The Transcriptome of Bathymodiolus azoricus Gill Reveals Expression of Genes from Endosymbionts and Free-Living Deep-Sea Bacteria

Abstract
:1. Introduction
2. Results and Discussion
2.1. Overall Patterns of Bacterial Gene Expression in Mussel Gills

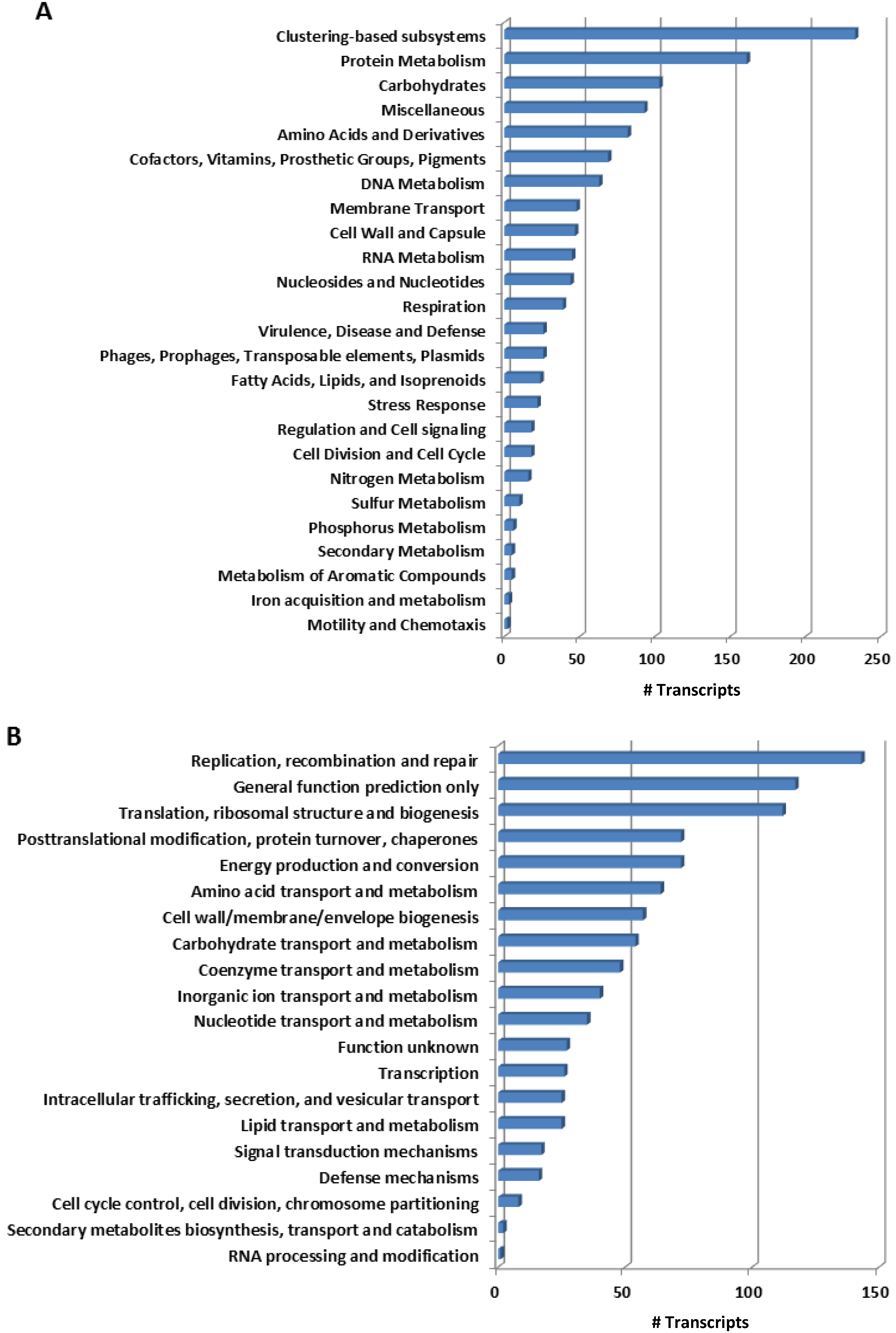{kind=link}
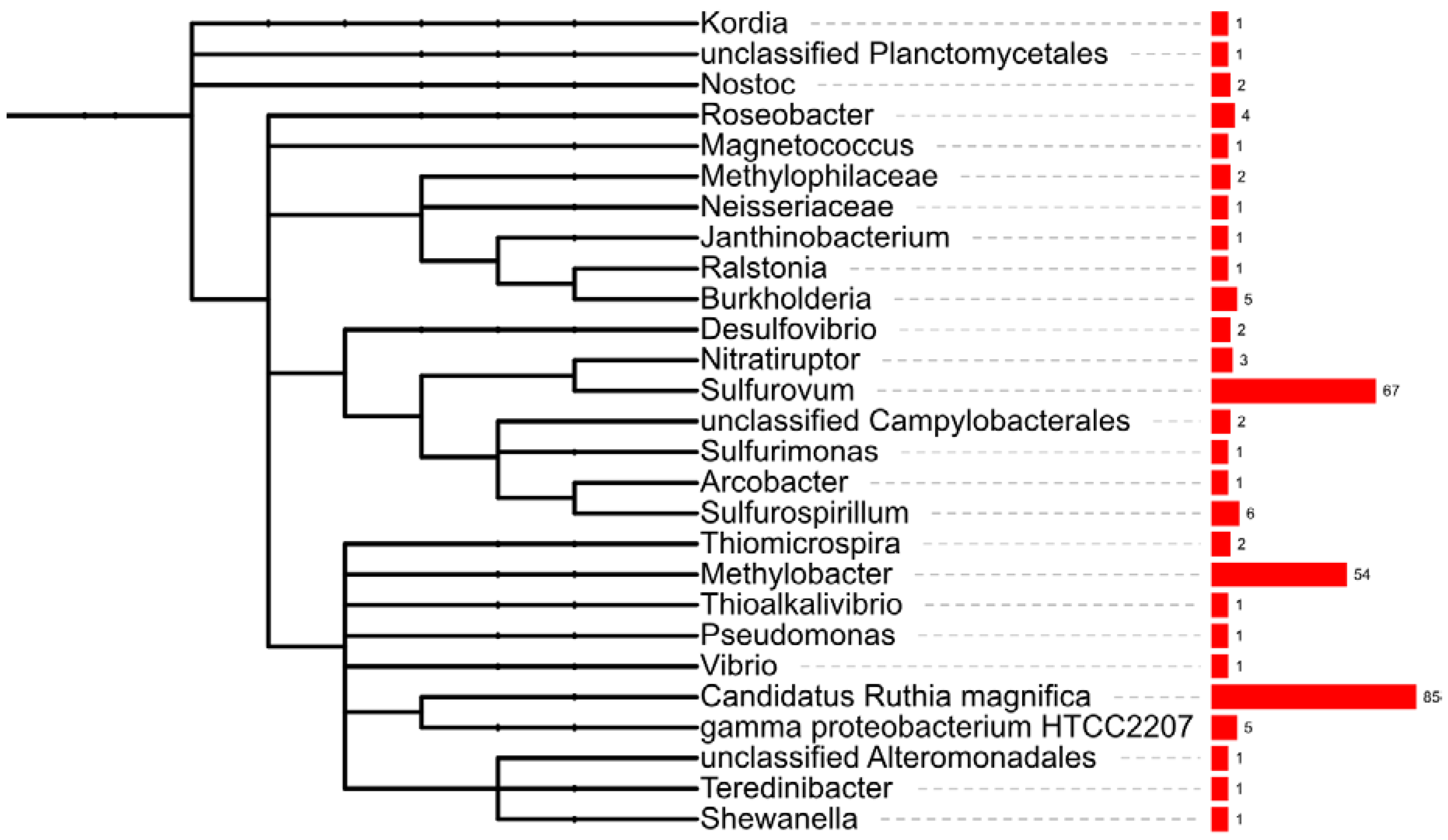{kind=link}
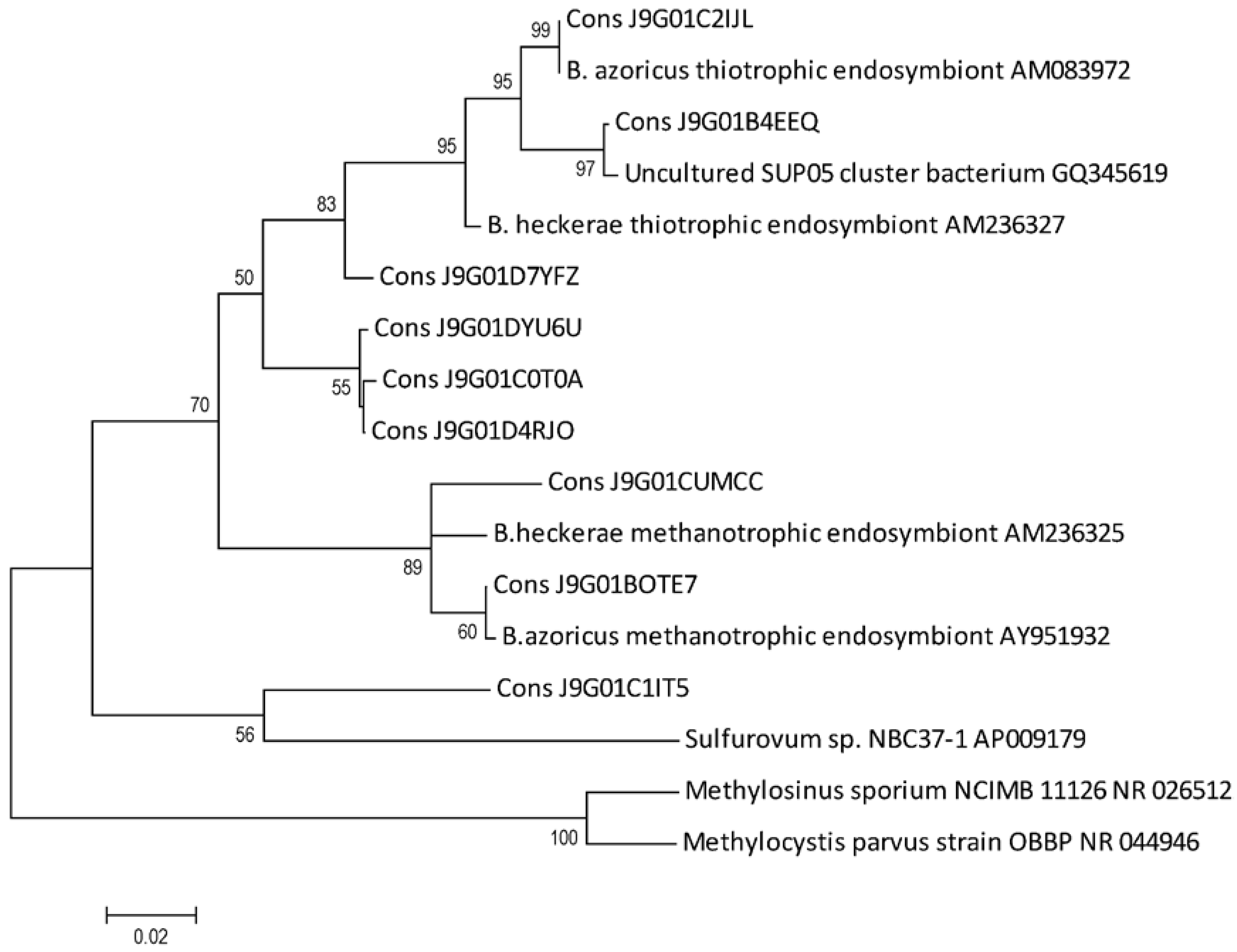{kind=link}
| Description | # Hits |
|---|---|
| # Transcripts submitted to MG-RAST | 3522 |
| Total sequence size submitted (bp) | 2,061,462 |
| Sequence length range (bp) | 100–3199 |
| Average transcripts length (bp) | 622.99 |
| # Transcripts after QC | 3099 |
| # Predicted Protein Features | 3223 |
| # Identified Protein Features | 1994 |
| # Identified Functional Categories | 1801 |
| # Transcripts annotated in SEED | 918 |
| # Transcripts annotated in KEGG | 570 |
| # Transcripts annotated in COG | 416 |
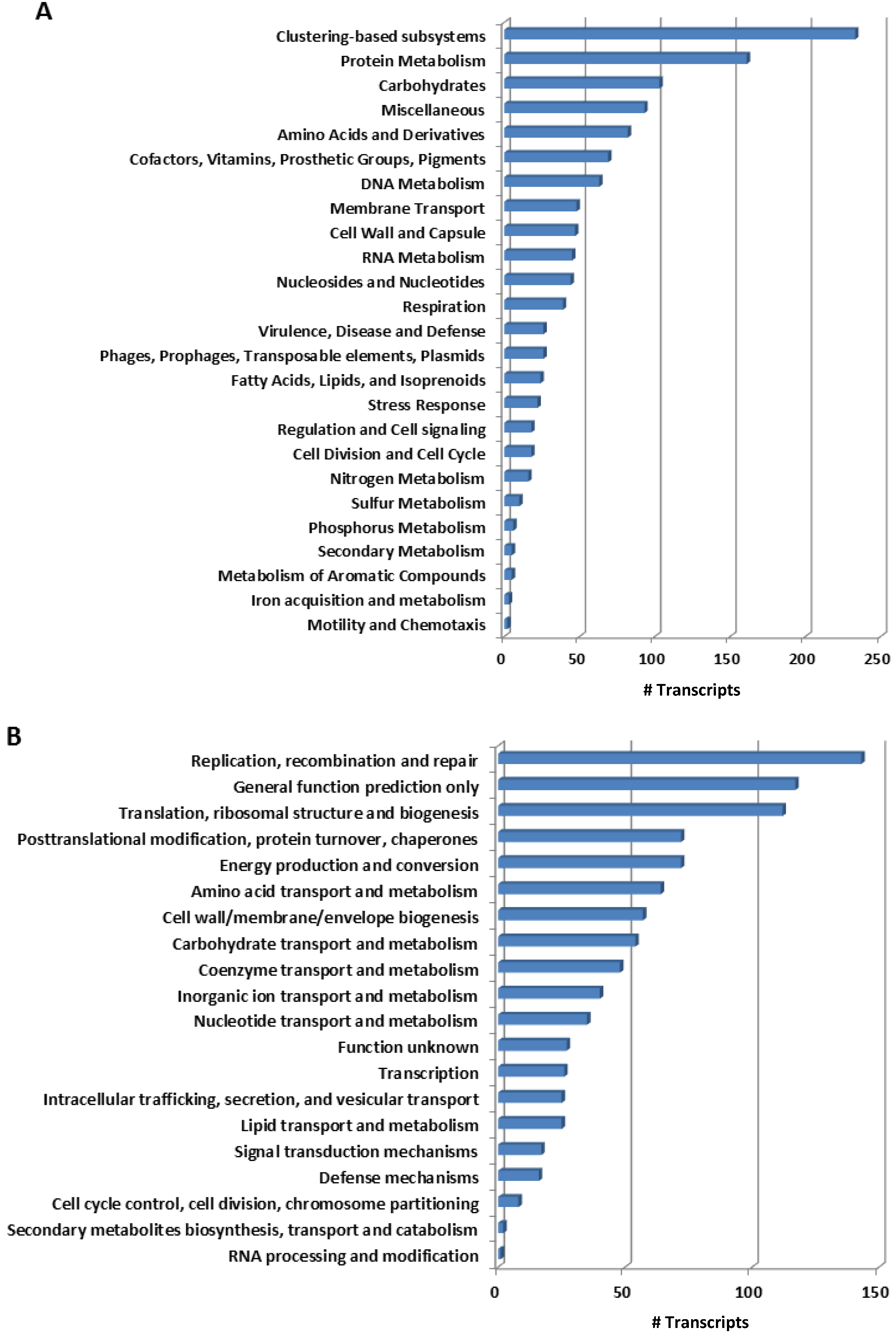
2.2. Functional Key Metabolisms in Mussel Gill Bacterial Community
