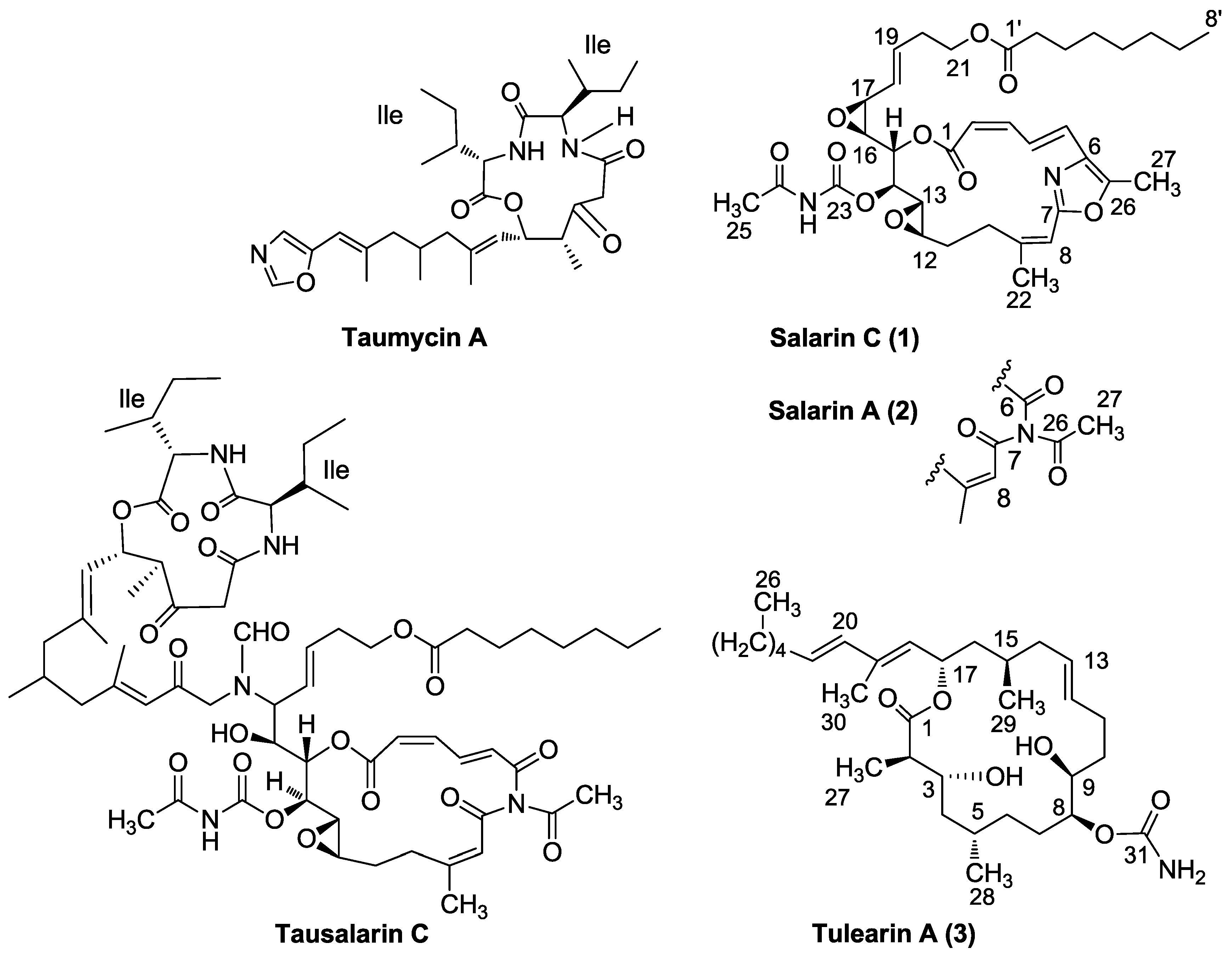
3.2. Salarin Derivatives
The NMR data of the salarin derivatives is given only for the transformed sites, for the rest of the functional moieties of the molecules the chemical shift changes of the various atoms were minimal (Δδ
H ± 0.1, Δδ
C ± 0.5). Thus for example the chemical shift of H-4 is very characteristic for the dienoate moiety and its surroundings (8.31 ppm). Full representative NMR spectra are given in the
Supporting Information. The NMR data of the changed sites are given for clarity in order of the atom numbers.
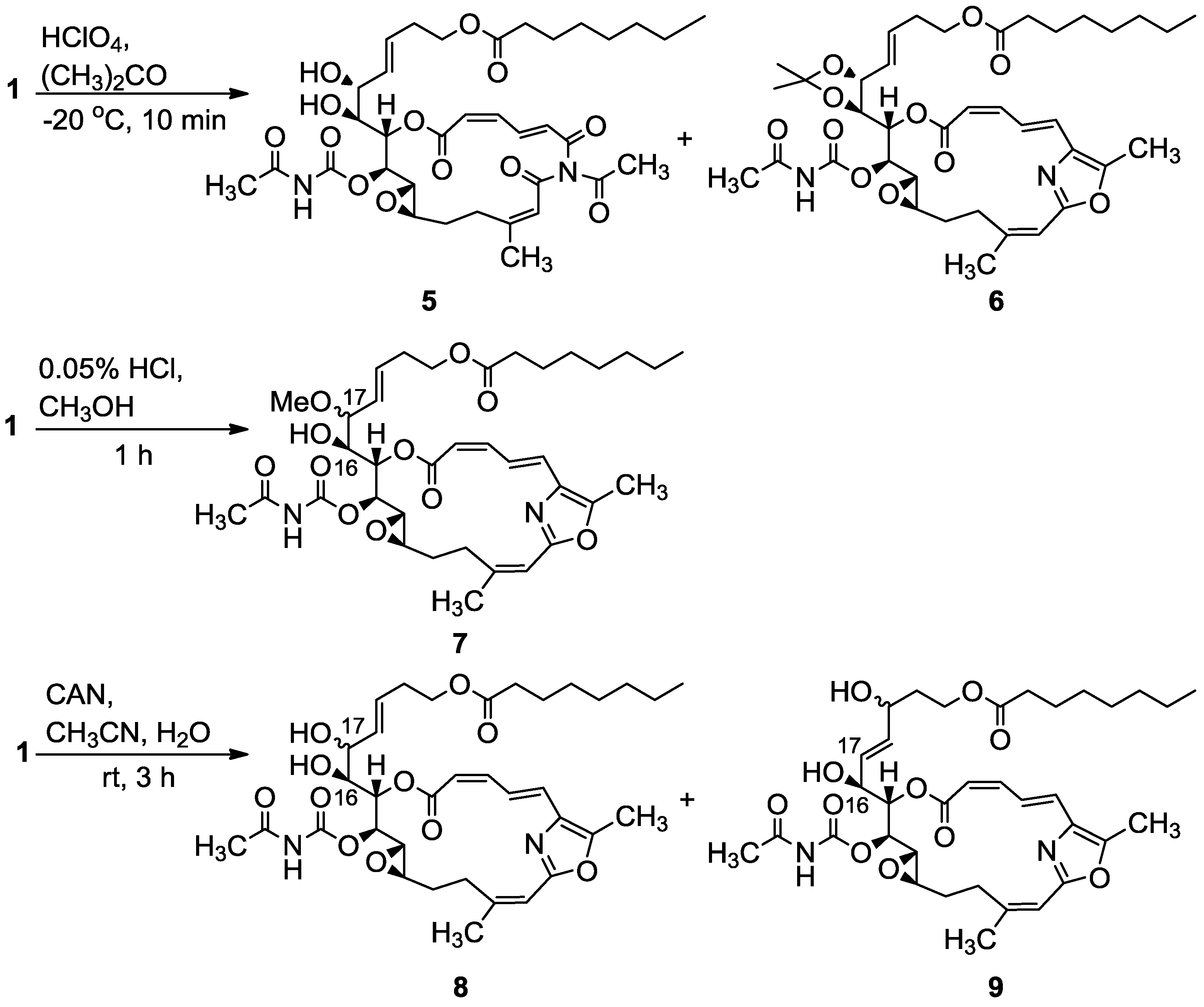
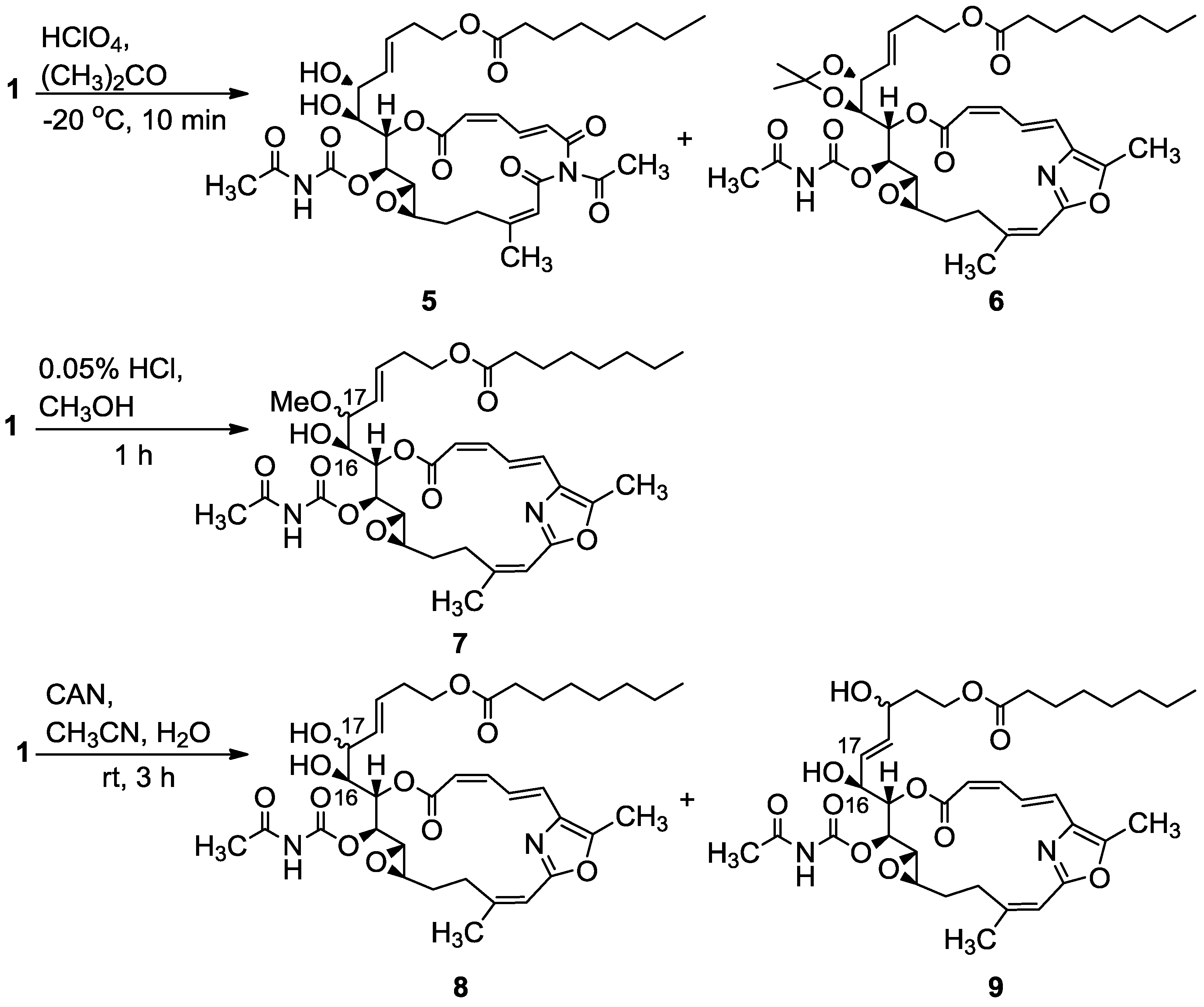
3.2.1. Compounds 5 and 6
To a mixture of salarin C (20 mg, 0.03 mmol) in acetone (5 mL), was added HClO4 (7%, 0.1 mL) at −20 °C and the reaction was stirred for 10 min. The reaction mixture was neutralized with aqueous NaHCO3 solution, evaporated and the residue diluted with water and extracted with DCM. The combined organic extract was dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (vacuum liquid chromatography, petroleum ether/ethyl acetate, 8:2) to afford 6, as a colorless oil, 3 mg (20%) and 5, a second colorless oil, 4.5 mg (21%). 6: [α]D23 +22 (c 0.1, CHCl3); NMR data for modified site (C14–C19): 1H-NMR (CDCl3, 400 MHz) δ 4.80 (dd, J = 8.7, 2.2 Hz, 1H, H-14), 5.61 (m, 1H, H-15), 4.00 (dd, J = 9.8, 7.8 Hz, 1H, H-16), 4.27 (t, J = 7.8 Hz, 1H, H-17), 5.41 (dd, J = 15.6, 8.1 Hz, 1H, H-18), 5.64 (m, 1H, H-19); acetonide moiety: 1.44 (s, 3H, H-29), 1.43 (s, 3H, H-30); 13C-NMR (CDCl3, 100 MHz) δ 76.4 d (C-14), 72.3 d (C-15), 78.3 d (C-16), 82.4 d (C-17), 127.5 (C-18), 131.8 d (C-19); acetonide moiety: 101.7 s (C-28), 28.7 q (Me-29), 27.3 q (Me-30). FABMS m/z 735.3, (C38H52N2O11Na, M + Na+); 5: [α]D23 +72 (c 0.05, CHCl3); NMR data for modified site (C14–C19): 1H-NMR (CDCl3, 400 MHz) δ 5.04 (t, J = 4.3 Hz, 1H, H-14), 5.44 (dd, J = 6.8, 4.3 Hz, 1H, H-15), 3.94 (t, J = 6.8 Hz, 1H, H-16), 4.37 (t, J = 7.9 Hz, 1H, H-17), 5.48 (dd, J = 15.4, 7.9 Hz, 1H, H-18), 5.77 (dt, J = 15.4, 7.9 Hz, 1H, H-19); 13C-NMR (CDCl3, 100 MHz) δ 73.1 d (C-14), 73.4 d (C-15), 78.3 d (C-16), 80.9 d (C-17), 128.3 d (C-18), 133.0 d (C-19). FABMS m/z 727.3, (C35H48N2O13Na, M + Na+).
3.2.2. Compound 7
A solution of salarin C (12 mg, 0.02 mmol) in MeOH (10 mL) was treated with methanolic HCl solution (0.05% v/v, 0.1 mL). The mixture was stirred at rt in the dark for 1 h. The reaction mixture was then neutralized with aqueous NaHCO3 solution, the solvent was evaporated and the residue diluted with water and extracted with DCM. The combined organic extract was dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (vacuum liquid chromatography, petroleum ether/ethyl acetate, 7:3) to afford 7, as a colorless oil, 8 mg (65%). [α]D24 +56 (c 0.26, CHCl3); NMR data for modified site (C14–C19): 1H-NMR (CDCl3, 500 MHz) δ 4.85 (dd, J = 10.8, 2.5 Hz, 1H, H-14), 5.53 (dd, J = 9.7, 2.5 Hz, 1H, H-15), 3.80 (dt, J = 9.7, 5.3 Hz, 1H, H-16), 3.40 (dd, J = 8.3, 5.3 Hz, 1H, H-17), 5.31 (dd, J = 15.5, 8.3 Hz, 1H, H-18), 5.57 (m, 1H, H-19), 3.18 (s, 3H, OMe); 13C-NMR (CDCl3, 125 MHz) δ 77.6 d (C-14), 71.4 d (C-15), 72.5 d (C-16), 82.1 d (C-17), 128.2 d (C-18), 131.9 d (C-19), 55.9 q (OMe). HR-ESIMS m/z calculated for C36H50N2O11Na (M + Na+) 709.3312, found 709.3322.
