyo) 1996, 49, 689–692. [Google Scholar] [CrossRef]
- Michael, A.P.; Grace, E.J.; Kotiw, M.; Barrow, R.A. Isochromophilone IX, a novel GABA-containing metabolite isolated from a cultured fungus, Penicillium sp. Aust. J. Chem. 2003, 56, 13–15. [Google Scholar] [CrossRef]
- Nam, J.Y.; Kim, H.K.; Kwon, J.Y.; Han, M.Y.; Son, K.H.; Lee, U.C.; Choi, J.D.; Kwon, B.M. 8-O-Methylsclerotiorinamine, antagonist of the Grb2-SH2 domain, isolated from Penicillium multicolor. J. Nat. Prod. 2000, 63, 1303–1305. [Google Scholar] [CrossRef]
- Yang, D.J.; Tomoda, H.; Tabata, N.; Masuma, R.; Omura, S. New isochromophilones VII and VIII produced by Penicillium sp. FO-4164. J. Antibiot. (Tokyo) 1996, 49, 223–229. [Google Scholar] [CrossRef]
- Wu, S.H.; Chen, Y.W.; Shao, S.C.; Wang, L.D.; Li, Z.Y.; Yang, L.Y.; Li, S.L.; Huang, R. Ten-membered lactones from Phomopsis sp., an endophytic fungus of Azadirachta indica. J. Nat. Prod. 2008, 71, 731–734. [Google Scholar] [CrossRef]
- Pilli, R.A.; Victor, M.M.; de Meijere, A. First total synthesis of aspinolide B, a new pentaketide produced by Aspergillus ochraceus. J. Org. Chem. 2000, 65, 5910–5916. [Google Scholar] [CrossRef]
- Steyn, P.S.; Vleggaar, R. The structure of dihydrodeoxy-8-epi-austdiol and the absolute configuration of the azaphilones. J. Chem. Soc. Perkin 1 1976, 204–206. [Google Scholar] [CrossRef]
- Whalley, W.B.; Ferguson, G.; Marsh, W.C.; Restivo, R.J. The chemistry of fungi. Part LXVIII. The absolute configuration of (+)-sclerotiorin and of the azaphilones. J. Chem. Soc. Perkin 1 Trans. 1976, 1366–1369. [Google Scholar] [CrossRef]
- Germain, A.R.; Bruggemeyer, D.M.; Zhu, J.; Genet, C.; O’Brien, P.; Porco, J.A. Synthesis of the azaphilones (+)-sclerotiorin and (+)-8-O-methylsclerotiorinamine utilizing (+)-sparteine surrogates in copper-mediated oxidative dearomatization. J. Org. Chem. 2011, 76, 2577–2584. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef]
- Bringmann, G.; Götz, D.; Bruhn, T. The Online Stereochemical Analysis of Chiral Compounds by HPLC-ECD Coupling in Combination with Quantum-Chemical Calculations. In Comprehensive Chiroptical Spectroscopy; Berova, N., Woody, R.W., Polavarapu, P., Nakanishi, K., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 2, pp. 355–420. [Google Scholar]
- Grebner, C.; Becker, J.; Stepanenko, S.; Engels, B. Efficiency of tabu-search-based conformational search algorithms. J. Comput. Chem. 2011, 32, 2245–2253. [Google Scholar] [CrossRef]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 17, 2717–2727. [Google Scholar]
- Arunpanichlert, J.; Rukachaisirikul, V.; Sukpondma, Y.; Phongpaichit, S.; Tewtrakul, S.; Rungjindamai, N.; Sakayaroj, J. Azaphilone and isocoumarin derivatives from the endophytic fungus Penicillium sclerotiorum PSU-A13. Chem. Pharm. Bull. (Tokyo) 2010, 58, 1033–1036. [Google Scholar] [CrossRef]
- Schulz, D.; Beese, P.; Ohlendorf, B.; Erhard, A.; Zinecker, H.; Dorador, C.; Imhoff, J.F. Abenquines A–D: Aminoquinone derivatives produced by Streptomyces sp. strain DB634. J. Antibiot. (Tokyo) 2011, 64, 763–768. [Google Scholar] [CrossRef]