| Metabolism/Pathway | Function | Hits # | e-Value | Identity (%) | Taxonomic Affiliation |
|---|---|---|---|---|---|
| CO2 fixation | Rubisco activation protein CbbQ | 2 | 1e−23 | 83.00 | Methylococcus capsulatus |
| Ribose 5-phosphate isomerase A (EC 5.3.1.6) | 1 | 1e−50 | 70.07 | Nitrosomonas | |
| Transketolase (EC 2.2.1.1) | 4 | 1e−59 | 74.38 | Methylococcus flagelatus | |
| Transketolase, N-terminal section (EC 2.2.1.1) | 1 | 1e−16 | 61.43 | ||
| Methane oxidation | Methane monooxygenase B-subunit (EC 1.14.13.25) | 1 | 1e−34 | 75.58 | Methylococcus capsulatus |
| Particulate methane monooxygenase C-subunit (EC 1.14.13.25) | 1 | 1e−44 | 85.01 | Methylococcus capsulatus | |
| Denitrification | Nitrous-oxide reductase (EC 1.7.99.6) | 1 | 1e−15 | 65.52 | |
| Nitrate and nitrite ammonification | Nitrite reductase (NAD(P)H) large subunit (EC 1.7.1.4) | 1 | 1e−44 | 66.93 | Burkholderia |
| Respiratory nitrate reductase alpha chain (EC 1.7.99.4) | 1 | 1e−46 | 78.90 | Hallela | |