3.2.3. Compounds 8 and 9
A solution of salarin C (25 mg, 0.038 mmol) in CH3CN:H2O (3:1, 0.5 mL), was stirred with cerium ammonium nitrate (41 mg, 0.076 mmol) at rt for 3 h. The solvent was then concentrated in vaccum, H2O and DCM were added and the organic layer was dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford 8, as a colorless oil, 7 mg (24%) and 9, a second colorless oil, 8 mg (35%). 8: [α]D24 +68 (c 0.26, CHCl3); NMR data for modified site (C14–C19): 1H-NMR (CDCl3, 500 MHz) δ 4.95 (dd, J = 9.3, 2.4 Hz, 1H, H-14), 5.55 (dd, J = 9.3, 3.9 Hz, 1H, H-15), 3.77 (ddd, J = 9.8, 6.2, 3.9 Hz, 1H, H-16), 4.07 (t, J = 6.2 Hz, 1H, H-17), 5.56 (dd, J = 15.8, 6.2 Hz, 1H, H-18), 5.72 (m, 1H, H-19); 13C-NMR (CDCl3, 100 MHz) δ 77.2 d (C-14), 71.6 d (C-15), 72.5 d (C-16), 70.7 d (C-17), 130.4 d (C-18), 129.5 d (C-19); FABMS m/z 673.3, (C35H49N2O11, MH+). 9: [α]D23 +70 (c 0.2, CHCl3); NMR data for modified site (C14–C19): 1H-NMR (CDCl3, 400 MHz) δ 4.89 (dd, J = 8.6, 2.2 Hz, 1H, H-14), 5.48 (dd, J = 8.6, 2.2 Hz, 1H, H-15), 4.45 (m, 1H, H-16), 5.67 (dd, J = 15.5, 6.1 Hz, 1H, H-17), 5.74 (m, 1H, H-18), 4.15 (m, 1H, H-19); 13C-NMR (CDCl3, 100 MHz) δ 76.4 d (C-14), 72.9 d (C-15), 71.2 d (C-16), 127.3 d (C-17), 136.4 d (C-18), 67.6 d (C-19); HR-ESIMS m/z calculated for C35H48N2O11Na (M + Na+) 695.3150, found 695.3135.
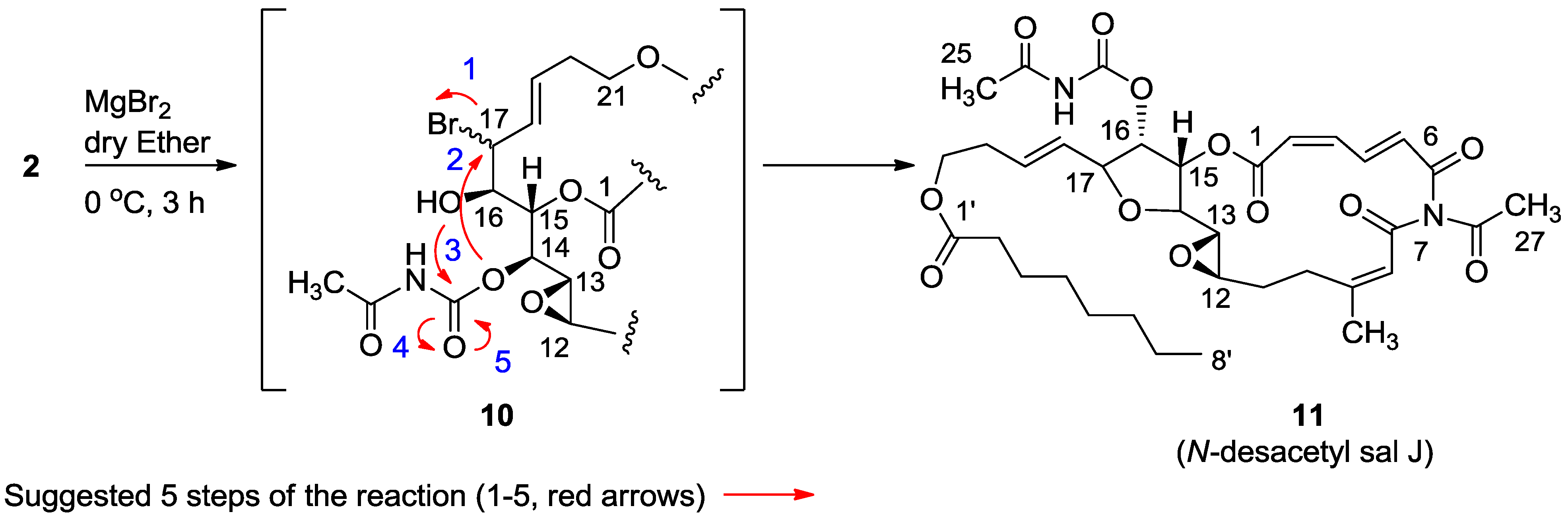
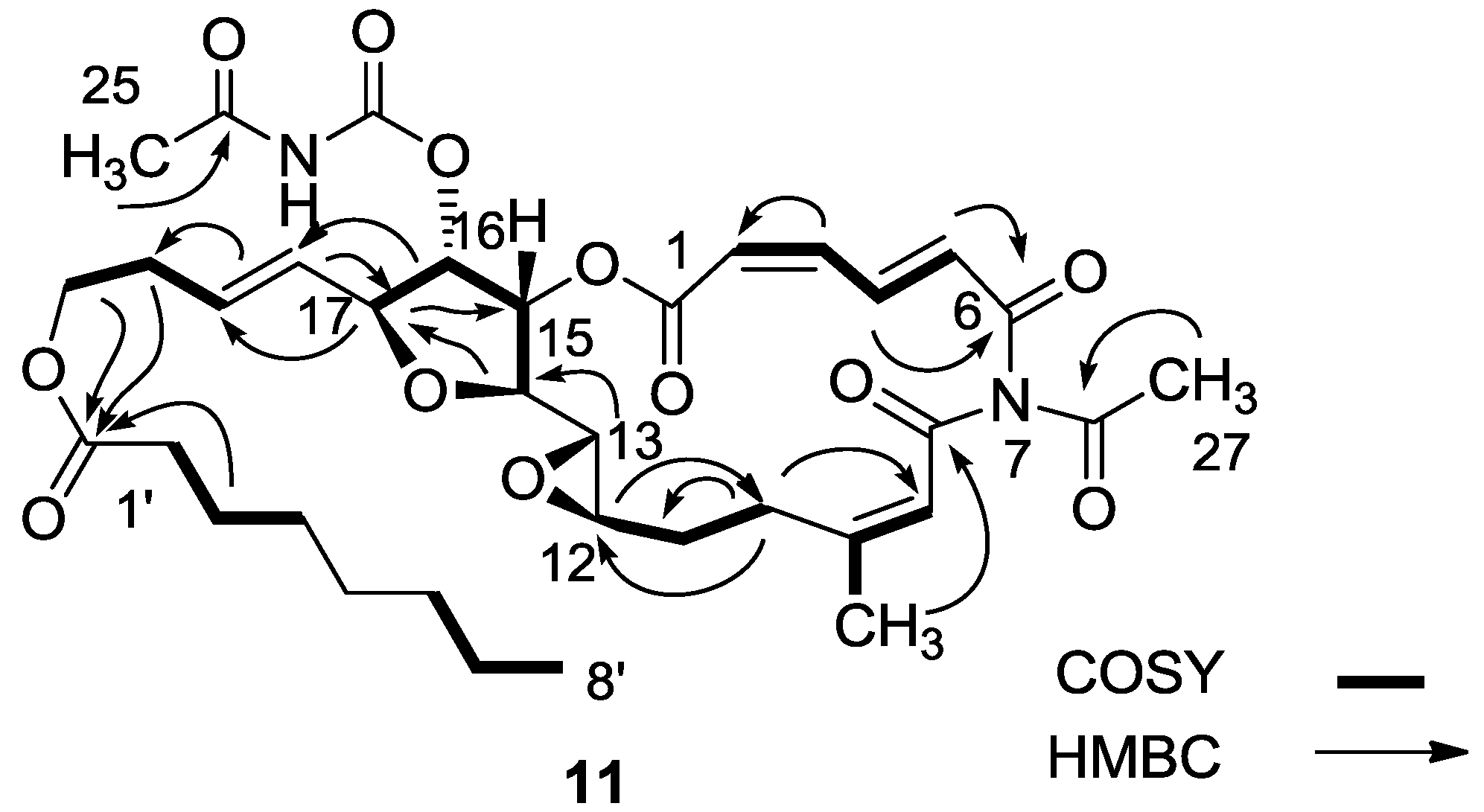
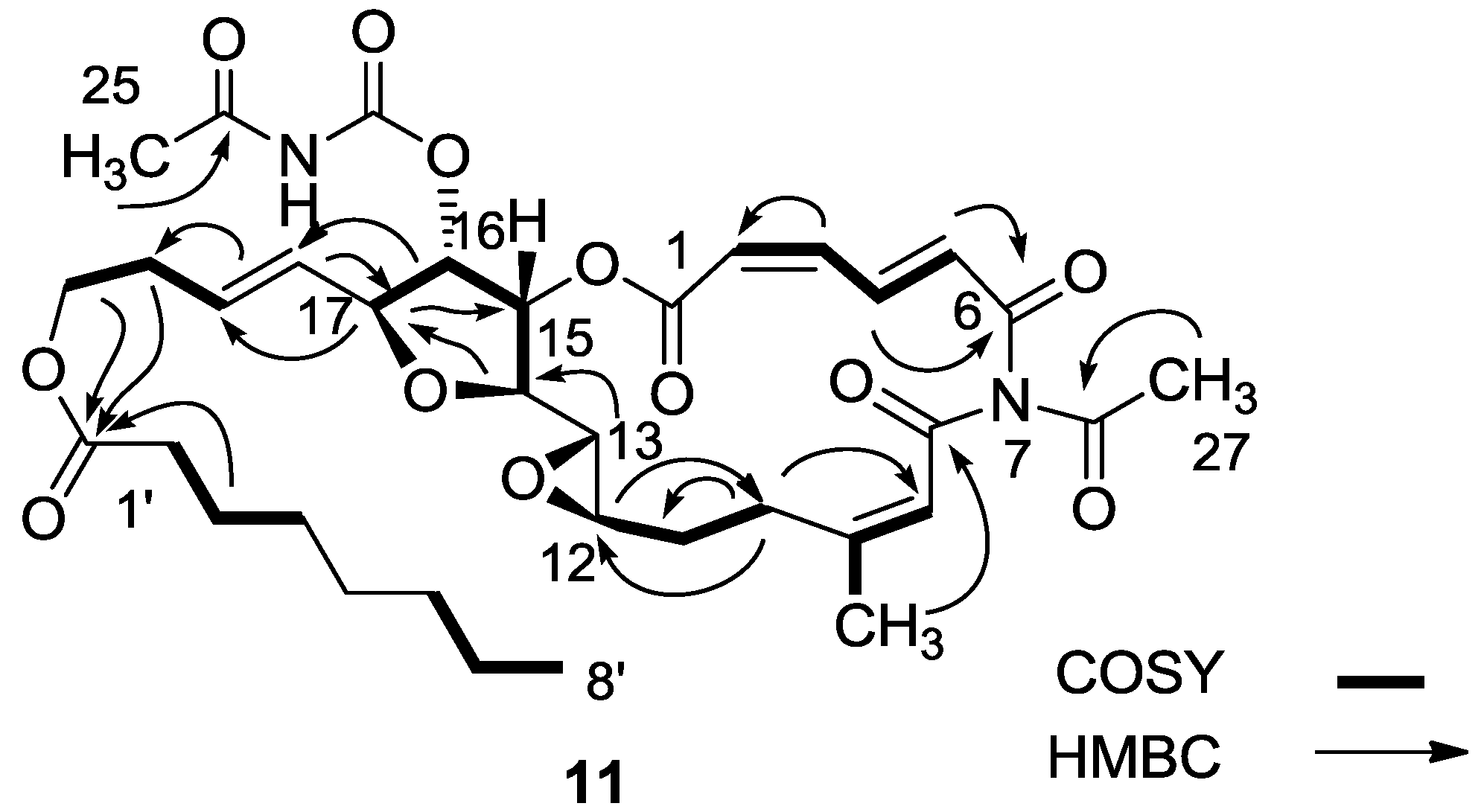
3.2.4. Compound 11
MgBr2 was prepared from magnesium turnings (100 mg) in dry diethyl ether (10 mL) and dibromoethane (1.5 mL). A solution of salarin A (21 mg, 0.03 mmol) in dry diethyl ether (1 mL), was added dropwise to the 0 °C mixture of the MgBr2. The solution was stirred for 2.5 h, then water added and the solution extracted with diethyl ether. The organic layer was washed with brine, dried over anhydrous MgSO4 and the solvents were evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford a bromohydrin product which was unstable; ESIMS m/z 789.2, (C34H45BrN2O12Na, M + Na+). After 3 h, a second VLC (PE/EA, 1:1) afforded 11, 8 mg (40%). NMR data for modified site (C13–C19): 1H-NMR (CDCl3, 500 MHz) δ 3.03 (brt, J = 2.4 Hz, 1H, H-13), 4.21 (brt, J = 2.4 Hz, 1H, H-14), 5.34 (dd, J = 5.1, 2.4 Hz, 1H, H-15), 5.04 (dd, J = 7.0, 5.1, 1H, H-16); 4.40 (t, J = 6.8 Hz, 1H, H-17); 5.56 (dd, J = 14.9, 6.8 Hz, 1H, H-18); 5.79 (m, 1H, H-19); 13C-NMR (CDCl3, 125 MHz) δ 55.0 d (C-13), 80.0 d (C-14), 74.5 d (C-15), 75.4 d (C-16), 80.9 d (C-17), 128.9 d (C-18), 130.2 d (C-19). FABMS m/z 709.3, (C35H46N2O12Na, M + Na+).
