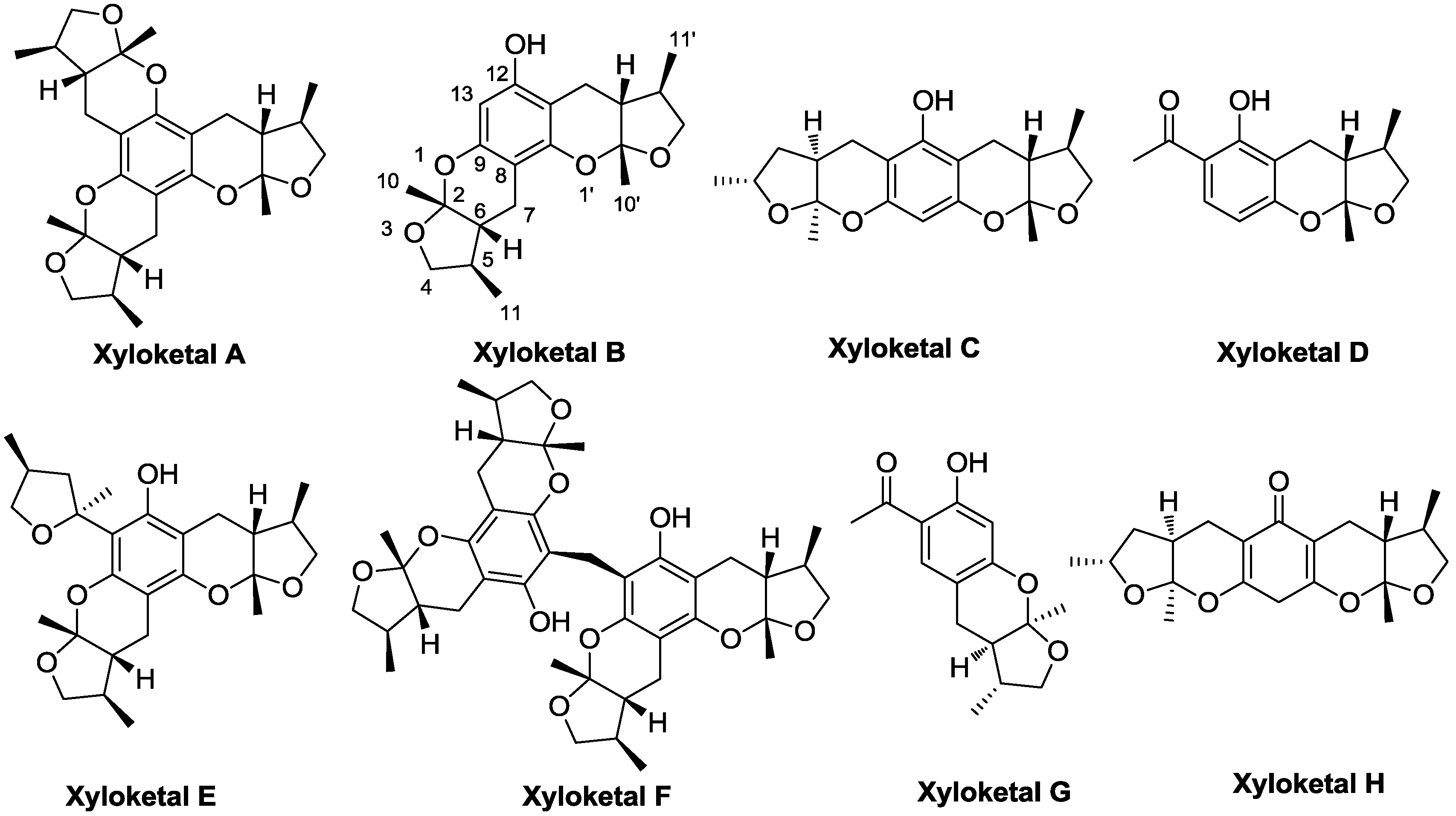
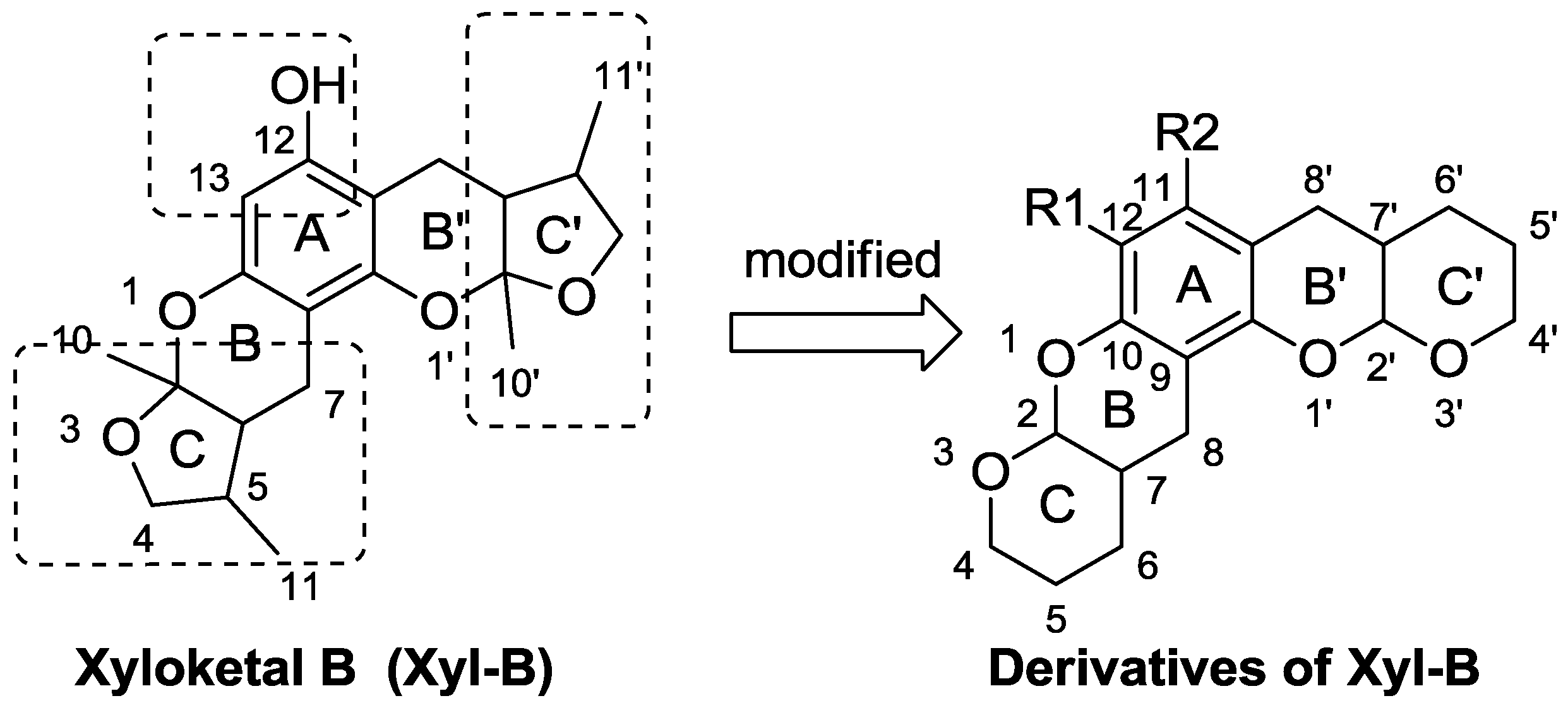
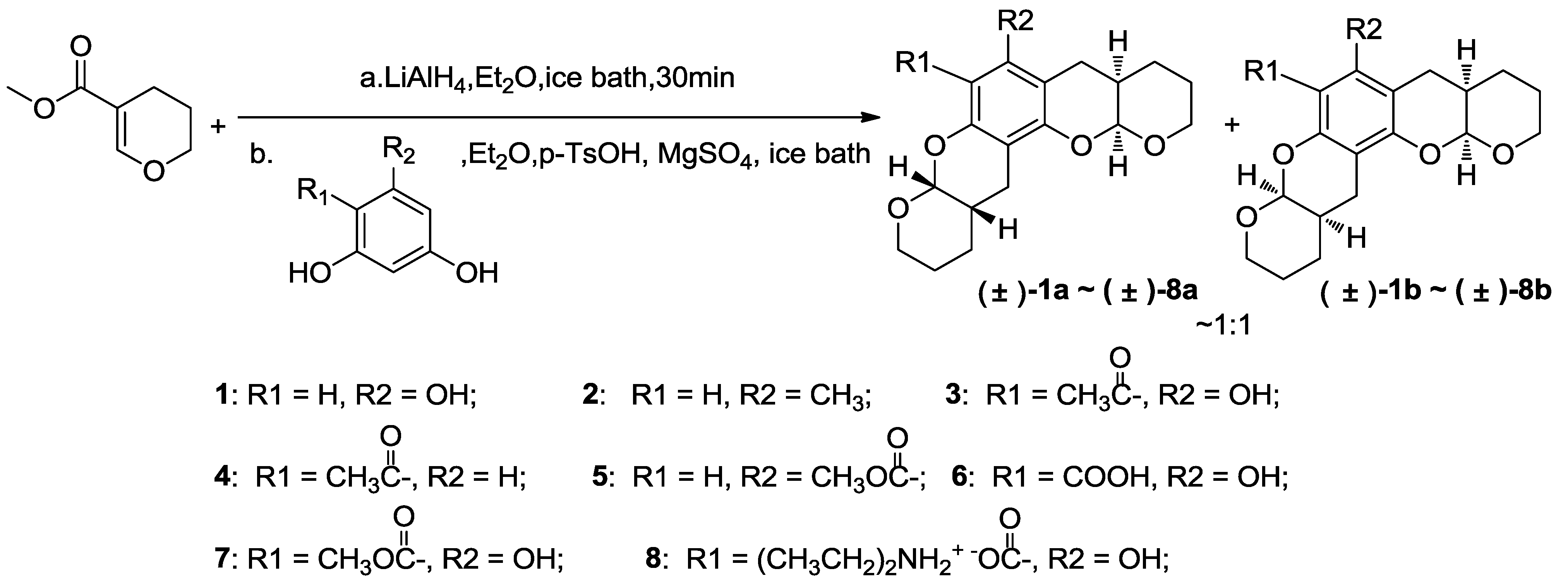
3.3. General Procedure for Synthesizing Compounds 1–8, Using 1 as an Example
3.3.1. Compound 1
To a suspension of lithium aluminum hydride (0.76 g, 20 mmol) in ether (30 mL) in an ice water bath was added dropwise a solution of methyl 3,4-dihydro-2H-pyran-5-carboxylate (1.88 g, 13.2 mmol, 3.2 molar equivalents with respect to phloroglucinol) in ether (20 mL). The resultant mixture was allowed to warm to room temperature and stirred for one additional hour. A 2 M aqueous sodium hydroxide (1.2 g) solution was then added at 0 °C. The resultant mixture was filtered and washed with ether (3 × 50 mL), and the combined filtrates were concentrated in vacuo below 10 °C to afford the reduction product as a colorless liquid. This material was diluted with ether (20 mL) and immediately used in subsequent experiments, because it is unstable.
