A Stable-Isotope Mass Spectrometry-Based Metabolic Footprinting Approach to Analyze Exudates from Phytoplankton


Abstract
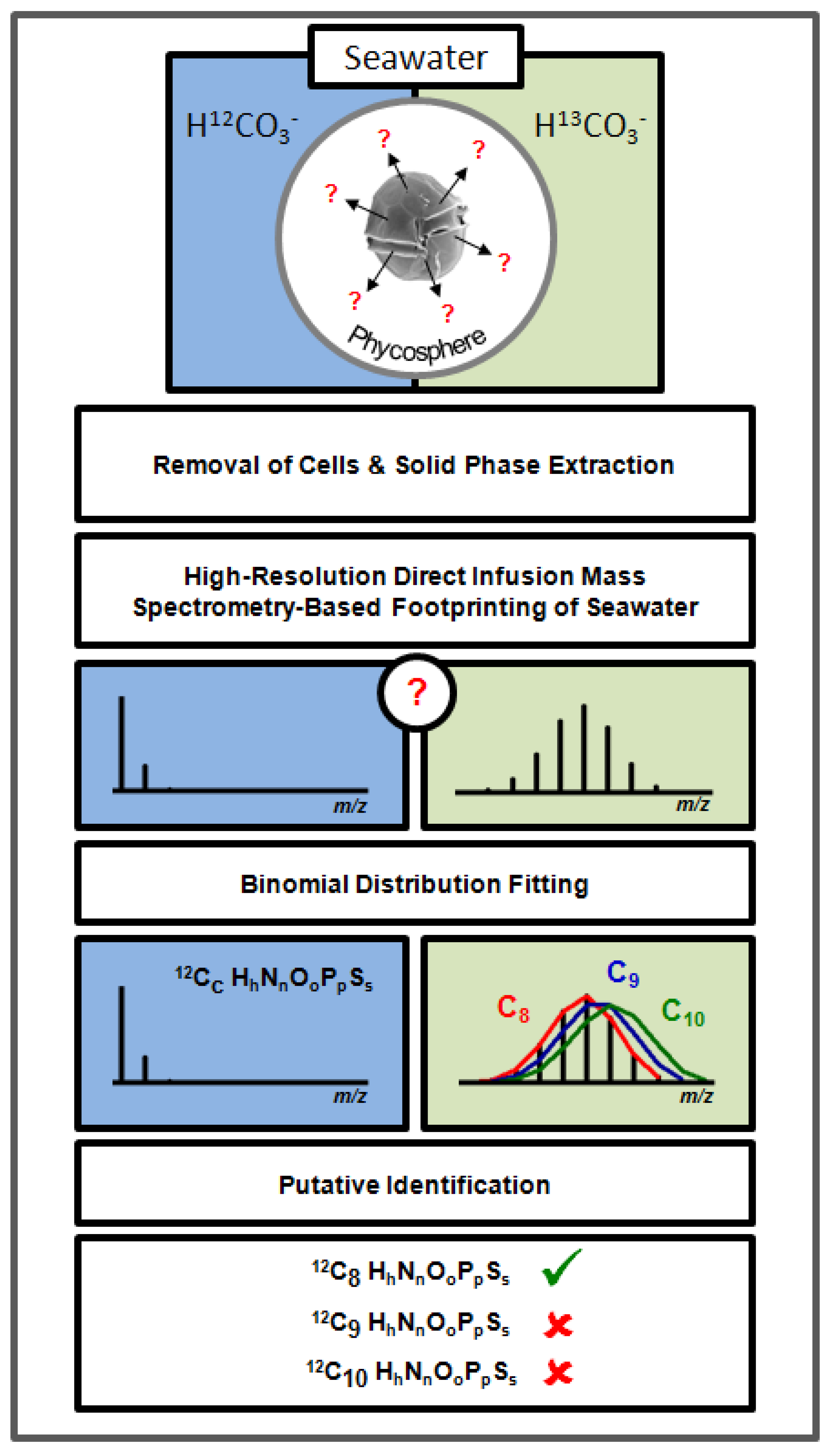
:1. Introduction

2. Results and Discussion
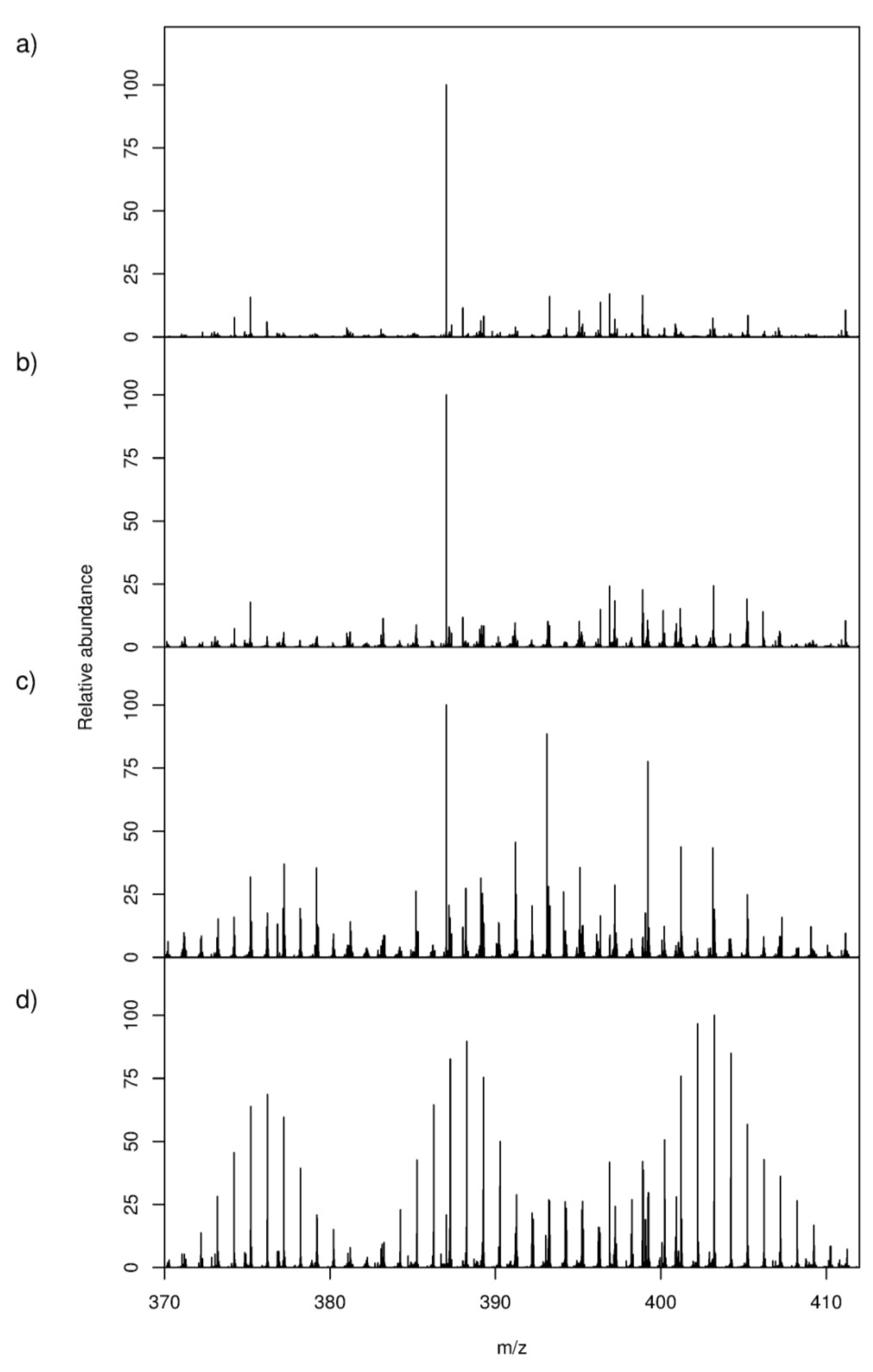
2.1. High-Resolution Mass Spectrometry-Based Metabolic Footprinting
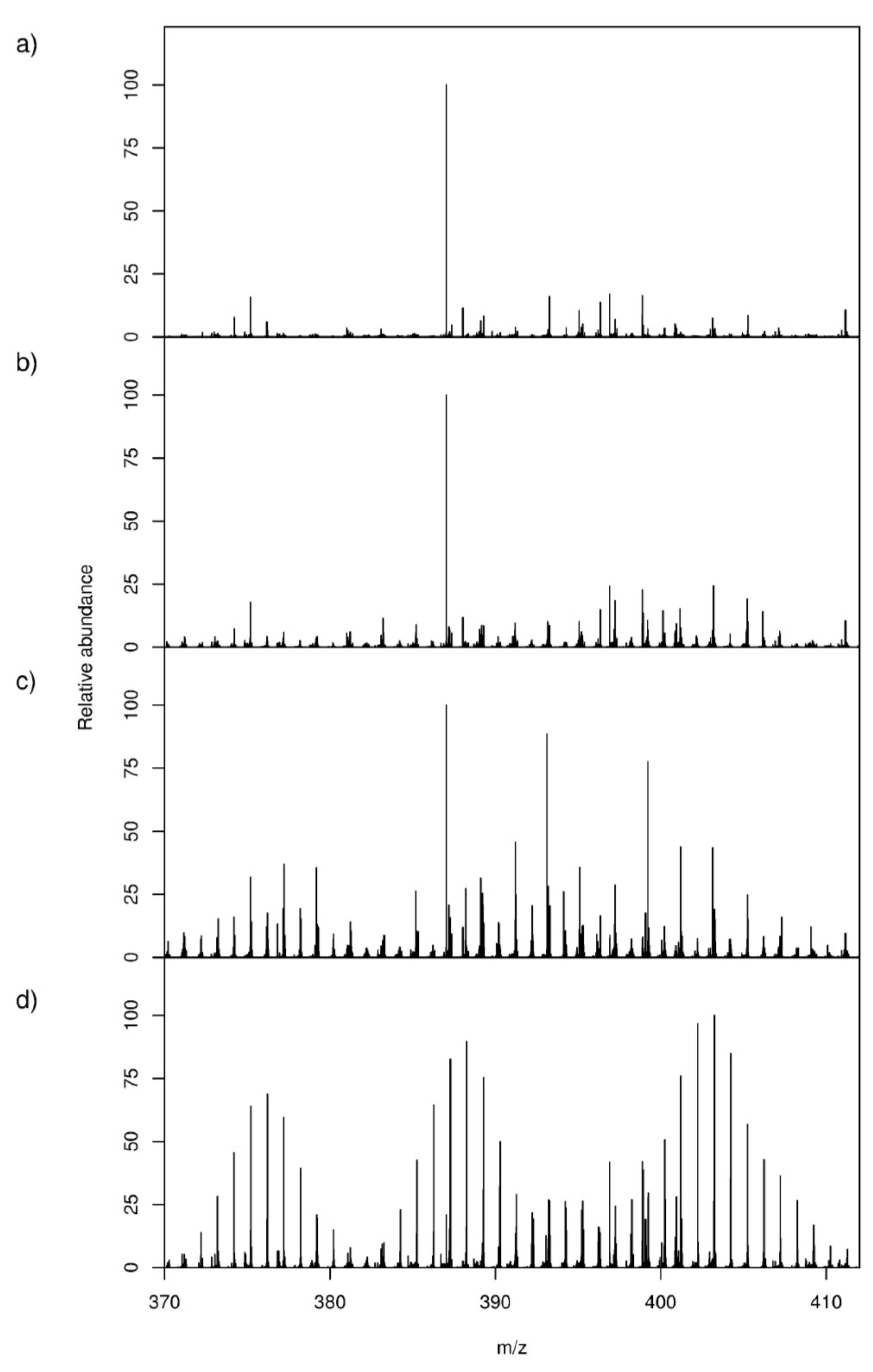
 ) unlabelled and 13C-labelled (
) unlabelled and 13C-labelled (  ) seawater without algal cells (as controls, n = 6 each), and (
) seawater without algal cells (as controls, n = 6 each), and (  ) unlabelled and (
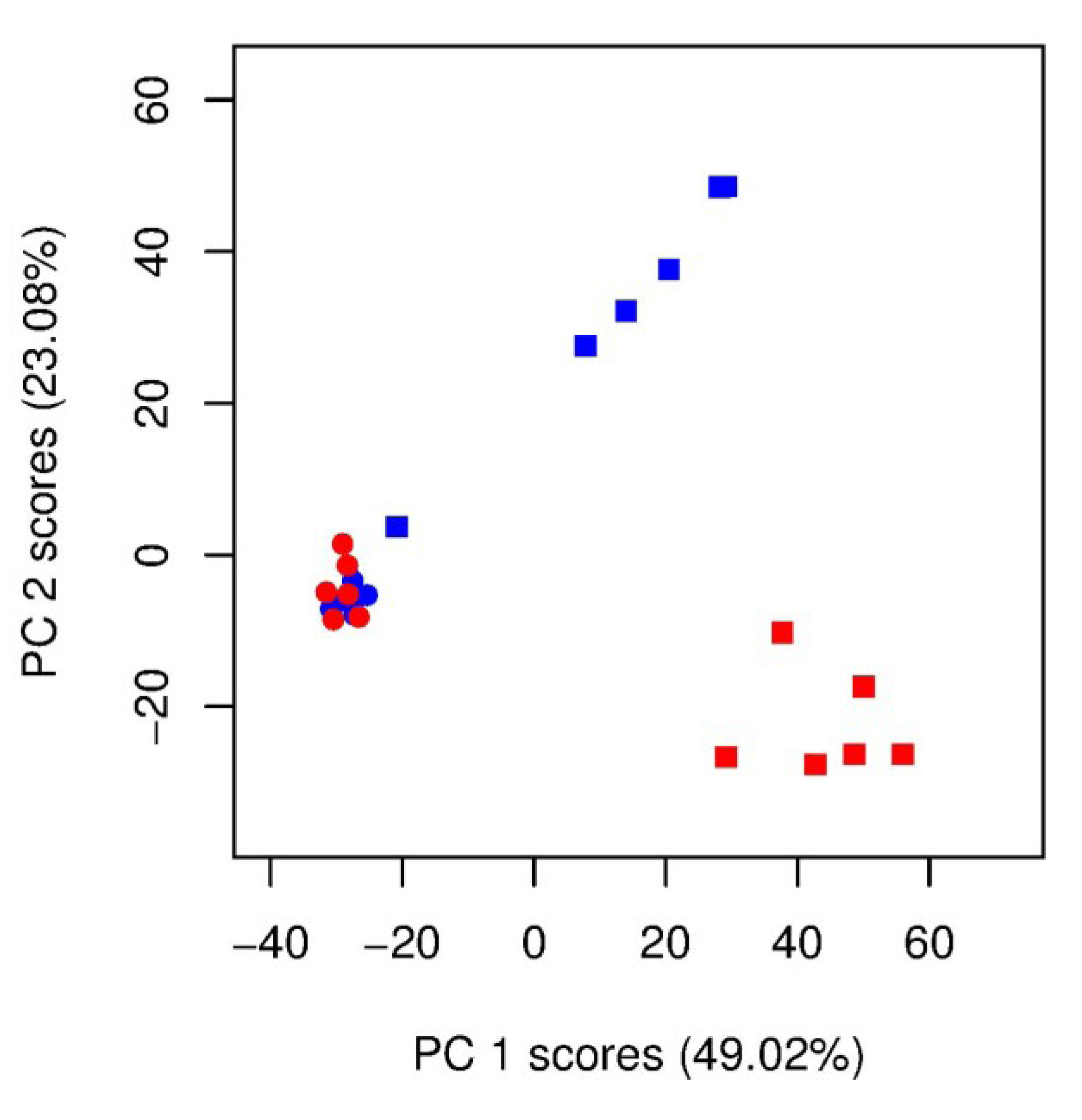
) unlabelled and (  ) 13C-labelled cultures of Alexandrium tamarense (n = 6 each). The major separation along the PC1 axis corresponds to the differences between the metabolic footprints of seawater samples with versus without algal cells present. Separation along PC2 corresponds to differences between the metabolic footprints of 13C-labelled vs. unlabelled A. tamarense cultures.
) unlabelled and 13C-labelled ( ) seawater without algal cells (as controls, n = 6 each), and ( ) unlabelled and ( ) 13C-labelled cultures of Alexandrium tamarense (n = 6 each). The major separation along the PC1 axis corresponds to the differences between the metabolic footprints of seawater samples with versus without algal cells present. Separation along PC2 corresponds to differences between the metabolic footprints of 13C-labelled vs. unlabelled A. tamarense cultures.
) 13C-labelled cultures of Alexandrium tamarense (n = 6 each). The major separation along the PC1 axis corresponds to the differences between the metabolic footprints of seawater samples with versus without algal cells present. Separation along PC2 corresponds to differences between the metabolic footprints of 13C-labelled vs. unlabelled A. tamarense cultures.