- Kim, B.-Y.; Willbold, S.; Kulik, A.; Helaly, S.E.; Zinecker, H.; Wiese, J.; Imhoff, J.F.; Goodfellow, M.; Süssmuth, R.D.; Fiedler, H.-P. Elaiomycins B and C, novel alkylhydrazides produced by Streptomyces sp. BK 190. J. Antibiot. (Tokyo) 2011, 64, 595–597. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC2012, version 12.157L; Stewart Computational Chemistry: Colorada Springs, CO, USA, 2012. [Google Scholar]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted gaussian-basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Grimme, S. Improved second-order Møller-Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Sinnecker, S.; Rajendran, A.; Klamt, A.; Diedenhofen, M.; Neese, F. Calculation of solvent shifts on electronic g-tensors with the conductor-like screening model (COSMO) and its self-consistent generalization to real solvents (Direct COSMO-RS). J. Phys. Chem. A 2006, 110, 2235–2245. [Google Scholar] [CrossRef]
- Karton, A.; Tarnopolsky, A.; Lamere, J.F.; Schatz, G.C.; Martin, J.M.L. Highly accurate first-principles benchmark data sets for the parametrization and validation of density functional and other approximate methods. Derivation of a robust, generally applicable, double-hybrid functional for thermochemistry and thermochemical kinetics. J. Phys. Chem. A 2008, 112, 12868–12886. [Google Scholar]
- Kossmann, S.; Neese, F. Efficient structure optimization with second-order many-body perturbation theory: The RIJCOSX-MP2 method. J. Chem. Theory Comput. 2010, 6, 2325–2338. [Google Scholar] [CrossRef]
- Petrenko, T.; Kossmann, S.; Neese, F. Efficient time-dependent density functional theory approximations for hybrid density functionals: Analytical gradients and parallelization. J. Chem. Phys. 2011, 134, 054116. [Google Scholar] [CrossRef]
- Neese, F.; Becker, U.; Ganyushin, D.; Hansen, A.; Liakos, D.; Kollmar, C.; Koßmann, S.; Petrenko, T.; Reimann, C.; Riplinger, C.; et al. ORCA—An Ab Initio, Density Functional and Semiempirical Program Package, version 2.9.1; Max Planck Institute for Bioinorganic Chemistry: Mühlheim, Germany, 2012. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis, version 1.53; University Würzburg: Würzburg, Germany, 2012. [Google Scholar]
- Dong, J.Y.; Zhou, Y.P.; Li, R.; Zhou, W.; Li, L.; Zhu, Y.H.; Huang, R.; Zhang, K.Q. New nematicidal azaphilones from the aquatic fungus Pseudohalonectria adversaria YMF101019. FEMS Microbiol. Lett. 2006, 264, 65–69. [Google Scholar] [CrossRef]
- Bell, P.J.L.; Karuso, P. Epicocconone, a novel fluorescent compound from the fungus Epicoccum nigrum. J. Am. Chem. Soc. 2003, 125, 9304–9305. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jansen, N.; Ohlendorf, B.; Erhard, A.; Bruhn, T.; Bringmann, G.; Imhoff, J.F. Helicusin E, Isochromophilone X and Isochromophilone XI: New Chloroazaphilones Produced by the Fungus Bartalinia robillardoides Strain LF550. Mar. Drugs 2013, 11, 800-816. https://doi.org/10.3390/md11030800
Jansen N, Ohlendorf B, Erhard A, Bruhn T, Bringmann G, Imhoff JF. Helicusin E, Isochromophilone X and Isochromophilone XI: New Chloroazaphilones Produced by the Fungus Bartalinia robillardoides Strain LF550. Marine Drugs. 2013; 11(3):800-816. https://doi.org/10.3390/md11030800
Chicago/Turabian StyleJansen, Nils, Birgit Ohlendorf, Arlette Erhard, Torsten Bruhn, Gerhard Bringmann, and Johannes F. Imhoff. 2013. "Helicusin E, Isochromophilone X and Isochromophilone XI: New Chloroazaphilones Produced by the Fungus Bartalinia robillardoides Strain LF550" Marine Drugs 11, no. 3: 800-816. https://doi.org/10.3390/md11030800
APA StyleJansen, N., Ohlendorf, B., Erhard, A., Bruhn, T., Bringmann, G., & Imhoff, J. F. (2013). Helicusin E, Isochromophilone X and Isochromophilone XI: New Chloroazaphilones Produced by the Fungus Bartalinia robillardoides Strain LF550. Marine Drugs, 11(3), 800-816. https://doi.org/10.3390/md11030800