To a suspension of phloroglucinol (0.52 g, 4.1 mmol) and anhydrous magnesium sulfate (2 g) in 40 mL ether at 0 °C was added the ether solution prepared above. p-Toluene sulfonic acid (0.1 g) was added after stirring for 1 minute. The resultant mixture was warmed to room temperature and stirred for 30 min. The reaction mixture was then filtered, and the filter cake was washed with ether (3 × 50 mL). The combined filtrates were washed with water (100 mL) and saturated brine (100 mL) before drying over anhydrous magnesium sulfate and concentrating in vacuo. Purification via flash chromatography eluting with ether:dichloromethane (2:25) or ethyl acetate:petroleum ether (1:20–1:5) afforded a colorless crystal (0.95 g, 3.0 mmol, 73% yield) as a diastereoisomeric mixture of (±)-1a and (±)-1b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.03 (s, 1H), 5.42 (s, 1H), 5.30 (t, J = 3.1 Hz, 1H), 5.23 (t, J = 3.1 Hz, 1H), 4.07–3.93 (m, 2H), 3.79–3.63 (m, 2H), 2.79–2.51 (m, 4H), 2.23–2.07 (m, 2H), 1.73–1.59 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 152.75, 152.71, 151.62, 151.54, 151.16, 151.12, 100.50, 100.41, 100.05, 100.00, 96.74, 96.68, 96.65, 95.70, 63.13, 62.96, 62.84, 62.57, 31.24, 31.20, 31.11, 31.08, 24.56, 24.43, 24.25, 24.14, 23.55, 23.41, 23.31, 22.93, 22.69, 22.54. EI-MS m/z 318 (M). EI-HR-MS m/z found: 318.1457; calcd. for C18H22O5: 318.1462.
3.3.2. Compound 2
The title compound was obtained as a colorless crystal in 55% yield as a diastereoisomeric mixture of (±)-2a and (±)-2b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.34 (s, 1H), 5.26–5.16 (m, 2H), 4.07–3.92 (m, 2H), 3.75–3.60 (m, 2H), 2.81–2.65 (m, 2H), 2.62–2.41 (m, 2H), 2.19–2.10 (m, 2H), 2.04 (s, 3H), 1.75–1.53 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 151.57, 135.30, 111.44, 111.42, 101.78, 95.95, 62.64, 62.51, 60.17, 31.73, 31.67, 26.67, 26.56, 24.45, 24.35, 23.35, 23.25, 14.37, 14.03. EI-MS m/z 316 (M). EI-HR-MS m/z found: 316.1667; calcd. for C19H24O4: 316.1669.
3.3.3. Compound 3
The title compound was obtained as a colorless crystal in 55% yield as a diastereoisomeric mixture of (±)-3a and (±)-3b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.03 (s, 1H), 5.40–5.33 (m, 2H), 4.05–3.87 (m, 2H), 3.82–3.65 (m, 2H), 2.77–2.51 (m, 7H), 2.23–2.11 (m, 2H), 1.79–1.53 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 203.34, 162.43, 162.39, 157.04, 156.97, 153.71, 153.59, 105.18, 100.37, 100.28, 98.58, 98.48, 97.47, 97.20, 62.71, 62.62, 62.46, 62.34, 33.08, 30.98, 30.91, 30.74, 30.66, 23.95, 23.89, 23.79, 23.69, 23.49, 23.38, 23.28, 22.75, 22.42. EI-MS m/z 360 (M). EI-HR-MS m/z found: 360.1565; calcd. for C20H24O6: 360.1567.
3.3.4. Compound 4
The title compound was obtained as a colorless crystal in 60% yield as a diastereoisomeric mixture of (±)-4a and (±)-4b (~1:1). 1H NMR (400 MHz, CDCl3) δ 13.02 (s, 1H), 7.52 (d, J = 8.9 Hz, 1H), 6.43 (d, J = 8.9 Hz, 1H), 5.36 (d, J = 2.6 Hz, 1H), 4.03–3.93 (m, 1H), 3.80–3.69 (m, 1H), 2.80–2.65 (m, 2H), 2.54 (s, 3H), 2.26–2.15 (m, 1H), 1.78–1.53 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 202.73, 162.65, 159.49, 129.83, 113.53, 108.24, 108.12, 97.10, 62.54, 30.82, 26.08, 23.98, 23.61, 22.78. EI-MS m/z 248 (M). EI-HR-MS m/z found: 248.1041; calcd. for C14H16O4: 248.1043.
3.3.5. Compound 5
The title compound was obtained as a colorless crystal in 71% yield as a diastereoisomeric mixture of (±)-5a and (±)-5b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.51 (s, 1H), 5.28 (d, J = 2.6 Hz, 2H), 5.26 (d, J = 2.6 Hz, 1H), 4.05–3.92 (m, 2H), 3.88 (s, 3H), 3.75–3.57 (m, 2H), 2.93–2.39 (m, 4H), 2.19–2.06 (m, 2H), 1.71–1.57 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 168.96, 152.36, 152.17, 133.34, 130.83, 128.83, 110.87, 110.79, 105.76, 96.43, 96.33, 65.50, 62.76, 62.34, 60.30, 51.83, 31.54, 31.45, 26.83, 26.27, 24.19, 23.96, 23.55, 23.27. EI-MS m/z 360 (M). EI-HR-MS m/z found: 360.1566; calcd. for C20H24O6: 360.1567.