) unlabelled and 13C-labelled ( ) seawater without algal cells (as controls, n = 6 each), and ( ) unlabelled and ( ) 13C-labelled cultures of Alexandrium tamarense (n = 6 each). The major separation along the PC1 axis corresponds to the differences between the metabolic footprints of seawater samples with versus without algal cells present. Separation along PC2 corresponds to differences between the metabolic footprints of 13C-labelled vs. unlabelled A. tamarense cultures.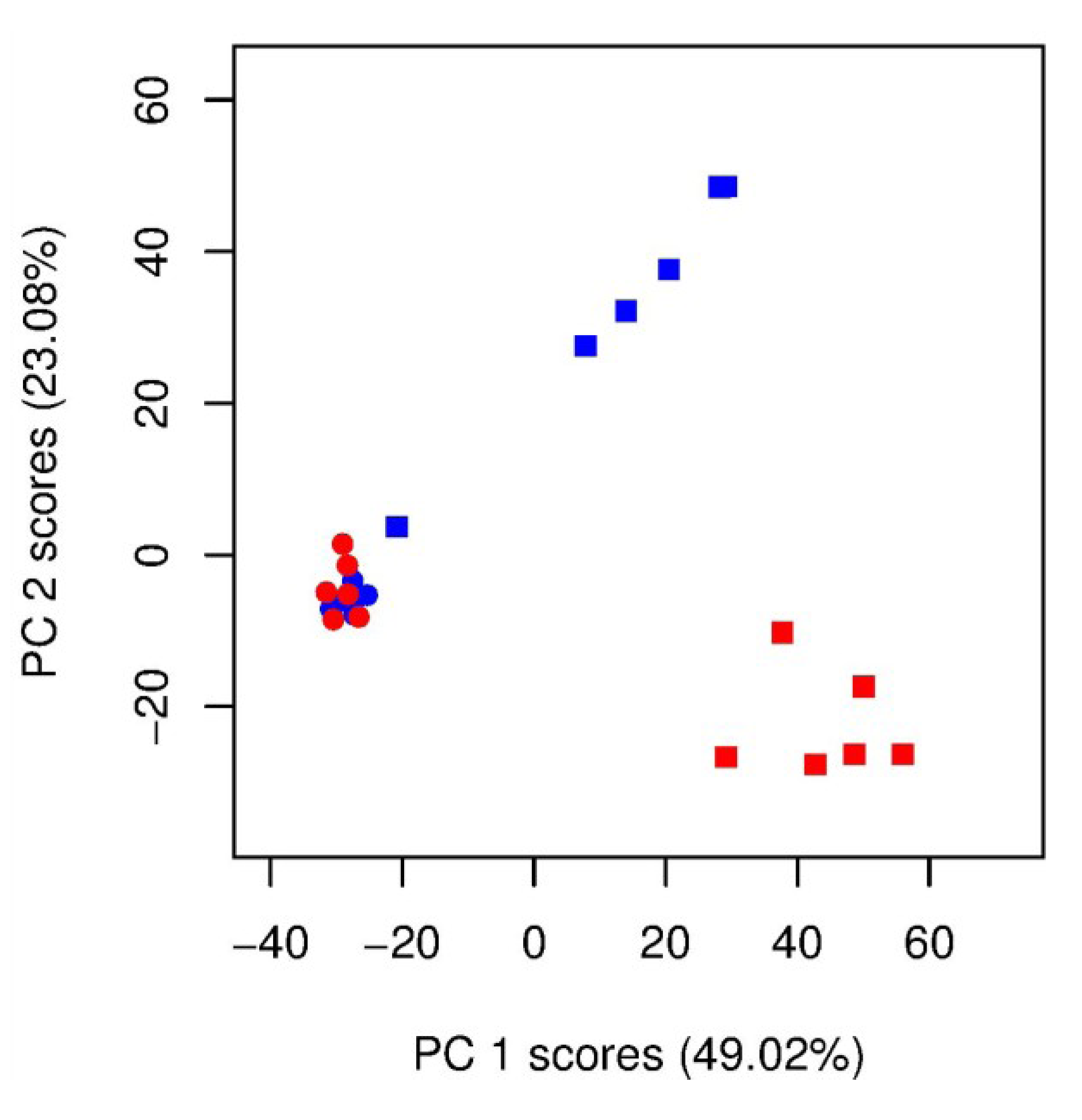
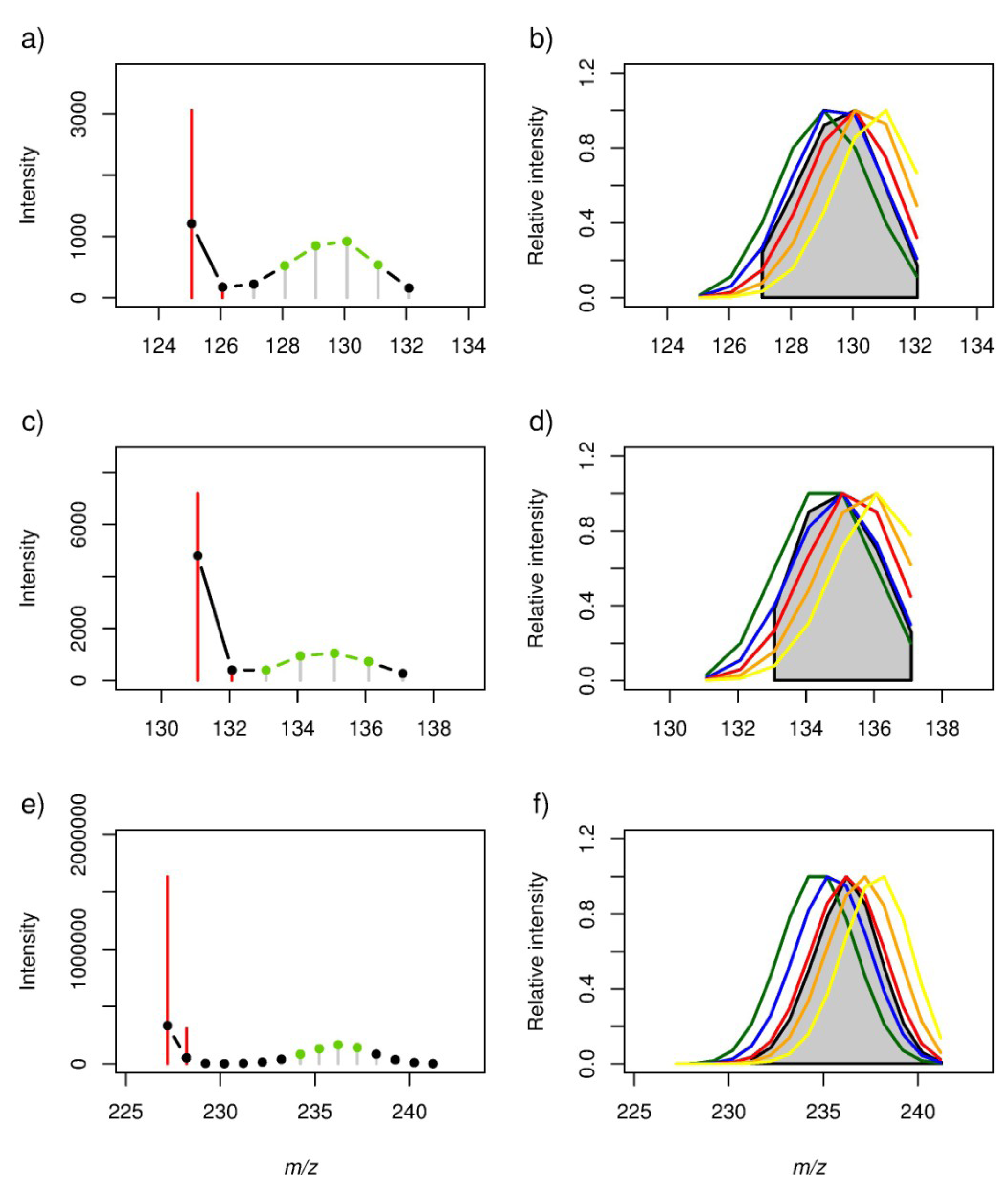
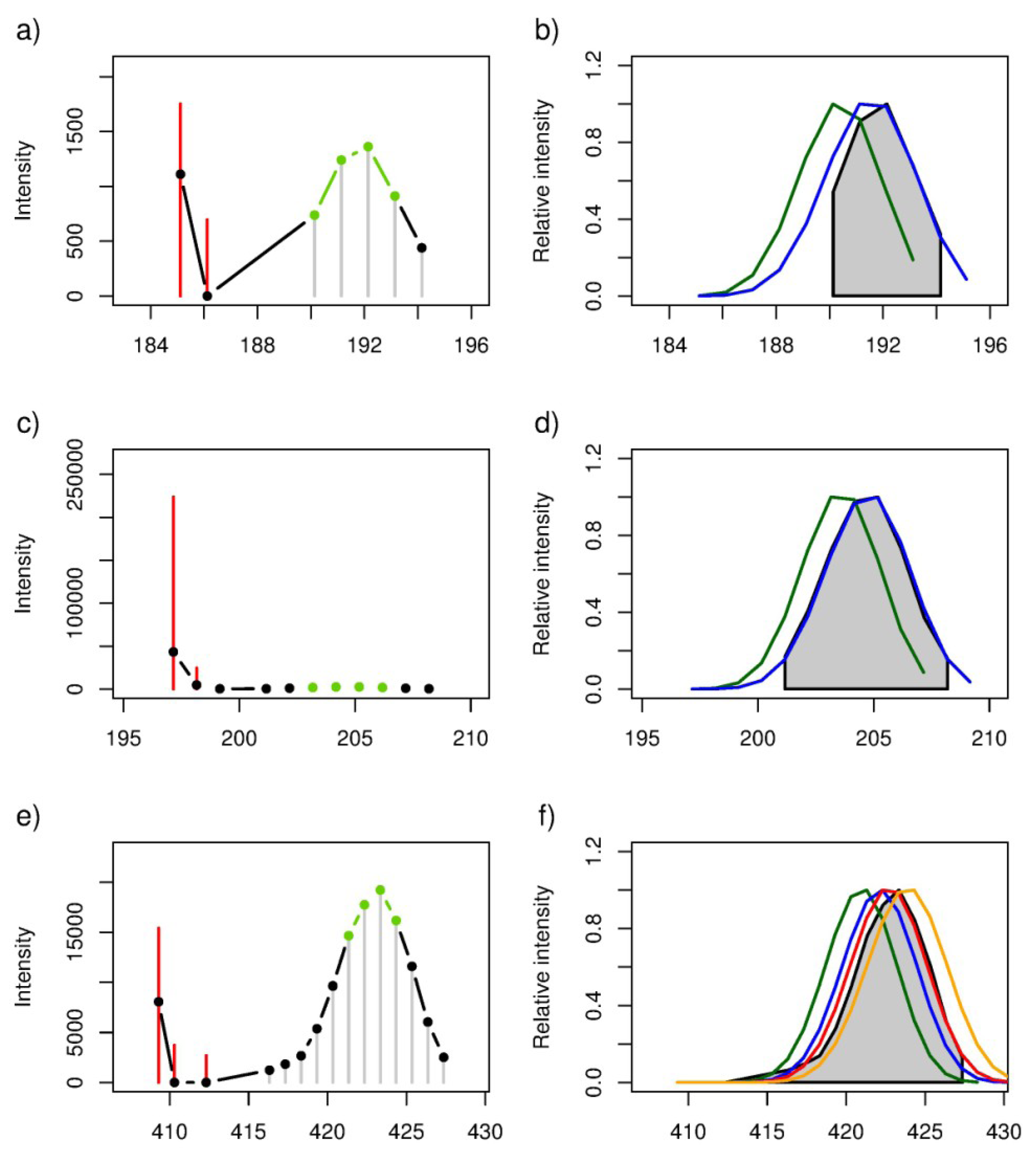
2.2. Locating Stable Isotope Patterns
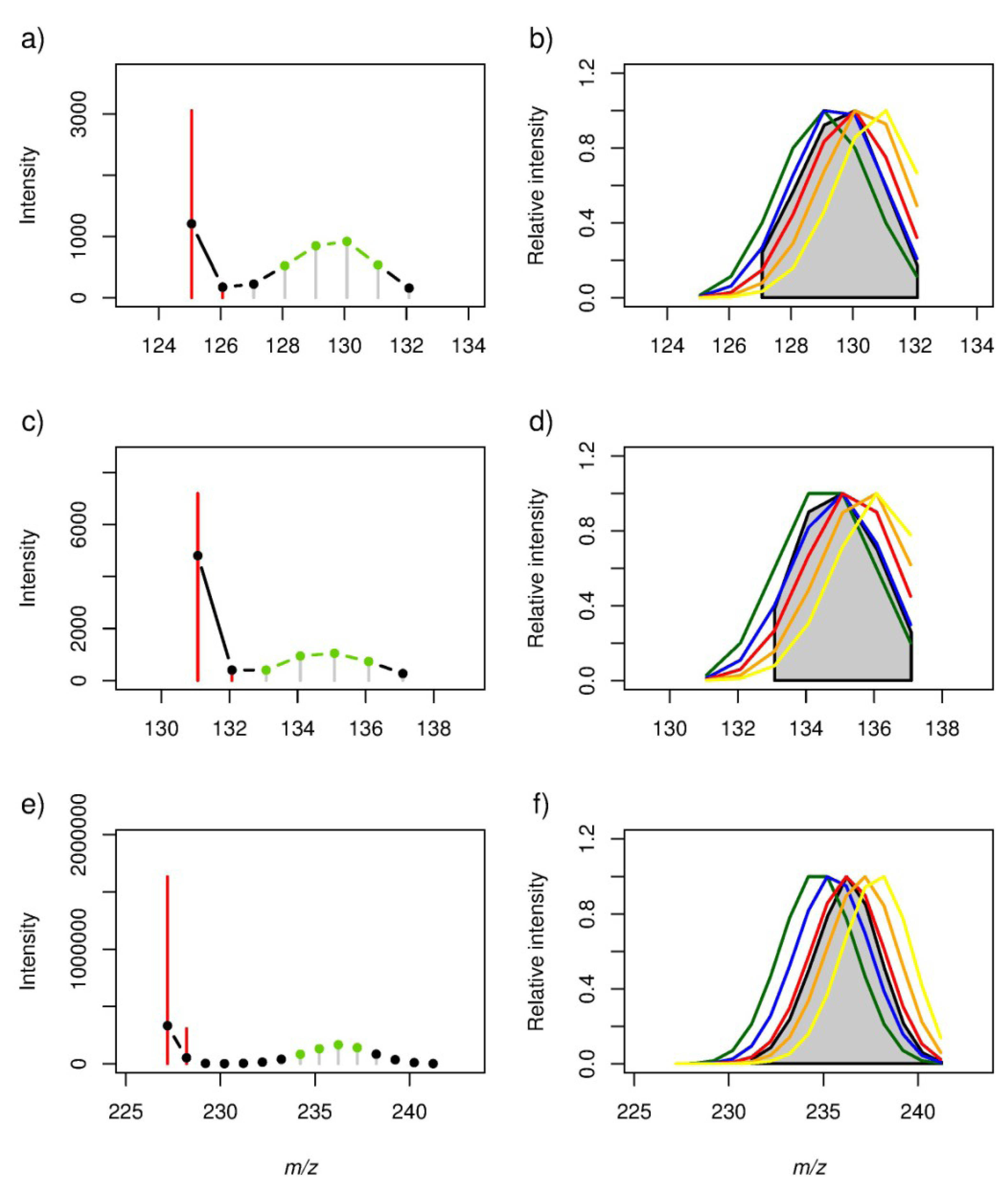

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m/z (all-12C containing peak as [M − H]− ion) | Empirical formula | Labelling Efficiency (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| r-value | p-value | ||||||||||
| 50% | 55% | 60% | 65% | 70% | 50% | 55% | 60% | 65% | 70% | ||
| 125.06084 | 12C7H10O2 | 0.86 | 0.99 | 0.93 | 0.68 | 0.35 | 0.0289 | 0.0001 | 0.0080 | 0.1347 | 0.4913 |
| 131.07140 | 12C6H12O3 | 0.92 | 0.99 | 0.82 | 0.48 | 0.12 | 0.0244 | 0.0010 | 0.0918 | 0.4181 | 0.8532 |
| 227.20143 | 12C14H28O2 | 0.63 | 0.90 | 1.00 | 0.88 | 0.58 | 0.0369 | 0.0002 | 0.0000 | 0.0004 | 0.0615 |
2.3. Estimation of the Stable Isotope Labelling Efficiency
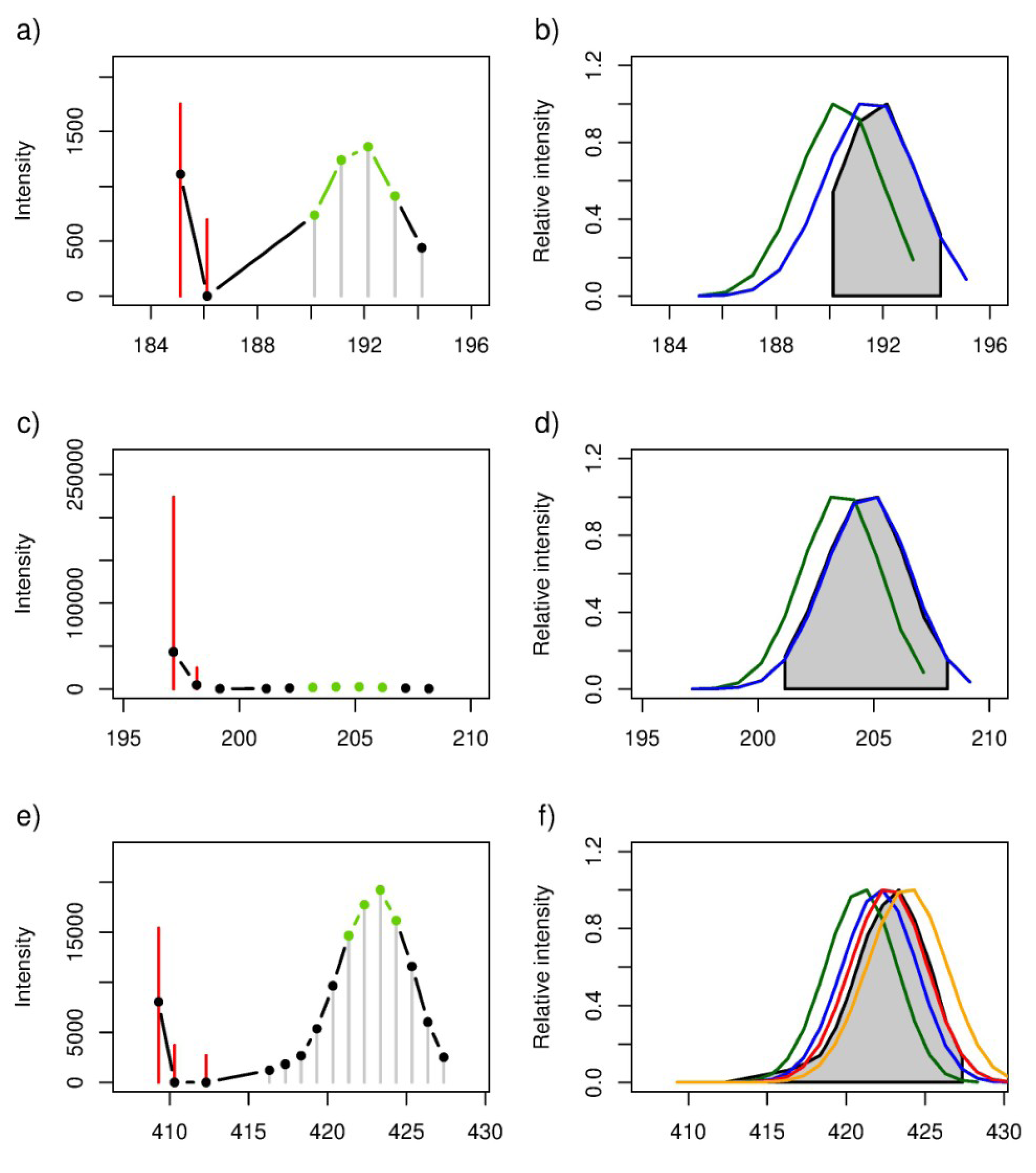
2.4. Putative Annotation of Exuded Metabolites
| m/z | Colour (in Figure 5) | Empirical Formula (peak) | Ion Form | Theoretical Mass (Da) | Mass Error (ppm) | KEGG Compound | r-value | p-value |
|---|---|---|---|---|---|---|---|---|