| Respiratory nitrate reductase beta chain (EC 1.7.99.4) | 3 | 1e−56 | 74.63 | Chromobacterium | |
| Respiratory dehydrogenases | Methanol dehydrogenase large subunit protein (EC 1.1.99.8) | 1 | 1e−55 | 74.80 | Rhodopseudomonas |
| Sulfate reduction-associated complexesSulfur oxidation | Sulfite reductase beta subunit (EC 1.8.99.1) | 1 | 1e−42 | 91.46 | Calyptogena endosymbionts |
| Sulfite dehydrogenase cytochrome subunit SoxD | 3 | 1e−14 | 63.04 | Manganese-oxidizing bacterium (strain SI85-9A1) | |
| Sulfite oxidase | 1 | 1e−26 | 65.38 | Calyptogena endosymbionts | |
| Sulfur oxidation protein SoxB | 1 | 1e−27 | 57.73 | Thiobacillus denitrificans | |
| Sulfur oxidation protein SoxY | 2 | 1e−15 | 76.16 | Calyptogena endosymbionts |
2.3. Taxonomic Affiliation of Bacterial Genes

2.4. 16S rRNA Analysis of the Microbial Community
| Kingdom or Phylum | Class | Genus/Description | OTU | Sequences |
|---|---|---|---|---|
| Bacteria | Uncultured bacterium | 3 | 13 | |