3.3.6. Compound 6
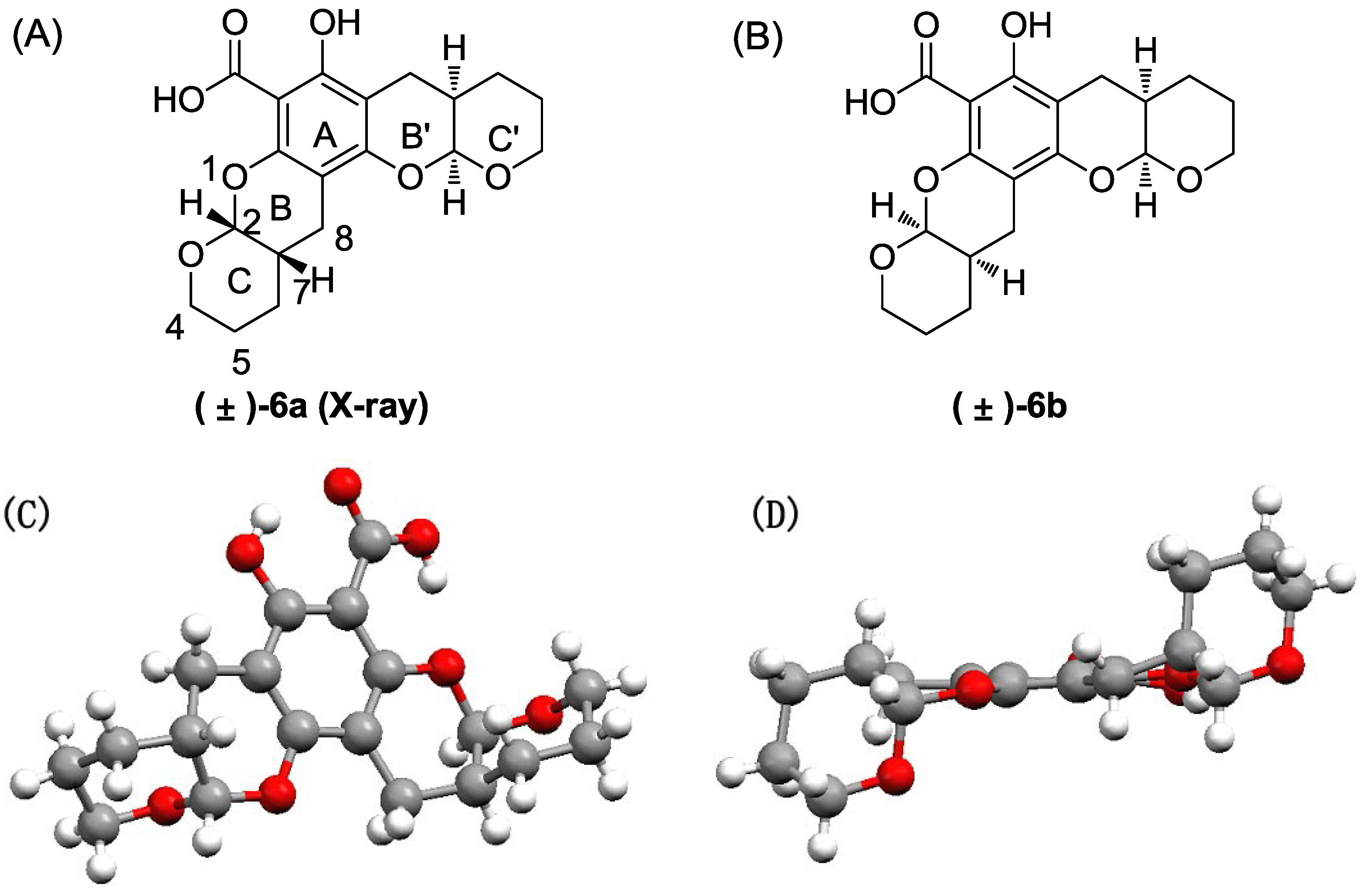
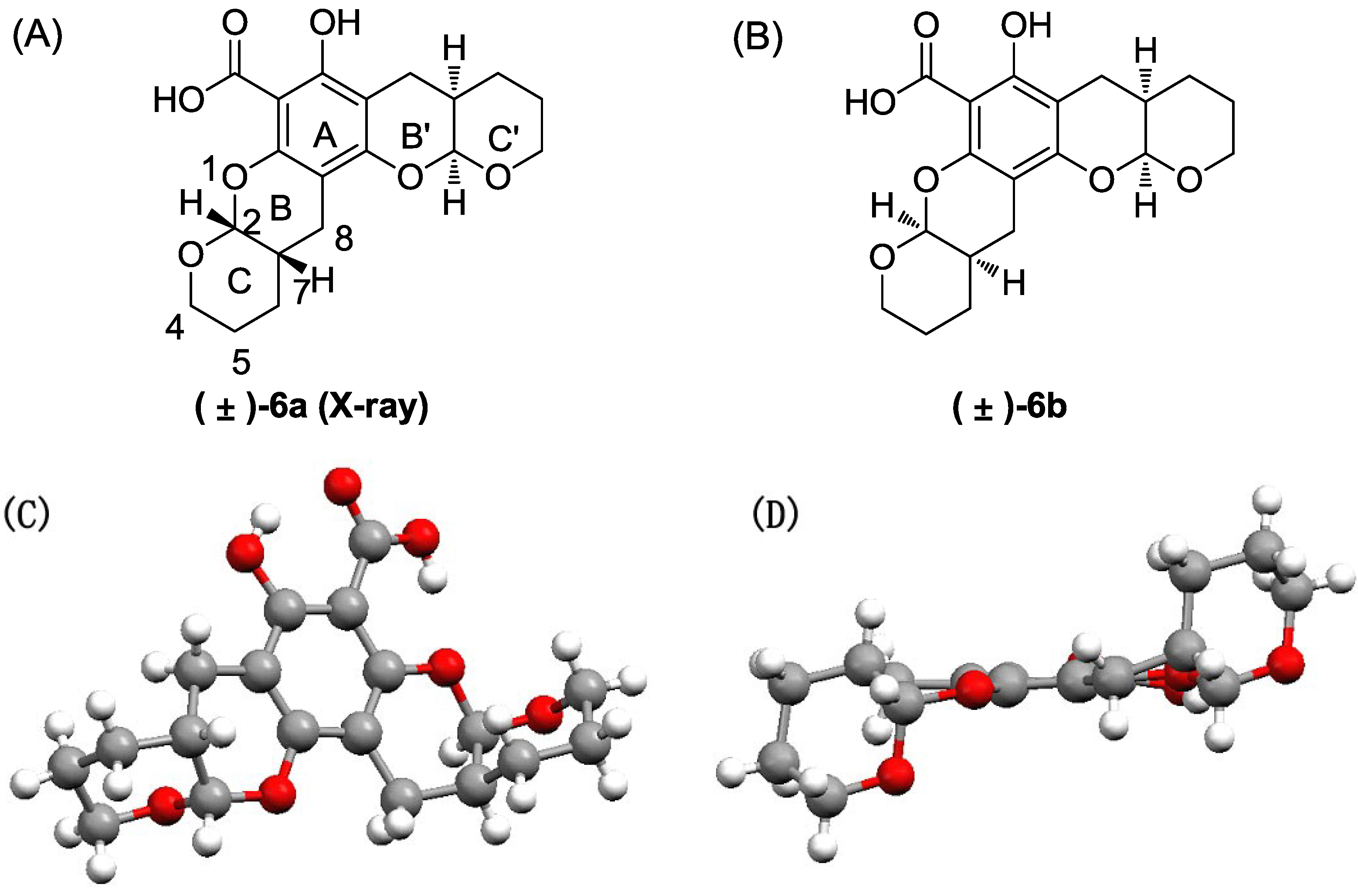
The title compound was obtained as a colorless crystal in 80% yield as a diastereoisomeric mixture of (±)-6a and (±)-6b (~1:1). 1H NMR (400 MHz, CDCl3) δ 12.48 (s, 1H), 1H NMR (400 MHz, CDCl3) δ 12.48 (s, 2H), 5.53 (d, J = 2.5 Hz, 1H), 5.52 (d, J = 2.5 Hz, 1H), 5.37 (d, J = 2.6 Hz, 1H), 5.35 (d, J = 2.6 Hz, 1H), 4.05–3.64 (m, 8H), 2.88–2.51 (m, 8H), 2.37–2.10 (m, 4H), 1.79–1.51 (m, 16H). 13C NMR (101 MHz, CDCl3) δ 171.17, 161.28, 161.24, 156.46, 156.38, 151.02, 150.91, 101.93, 101.82, 98.94, 98.85, 98.83, 97.55, 97.49, 94.06, 94.03, 62.90, 62.54, 62.48, 62.42, 30.89, 30.81, 30.75, 30.66, 23.98, 23.81, 23.78, 23.74, 23.55, 23.41, 23.40, 23.32, 22.77, 22.39.
Compound 6 was purified via flash chromatography (petro ether:ethyl acetate = 50:1–30:1) followed by crystallization to afford the pure isomers (±)-6a and (±)-6b:
(±)-6a: m.p. 171.2–172.1 °C. 1H NMR (400 MHz, CDCl3) δ 12.47 (s, 1H), 11.31 (s, 1H), 5.51 (d, J = 2.4 Hz, 1H), 5.35 (d, J = 2.4 Hz, 1H), 4.05–3.67 (m, 4H), 2.87–2.53 (m, 4H), 2.39–2.10 (m, 2H), 1.87–1.48 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 171.14, 161.24, 156.37, 150.91, 101.93, 98.94, 98.84, 97.55, 94.06, 62.89, 62.54, 30.89, 30.67, 23.99, 23.73, 23.56, 23.39, 23.32, 22.39.
(±)-6b: m.p. 178.4–179.5 °C. 1H NMR (400 MHz, CDCl3) δ 12.47 (s, 1H), 11.31 (s, 1H), 5.53 (d, J = 2.4 Hz, 1H), 5.37 (d, J = 2.8 Hz, 1H), 3.99–3.68 (m, 4H), 2.82–2.56 (m, 4H), 2.32–2.13 (m, 2H), 1.81–1.52 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 171.15, 161.28, 156.45, 151.01, 101.82, 98.86, 97.50, 94.04, 62.48, 62.42, 30.81, 30.76, 23.79, 23.54, 23.40, 22.76.
Crystallographic data for (±)-
6a: Single-crystal growth was performed in EtOAc at room temperature. C
19H
22O
7, crystal dimension 0.42 mm × 0.40 mm × 0.37 mm, space group monoclinic,
C2/c; unit cell dimensions,
a = 25.4092 (3) Å,
b = 12.9938 (2) Å,
c = 10.67030 (10) Å, volume = 3279.17 (7) Å
3,
Z = 8,
Dcalcd = 1.468 Mg/m
3,
m = 0.939 mm
−1,
F000 = 1,536. All single-crystal data were collected via the hemisphere technique on a Bruker SMART 1000 CCD system diffractometer with graphite-monochromated Mo Ká radiation
ë = 1.54178 at 150(2) K. The structure was solved using the direct method. The final
R value was 0.0322, wR2 = 0.0791 [
I > 2
ó (
I)]. More details are provided in the
Supplementary Information.
3.3.7. Compound 7
The title compound was obtained as a colorless crystal in 90% yield as a diastereoisomeric mixture of (±)-7a and (±)-7b (~1:1). 1H NMR (400 MHz, CDCl3) δ 11.97 (s, 1H), 11.96 (s, 1H), 5.34 (apparent t, 2H), 5.32 (d, J = 2.6 Hz, 1H), 5.30 (d, J = 2.6 Hz, 1H), 4.17–3.93 (m, 4H), 3.91 (s, 6H), 3.83–3.62 (m, 4H), 2.87–2.43 (m, 8H), 2.25–2.01 (m, 4H), 1.77–1.58 (m, 16H). 13C NMR (101 MHz, CDCl3) δ 172.01, 160.67, 160.65, 156.01, 155.95, 152.77, 152.64, 100.16, 100.08, 99.43, 99.32, 97.38, 97.32, 97.00, 96.97, 95.79, 95.77, 62.93, 62.69, 62.37, 52.03, 31.05, 30.99, 30.47, 30.44, 24.20, 24.02, 23.90, 23.69, 23.51, 23.40, 23.25, 23.01, 22.76, 22.71, 22.55. EI-MS m/z 376 (M). EI-HR-MS m/z found: 376.1513; calcd. for C20H24O7: 376.1517.
3.3.8. Compound 8
A solution of 6 (0.2 g, 0.6 mmol) and diethylamine (0.2 g, 6 mmol) in acetone (20 mL) was stirred at room temperature for 30 min. The acetone and excess diethylamine were removed under reduced pressure on a rotary evaporator to afford the title compound as a white foam as a diastereoisomeric mixture of (±)-8a and (±)-8b (~1:1). 1H NMR (400 MHz, CDCl3) δ 5.31 (d, J = 2.5 Hz, 1H), 5.25 (t, J = 2.8 Hz, 1H), 4.13–3.90 (m, 2H), 3.76–3.54 (m, 2H), 3.00 (q, J = 7.3 Hz, 4H), 2.74–2.54 (m, 4H), 2.16 (s, 1H), 2.15–2.08 (m, 2H), 1.79–1.54 (m, 8H), 1.29 (t, J = 7.3 Hz, 6H).