| 185.11855 | blue | C10H18O3 | [M − H]− | 185.11832 | 1.25 | [(3R)-6-Hydroxy-3-isopropenyl-heptanoate, (3S)-6-Hydroxy-3-isopropenyl-heptanoate, (5R)-6-Hydroxy-5-isopropenyl-2-methylhexanoate, (5S)-6-Hydroxy-5-isopropenyl-2-methylhexanoate, 10-Oxodecanoate, 2-Oxodecanoic acid, Epomediol] | 0.95 | 0.0115 |
| green | C8H14O | [M + Acetate]− | 185.11832 | 1.25 | [Sulcatone] | −0.09 | 0.9149 | |
| 197.15491 | blue | C12H22O2 | [M − H]− | 197.15470 | 1.05 | [(−)-Menthyl acetate, Citronellyl acetate, Decanoyl acetaldehyde, Neomenthyl acetate] | 1.00 | 0.0000 |
| green | C10H18 | [M + Acetate]− | 197.15470 | 1.05 | 0.58 | 0.1735 | ||
| 409.29594 | red | C22H38O3 | [M + Acetate]− | 409.29595 | −0.02 | 0.99 | 0.0000 | |
| blue | C21H45N2O2P | [M + Acetate]− | 409.29653 | −1.45 | 0.91 | 0.0000 | ||
| orange | C24H42O5 | [M − H]− | 409.29595 | −0.02 | 0.91 | 0.0000 | ||
| green | C19H39N8P | [M − H]− | 409.29625 | −0.77 | 0.55 | 0.0506 |
3. Experimental Section
3.1. Cell Culture and Stable Isotope Labelling
3.2. Metabolite Extraction from Media
3.3. FT-ICR MS Analysis, Spectral Processing and Unsupervised Multivariate Analyses
3.4. Locating and Putative Identification of SIPs
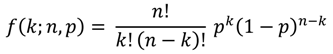
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bjornsen, P.K. Phytoplankton exudation of organic-matter—Why do healthy cells do it. Limnol. Oceanogr. 1988, 33, 151–154. [Google Scholar] [CrossRef]
- Kind, T.; Meissen, J.K.; Yang, D.; Nocito, F.; Vaniya, A.; Cheng, Y.-S.; VanderGheynst, J.S.; Fiehn, O. Qualitative analysis of algal secretions with multiple mass spectrometric platforms. J. Chromatogr. A 2012, 1244, 139–147. [Google Scholar] [CrossRef]
- Van Donk, E. Chemical information transfer in freshwater plankton. Ecol. Inf. 2007, 2, 112–120. [Google Scholar] [CrossRef]