| Proteobacteria | Gammaproteobacteria | Thiotrophic endosymbiont of B. azoricus | 1 | 4180 |
| Proteobacteria | Methanotrophic endosymbiont of B. azoricus | 1 | 1030 | |
| Proteobacteria | Psychromonas | 1 | 2 | |
| Proteobacteria | Unculturedgammaproteobacterium | 4 | 161 | |
| Proteobacteria | Epsilonproteobacteria | Sulfurovum | 1 | 1 |
| Spirochaetes | Uncultured Spirochaetes | 2 | 2 | |
| Unidentified | 18 | 224 |
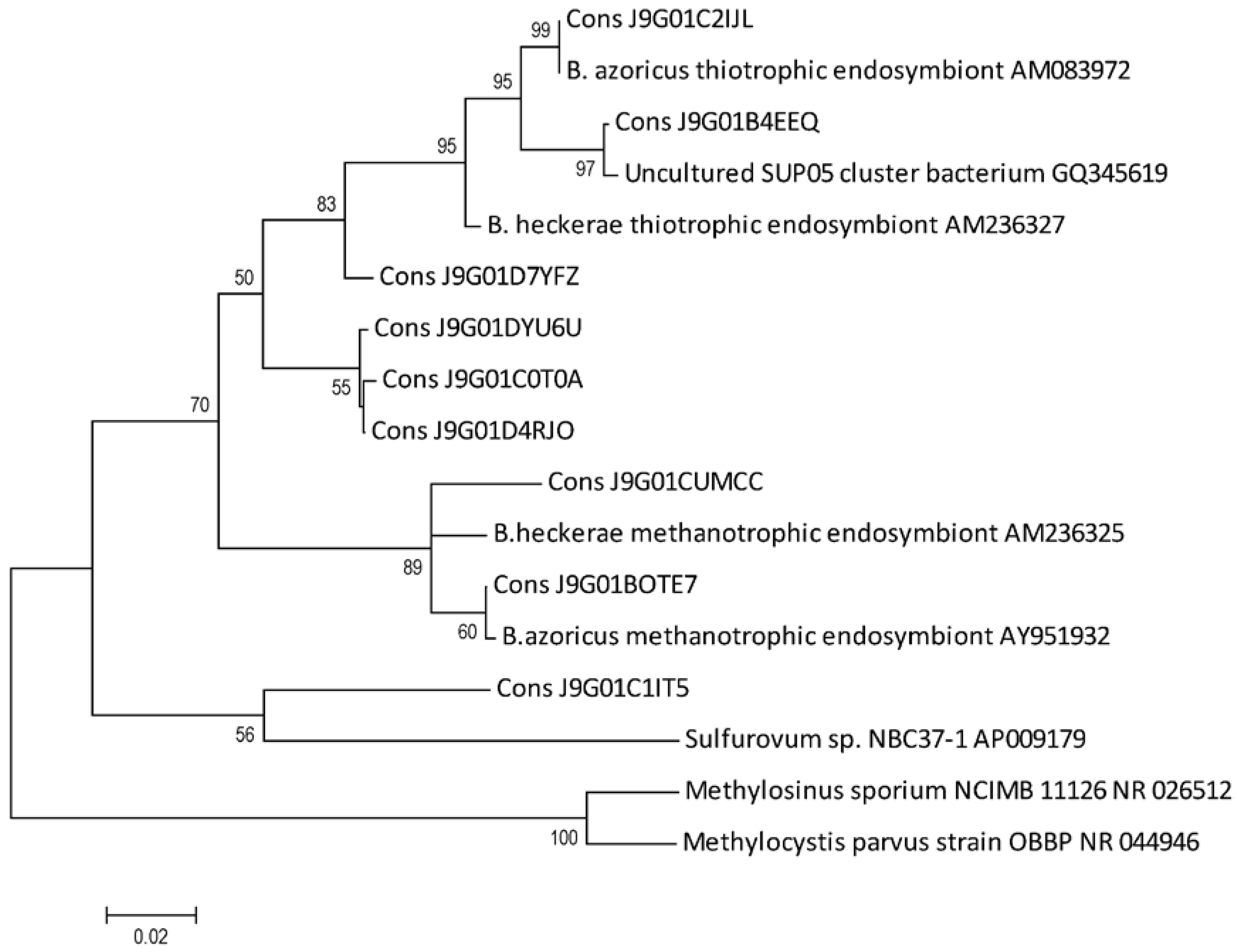
2.5. B. azoricus Gill Tissue Microbial Community
3. Experimental Section
3.1. Animal Collection
3.2. Identification of Transcripts of Bacterial Origin in the Transcriptome of Mussel Gills
3.3. Annotation of Transcripts of Bacterial Origin and Transcript Binning
3.4. 16S rRNA Amplicon Sequencing of the Gill’s Microbiome
3.5. Phylogenetic Analysis
3.6. Accession Numbers
4. Conclusions
Acknowledgments
References