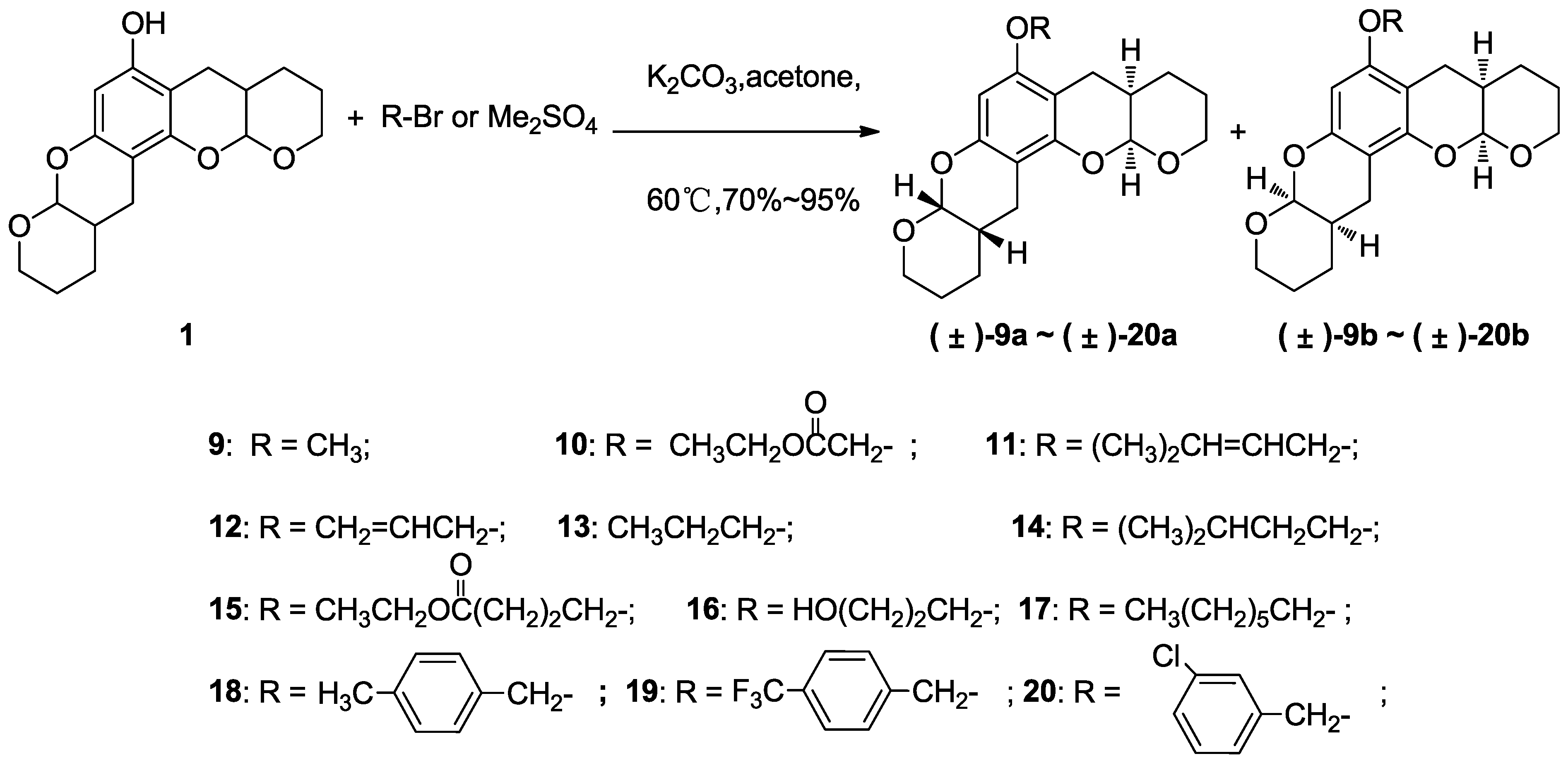
3.4.Typical Method of Etherification of 1 to Prepare 9–20, Using 9 as an Example
3.4.1. Compound 9
A solution of 1 (0.1 g, 0.31 mmol), potassium carbonate (87 mg, 0.62 mmol) and dimethyl sulfate (78 mg, 0.62 mmol) was stirred in acetone (15 mL) at 60 °C and monitored via thin layer chromatography. The resultant mixture was allowed to cool to room temperature before pouring into water (50 mL). The aqueous layer was extracted with ethyl acetate (3 × 40 mL). The combined organic layers were washed with saturated brine (50 mL), dried over anhydrous magnesium sulfate and concentrated in vacuo. Purification via flash chromatography using ethyl acetate:petroleum ether (1:20) as the eluant afforded colorless crystals (87 mg, 0.26 mmol, 85% yield) that were a mixture of diastereoisomers (±)-9a and (±)-9b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.07 (s, 1H), 5.28 (t, J = 2.9 Hz, 1H), 5.24 (t, J = 3.3 Hz, 1H), 4.09–3.91 (m, 2H), 3.74 (s, 3H), 3.74–3.66 (m, 2H), 2.80–2.47 (m, 4H), 2.23–2.07 (m, 2H), 1.74–1.58 (m, 8H).13C NMR (101 MHz, CDCl3) δ 156.56, 156.52, 151.66, 151.58, 150.82, 101.10, 101.04, 100.37, 100.28, 96.74, 96.72, 96.66, 91.81, 63.09, 62.92, 62.83, 62.52, 55.33, 31.22, 31.18, 31.07, 31.03, 24.57, 24.44, 24.25, 24.14, 23.57, 23.41, 23.33, 23.04, 22.74, 22.69, 22.54. EI-MS m/z 332 (M). EI-HR-MS m/z found: 332.1621; calcd. for C19H24O5: 332.1618.
3.4.2. Compound 10
The title compound was obtained as a colorless crystal in 82% yield as a diastereoisomeric mixture of (±)-10a and (±)-10b (~1:1). 1H NMR (400 MHz, CDCl3) δ 5.94 (s, 1H), 5.29 (t, J = 3.0 Hz, 1H), 5.23 (dd, J = 4.7, 2.5 Hz, 1H), 4.54 (s, 2H), 4.25 (q, J = 7.1 Hz, 2H), 4.08–3.90 (m, 2H), 3.80–3.60 (m, 2H), 2.83–2.50 (m, 4H), 2.18 (d, J = 27.6 Hz, 2H), 1.78–1.59 (m, 8H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.83, 154.88, 154.83, 151.60, 151.51, 151.12, 151.09, 101.87, 101.81, 101.50, 101.41, 96.83, 96.76, 96.72, 96.66, 92.81, 65.49, 63.06, 62.87, 62.81, 62.52, 61.16, 31.19, 31.15, 31.06, 31.03, 24.56, 24.41, 24.22, 24.10, 23.56, 23.41, 23.32, 23.01, 22.74, 22.58, 14.10. EI-MS m/z 404 (M). EI-HR-MS m/z found: 404.1833; calcd. for C22H28O7: 404.1830.
3.4.3. Compound 11
The title compound was obtained as a colorless crystal in 82% yield as a diastereoisomeric mixture of (±)-11a and (±)-11b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.11 (s, J = 7.6 Hz, 1H), 5.57–5.41 (m, 1H), 5.30 (t, J = 2.8 Hz, 1H), 5.28–5.24 (m, 1H), 4.46 (d, J = 6.5 Hz, 2H), 4.12–3.93 (m, 2H), 3.81–3.61 (m, 2H), 2.85–2.52 (m, 4H), 2.24–2.08 (m, 2H), 1.73–1.61 (m, 8H), 1.58 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 155.88, 155.83, 151.57, 151.50, 150.84, 150.81, 137.13, 120.10, 101.52, 101.46, 100.32, 100.23, 96.79, 96.73, 96.67, 92.98, 65.04, 63.07, 62.92, 62.82, 62.53, 31.29, 31.25, 31.13, 25.70, 24.62, 24.50, 24.30, 24.19, 23.59, 23.43, 23.35, 23.13, 22.84, 22.70, 22.56, 18.16. EI-MS m/z 386 (M). EI-HR-MS m/z found: 386.2086; calcd. for C23H30O5: 386.2088.
3.4.4. Compound 12
The title compound was obtained by reaction in a sealed tube as a colorless crystal in 74% yield as a diastereoisomeric mixture of (±)-12a and (±)-12b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.06 (s, J = 7.1 Hz, 1H), 6.09–5.97 (m, 1H), 5.40 (ddd, J = 17.3, 3.2, 1.6 Hz, 1H), 5.29 (t, J = 2.8 Hz, 1H), 5.28–5.22 (m, 1H), 5.24 (t, J = 3.0 Hz, 1H), 4.46 (dt, J = 5.0, 1.4 Hz, 2H), 4.10–3.91 (m, 2H), 3.81–3.63 (m, 2H), 2.86–2.46 (m, 4H), 2.22–2.08 (m, 2H), 1.80–1.59 (m, 8H).13C NMR (101 MHz, CDCl3) δ 155.51, 155.47, 151.56, 151.48, 150.89, 150.85, 133.41, 117.03, 101.48, 101.42, 100.56, 100.47, 96.78, 96.71, 96.68, 92.94, 68.67, 63.16, 63.00, 62.85, 62.55, 31.21, 31.17, 31.11, 31.07, 24.60, 24.48, 24.28, 24.16, 23.59, 23.42, 23.39, 23.31, 23.13, 22.84, 22.66, 22.51. EI-MS m/z 358 (M). EI-HR-MS m/z found: 358.1767; calcd. for C21H26O5: 358.1775.