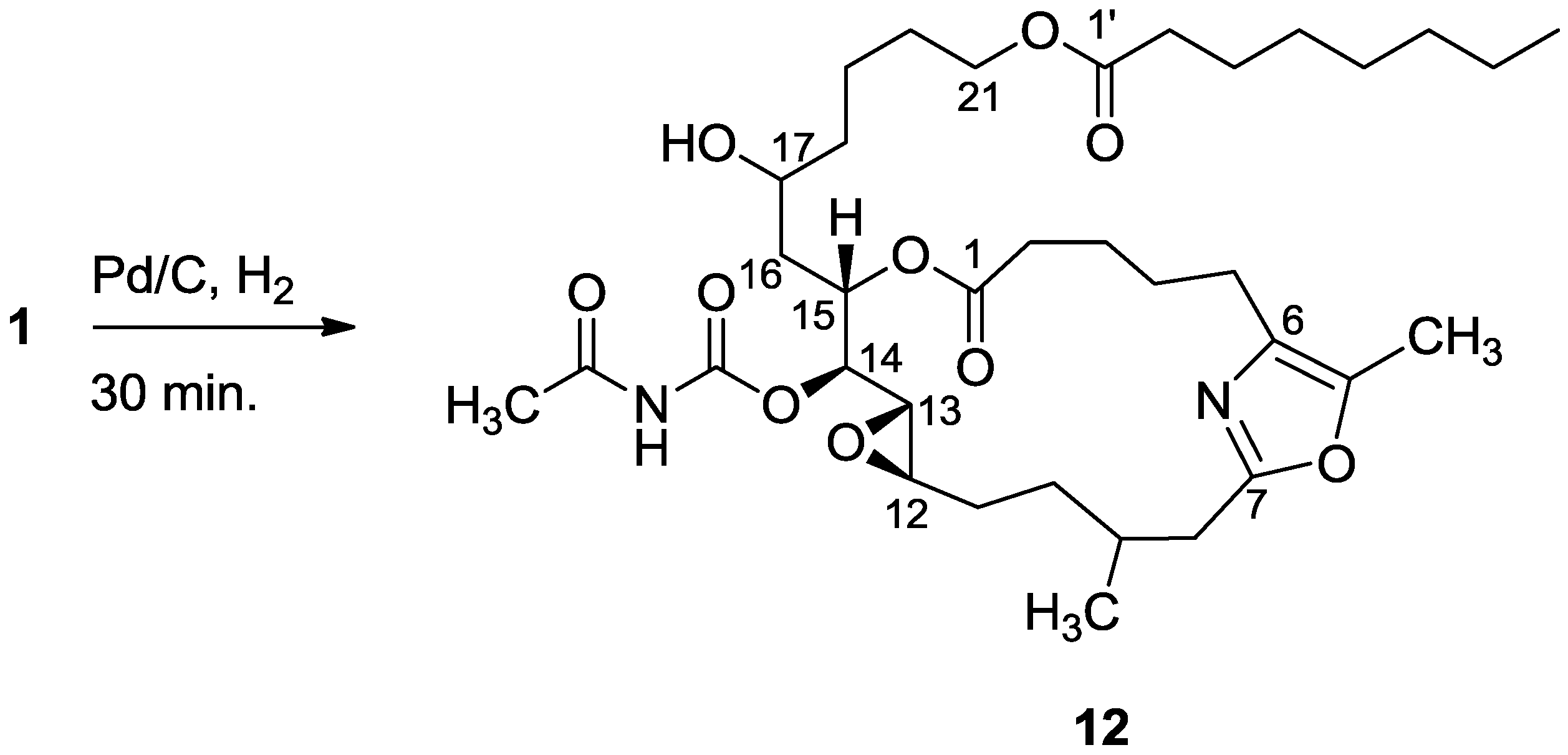
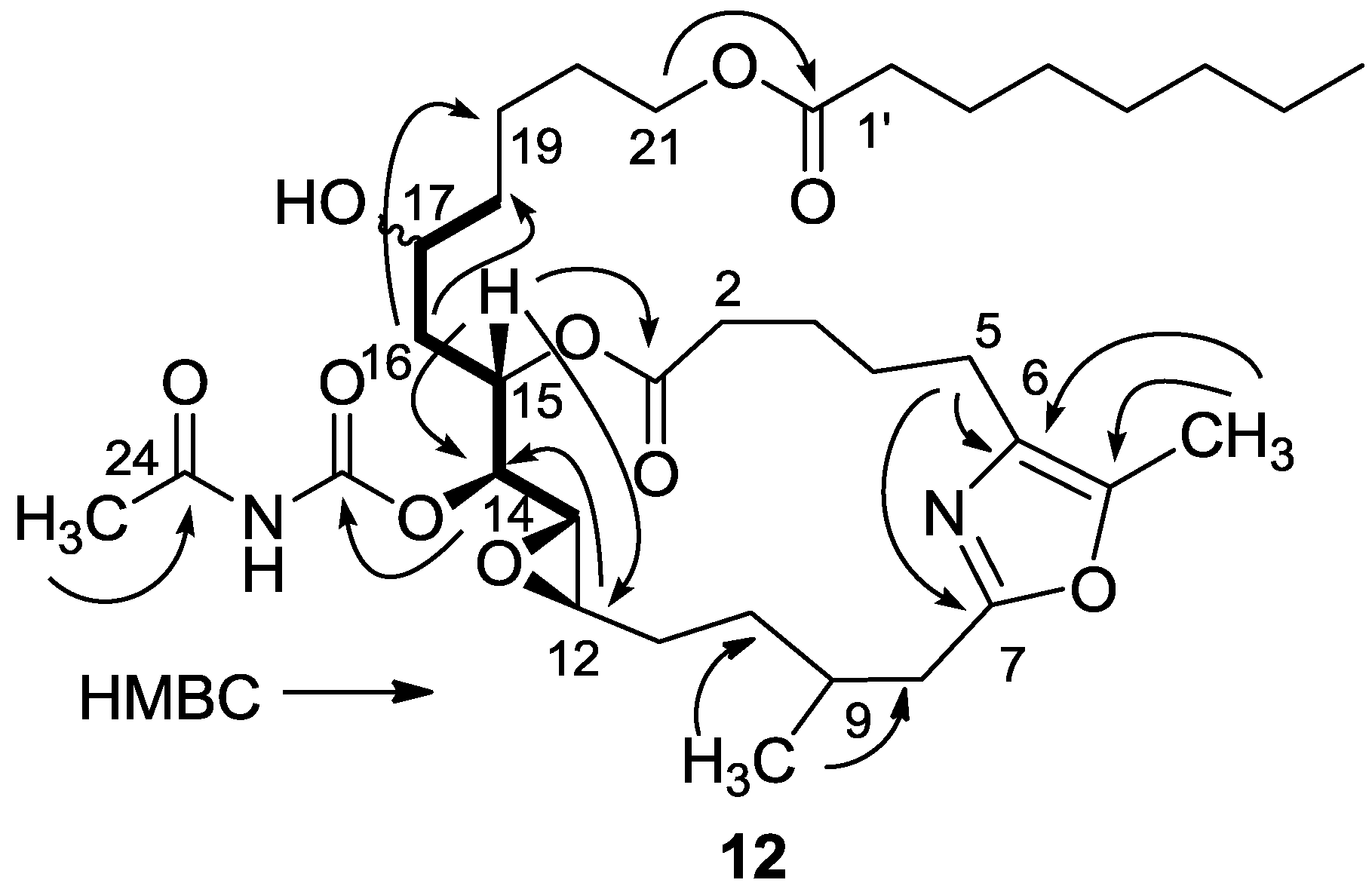
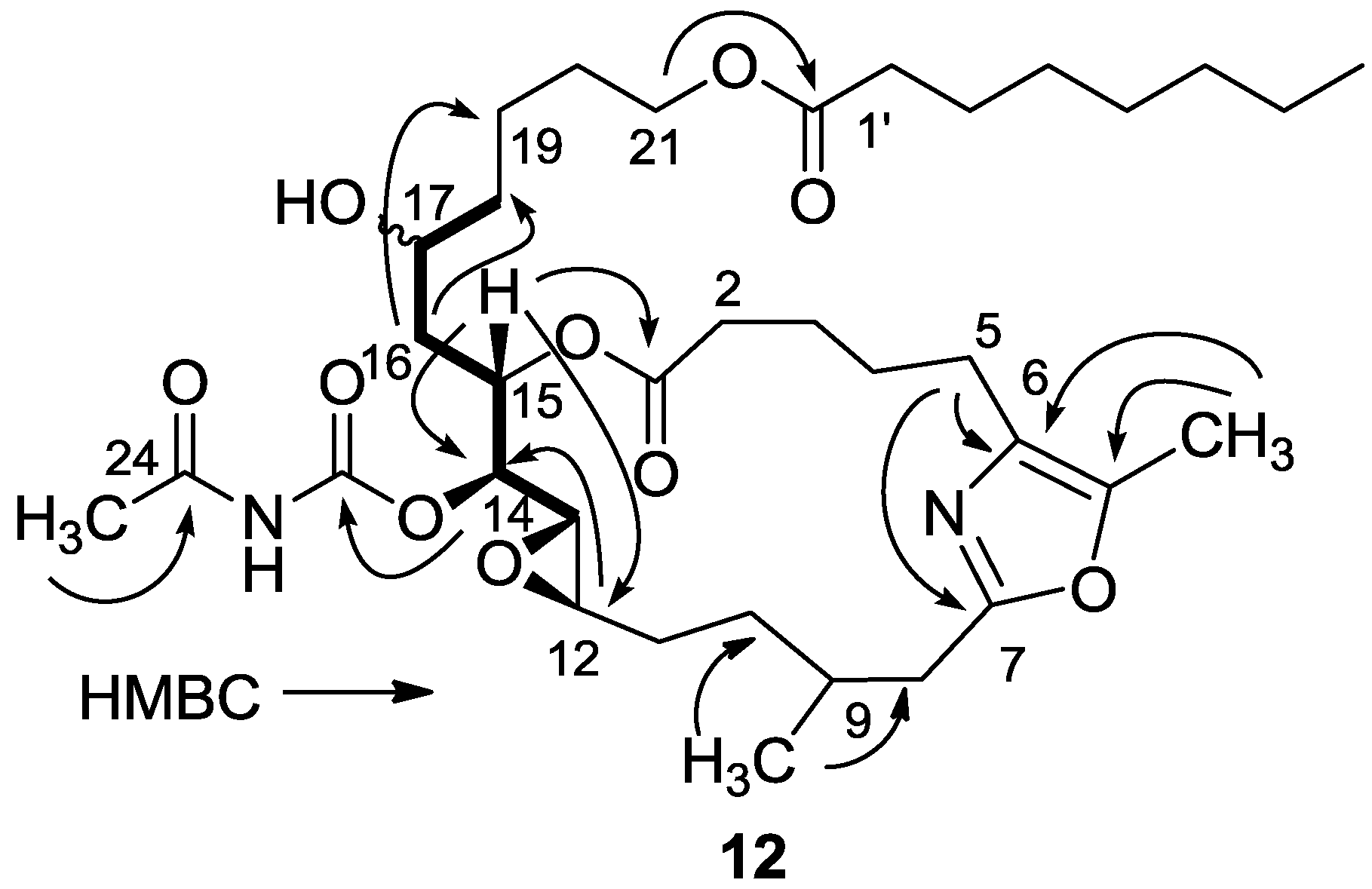
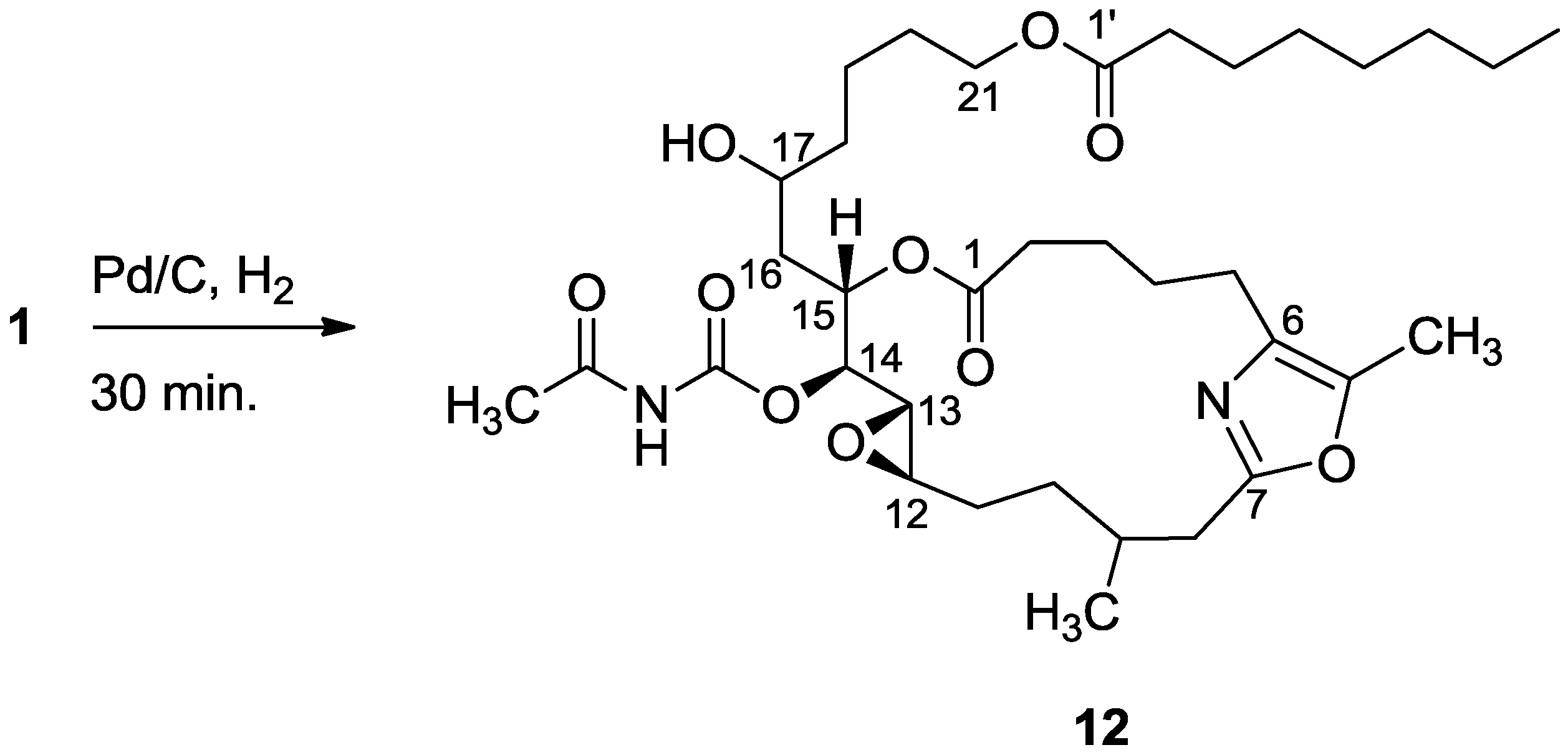
3.2.5. Compound 12
A solution of salarin C (10 mg, 0.015 mmol) in EtOH (5 mL) with catalytic amounts of Pd/C (5%) was hydrogenated for 30 min at 1 atmosphere to afford 12, as a colorless oil, 8 mg (80%). [α]D23 −22 (c 0.14, CHCl3); NMR data for modified site (C14–20, C26): 1H-NMR (CDCl3, 500 MHz) δ 2.50 (m, 2H, H-8), 2.13 (m, 1H, H-9), 1.84 (m, 2H, H-10), 3.32 (dd, J = 4.4, 2.2 Hz, H-13), 4.77 (dd, J = 7.5, 2.9 Hz, 1H, H-14), 5.34 (dd, J = 7.5, 3.2 Hz, 1H, H-15), 2.74 (dd, J = 14.9, 3.2 Hz, 1Ha, H-16), 2.49 (m, 1Hb, H-16), 3.71 (m, 1H, H-17), 2.39 (m, 2H, H-18), 1.73 (m, 2H, H-19), 1.64 (m, 2H, H-20); COSY correlations and J values supported the identification of the later protons.
13C-NMR (CDCl3, 125 MHz) δ 172.2 s (C-1), 33.1 t (C-2), 25.7 t (C-3), 31.6 t (C-4), 23.9 t (C-5), 133.2 s (C-6), 161.2 s (C-7), 35.4 t (C-8), 29.8 d (C-9), 30.6 t (C-10), 76.3 d (C-14), 75.8 d (C-15), 34.3 t (C-16), 71.1 d (C-17), 35.1 t (C-18), 22.6 t (C-19), 28.5 t (C-20), 142.3 s (C-26). FABMS m/z 687.3, (C35H56N2O10Na, M + Na+).