- Gillard, J.; Frenkel, J.; Devos, V.; Sabbe, K.; Paul, C.; Rempt, M.; Inze, D.; Pohnert, G.; Vuylsteke, M.; Vyverman, W. Metabolomics enables the structure elucidation of a diatom sex pheromone. Angew. Chem. Int. Ed. 2013, 52, 854–857. [Google Scholar] [CrossRef]
- Fistarol, G.O.; Legrand, C.; Selander, E.; Hummert, C.; Stolte, W.; Graneli, E. Allelopathy in Alexandrium spp.: Effect on a natural plankton community and on algal monocultures. Aquat. Microb. Ecol. 2004, 35, 45–56. [Google Scholar] [CrossRef]
- Stocker, R. Marine microbes see a sea of gradients. Science 2012, 338, 628–633. [Google Scholar] [CrossRef]
- Kiorboe, T. A Mechanistic Aproach to Plankton Ecology; Princeton University Press: Princeton, NJ, USA, 2008; pp. 1–209. [Google Scholar]
- Azam, F.; Fenchel, T.; Field, J.G.; Gray, J.S.; Meyerreil, L.A.; Thingstad, F. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar]
- Falkowski, P.G.; Barber, R.T.; Smetacek, V. Biogeochemical controls and feedbacks on ocean primary production. Science 1998, 281, 200–206. [Google Scholar] [CrossRef]
- Jonsson, P.R.; Pavia, H.; Toth, G. Formation of harmful algal blooms cannot be explained by allelopathic interactions. Proc. Natl. Acad. Sci. USA 2009, 106, 11177–11182. [Google Scholar] [CrossRef]
- Nielsen, T.G.; Kiorboe, T.; Bjornsen, P.K. Effects of a chrysochromulina-polylepis subsurface bloom on the planktonic community. Mar. Ecol. Prog. Ser. 1990, 62, 21–35. [Google Scholar] [CrossRef]
- Underdal, B.; Skulberg, O.M.; Dahl, E.; Aune, T. Disastrous bloom of Chrysochromulina polylepis (prymnesiophyceae) in Norwegian Coastal Waters 1988—Mortality in Marine Biota. AMBIO 1989, 18, 265–270. [Google Scholar]
- Nevitt, G.A. Olfactory foraging by Antarctic procellariiform seabirds: Life at high Reynolds numbers. Biol. Bull. 2000, 198, 245–253. [Google Scholar] [CrossRef]