- Boutet, I.; Ripp, R.; Lecompte, O.; Dossat, C.; Corre, E.; Tanguy, A.; Lallier, F.H. Conjugating effects of symbionts and environmental factors on gene expression in deep-sea hydrothermal vent mussels. BMC Genomics 2011, 12. [Google Scholar]
- De Chaine, E.G.; Bates, A.E.; Shank, T.M.; Cavanaugh, C.M. Off-axis symbiosis found: Characterization and biogeography of bacterial symbionts of Bathymodiolus mussels from Lost City hydrothermal vents. Environ. Microbiol. 2006, 8, 1902–1912. [Google Scholar] [CrossRef]
- Huber, J.A.; Mark Welch, D.B.; Morrison, H.G.; Huse, S.M.; Neal, P.R.; Butterfield, D.A.; Sogin, M.L. Microbial population structures in the deep marine biosphere. Science 2007, 318, 97–100. [Google Scholar]
- Barry, J.P.; Buck, K.R.; Kochevar, R.K.; Nelson, D.C.; Fujiwara, Y.; Goffredi, S.K.; Hashimoto, J. Methane-based symbiosis in a mussel, Bathymodiolus platifrons, from cold seeps in Sagami Bay, Japan. Invertebr. Biol. 2002, 121, 47–54. [Google Scholar]
- Dover, C.V. The Ecology of Deep-Sea Hydrothermal Vents; Princeton University Press: Princeton, NJ, USA, 2000. [Google Scholar]