3.2.6. Compound 13
A mixture of salarin C (30 mg, 0.043 mmol), morpholine (6 mg, 0.08 mmol) and Zn(ClO4)2·6H2O (0.3 mg, 2 mol%) in dry DCM (2 mL) was stirred at rt under argon in the dark for 48 h. After completion of the reaction (TLC), DCM (5 mL) was added and the mixture washed with water, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 3:2) to afford 13, as a yellow oil, 28 mg (78%). [α]D27 +14 (c 0.13, CHCl3); NMR data for modified site (C14–C19, C25): 1H-NMR (CDCl3, 500 MHz) δ 4.93 (dd, J = 8.9, 2.5 Hz, 1H, H-14), 5.48 (m, 1H, H-15), 3.85 (t, J = 9.2 Hz, 2H, H-16), 2.80 (t, J = 9.2 Hz, 1H, H-17), 5.27 (dd, J = 15.4 Hz, 9.2, 2H, H-18), 5.47 (m, 2H, H-19), 2.44 (s, 3H, H-25); morpholine moiety: 2.52 (m, 4H, CH2N), 3.68 (m, 4H, CH2O); 13C-NMR (CDCl3, 125 MHz) δ 77.8 d (C-14), 73.7 d (C-15), 66.9 d (C-16), 71.5 d (C-17), 125.7 d (C-18), 132.8 d (C-19), 23.4 d (C-25); morpholine moiety: 48.6 t (CH2N), 66.9 t (CH2O). HR-ESIMS m/z calculated for C39H56N3O11 (MH+) 742.3915, found 742.3917.
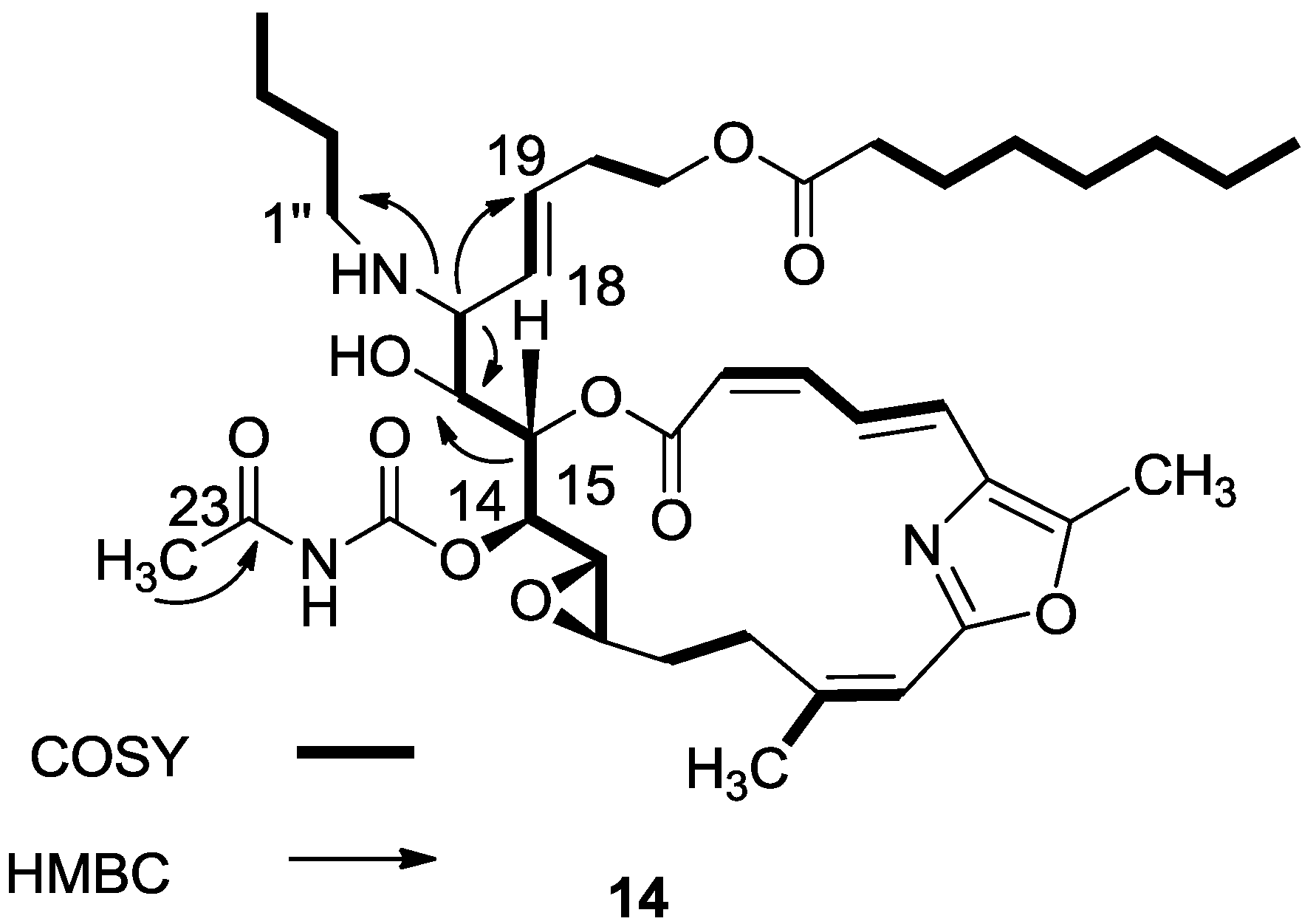
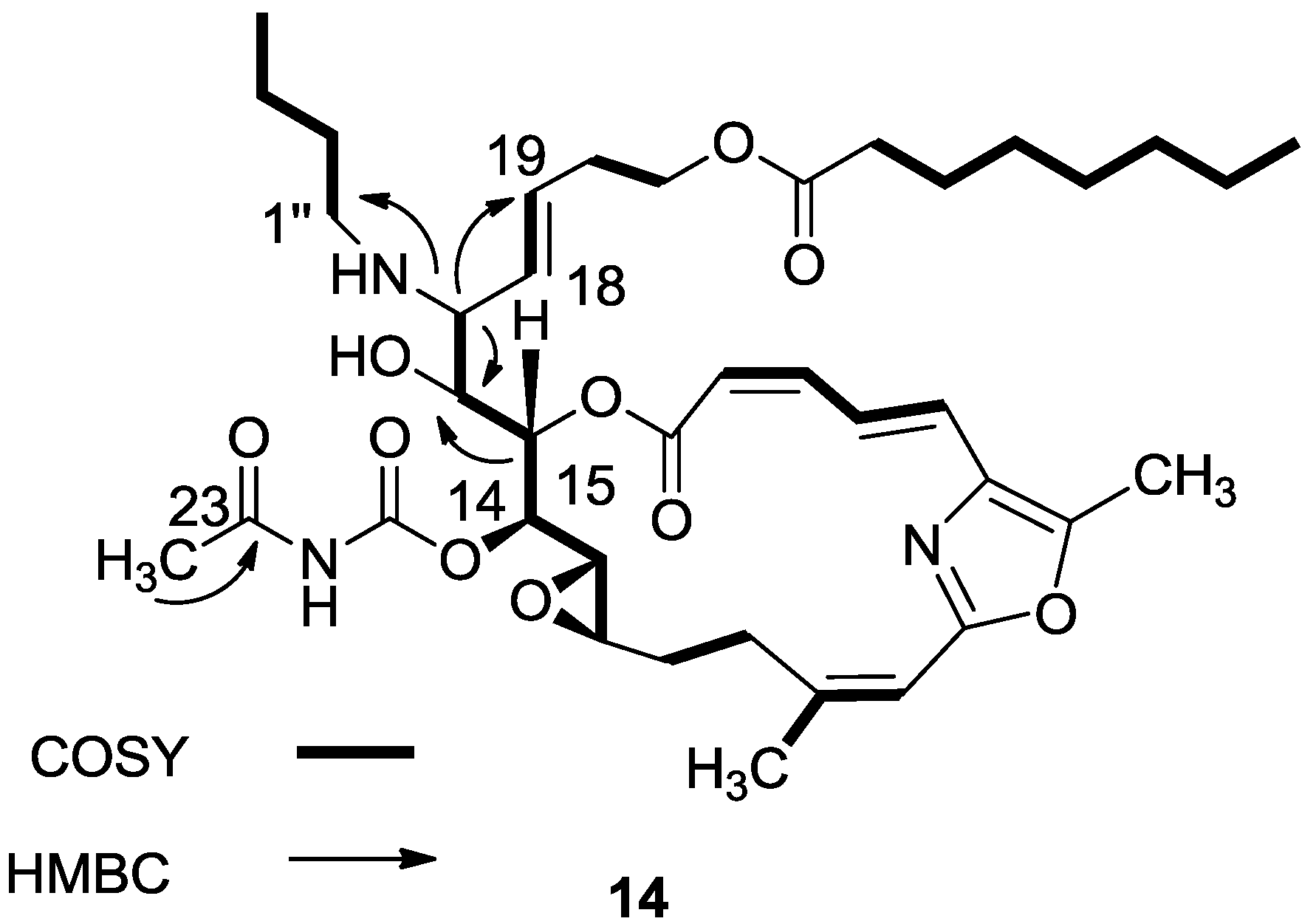
3.2.7. Compound 14
A mixture of salarin C (50 mg, 0.043 mmol), n-butylamine (18 mg, 0.2 mmol) and Zn(ClO4)2·6H2O (0.6 mg, 2 mol%) in dry DCM (4 mL) was stirred at rt under argon in the dark for 48 h. After completion of the reaction (TLC), DCM was added (5 mL), and the mixture was washed with water, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford 14, as a yellow oil, 7 mg (12%). [α]D27 +30 (c 0.17, CHCl3); NMR data for modified site (C14–C19, C25): 1H-NMR (CDCl3, 500 MHz) δ 4.91 (dd, J = 8.8, 2.0 Hz, 1H, H-14), 5.45 (dd, J = 7.7, 2.0 Hz, 1H, H-15), 3.60 (t, J = 7.7 Hz, 1H, H-16), 2.83 (t, J = 7.7 Hz, 1H, H-17), 5.21 (dd, J = 15.0, 8.8 Hz, 1H, H-18), 5.40 (dd, J = 15.0, 6.5 Hz, 1H, H-19), 2.42 (s, 3H, H-25); butylamine moiety: 2.61 (m, CH2NH), 2.34 (m, CH2NH), 1.41 (m, 2H, H-2″); 1.31 (m, 2H, H-3″), 0.92 (t, J = 7.3 Hz, CH3); 13C-NMR (CDCl3, 125 MHz) δ 78.4 d (C-14), 73.8 d (C-15), 71.6 d (C-16), 64.8 d (C-17), 131.0 d (C-18), 129.9 d (C-19), 23.9 d (C-25); butylamine moiety: 46.5 t (CH2NH), 32.3 t (CH2, C-2″), 20.2 t (CH2, C-3″), 14.8 q (CH3). HR-ESIMS m/z calculated for C39H58N3O10 (MH+) 728.4122, found 728.4130.
3.2.8. Compound 15
A mixture of salarin C (20 mg, 0.03 mmol), benzylamine (12 mg, 0.09 mmol) and Zn(ClO4)2·6H2O (0.3 mg, 2 mol%) in dry DCM (4 mL) was stirred at rt under argon in the dark for 72 h. After completion of the reaction (TLC), DCM was added and the mixture was washed with water, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 3:7) to afford 15, as a yellow oil, 9 mg (36%). [α]D27 30 (c 0.17, CHCl3); NMR data for modified site (C14–C19): 1H-NMR (CDCl3, 500 MHz) δ 4.81 (dd, J = 8.6, 2.0 Hz, 1H, H-14), 5.47 (m, 1H, H-15), 3.71 (m, 1H, H-16), 2.93 (t, J = 7.6 Hz, 1H, H-17), 5.26 (dd, J = 16.0, 8.1 Hz, 1H, H-18), 5.50 (m, 1H, H-19); benzylamine moiety: δ 3.78 (d, J = 12.8 Hz, 1Ha, H-26), 3.54 (d, J = 12.8 Hz, 1Hb, H-26), 7.31 (m, 2H, H-28,30), 7.31 (m, 2H, H-29,31), 7.31 (m, 1H, H-30); 13C-NMR (CDCl3, 125 MHz) δ 76.7 d (C-14), 74.3 d (C-15), 72.4 d (C-16), 64.8 d (C-17), 130.7 d (C-18), 130.9 d (C-19), 156.3 s (C-23); benzylamine moiety: 50.9 t (C-26), 140.0 s (C-27), 129.2 d (C-28,32), 129.3 d (C-29,31), 127.8 d (C-30). HR-ESIMS m/z calculated for C40H54N3O9 (MH+) 720.3860, found 720.3865.
3.2.9. Compounds 16 and 18
A mixture of salarin C (25 mg, 0.038 mmol), NH-Boc-hexane-amine (30 mg, 0.11 mmol) and Zn(ClO4)2·6H2O (0.3 mg, 2 mol%) in dry DCM (2 mL) was stirred at rt under argon in the dark for 72 h. After completion of the reaction (TLC), DCM was added and the mixture was washed with water, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 3:7) to afford 16, as a colorless oil, 8 mg (25%) and 18, a second colorless oil, 4 mg (13%). 16: [α]D27 +50 (c 0.15, CHCl3); NMR data for modified site (C14–C19, C23): 1H-NMR (CDCl3, 500 MHz) δ 4.85 (d, J = 9.0 Hz, 1H, H-14), 5.43 (m, 1H, H-15), 3.61 (brt, J = 7.0 Hz, 1H, H-16), 2.86 (brt, J = 7.0 Hz, 1H, H-17), 5.27 (m, 1H, H-18), 5.541 (m, 1H, H-19); NH-Boc-diaminehexane moiety: 2.62 (m, 2H, CH2NH), 2.35 (m, 2H, CH2NH), 1.40 (m, 2H), 1.32 (m, 2H), 1.30 (m, 2H), 1.40 (m, 2H), 3.10 (m, 2H, CH2NH-Boc), 1.44 (s, 9H, CH3); 13C-NMR (CDCl3, 125 MHz) δ 75.1 d (C-14), 72.5 d (C-15), 70.4 d (C-16), 63.2 d (C-17), 130.6 d (C-18), 129.4 d (C-19); 155.1 s (C-23); NH-Boc-diaminehexane moiety: δ 45.4 t (CH2NH), 29.6 t (CH2), 25.5 t (CH2), 25.4 t (CH2), 29.6 t (CH2), 39.2 t (CH2NH-Boc), 155.6 s (CO), 78.9 s (C), δC 26.7 q (3CH3). HR-ESIMS m/z calculated for C44H70N4O11 (MH+) 829.4963, found 829.4969; 18: [α]D27 +53 (c 0.13, CHCl3); NMR data for modified site (C14–C19, C23): 1H-NMR (CDCl3, 500 MHz) δ 3.94 (m, 1H, H-14), 5.27 (m, 1H, H-15), 3.69 (brt, J = 7.1 Hz, 1H, H-16), 2.92 (brt, J = 7.5 Hz, 1H, H-17), 5.31 (dd, J = 15.1, 7.5 Hz, 1H, H-18), 5.45 (dt, J = 15.1, 6.3 Hz, 1H, H-19); NH-Boc-diaminehexane moiety: 2.61 (m, 1Ha, CH2NH), 2.34 (m, 1Hb, CH2NH), 1.48 (m, 2H), 1.34 (m, 2H), 1.30 (m, 2H), 1.40 (m, 2H), 3.11 (m, 2H, CH2NH-Boc), 1.48 (s, 9H, CH3); 13C-NMR (CDCl3, 125 MHz) δ 71.5 d (C-14), 75.8 d (C-15), 71.8 d (C-16), 63.3 d (C-17), 125.5 d (C-18), 130.6 d (C-19); NH-Boc-diaminehexane moiety: 45.9 t (CH2NH), 30.0 t (CH2), 25.5 t (CH2), 25.5 t (CH2), 30.3 t (CH2), 39.8 t (CH2NH-Boc), 156.0 s (CO), 79.0 s (C), δC 27.0 q (3CH3). HR-ESIMS m/z calculated for C43H68N3O10 (MH+) 786.4905, found 786.4918.