- Hockelmann, C.; Moens, T.; Juttner, F. Odor compounds from cyanobacterial biofilms acting as attractants and repellents for free-living nematodes. Limnol. Oceanogr. 2004, 49, 1809–1819. [Google Scholar] [CrossRef]
- Hay, M.E. Marine chemical ecology: Chemical signals and cues structure marine populations, communities, and ecosystems. Ann. Rev. Mar. Sci. 2009, 1, 193–212. [Google Scholar] [CrossRef]
- Viant, M.; Sommer, U. Mass spectrometry based environmental metabolomics: A primer and review. Metabolomics 2013, 9, 144–158. [Google Scholar] [CrossRef]
- Dunn, W.; Erban, A.; Weber, R.M.; Creek, D.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66. [Google Scholar]
- Draper, J.; Lloyd, A.; Goodacre, R.; Beckmann, M. Flow infusion electrospray ionisation mass spectrometry for high throughput, non-targeted metabolite fingerprinting: A review. Metabolomics 2013, 9, 4–29. [Google Scholar] [CrossRef]
- Kell, D.B.; Brown, M.; Davey, H.M.; Dunn, W.B.; Spasic, I.; Oliver, S.G. Metabolic footprinting and systems biology: The medium is the message. Nat. Rev. Microbiol. 2005, 3, 557–565. [Google Scholar]
- Taylor, N.; Weber, R.M.; Southam, A.; Payne, T.; Hrydziuszko, O.; Arvanitis, T.; Viant, M. A new approach to toxicity testing in Daphnia magna: Application of high throughput FT-ICR mass spectrometry metabolomics. Metabolomics 2009, 5, 44–58. [Google Scholar]
- Oikawa, A.; Nakamura, Y.; Ogura, T.; Kimura, A.; Suzuki, H.; Sakurai, N.; Shinbo, Y.; Shibata, D.; Kanaya, S.; Ohta, D. Clarification of pathway-specific inhibition by Fourier transform ion cyclotron resonance/mass spectrometry-based metabolic phenotyping studies. Plant Physiol. 2006, 142, 398–413. [Google Scholar] [CrossRef]
- Brown, S.C.; Kruppa, G.; Dasseux, J.L. Metabolomics applications of FT-ICR mass spectrometry. Mass Spectrom. Rev. 2005, 24, 223–231. [Google Scholar] [CrossRef]