3.4.5. Compound 13
The title compound was obtained by reaction in a sealed tube as a colorless crystal in 97% yield as a diastereoisomeric mixture of (±)-13a and (±)-13b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.05 (s, 1H), 5.28 (t, J = 2.8 Hz, 1H), 5.24 (t, J = 3.1 Hz, 1H), 4.06–3.95 (m, 2H), 3.85 (t, J = 6.4 Hz, 2H), 3.77–3.62 (m, 2H), 2.83–2.47 (m, 4H), 2.23–2.07 (m, 2H), 1.84–1.72 (m, 2H), 1.73–1.59 (m, 8H), 1.01 (t, J = 7.4 Hz, 3H).13C NMR (101 MHz, CDCl3) δ 155.93, 155.89, 151.52, 151.45, 150.73, 150.69, 101.29, 101.23, 100.05, 99.96, 96.70, 96.62, 96.56, 92.53, 69.29, 62.96, 62.81, 62.44, 31.19, 31.14, 31.07, 31.03, 24.49, 24.37, 24.23, 24.11, 23.55, 23.38, 23.31, 23.03, 22.72, 22.65, 22.51, 22.45, 10.53. EI-MS m/z 360 (M). EI-HR-MS m/z found: 360.1935; calcd. for C21H28O5: 360.1931.
3.4.6. Compound 14
The title compound was obtained as a colorless crystal in 61% yield as a diastereoisomeric mixture of (±)-14a and (±)-14b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.07 (s, 1H), 5.28 (t, J = 2.8 Hz, 1H), 5.24 (t, J = 2.8 Hz, 1H), 4.11–3.96 (m, 2H), 3.91 (t, J = 6.6 Hz, 2H), 3.76–3.64 (m, 2H), 2.78–2.49 (m, 4H), 2.24–2.05 (m, 2H), 1.87–1.73 (m, 1H), 1.74–1.59 (m, 10H), 0.95 (dd, J = 6.6, 0.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 170.61, 161.36, 161.32, 154.30, 154.22, 150.78, 150.68, 141.49, 126.87, 125.28, 123.72, 101.33, 101.24, 98.04, 97.94, 97.50, 97.25, 97.18, 96.48, 62.84, 62.42, 62.34, 62.21, 40.71, 31.01, 30.92, 30.71, 30.63, 29.78, 29.64, 24.10, 23.90, 23.78, 23.62, 23.49, 23.38, 23.33, 22.88, 22.50. EI-MS m/z 388 (M). EI-HR-MS m/z found: 388.2245; calcd. for C23H32O5: 388.2244.
3.4.7. Compound 15
The title compound was obtained as a colorless crystal in 75% yield as a diastereoisomeric mixture of (±)-15a and (±)-15b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.04 (s, 1H), 5.28 (t, J = 2.7 Hz, 1H), 5.24 (t, J = 3.1 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 4.08–3.97 (m, 2H), 3.92 (t, J = 6.1 Hz, 2H), 3.78–3.65 (m, 2H), 2.82–2.52 (m, 4H), 2.49 (t, J = 7.4 Hz, 2H), 2.24–2.05 (m, 4H), 1.65 (m, 8H), 1.26 (t, J = 7.1 Hz, 3H).13C NMR (101 MHz, CDCl3) δ 173.13, 155.70, 151.68, 151.61, 150.91, 101.38, 101.33, 100.49, 100.40, 96.80, 96.73, 96.68, 92.65, 66.72, 63.04, 62.89, 62.55, 60.38, 31.28, 31.24, 31.16, 30.98, 24.65, 24.59, 24.47, 24.33, 24.22, 23.62, 23.46, 23.39, 23.10, 22.82, 22.74, 22.61, 14.20. EI-MS m/z 432 (M). EI-HR-MS m/z found: 432.2151; calcd. for C24H32O7: 432.2143.
3.4.8. Compound 16
The title compound was obtained as a colorless crystal in 71% yield as a diastereoisomeric mixture of (±)-16a and (±)-16b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.08 (s, 1H), 5.27 (t, J = 2.8 Hz, 1H), 5.24 (t, J = 3.1 Hz, 1H), 4.05 (t, J = 5.8 Hz, 2H), 4.00 (m, 2H), 3.84 (t, J = 5.9 Hz, 2H), 3.78–3.62 (m, 2H), 2.82–2.45 (m, 4H), 2.25–2.07 (m, 2H), 2.06–2.01 (m, 2H), 1.76–1.54 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 155.61, 155.57, 151.68, 151.61, 150.90, 150.85, 145.18, 128.49, 128.08, 101.14, 101.07, 100.67, 100.58, 96.74, 96.72, 96.67, 92.59, 66.06, 63.08, 62.93, 62.61, 60.93, 31.89, 31.20, 31.15, 31.05, 31.01, 29.67, 28.38, 28.27, 24.53, 24.42, 24.31, 24.18, 23.54, 23.41, 23.36, 23.15, 22.85, 22.69, 22.57. EI-MS m/z 376 (M). EI-HR-MS m/z found: 376.1885; calcd. for C21H28O6: 376.1880.
3.4.9. Compound 17
The title compound was obtained as a colorless crystal in 72% yield as a diastereoisomeric mixture of (±)-17a and (±)-17b (~1:1). 1H NMR (400 MHz, CDCl3) δ 6.05 (s, 1H), 5.28 (t, J = 2.8 Hz, 1H), 5.24 (t, J = 3.1 Hz, 1H), 4.12–3.95 (m, 2H), 3.88 (t, J = 6.5 Hz, 2H), 3.78–3.60 (m, 2H), 2.85–2.48 (m, 4H), 2.14 (m, 2H), 1.75–1.60 (m, 8H), 1.43–1.17 (m, 10H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 156.08, 156.05, 151.65, 151.57, 150.85, 150.81, 101.40, 101.34, 100.16, 100.06, 96.80, 96.73, 96.67, 92.67, 67.96, 63.03, 62.88, 62.79, 62.49, 31.77, 31.33, 31.28, 31.22, 31.18, 29.18, 28.99, 26.06, 24.61, 24.48, 24.32, 24.21, 23.66, 23.49, 23.40, 23.18, 22.89, 22.76, 22.58, 14.03. EI-MS m/z 416 (M). EI-HR-MS m/z found: 416.2547; calcd. for C25H36O5: 416.2557.
3.4.10. Compound 18
The title compound was obtained as a colorless crystal in 70% yield as a diastereoisomeric mixture of (±)-18a and (±)-18b (~1:1). 1H NMR (400 MHz, CDCl3) δ 7.30 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 7.9 Hz, 2H), 6.15 (s, 1H), 5.29 (t, J = 3.0 Hz, 1H), 5.27–5.22 (m, 1H), 4.94 (s, 2H), 4.11–3.93 (m, 2H), 3.80–3.61 (m, 2H), 2.82–2.51 (m, 4H), 2.36 (s, 3H), 2.19–2.08 (m, 2H), 1.77–1.58 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 155.82, 155.79, 151.67, 151.59, 150.93, 150.90, 137.47, 134.26, 129.13, 127.33, 101.64, 101.59, 100.66, 100.57, 96.84, 96.75, 96.70, 93.19, 69.88, 63.08, 62.93, 62.86, 62.57, 31.29, 31.26, 31.14, 24.62, 24.50, 24.32, 24.21, 23.61, 23.45, 23.38, 23.24, 22.96, 22.75, 22.61, 21.15. EI-MS m/z 422 (M). EI-HR-MS m/z found: 422.2086; calcd. for C26H30O5: 422.2088.
3.4.11. Compound 19
The title compound was obtained as a colorless crystal in 66% yield as a diastereoisomeric mixture of (±)-19a and (±)-19b (~1:1). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 6.10 (s, 1H), 5.31 (t, J = 2.9 Hz, 1H), 5.24 (t, J = 3.3 Hz, 1H), 5.05 (s, 2H), 4.09–3.93 (m, 2H), 3.77–3.64 (m, 2H), 2.80–2.58 (m, 4H), 2.23–2.07 (m, 2H), 1.75–1.60 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 155.32, 151.73, 151.65, 151.10, 141.37, 127.12, 125.47, 125.44, 101.58, 101.54, 101.16, 101.08, 96.85, 96.78, 96.74, 93.12, 69.03, 63.19, 63.04, 62.91, 62.63, 31.18, 31.12, 29.67, 24.64, 24.53, 24.35, 24.25, 23.55, 23.37, 23.29, 23.21, 22.66. EI-MS m/z 476 (M). EI-HR-MS m/z found: 476.1809; calcd. for C26H27O5F3: 476.1805.
3.4.12. Compound 20
The title compound was obtained as a colorless crystal in 71% yield as a diastereoisomeric mixture of (±)-20a and (±)-20b (~1:1). 1H NMR (400 MHz, CDCl3) δ 7.40 (s, 1H), 7.32–7.26 (m, 3H), 6.10 (s, 1H), 5.31 (t, J = 2.9 Hz, 1H), 5.28–5.20 (m, 1H), 4.96 (s, 2H), 4.06–3.94 (m, 2H), 3.80–3.63 (m, 2H), 2.84–2.52 (m, 4H), 2.24–2.08 (m, 2H), 1.80–1.59 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 155.36, 155.33, 151.62, 151.54, 151.01, 150.98, 139.33, 134.40, 129.77, 127.89, 127.09, 125.08, 101.57, 101.51, 100.99, 100.90, 96.81, 96.75, 96.69, 93.06, 69.02, 63.18, 63.02, 62.86, 62.56, 31.17, 31.13, 31.07, 31.04, 24.60, 24.47, 24.25, 24.14, 23.59, 23.43, 23.37, 23.28, 22.96, 22.66, 22.51. EI-MS m/z 442 (M). EI-HR-MS m/z found: 442.1545; calcd. for C25H27O5Cl1: 442.1542.