3.2.10. Compound 17
A mixture of salarin C (16 mg, 0.024 mmol), 1,6-diaminohexane (8.5 mg, 0.07 mmol) and Zn(ClO4)2·6H2O (0.2 mg, 2 mol%) at 60 °C, neat, was stirred under argon in the dark for 20 min. After completion of the reaction (TLC), DCM was added and the mixture was washed with water, dried over anhydrous MgSO4, and evaporated to afford 17, as a colorless oil, 13 mg (70%). [α]D27 +50 (c 0.3, CHCl3); NMR data for modified site (C14–C19, C25): 1H-NMR (CDCl3, 500 MHz) δ 4.84 (d, J = 9.3 Hz, 1H, H-14), 5.47 (d, J = 9.3 Hz, 1H, H-15), 3.60 (brt, J = 7.6 Hz, 1H, H-16), 2.82 (brt, J = 7.6 Hz, 1H, H-17), 5.23 (dd, J = 15.3, 9.1 Hz, 1H, H-18), 5.42 (m, 1H, H-19), 2.01 (s, 3H, H-25); 1,6-diaminehexane moiety: δ 2.62 (m, 1Ha, CH2NH), 2.31 (m, 1Hb, CH2NH), 1.44 (m, 2H), 1.32 (m, 2H), 1.30 (m, 2H), 1.44 (m, 2H), 2.69 (m, 2H, CH2NH2); 13C-NMR (CDCl3, 125 MHz) δ 75.4 d (C-14), 73.1 d (C-15), 70.1 d (C-16), 63.1 d (C-17), 130.8 d (C-18), 129.0 d (C-19), 155.7 s (C-23), 170.2 s (C-24), 24.8 q (C-25); 1,6-diaminehexane moiety: δ 45.8 t (CH2NH), 32.5 t (CH2), 26.1 t (CH2), 26.1 (CH2), 32.5 t (CH2), 41.3 t (CH2NH2); HR-ESIMS m/z calculated for C41H63N4O10 (MH+) 771.4544, found 771.4547.
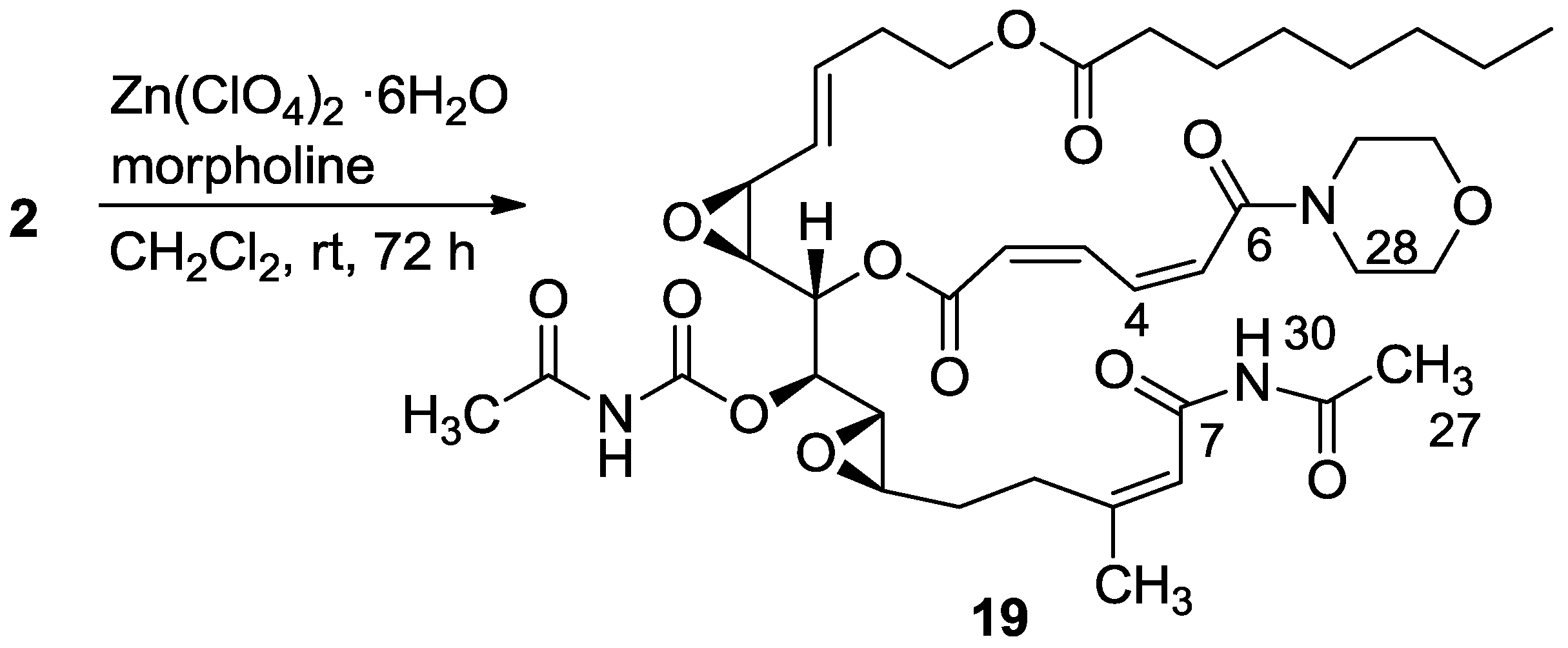
3.2.11. Compound 19
A mixture of salarin A (30 mg, 0.043 mmol), morpholine (6 mg, 0.08 mmol) and Zn(ClO4)2·6H2O (0.3 mg, 2 mol%) in dry DCM (2 mL) was stirred at rt under argon. After completion of the reaction (72 h, TLC), the mixture was diluted with DCM (5 mL), washed with water, dried over anhydrous MgSO4 and then evaporated. The crude residue was purified by VLC (petroleum ether/ethyl acetate, 4:6) to afford 19, as a yellow oil, 28 mg (78%). [α]D23 −30 (c 0.16, CHCl3); NMR data for modified site (C1–7, C26,27): 1H-NMR (CDCl3, 500 MHz) δ 5.90 (m, 1H, H-2), 6.70 (t, J = 11.8 Hz, 1H, H-3), 8.19 (dd, J = 15.0, 11.8 Hz, 1H, H-4), 6.57 (d, J = 15.0 Hz, 1H, H-5), 2.34 (s, 3H, H-27), 8.15 (s, 1H, NH); Morpholine: 3.71 (m, 4H, 2 × CH2O), 3.74 (m, 4H, 2 × CH2NH); 13C-NMR (CDCl3, 100 MHz) δ 164.1 s (C-1), 121.9 d (C-2), 140.0 d (C-3), 137.4 d (C-4), 126.6 d (C-5), 164.0 s (C-6), 163.6 s (C-7), 171.9 s (C-26), 23.8 q (C-27); Morpholine: 66.0 t (CH2O), 42.5 t (CH2NH); FABMS m/z 796.1, (C39H55N3O13Na, M + Na+).
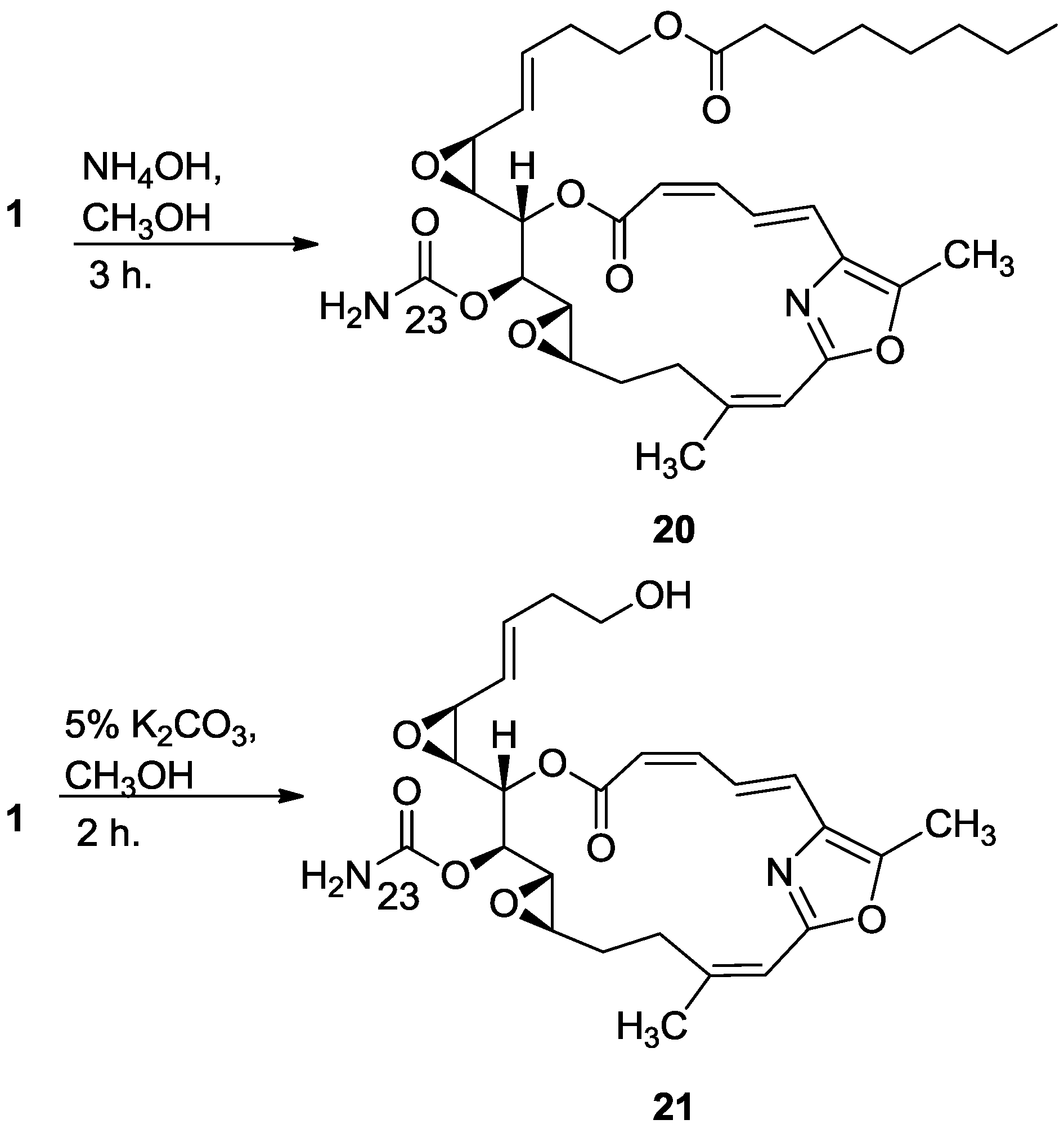
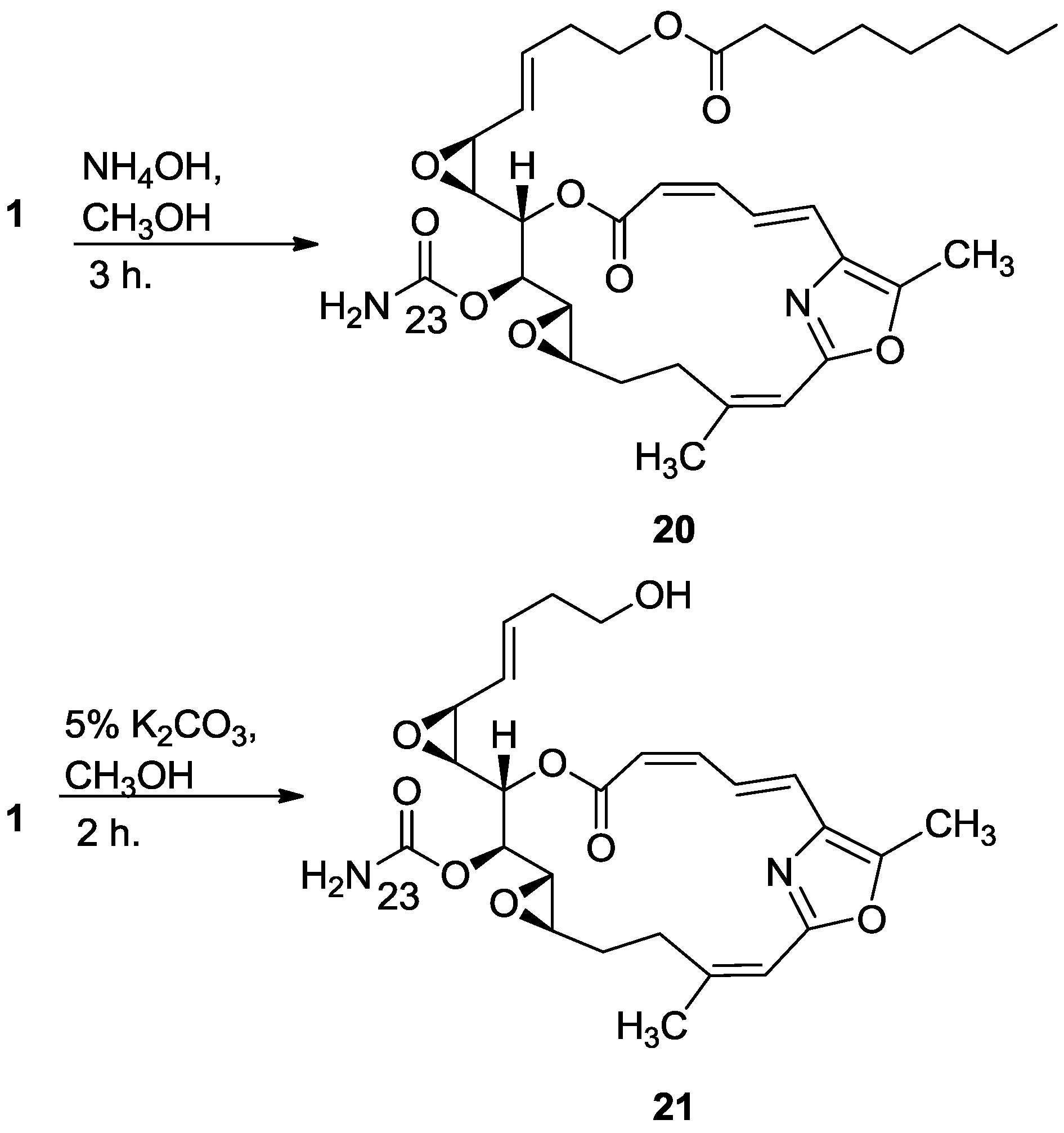
3.2.12. Compound 20
To a solution of salarin C (10 mg, 0.015 mmol) in MeOH (2 mL) was added aqueous ammonia (0.5 mL). The mixture was stirred 3 h at rt and then evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford 20, as a colorless oil, 7 mg (87%). [α]D25 −38 (c 0.17, CHCl3); NMR data for modified site (C13–C15, C23): 1H-NMR (CDCl3, 500 MHz) δ 3.43 (dd, J = 8.6, 1.8 Hz, 1H, H-13), 4.60 (m, 1H, H-14), 5.44 (dd, J = 8.6, 2.6 Hz, 1H, H-15); 13C-NMR (CDCl3, 125 MHz) δ 55.1 d (C-13), 76.4 d (C-14), 68.3 d (C-15), 155.2 s (C-23). HR-ESIMS m/z calculated for C33H44N2O9Na (M + Na+) 635.2945, found 635.2947.