- Giavalisco, P.; Hummel, J.; Lisec, J.; Inostroza, A.C.; Catchpole, G.; Willmitzer, L. High-resolution direct infusion-based mass spectrometry in combination with whole 13C metabolome isotope labeling allows unambiguous assignment of chemical sum formulas. Anal. Chem. 2008, 80, 9417–9425. [Google Scholar] [CrossRef]
- Giavalisco, P.; Kohl, K.; Hummel, J.; Seiwert, B.; Willmitzer, L. 13C isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 2009, 81, 6546–6551. [Google Scholar] [CrossRef]
- Mueller, D.; Heinz, E. Stable isotope-assisted metabolomics to detect metabolic flux changes in mammalian cell cultures. Curr. Opin. Biotechnol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Rodgers, R.; Blumer, E.; Hendrickson, C.; Marshall, A. Stable isotope incorporation triples the upper mass limit for determination of elemental composition by accurate mass measurement. J. Am. Soc. Spectrom. 2000, 11, 835–840. [Google Scholar] [CrossRef]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef]
- Birkemeyer, C.; Luedemann, A.; Wagner, C.; Erban, A.; Kopka, J. Metabolome analysis: The potential of in vivo labeling with stable isotopes for metabolite profiling. Trends Biotechnol. 2005, 23, 28–33. [Google Scholar] [CrossRef]
- Baran, R.; Bowen, B.P.; Bouskill, N.J.; Brodie, E.L.; Yannone, S.M.; Northen, T.R. Metabolite identification in Synechococcus sp. PCC 7002 using untargeted stable isotope assisted metabolite profiling. Anal. Chem. 2010, 82, 9034–9042. [Google Scholar] [CrossRef]
- Baran, R.; Bowen, B.P.; Northen, T.R. Untargeted metabolic footprinting reveals a surprising breadth of metabolite uptake and release by Synechococcus sp. PCC 7002. Mol. Biosyst. 2011, 7, 3200–3206. [Google Scholar] [CrossRef]