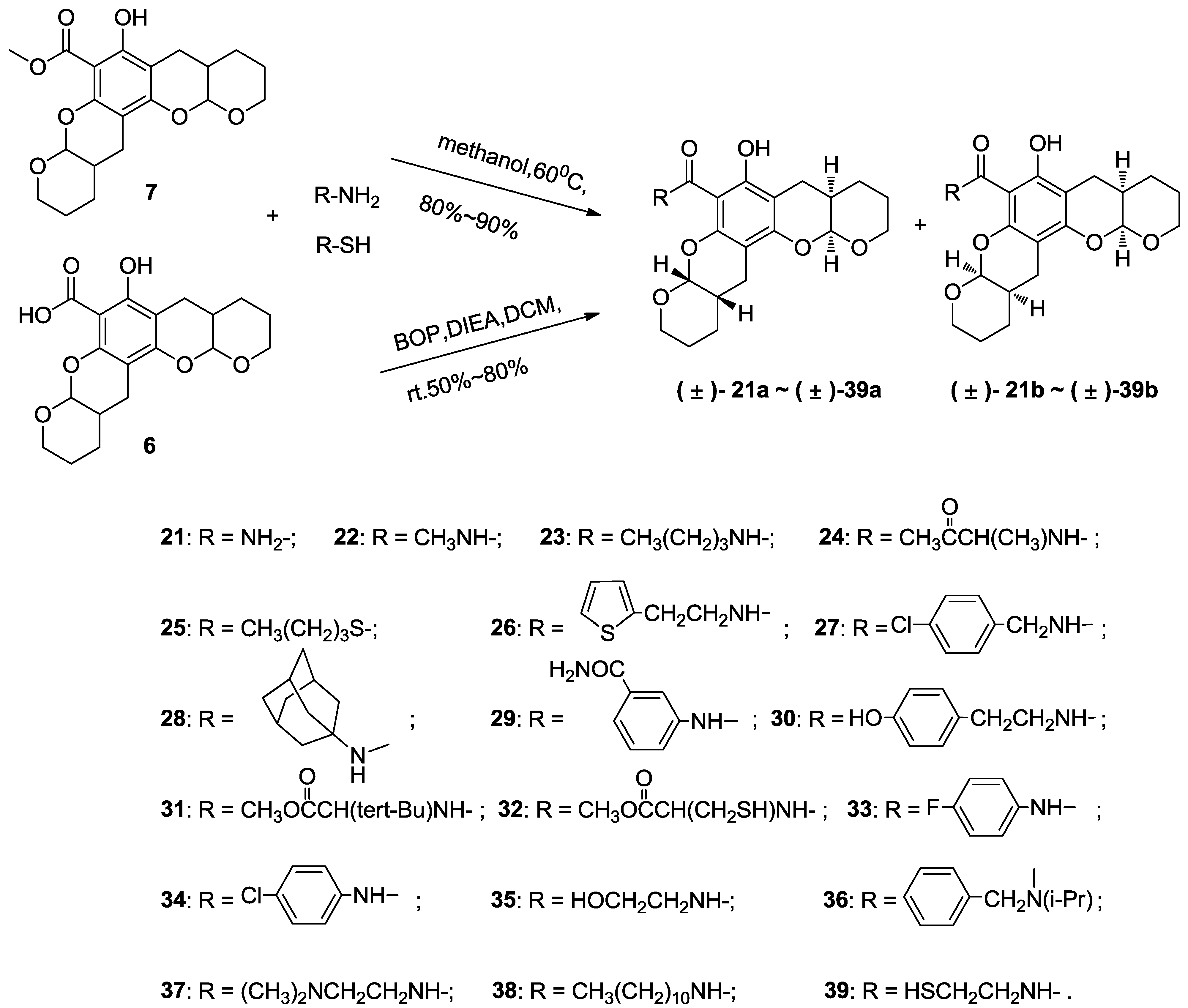
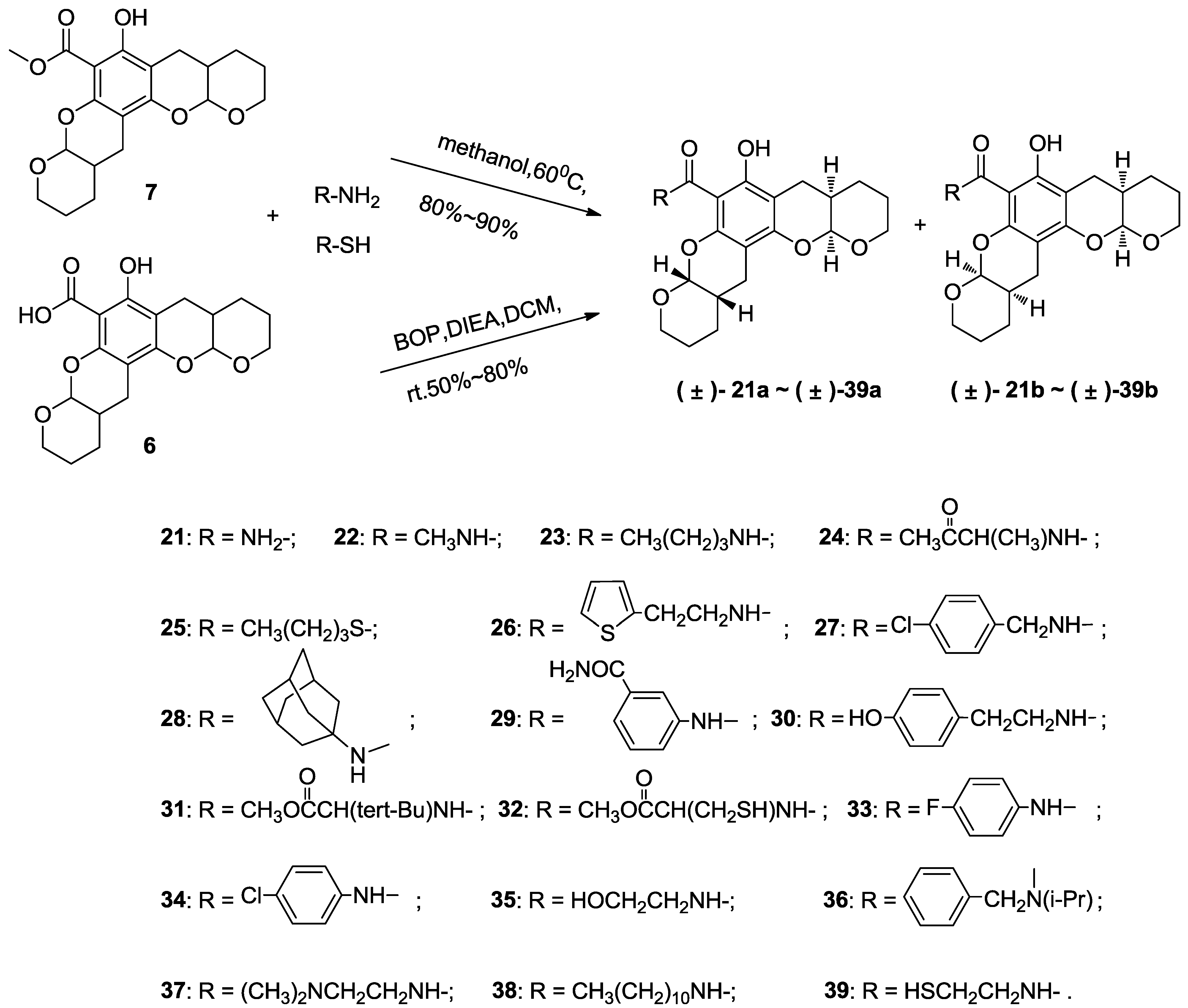
3.5. Typical Method to Prepare 21–23, Using 21 as an Example
3.5.1. Compound 21
A solution of compound 7 (0.1 g, 0.27 mmol) and ammonia (5 mL) in acetone (10 mL) was stirred in a sealed tube at 60 °C for 5 h and monitored via thin layer chromatography. The resultant mixture was then poured into water (50 mL) and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with saturated brine (50 mL), dried over anhydrous magnesium sulfate and concentrated in vacuo. Purification via flash chromatography using petroleum ethyl acetate: ether (3:2) as the eluant afforded the title compound (80 mg, 0.22 mmol, 82%) as a colorless crystal consisting of a diastereoisomeric mixture of (±)-21a and (±)-21b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.11 (s, 1H), 14.11 (s, 1H), 8.17 (s, 2H), 5.71 (s, 2H), 5.54–5.38 (m, 2H), 5.38–5.17 (m, 2H), 4.09–3.85 (m, 4H), 3.85–3.63 (m, 4H), 2.85–2.51 (m, 8H), 2.29–2.08 (m, 4H), 1.80–1.54 (m, 16H). 13C NMR (101 MHz, CDCl3) δ 172.74, 161.84, 161.80, 155.03, 154.96, 151.21, 151.11, 101.30, 101.21, 98.21, 98.12, 97.64, 97.36, 97.29, 96.00, 62.82, 62.42, 62.35, 62.24, 31.02, 30.94, 30.82, 30.74, 24.09, 23.97, 23.90, 23.64, 23.51, 23.45, 22.87, 22.51. EI-MS m/z 361 (M). EI-HR-MS m/z found: 361.1520; calcd. for C19H23O6N1: 361.1520.