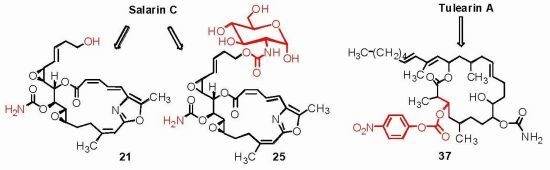
3.2.13. Compound 21
To a solution of salarin C (20 mg, 0.03 mmol) in MeOH (0.5 mL) was added methanolic K2CO3 (5% v/v, 1 mL). The reaction was stirred for 2 h at rt and then EA (5 mL) was added, the organic layer was washed with saturated NH4Cl, dried over anhydrous Na2SO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 3:7) to afford 21, as a major product, colorless oil, 9 mg (62%) accompanied with traces of 20 and (2Z,4E)-dimethyl hexa-2,4-dienedioate; NMR data for modified site (C18–C21, C23): 1H-NMR (CDCl3, 400 MHz) δ 5.54 (dd, J = 4.1, 15.6 Hz, 1H, H-18), 5.86 (ddd, J = 15.6, 8.8, 5.2 Hz, 1H, H-19), 2.33 (m, 2H, H-20), 3.67 (m, 1Ha, H-21), 3.59 (ddd, J = 11.7, 6.8, 4.1 Hz, 1Hb, H-21); 13C-NMR (CDCl3, 100 MHz) δ 123.9 d (C-18), 133.7 d (C-19), 36.2 t (C-20), 61.5 t (C-21), 159.1 s (C-23). HR-ESIMS m/z calculated for C25H30N2O8Na (M + Na+) 509.1900, found 509.1898.
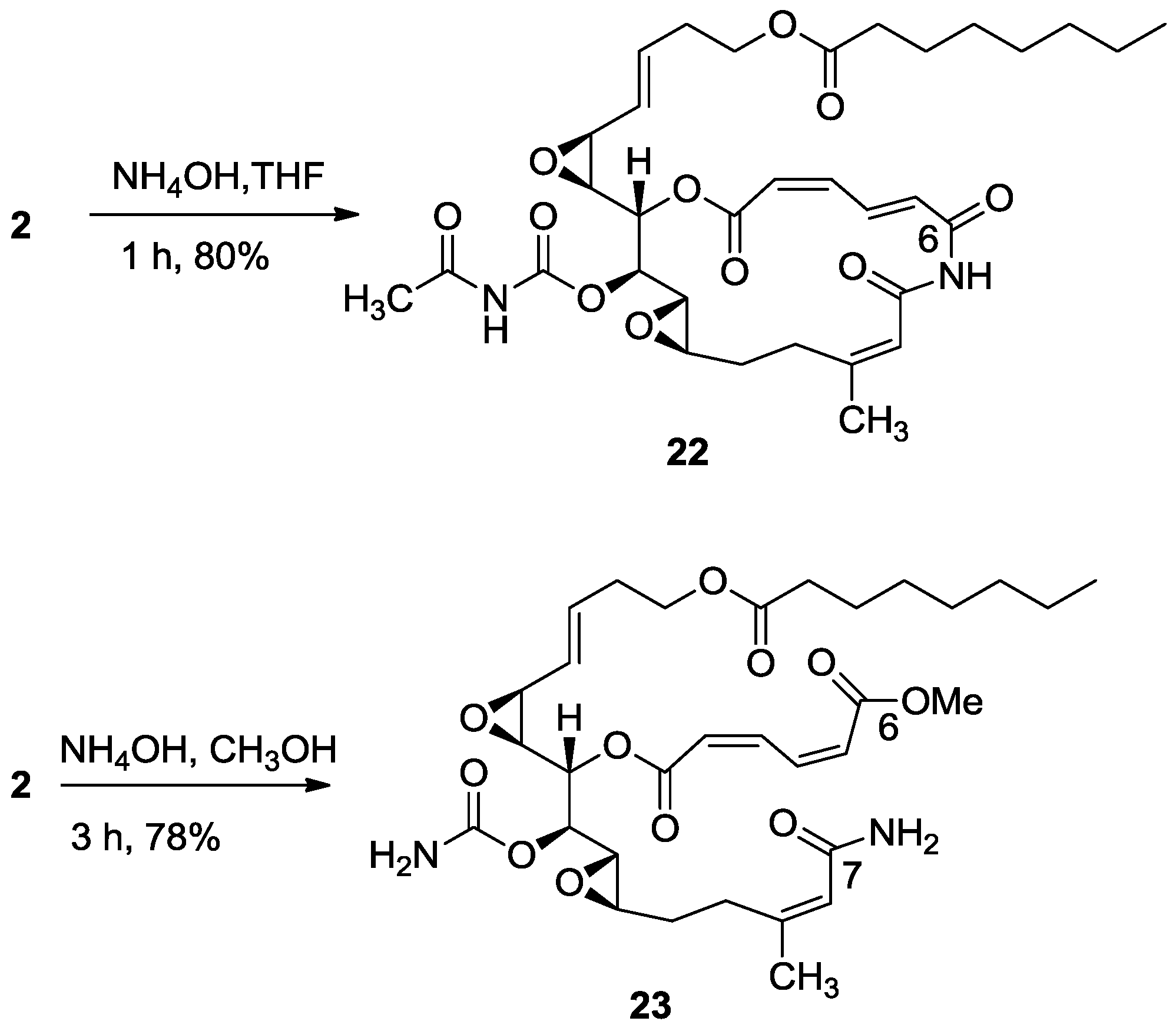
3.2.14. Compound 22
To a solution of salarin A (11 mg, 0.016 mmol) in EtOH (5 mL) was added NH
4Cl (2.5 mg, 0.048 mmol) and NaN
3 (3.1 mg, 0.048 mmol). The solution was slowly warmed up to 60 °C for 1 h, and then allowed to cool down to room temperature. The reaction mixture was filtered off, the solid residue washed with EtOH, and the solvent was evaporated. The residue was dissolved in DCM (50 mL) and washed with H
2O (30 mL). The organic layer was dried over anhydrous Na
2SO
4 and then evaporated to afford
22, as a yellow oil, 8 mg (80%). The NMR-data is identical to natural salarin E [
6].
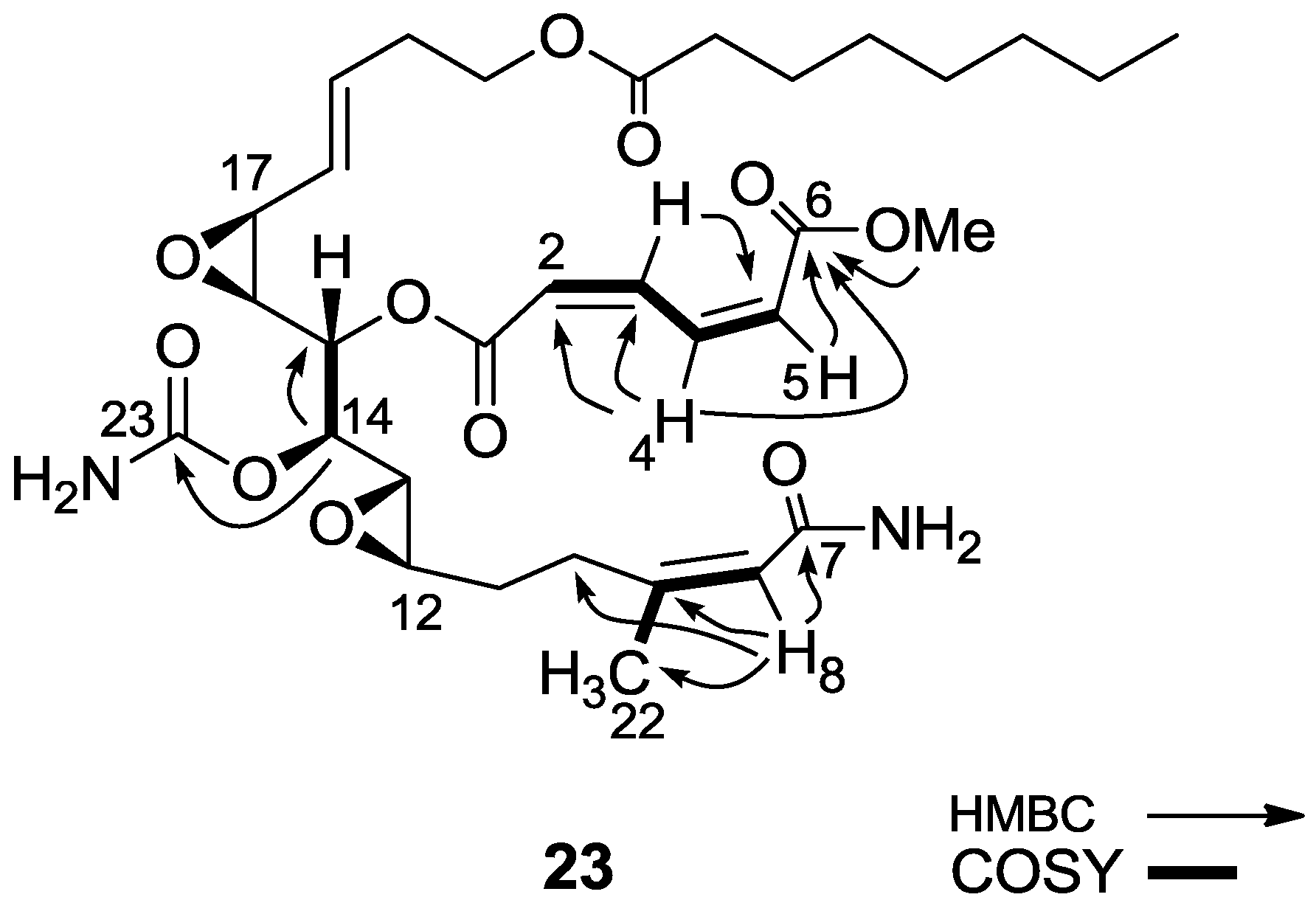
3.2.15. Compound 23
To a solution of salarin A (10 mg, 0.015 mmol) in MeOH (2 mL), was added NH4OH (0.5 mL, 0.1 mmol). The reaction mixture was stirred for 3 h at room temperature and then evaporated. The residue was purified by LH-20 chromatography (petroleum ether/DCM/MeOH, 2:1:1) to afford 23, as a yellow oil, 7.5 mg (78%). [α]D23 −21 (c 0.2, CHCl3); NMR data for modified site (C1–9, C14, C22,23): 1H-NMR (CDCl3, 500 MHz) δ 5.98 (d, J = 11.2 Hz, 1H, H-2), 6.68 (t, J = 11.2 Hz, 1H, H-3), 8.29 (dd, J = 15.3, 11.2 Hz, 1H, H-4), 6.13 (d, J = 15.3 Hz, 1H, H-5), 5.66 (d, J = 8.4 Hz, 1H, H-8), 4.90 (dd, J = 6.6, 3.6 Hz, 1H, H-14), 1.88 (s, 3H, H-22), 3.78 (s, 3H, OMe); δ 13C-NMR (CDCl3, 125 MHz) δ 163.9 s (C-1), 124.1 d (C-2), 141.5 d (C-3), 138.9 d (C-4), 129.3 d (C-5), 166.8 s (C-6), 168.7 s (C-7), 118.7 d (C-8), 155.7 s (C-9), 74.3 d (C-14), 25.1 q (C-22), 155.8 s (C-23), 52.3 q (OMe). HR-ESIMS m/z calculated for C32H46N2O11Na (M + Na+) 657.2999, found 657.3004.
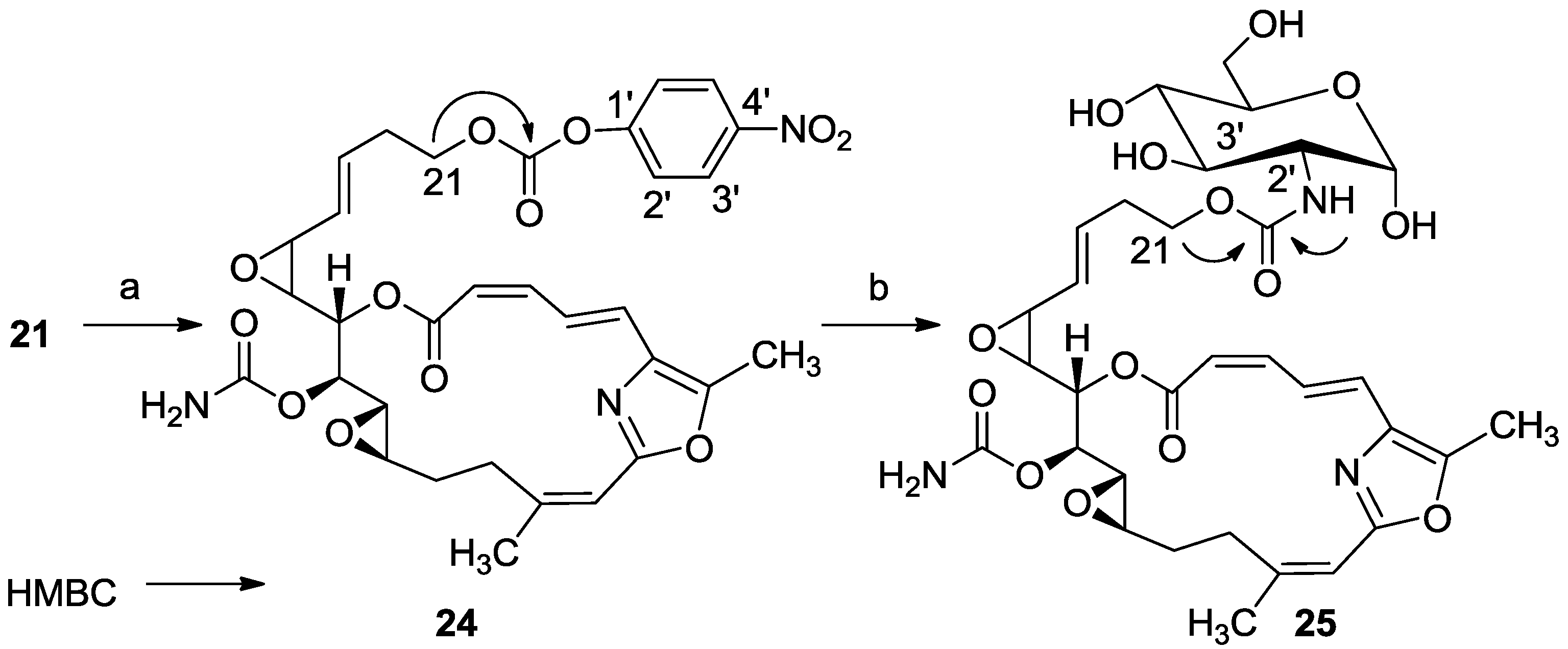
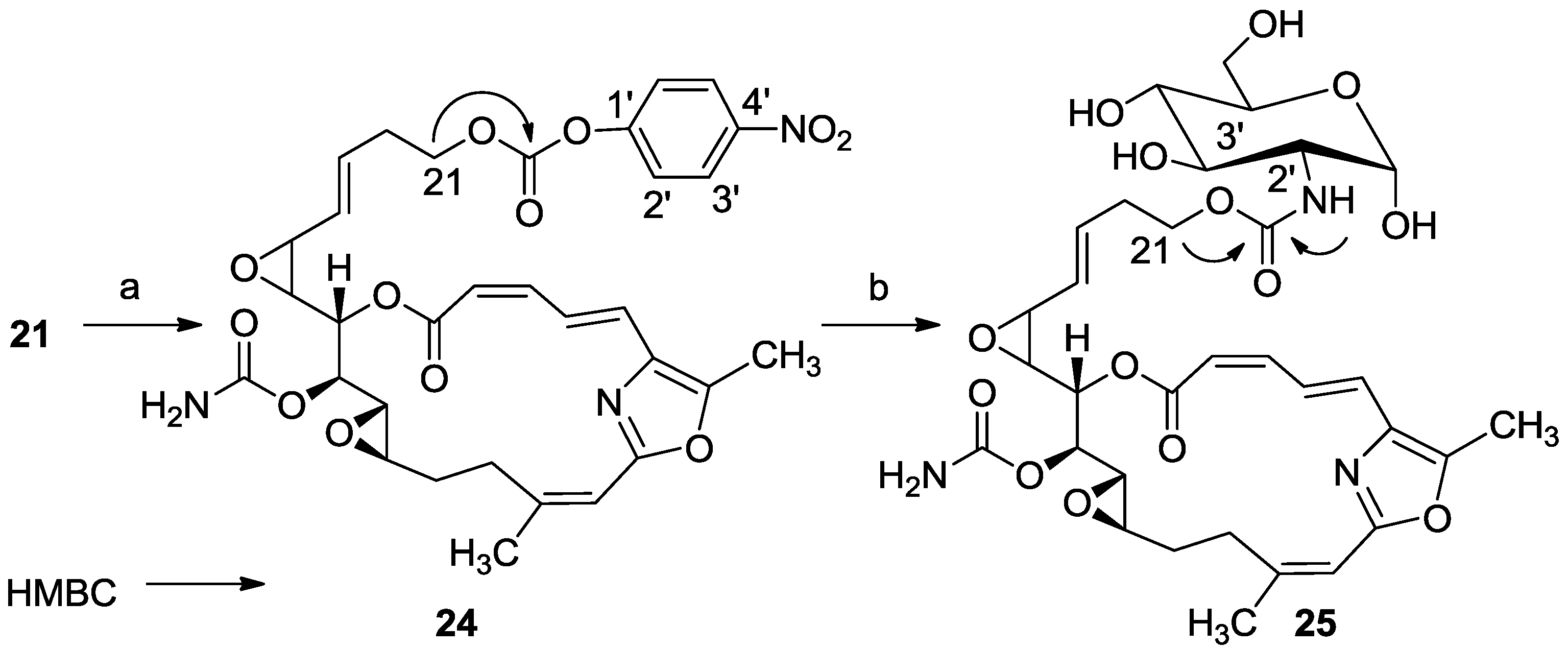
3.2.16. Compound 24
To a solution of 21 (20 mg, 0.04 mmol) in DCM (2 mL) was added triethylamine (20 μL, 0.12 mmol), DMAP (0.48 mg, 0.004 mmol) and bis(4-nitrophenyl) carbonate (20 mg, 0.06 mmol). The mixture was stirred for 2 h at rt and then DCM was added and the mixture was washed with brine. The organic layer was dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 4:6) to afford 24, as a colorless oil, 12 mg (46%). [α]D25 +117 (c 0.07, CHCl3); NMR data for modified site (C21-PNP): 1H-NMR (CDCl3, 400 MHz) δ 4.38 (t, J = 6.6 Hz, 2H, H-21); PNP moiety: δ 8.27 (m, J = 8.8 Hz, 1H, H-2′), 7.34 (m, J = 8.8 Hz, 1H, H-3′); 13C-NMR (CDCl3, 100 MHz) δ 68.2 t (C-21); PNP moiety: δ 152.15 s (CO), 155.3 s (C-1′), 121.9 d (C-2′), 125.2 d (C-3′), 145.9 s (C-4′); ESIMS m/z 674.45, (C32H33N3O12Na, M + Na+).