- Wakimoto, T.; Kondo, H.; Nii, H.; Kimura, K.; Egami, Y.; Oka, Y.; Yoshida, M.; Kida, E.; Ye, Y.; Akahoshi, S.; et al. Furan fatty acid as an anti-inflammatory component from the green-lipped mussel Perna canaliculus. Proc. Natl. Acad. Sci. USA 2011, 108, 17533–17537. [Google Scholar] [CrossRef]
- Ingvarsdottir, A.; Birkett, M.A.; Duce, I.; Genna, R.L.; Mordue, W.; Pickett, J.A.; Wadhams, L.J.; Mordue, A.J. Semiochemical strategies for sea louse control: Host location cues. Pest Manag. Sci. 2002, 58, 537–545. [Google Scholar] [CrossRef]
- Scandinavian Culture Collection of Algae and Protozoa. Available online: http://www.sccap.dk/ (accessed on 18 October 2013).
- Schantz, E.J.; Lynch, J.M.; Vayvada, G.; Matsumoto, K.; Rapoport, H. The purification and characterization of the poison produced Gonyaulax catenella in axenic cultures. Biochemistry 1966, 5, 1191–1195. [Google Scholar] [CrossRef]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R.L. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 1987, 23, 633–638. [Google Scholar]
- Dittmar, T.; Koch, B.; Hertkorn, N.; Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr. Meth. 2008, 6, 230–235. [Google Scholar] [CrossRef]
- Weber, R.J.; Southam, A.D.; Sommer, U.; Viant, M.R. Characterization of isotopic abundance measurements in high resolution FT-ICR and Orbitrap mass spectra for improved confidence of metabolite identification. Anal. Chem. 2011, 83, 3737–3743. [Google Scholar] [CrossRef]
- Southam, A.D.; Payne, T.G.; Cooper, H.J.; Arvanitis, T.N.; Viant, M.R. Dynamic range and mass accuracy of wide-scan direct infusion nanoelectrospray fourier transform ion cyclotron resonance mass spectrometry-based metabolomics increased by the spectral stitching method. Anal. Chem. 2007, 79, 4595–4602. [Google Scholar]