3.5.2. Compound 22
The title compound was obtained with compound 7 similar to the method for synthesizing compound 21 by replacing ammonia with methylamine as a colorless crystal in 90% yield as a diastereoisomeric mixture of (±)-22a and (±)-22b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.39 (s, 1H), 14.38 (s, 1H), 8.33 (s, 2H), 5.47–5.38 (m, 2H), 5.38–5.26 (m, 2H), 4.04–3.87 (m, 4H), 3.84–3.64 (m, 4H), 2.97 (s, 3H), 2.95 (s, 3H), 2.85–2.51 (m, 8H), 2.23–2.09 (m, 4H), 1.76–1.59 (m, 16H). 13C NMR (101 MHz, CDCl3) δ 171.22, 161.26, 161.22, 154.18, 154.12, 150.70, 150.60, 101.40, 101.31, 98.01, 97.92, 97.60, 97.29, 97.22, 96.64, 62.85, 62.59, 62.47, 60.33, 31.07, 30.99, 30.79, 30.71, 26.03, 24.14, 23.97, 23.80, 23.73, 23.63, 23.60, 23.43, 23.30, 23.25, 22.89, 22.54, 14.16. EI-MS m/z 375 (M). EI-HR-MS m/z found: 375.1666; calcd. for C20H25O6N1: 375.1676.
3.5.3. Compound 23
The title compound was obtained with compound 7 similar to the method for synthesizing compound 21 by replacing ammonia with butylamine as a colorless crystal in 81% yield as a diastereoisomeric mixture of (±)-23a and (±)-23b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.43 (s, 1H), 8.43 (s, 1H), 5.44–5.38 (m, 1H), 5.34–5.27 (m, 1H), 4.04–3.85 (m, 2H), 3.84–3.63 (m, 2H), 3.51–3.27 (m, 2H), 2.90–2.47 (m, 4H), 2.31–2.07 (m, 2H), 1.77–1.51 (m, 10H), 1.49–1.38 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.43, 161.33, 161.29, 154.16, 154.09, 150.86, 150.77, 101.36, 101.26, 97.87, 97.78, 97.57, 97.27, 97.20, 96.63, 96.61, 62.84, 62.43, 61.99, 61.91, 39.36, 38.88, 31.34, 31.24, 31.08, 30.98, 30.89, 30.80, 24.13, 23.94, 23.78, 23.64, 23.50, 23.42, 23.37, 22.91, 22.52, 20.15, 19.94, 13.67, 13.57. EI-MS m/z 417 (M). EI-HR-MS m/z found: 417.2141; calcd. for C23H31O6N1: 417.2146.
3.6. Typical Method to Prepare 24–39, Using 24 as an Example
3.6.1. Compound 24
To a solution of compound 6 (0.1 g, 0.28 mmol), Ala-OMe.HCl (70 mg, 0.56 mmol) and BOP (0.18 g, 0.42 mmol) in dichloromethane (5 mL) was added DIEA (0.5 g, 3.9 mmol). This solution was stirred at room temperature and monitored by thin layer chromatography. The resultant mixture was then diluted with 100 mL of ethyl acetate and washed with 1 M HCl (50 mL) and saturated brine (50 mL) before drying over anhydrous magnesium sulfate and concentrating in vacuo. Purification by flash chromatography using ethyl acetate:petroleum ether (1:10) as the eluant afforded the title compound (0.11 g, 0.25 mmol, 90% yield) as a colorless crystal consisting of a diastereoisomeric mixture of (±)-24a and (±)-24b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.06 (s, 1H), 9.01 (s, 1H), 5.53–5.42 (m, 1H), 5.38–5.28 (m, 1H), 4.89–4.62 (m, 1H), 4.14–4.03 (m, 1H), 4.02–3.90 (m, 1H), 3.88–3.80 (m, 1H), 3.78 (s, 3H), 3.75–3.66 (m, 1H), 2.80–2.58 (m, 4H), 2.25–2.12 (m, 2H), 1.84–1.56 (m, 8H), 1.51 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.34, 169.88, 161.34, 154.61, 151.22, 101.08, 97.98, 97.70, 97.17, 96.21, 62.39, 61.74, 52.42, 48.14, 30.97, 30.84, 29.68, 24.29, 24.07, 23.83, 23.64, 23.17, 22.90, 18.53. EI-MS m/z 447 (M). EI-HR-MS m/z found: 447.1886; calcd. for C23H29O8N1: 447.1888.
3.6.2. Compound 25
The title compound was obtained as a colorless crystal in 54% yield as a diastereoisomeric mixture of (±)-25a and (±)-25b (~1:1). 1H NMR (400 MHz, CDCl3) δ 12.93 (s, 1H), 5.44 (dd, J = 6.0, 2.5 Hz, 1H), 5.34 (dd, J = 5.1, 2.7 Hz, 1H), 4.21–3.88 (m, 2H), 3.84–3.68 (m, 2H), 2.94 (t, J = 7.3 Hz, 2H), 2.76–2.50 (m, 4H), 2.24–2.10 (m, 2H), 1.72–1.59 (m, 8H), 1.56–1.30 (m, 4H), 0.94 (t, J = 7.3 Hz, 3H). EI-MS m/z 434 (M). EI-HR-MS m/z found: 434.1759; calcd. for C23H30O6S1: 434.1758.
3.6.3. Compound 26
The title compound was obtained as a light yellow crystal in 98% yield as a diastereoisomeric mixture of (±)-26a and (±)-26b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.36 (s, 1H), 8.53 (s, 1H), 7.14 (dd, J = 5.1, 1.2 Hz, 1H), 6.92 (dd, J = 5.1, 3.4 Hz, 1H), 6.88 (dd, J = 3.4, 0.9 Hz, 1H), 5.38–5.27 (m, 2H), 4.03–3.90 (m, 1H), 3.81 (m, 1H), 3.76–3.56 (m, 4H), 3.30–3.01 (m, 2H), 2.83–2.51 (m, 4H), 2.24–2.08 (m, 2H), 1.85–1.57 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 170.59, 161.31, 154.27, 154.18, 150.74, 150.64, 141.46, 126.87, 125.31, 123.75, 101.30, 101.20, 98.03, 97.93, 97.47, 97.22, 97.14, 96.43, 62.42, 62.34, 62.20, 40.70, 30.95, 30.86, 30.65, 30.57, 29.76, 29.65, 24.06, 23.85, 23.76, 23.59, 23.44, 23.32, 22.87, 22.46. EI-MS m/z 471 (M). EI-HR-MS m/z found: 471.1712; calcd. for C25H29O6N1S1: 471.1710.
3.6.4. Compound 27
The title compound was obtained as a light yellow crystal in 70% yield as a diastereoisomeric mixture of (±)-27a and (±)-27b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.12 (s, 1H), 8.73 (s, 1H), 7.31–7.19 (m, 4H), 5.36 (s, 1H), 5.30 (s, 1H), 4.63 (dd, J = 15.2, 5.9 Hz, 1H), 4.48 (dd, J = 15.1, 4.8 Hz, 1H), 4.00–3.85 (m, 1H), 3.75–3.56 (m, 3H), 2.79–2.50 (m, 4H), 2.21–2.05 (m, 2H), 1.76–1.53 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 170.52, 161.40, 154.62, 154.54, 154.49, 150.90, 150.81, 136.89, 133.13, 128.85, 128.78, 101.49, 101.39, 98.15, 98.07, 97.66, 97.34, 97.27, 96.43, 62.85, 62.48, 62.16, 62.07, 42.58, 31.05, 30.97, 30.81, 30.73, 24.13, 23.98, 23.64, 23.59, 23.43, 22.91, 22.57. EI-MS m/z 485 (M). EI-HR-MS m/z found: 485.1606; calcd. for C26H28O6N1Cl1: 485.1600.
3.6.5. Compound 28
The title compound was obtained as a colorless crystal in 100% yield as a diastereoisomeric mixture of (±)-28a and (±)-28b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.59 (s, 1H), 8.41 (s, 1H), 5.44–5.37 (m, 1H), 5.37–5.27 (m, 1H), 4.00–3.88 (m, 2H), 3.83–3.75 (m, 1H), 3.75–3.65 (m, 1H), 2.82–2.53 (m, 4H), 2.23–2.14 (m, 2H), 2.14–2.07 (m, 9H), 1.80–1.69 (m, 8H), 1.68–1.57 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 169.93, 161.59, 161.54, 153.84, 153.76, 150.68, 150.59, 101.41, 101.29, 97.56, 97.26, 97.15, 62.85, 62.37, 61.69, 52.21, 41.69, 36.47, 31.13, 31.01, 30.97, 30.86, 29.68, 29.51, 24.37, 24.21, 24.13, 23.90, 23.72, 23.45, 23.30, 23.17, 23.01, 22.56, 14.18. EI-MS m/z 495 (M). EI-HR-MS m/z found: 495.2612; calcd. for C29H37O6N1: 495.2615.
3.6.6. Compound 29
The title compound was obtained as a colorless crystal in 42% yield as a diastereoisomeric mixture of (±)-29a and (±)-29b (~1:1). 1H NMR (300 MHz, CDCl3) δ 13.89 (s, 1H), 10.60 (s, 1H), 8.17 (s, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 5.54 (t, J = 3.2 Hz, 1H), 5.45–5.33 (m, 1H), 4.05–3.73 (m, 4H), 2.69–2.63 (m, 4H), 2.34–2.14 (m, 2H), 1.75–1.60 (m, 8H). EI-MS m/z 480 (M).
3.6.7. Compound 30
The title compound was obtained as a colorless crystal in 71% yield as a diastereoisomeric mixture of (±)-30a and (±)-30b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.42 (s, 1H), 8.41 (s, 1H), 7.09 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 8.5 Hz, 1H), 5.35–5.30 (m, 1H), 5.30–5.25 (m, 1H), 4.13–3.85 (m, 2H), 3.78–3.58 (m, 4H), 2.90–2.77 (m, 2H), 2.74–2.58 (m, 4H), 2.19–2.11 (m, 2H), 1.74–1.62 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 170.54, 161.34, 161.29, 154.33, 154.15, 150.77, 150.66, 131.05, 129.91, 115.37, 101.36, 98.06, 97.50, 97.27, 97.20, 97.07, 97.01, 96.56, 62.92, 62.35, 40.76, 34.55, 31.03, 30.94, 30.71, 30.65, 29.68, 26.90, 24.13, 23.91, 23.64, 23.49, 23.38, 23.27, 22.90, 22.63, 22.48. EI-MS m/z 481 (M). EI-HR-MS m/z found: 481.2087; calcd. for C27H31O7N1: 481.2095.