3.2.17. Compound 25
To a mixture of d(+)-glucosamine and NaOMe (7 mg, 0.035 mmol) in dry DMF (0.5 mL) was added Et3N (15 μL, 0.1 mmol) followed by 24 (10 mg, 0.014 mmol) in dry DMF (0.2 mL) and the mixture was stirred for 2 h at rt. The solvent was evaporated and the residue was purified by LH-20 chromatography (petroleum ether/DCM/MeOH, 2:1:1) to afford 25, as a colorless oil, 8 mg (80%). [α]D25 +206 (c 0.2, MeOH); NMR data for modified site (C14–C19): 1H-NMR (d4-MeOH, 500 MHz) δ 5.13 (d, J = 3.8 Hz, 1H, H-1′), 4.12 (m, 2H, H-21), 3.57 (m, J = 10.4, 3.8 Hz, H-2′), 3.64 (m, 1H, H-3′), 3.89 (m, 1H, H-4′), 3.39 (m, 1H, H-5′), 3.63 (m, 1Ha, H-6′), 3.69 (m, 1Hb, H-6′); 13C-NMR (d4-MeOH, 125 MHz) δ 63.2 t (C-21), 155.9 s (CO), 91.3 d (C-1′), 56.5 d (C-2′), 71.6 d (C-3′), 71.4 d (C-4′), 70.8 d (C-5′), 61.4 t (C-6′). HR-ESIMS m/z calculated for C32H41N3O14Na (M + Na+) 714.2486, found 714.2489.
3.3. Tulearin Derivatives
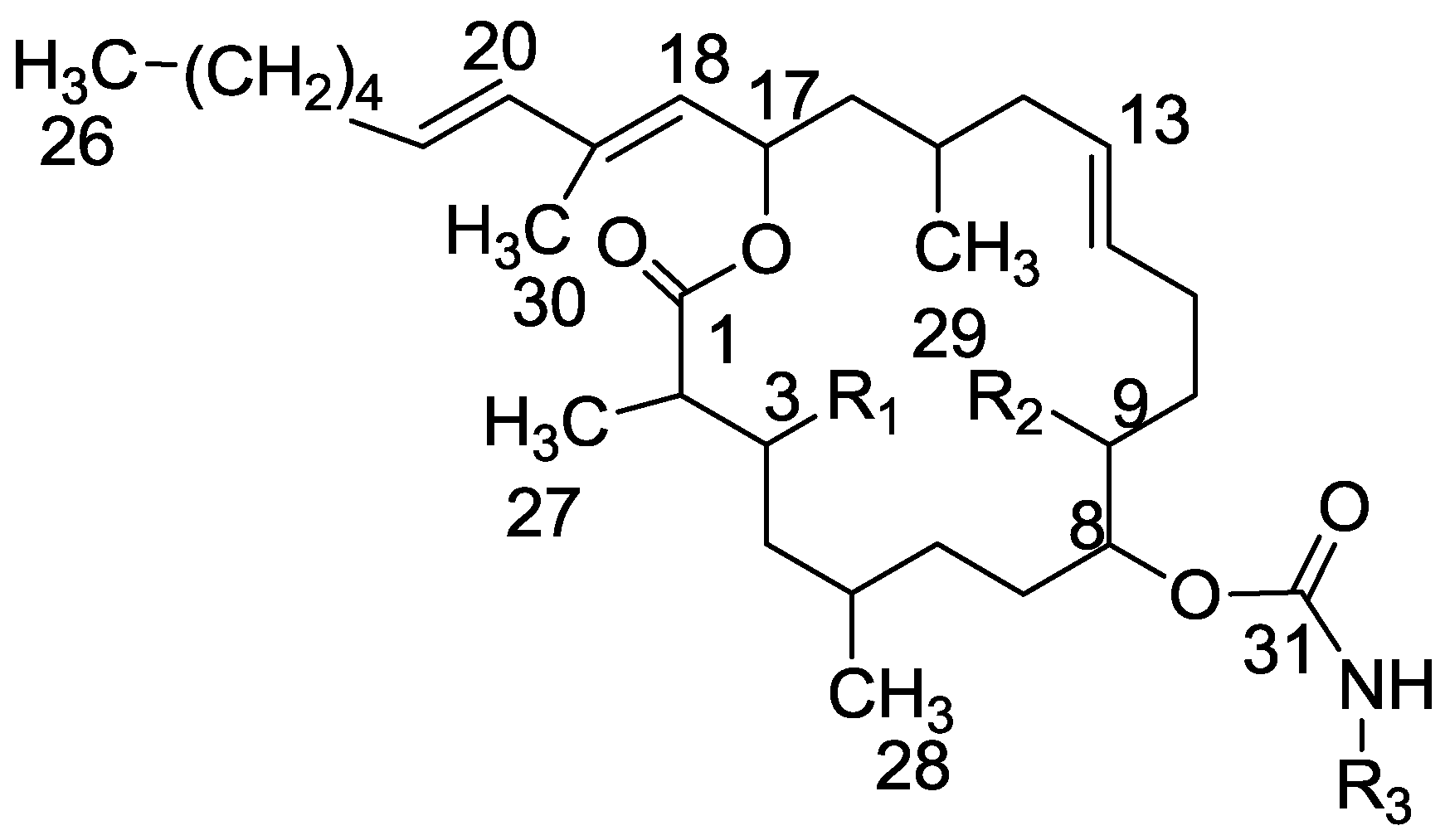
3.3.1. Compounds 26 and 27
To a mixture of tulearin A (222 mg, 0.415 mmol) and pyridine (1 mL) in chloroform (3 mL), was added TsCl (160 mg, 0.8 mmol) at 0 °C. After 48 h of stirring, EA was added and the organic phase was washed with saturated NH4Cl, water and brine, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford ditosyl 26, as a colorless oil, 52 mg (20%) and 9-monotosyl 27, a second colorless oil, 43 mg (35%). 26: [α]D25 +15 (c 0.4, CHCl3); NMR data for modified site (C3–C10): 1H-NMR (CDCl3, 500 MHz) δ 4.50 (m, 1H, H-3), 1.56 (m, 1H, H-4), 4.78 (m, 1H, H-8), 4.69 (m, 1H, H-9), 1.57 (m, 1H, H-10);. 3-Ts: δ 7.35 (d, 2H, H-33,34), 7.79 (d, 2H, H-35,36), 2.45 (s, 3H, H-38); 9-Ts: δ 7.35 (d, 2H, H-33,34), 7.79 (d, 2H, H-35,36), 2.45 (s, 3H, H-38); 13C-NMR (CDCl3, 125 MHz) δ 80.4 d (C-3), 35.3 t (C-4), 72.1 d (C-8), 81.5 d (C-9), 31.3 t (C-10); 3-Ts: δ 127.5 s (C-32), 129.9 d (C-33,34), 127.8 d (C-35,36), 137.0 s (C-37), 21.0 q (C-38); 9-Ts: δ 127.5 s (C-32), 129.9 d (C-33,34), 127.8 d (C-35,36), 137.0 s (C-37), 21.0 q (C-38). HR-ESIMS m/z calculated for C45H65NO10S2Na (M + Na+) 866.3948, found 866.3954. 27: [α]D25 +17 (c 0.2, CHCl3); NMR data for modified site (C8–C10): 1H-NMR (CDCl3, 500 MHz) δ 4.78 (m, 1H, H-8), 4.69 (m, 1H, H-9), 1.57 (m, 1H, H-10); 9-monoTs: δ 7.35 (d, 2H, H-33,34), 7.79 (d, 2H, H-35,36), 2.45 (s, 3H, H-38); 13C-NMR (CDCl3, 125 MHz) δ 72.1 d (C-8), 81.5 d (C-9), 31.3 t (C-10); 9-monoTs: δ 127.5 s (C-32), 129.9 d (C-33,34), 127.8 d (C-35,36), 137.0 s (C-37), 21.0 q (C-38); HR-ESIMS m/z calculated for C38H59NO8NaS (M + Na+) 712.3859, found 712.3860.
3.3.2. Compound 28
To a solution of tulearin A (10 mg, 0.025 mmol) in DCM (2 mL) were added Et3N (5.2 mg, 0.05 mmol) and p-bromobenzoylchloride (11.3 mg, 0.05 mmol). The mixture was stirred at room temperature for 24 h. DCM was then added (5 mL) and the mixture was washed with saturated NH4Cl, water and brine, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford 28, as a colorless oil, 7.2 mg (40%). [α]D25 +27 (c 0.2, CHCl3); NMR data for modified site (C8–C10): 1H-NMR (CDCl3, 500 MHz) δ 4.95 (q, J = 5.3 Hz, 1H, H-8), 5.29 (q, J = 5.3 Hz, 1H, H-9), 1.62 (m, 2H, H-10); Aryl group (Ar): δ 7.90 (d, J = 8.3 Hz, 2H, H-34,35), 7.57 (d, J = 8.3 Hz, H-36,37); 13C-NMR (CDCl3, 125 MHz) δ 74.3 d (C-8), 73.4 d (C-9), 31.4 t (C-10); Aryl group (Ar): δ 165.2 s (C-32), 137.0 s (C-33), 131.9 d (C-34,35), 131.4 d (C-36,37), 128.0 s (C-38). FABMS m/z 718.0, (C38H57BrNO7, MH+).
3.3.3. Compound 29
Mesyl chloride (80 mg, 0.7 mmol) was added to a cold solution (0 °C) of tulearin A (140 mg, 0.26 mmol) in pyridine (0.14 mL). The mixture was stirred at rt for 3 h. Then, water was added and the mixture extracted with DCM. The organic phase was dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 4:1) to afford 29, as a colorless oil, 110 mg (61%). [α]D25 +15 (c 0.5, CHCl3); NMR data for modified site: 1H-NMR (CDCl3, 500 MHz) δ 5.00 (brd, J = 9.0 Hz, 1H, H-3), 3.08 (s, CH3-Ms), 5.21 (brd, J = 8.9 Hz, 1H, H-9), 3.05 (s, 3H, CH3-Ms); 13C-NMR (CDCl3, 125 MHz) δ 80.6 d (C-3), 40.9 q (CH3-Ms), 80.9 d (C-9), 38.9 q (CH3-Ms); CIMS m/z 692.0, (C33H58NO10S2, MH+).
3.3.4. Compound 30
To a solution of tulearin A (22 mg, 0.044 mmol) in DCM (0.5 mL) were added Et3N (18 μL, 0.13 mmol) and catalytic amounts of DMAP at 0 °C. Benzoyl chloride was then added (20 μL, 0.17 mmol) and the mixture was warmed up slowly to rt. After 2.5 h, the reaction mixture was neutralized by acetic acid to pH 7 and partitioned between ethyl acetate/H2O. The organic layer was washed with sat. NH4Cl, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (PE/EA, 1:1) to afford 30, as a colorless oil, 19 mg (70%). NMR data for modified site (C8–C10): 1H-NMR (CDCl3, 500 MHz) δ 5.02 (dt, J = 5.6, 6.2 Hz, 1H, 1H, H-8), 5.37 (dt, J = 5.6, 5.9 Hz, 1H, H-9), 1.78 (m, 2H, H-10); Benzoyl group: δ 7.57 (d, J = 7.8 Hz, 1H), 8.06 (d, J = 7.8 Hz, 2H), 7.45 (t, J = 7.8 Hz, 2H); 13C-NMR (CDCl3, 125 MHz) δ 73.9 d (C-8), 72.55 d (C-9), 28.9 t (C-10); Benzoyl group: δ 166.6 s (CO), 132.6 d (CH), 129.4 d (CH, 2H), 127.9 d (CH, 2H); HR-ESIMS m/z calculated for C38H57NO7Na (M + Na+) 662.4033, found 662.4030.
3.3.5. Compound 31
To a mixture of pyridine (0.1 mL, 1.06 mmol) and acetic anhydride (0.1 mL, 1.2 mmol) was added tulearin A (43 mg, 0.086 mmol) at rt. After 20 min the solvents were evaporated and the residue was purified by VLC (petroleum ether/ethyl acetate, 3:2) to afford 31, as major product, accompanied by 10% of 32, and 10% tulearin A. 31: colorless oil, 30 mg (61%); [α]D25 +27 (c 0.3, CHCl3); NMR data for modified site (C7–C10): 1H-NMR (CDCl3, 500 MHz) δ 1.62 (m, 2H, H-7), 4.84 (dt, J = 10.5, 5.8 Hz, 1H, H-8), 5.13 (dd, J = 6.5, 10.5 Hz, 1H, H-9), 1.47 (m, 2H, H-10); acetate group: δ 2.09 (s, CH3); 13C-NMR (CDCl3, 125 MHz) δ 29.5 t (C-7), 73.9 d (C-8), 71.2 d (C-9), 29.7 t (C-10); acetate group: δ 171.0 s (CO), 19.9 q (CH3); HR-ESIMS m/z calculated for C33H55NO7Na (M + Na+) 600.3876, found 600.3876.