- Payne, T.G.; Southam, A.D.; Arvanitis, T.N.; Viant, M.R. A signal filtering method for improved quantification and noise discrimination in fourier transform ion cyclotron resonance mass spectrometry-based metabolomics data. J. Am. Soc. Mass Spectr. 2009, 20, 1087–1095. [Google Scholar] [CrossRef]
- Hrydziuszko, O.; Viant, M. Missing values in mass spectrometry based metabolomics: An undervalued step in the data processing pipeline. Metabolomics 2012, 8, 161–174. [Google Scholar] [CrossRef]
- Parsons, H.M.; Ludwig, C.; Gunther, U.L.; Viant, M.R. Improved classification accuracy in 1- and 2-dimensional NMR metabolomics data using the variance stabilising generalised logarithm transformation. BMC Bioinf. 2007, 8, 234. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Weber, R.J.M.; Viant, M.R. MI-Pack: Increased confidence of metabolite identification in mass spectra by integrating accurate masses and metabolic pathways. Chemom. Intell. Lab. Syst. 2010, 104, 75–82. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinf. 2007, 8, 105. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Weber, R.J.M.; Selander, E.; Sommer, U.; Viant, M.R. A Stable-Isotope Mass Spectrometry-Based Metabolic Footprinting Approach to Analyze Exudates from Phytoplankton. Mar. Drugs 2013, 11, 4158-4175. https://doi.org/10.3390/md11114158
Weber RJM, Selander E, Sommer U, Viant MR. A Stable-Isotope Mass Spectrometry-Based Metabolic Footprinting Approach to Analyze Exudates from Phytoplankton. Marine Drugs. 2013; 11(11):4158-4175. https://doi.org/10.3390/md11114158
Chicago/Turabian StyleWeber, Ralf J. M., Erik Selander, Ulf Sommer, and Mark R. Viant. 2013. "A Stable-Isotope Mass Spectrometry-Based Metabolic Footprinting Approach to Analyze Exudates from Phytoplankton" Marine Drugs 11, no. 11: 4158-4175. https://doi.org/10.3390/md11114158