3.6.8. Compound 31
The title compound was obtained as a colorless crystal in 51% yield as a diastereoisomeric mixture of (±)-31a and (±)-31b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.02 (s, 1H), 8.87 (s, 1H), 5.46 (s, 1H), 5.34 (s, 1H), 4.93–4.62 (m, 1H), 4.12–3.89 (m, 2H), 3.90–3.39 (m, 5H), 2.87–2.49 (m, 4H), 2.28–2.07 (m, 2H), 1.85–1.45 (m, 12H), 0.97 (d, J = 5.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 173.31, 170.16, 161.39, 154.64, 154.59, 151.26, 151.17, 101.27, 101.17, 98.05, 97.97, 97.76, 97.30, 97.22, 96.29, 62.79, 62.40, 61.61, 52.17, 50.81, 41.61, 31.03, 30.93, 25.03, 24.40, 24.25, 24.17, 24.05, 23.90, 23.67, 23.46, 23.28, 23.16, 22.93, 22.79, 22.57, 22.08. EI-MS m/z 489 (M). EI-HR-MS m/z found: 489.2351; calcd. for C26H35O8N1: 489.2357.
3.6.9. Compound 32
The title compound was obtained as a light yellow crystal in 80% yield as a diastereoisomeric mixture of (±)-32a and (±)-32b (~1:1). 1H NMR (400 MHz, CDCl3) δ 13.97–13.74 (m, 1H), 9.28–9.06 (m, 1H), 5.51–5.40 (m, 1H), 5.40–5.28 (m, 1H), 5.11–4.91 (m, 1H), 4.13–3.88 (m, 2H), 3.82–3.59 (m, 5H), 3.51–3.04 (m, 2H), 2.84–2.47 (m, 4H), 2.27–2.09 (m, 2H), 1.77–1.54 (m, 8H). EI-MS m/z 479 (M). EI-HR-MS m/z found: 479.1605; calcd. for C23H29O8N1S1: 479.1608.
3.6.10. Compound 33
The title compound was obtained as a light yellow crystal in 69% yield as a diastereoisomeric mixture of (±)-33a and (±)-33b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.00 (s, 1H), 10.45 (s, 1H), 7.67–7.45 (m, 2H), 7.13–6.94 (m, 2H), 5.51 (d, J = 2.6 Hz, 1H), 5.34 (d, J = 2.6 Hz, 1H), 4.10–3.61 (m, 4H), 2.92–2.49 (m, 4H), 2.33–2.11 (m, 2H), 1.89–1.56 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 168.80, 161.71, 160.63, 158.21, 154.78, 150.48, 133.82, 122.58, 122.50, 115.71, 115.49, 101.74, 98.33, 97.97, 97.35, 96.55, 62.93, 62.09, 30.82, 24.09, 23.88, 23.38, 23.33, 22.42. EI-MS m/z 455 (M). EI-HR-MS m/z Found: 455.1741; calcd. for C25H26O6N1 F1: 455.1739.
3.6.11. Compound 34
The title compound was obtained as a light yellow crystal in 72% yield as a diastereoisomeric mixture of (±)-34a and (±)-34b (~1:1). 1H NMR (400 MHz, CDCl3) δ 13.94 (s, 1H), 10.52 (s, 1H), 7.55 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 8.8 Hz, 2H), 5.58–5.47 (m, 1H), 5.39–5.30 (m, 1H), 4.06–3.62 (m, 4H), 2.84–2.56 (m, 4H), 2.31–2.12 (m, 2H), 1.90–1.53 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 168.88, 161.82, 161.77, 154.99, 154.91, 150.57, 150.48, 136.48, 129.24, 129.00, 128.86, 122.22, 121.96, 101.79, 101.68, 98.39, 98.30, 98.03, 97.38, 97.31, 96.83, 96.75, 96.60, 62.95, 62.50, 62.09, 62.01, 60.36, 30.95, 30.84, 30.73, 24.12, 23.93, 23.59, 23.37, 23.24, 22.85, 22.43, 14.16. EI-MS m/z 471 (M). EI-HR-MS m/z found: 471.1441; calcd. for C25H26O6N1Cl1: 471.1443.
3.6.12. Compound 35
The title compound was obtained as a colorless crystal in 64% yield as a diastereoisomeric mixture of (±)-35a and (±)-35b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.11 (s, 1H), 8.77 (s, 1H), 5.46–5.38 (m, 1H), 5.39–5.30 (m, 1H), 4.02–3.89 (m, 2H), 3.86–3.80 (m, 2H), 3.79–3.50 (m, 4H), 2.78–2.55 (m, 4H), 2.45 (s, 1H), 2.25–2.11 (m, 2H), 1.81–1.60 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 171.48, 161.28, 154.49, 150.92, 150.82, 101.45, 98.20, 97.69, 97.34, 97.27, 96.47, 62.85, 62.56, 62.46, 62.36, 62.26, 42.20, 31.07, 30.99, 30.84, 30.76, 24.12, 23.95, 23.88, 23.61, 23.53, 23.47, 22.57, 14.18. EI-MS m/z 405 (M). EI-HR-MS m/z found: 405.1780; calcd. for C21H27O7N1: 405.1782.
3.6.13. Compound 36
The title compound was obtained as a colorless crystal in 77% yield as a diastereoisomeric mixture of (±)-36a and (±)-36b (~1:1). 1H NMR (400 MHz, CDCl3) δ 7.25–7.11 (m, 5H), 5.41–5.06 (m, 2H), 4.05–3.85 (m, 2H), 3.72 (s, 2H), 3.71–3.53 (m, 2H), 2.91–2.75 (m, 1H), 2.75–2.42 (m, 4H), 2.25–2.00 (m, 2H), 1.81–1.33 (m, 8H), 1.04 (d, J = 6.3 Hz, 6H). EI-MS m/z 493 (M). EI-HR-MS m/z found: 493.2453; calcd. for C29H35O6N1: 493.2459.
3.6.14. Compound 37
The title compound was obtained as a colorless crystal in 60% yield as a diastereoisomeric mixture of (±)-37a and (±)-37b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.43 (s, 1H), 5.42–5.33 (m, 1H), 5.32–5.25 (m, 1H), 4.04–3.85 (m, 2H), 3.79–3.64 (m, 2H), 3.57–3.27 (m, 2H), 2.77–2.42 (m, 6H), 2.24 (s, 6H), 2.17–2.07 (m, 2H), 1.70–1.52 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 170.28, 161.19, 161.15, 154.09, 154.02, 153.96, 150.95, 150.87, 150.73, 150.65, 101.21, 101.11, 101.05, 100.95, 97.79, 97.75, 97.71, 97.67, 97.44, 97.13, 97.06, 96.58, 96.45, 77.32, 77.00, 76.68, 62.74, 62.31, 61.93, 61.86, 61.71, 61.64, 57.47, 44.98, 39.10, 36.91, 31.77, 30.94, 30.89, 30.85, 30.80, 30.75, 30.66, 29.48, 29.45, 29.19, 26.94, 24.16, 24.12, 24.02, 23.82, 23.62, 23.53, 23.40, 23.28, 22.78, 22.53, 22.38, 13.96. EI-MS m/z 432 (M). EI-HR-MS m/z found: 432.2262; calcd. for C23H32O6N2: 432.2255.
3.6.15. Compound 38
The title compound was obtained as a colorless crystal in 71% yield as a diastereoisomeric mixture of (±)-38a and (±)-38b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.46 (s, 1H), 8.44 (s, 1H), 5.41 (d, J = 2.6 Hz, 1H), 5.31 (d, J = 2.5 Hz, 1H), 4.03–3.85 (m, 2H), 3.84–3.62 (m, 2H), 3.55–3.22 (m, 2H), 2.91–2.47 (m, 4H), 2.25–2.09 (m, 2H), 1.77–1.54 (m, 10H), 1.40–1.21 (m, 18H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.72, 161.59, 154.45, 154.38, 151.16, 151.07, 102.95, 101.66, 101.56, 100.31, 98.17, 97.87, 97.57, 97.50, 96.93, 67.56, 63.18, 62.84, 62.75, 62.35, 62.28, 39.65, 39.54, 35.12, 32.20, 31.36, 31.27, 31.18, 31.09, 29.91, 29.88, 29.62, 28.81, 27.37, 25.67, 24.44, 24.41, 24.30, 24.24, 24.09, 23.80, 23.72, 23.22, 23.13, 22.96, 22.82, 14.38. EI-MS m/z 529 (M). EI-HR-MS m/z found: 529.3404; calcd. for C31H47O6N1: 529.3398.
3.6.16. Compound 39
The title compound was obtained as a colorless crystal in 61% yield as a diastereoisomeric mixture of (±)-39a and (±)-39b (~1:1). 1H NMR (400 MHz, CDCl3) δ 14.21 (s, 1H), 8.74 (s, 1H), 5.49–5.38 (m, 1H), 5.38–5.27 (m, 1H), 4.06–3.88 (m, 2H), 3.84–3.64 (m, 3H), 3.62–3.48 (m, 1H), 2.86–2.51 (m, 6H), 2.25–2.11 (m, 2H), 2.04 (s, 1H), 1.82–1.57 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 170.70, 161.42, 161.38, 154.51, 154.45, 150.96, 150.87, 101.44, 101.35, 98.16, 98.07, 97.73, 97.33, 97.27, 96.49, 62.84, 62.45, 62.36, 42.37, 31.07, 30.99, 30.88, 30.80, 24.52, 24.12, 24.02, 23.97, 23.67, 23.60, 23.53, 23.47, 22.92, 22.59. EI-MS m/z 421 (M). EI-HR-MS m/z found: 421.1551; calcd. for C21H27O6N1S1: 421.1554.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}