3.3.6. Compound 32
To a mixture of pyridine (0.5 mL, 5.3 mmol) and acetic anhydride (0.5 mL, 6.0 mmol), was added tulearin A (20 mg, 0.04 mmol) at rt. After 20 min the solvents were evaporated and the residue was purified by VLC (petroleum ether/ethyl acetate, 4:1) to afford 32, as a colorless oil, 22 mg, (90%); [α]D25 +26 (c 0.3, CHCl3); NMR data for modified site C3 and C9: 1H-NMR (CDCl3, 500 MHz) δ 5.19 (m, 1H, H-3); 3-acetate group: δ 2.10 (s, 3H, CH3), 5.10 (q, J = 6.7 Hz, 1H, H-9); 9-acetate group: δ 2.11 (s, 3H, CH3); 13C-NMR (CDCl3, 125 MHz) δ 71.6 d (C-3); 3-acetate group: δ 170.5 s (CO), 21.3 q (CH3); 71.3 d (C-9); 9-acetate group: δ 170.1 s (CO), 21.1 q (CH3); FABMS m/z 620.9, (C35H58NO8, MH+).
3.3.7. Compound 33
To a mixture of pyridine (0.1 mL, 1.06 mmol) and acetic anhydride (0.1 mL, 1.02 mmol) with catalytic amounts of DMAP (0.43 mg, 0.004 mmol), was added tulearin A (18 mg, 0.036 mmol) at rt. After 7 days the solvents were evaporated and the residue was purified by VLC (petroleum ether/ethyl acetate, 4:1) to afford 33, as major product, accompanied by 10% of 32; 33: colorless oil, 9 mg (40%). NMR data for modified site: 1H-NMR (CDCl3, 500 MHz) δ 5.18 (m, 1H, H-3), 5.1 (m, 1H, H-9); 9-acetate group: δ 2.03 (s, 3H, CH3); 3-acetate group: δ 2.01 (s, 3H, CH3); acetyl carbamate: δ 2.36 (s, 3H, CH3); 13C-NMR (CDCl3, 125 MHz) δ 71.4 d (C-3), 76.1 d (C-9); 9-acetate group: δ 172.1 s (CO), 21.0 q (CH3); 3-acetate group: δ 171.6 s (CO), 20.9 q (CH3); acetyl carbamate: δ 170.8 s (CO), 22.5 q (CH3); HR-ESIMS m/z calculated for C37H59NO9Na (M + Na+) 684.4088, found 684.4084.
3.3.8. Compound 34
To a mixture of Et3N (13 μL, 0.096 mmol) with catalytic amounts of DMAP (0.3 mg, 0.003 mmol) in DCM (0.2 mL), was added tulearin A (12 mg, 0.024 mmol) at 0 °C. Hexanoyl chloride was then slowly added (13 μL, 0.096 mmol) and the mixture warmed up slowly to rt. After 2.5 h, the reaction mixture was neutralized by acetic acid to pH 7 and the residue was partitioned between ethyl acetate/H2O. The organic layer washed by sat. NH4Cl, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 4:1) to afford 34, as a colorless oil, 6 mg (35%). NMR data for modified site: 1H-NMR (CDCl3, 500 MHz) δ 5.15 (dt, J = 5.8, 6.7 Hz, 1H, H-9), 5.26 (m, 1H, H-3); 14 methylenes according to HSQC experiments in the range of 1–2.5 ppm; 13C-NMR (CDCl3, 125 MHz) δ 71.0 d (C-9), 71.2 d (C-3); Hexanoyl carbonyls: 172.0 s (C3-CO), 172.3 s (C9-CO), 173.3 s (carbamate), HR-ESIMS m/z calculated for C49H83NO9Na (M + Na+) 852.5966, found 852.5969.
3.3.9. Compound 35
To a solution of tulearin A (35 mg, 0.065 mmol) in dry DCM (3 mL) were added of p-nitrophenyl chlorocarbonate (PNPCl, 15 mg, 0.074 mmol) and DMAP (18 mg, 0.14 mmol). The reaction was stirred at rt for 48 h. DCM was then added (5 mL) and the mixture was washed with water, dried over anhydrous MgSO4 and evaporated. The residue was purified by VLC (petroleum ether/ethyl acetate, 1:1) to afford 35, as a colorless oil, 15 mg (35%). [α]D25 +10 (c 0.3, CHCl3); NMR data for modified site (C2, C3): 1H-NMR (CDCl3, 500 MHz) δ 2.70 (qd, J = 7.6, 2.4 Hz, 1H, H-2), 5.09 (dd, J = 10.1, 2.4 Hz, 1H, H-3); PNP-group: δ 6.48 (d, J = 7.6 Hz, 2H, CH), 8.23 (d, J = 7.6 Hz, 2H, CH); 13C-NMR (CDCl3, 100 MHz) δ 44.5 d (C-2), 72.6 d (C-3); PNP-group: δ 156.6 s (CO), 130.2 q (C), 106.6 d (CH), 149.6 d (CH), 136.2 s (C). ESIMS m/z 723.3, (C38H56N2O10Na, M + Na+).
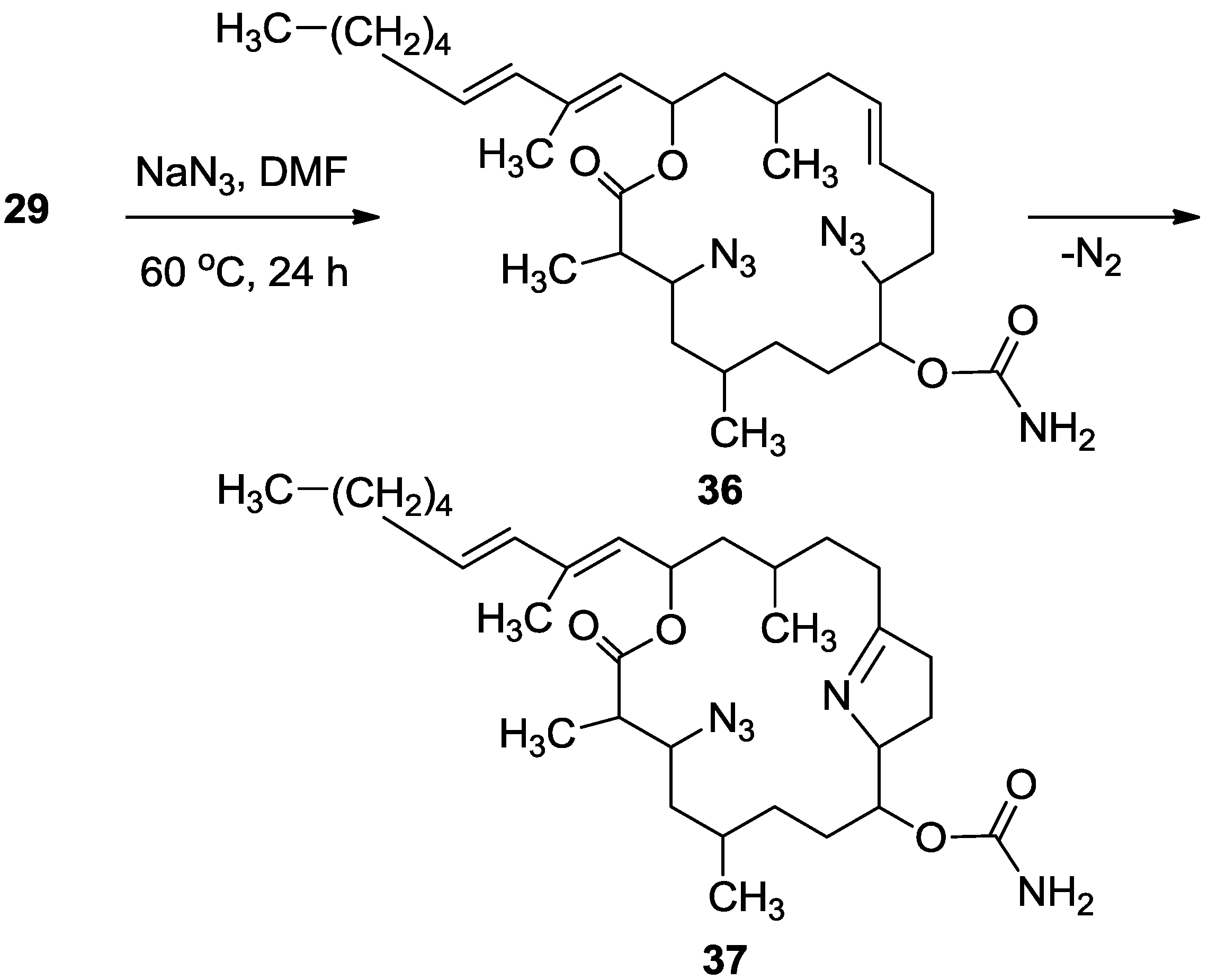
3.3.10. Compounds 36 and 37
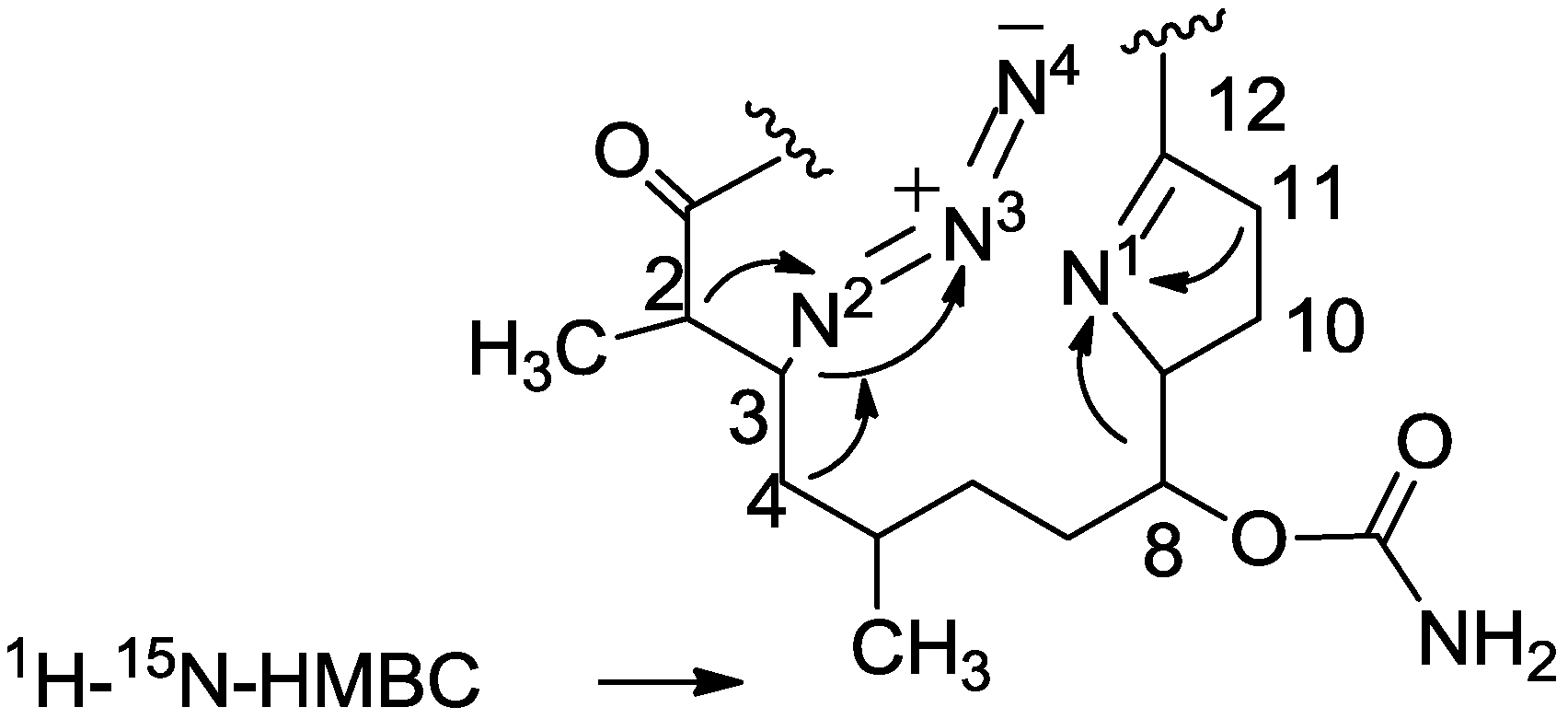
To a solution of 29 (22 mg, 0.03 mmol) in DMF (0.5 mL) was added sodium azide (10 mg, 0.15 mmol), and the mixture warmed up to 60 °C for. After 24 h, the reaction mixture was cooled down to room temperature. The mixture was then partitioned between ethyl acetate/H2O and the organic phase dried over anhydrous MgSO4, and evaporated. The DMF was removed under high vacuum and the residue was purified by VLC (petroleum ether/ethyl acetate) to afford three products of azide substituted tulearin A; two epmeric 3,9-diazide tulearins: 36a, as a colorless oil, 3.8 mg (21%); 36b, a second colorless oil, 5.1 mg (29%) and a cycloaddition product 37, another colorless oil, 6.3 mg (37%); 36a: [α]D25 +20 (c 0.2, CHCl3); NMR data for modified site: 1H-NMR (CDCl3, 500 MHz) δ 3.59 (ddd, J = 8.7, 6.8, 2.0 Hz, 1H, H-3), 3.29 (td, J = 7.5, 3.7 Hz, 1H, H-9); 13C-NMR (CDCl3, 125 MHz) δ 62.3 d (C-3), 62.7 d (C-9); HR-ESIMS m/z calculated for C31H51N7O4Na (M + Na+) 608.3900, found 608.3898; 36b: [α]D25 −26 (c 0.24, CHCl3); NMR data for modified site: 1H-NMR (CDCl3, 500 MHz) δ 3.63 (ddd, J = 8.7, 6.6, 1.5 Hz, 1H, H-3), 3.47 (dt, J = 9.5, 3.5 Hz, 1H, H-9); 13C-NMR (CDCl3, 125 MHz) δ 62.8 d (C-3), 60.1 d (C-9); HR-ESIMS m/z calculated for C31H51N7O4Na (M + Na+) 608.3900, found 608.3904; 37: [α]D25 +69 (c 0.7, CHCl3); NMR data for modified site: 1H-NMR (CDCl3, 500 MHz) δ 3.77 (ddd, J = 6.7, 4.6, 2.0 Hz, 1H, H-3), 1.88 (m, 2H, H-4), 5.02 (ddd, J = 7.4, 4.3, 2.7 Hz, 1H, H-8), 4.24 (m, 1H, H-9), 1.92 (m, 1Ha, H-10), 1.81 (m, 1Hb, H-10), 2.36 (m, 2H, H-11), 2.32 (m, 2H, H-13); 13C-NMR (CDCl3, 125 MHz) δ 61.8 d (C-3), 39.8 t (C-4), 76.0 d (C-8), 74.9 d (C-9), 24.2 t (C-10), 38.3 t (C-11), 179.2 s (C-12), 30.1 t (C-13); HR-ESIMS m/z calculated for C31H52N5O4 (MH+) 558.4019, found 558.4011.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}