- Fiala-Médioni, A.; McKiness, Z.; Dando, P.; Boulegue, J.; Mariotti, A.; Alayse-Danet, A.; Robinson, J.; Cavanaugh, C. Ultrastructural, biochemical, and immunological characterization of two populations of the mytilid mussel Bathymodiolus azoricus from the Mid-Atlantic Ridge: Evidence for a dual symbiosis. Mar. Biol. 2002, 141, 1035–1043. [Google Scholar] [CrossRef]
- Won, Y.-J.; Hallam, S.J.; O’Mullan, G.D.; Pan, I.L.; Buck, K.R.; Vrijenhoek, R.C. Environmental acquisition of thiotrophic endosymbionts by deep-sea mussels of the genus Bathymodiolus. Appl. Environ. Microbiol. 2003, 69, 6785–6792. [Google Scholar]
- Duperron, S.; Lorion, J.; Samadi, S.; Gros, O.; Gaill, F. Symbioses between deep-sea mussels (Mytilidae: Bathymodiolinae) and chemosynthetic bacteria: Diversity, function and evolution. C. R. Biol. 2009, 332, 298–310. [Google Scholar] [CrossRef]
- Page, H.M.; Fiala-Medioni, A.; Fisher, C.R.; Childress, J.J. Experimental evidence for filter-feeding by the hydrothermal vent mussel, Bathymodiolus thermophilus. Deep Sea Res. A Oceanogr. Res. Pap. 1991, 38, 1455–1461. [Google Scholar]
- Distel, D.L.; Lee, H.K.; Cavanaugh, C.M. Intracellular coexistence of methano- and thioautotrophic bacteria in a hydrothermal vent mussel. Proc. Natl. Acad. Sci. USA 1995, 92, 9598–9602. [Google Scholar] [CrossRef]
- De Long, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.-U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; Chisholm, S.W.; Karl, D.M. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar]
- Konstantinidis, K.T.; Braff, J.; Karl, D.M.; DeLong, E.F. Comparative metagenomic analysis of a microbial community residing at a depth of 4,000 meters at station ALOHA in the North Pacific subtropical gyre. Appl. Environ. Microbiol. 2009, 75, 5345–5355. [Google Scholar]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; Delong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA 2008, 105, 3805–3810. [Google Scholar]
- Gifford, S.M.; Sharma, S.; Rinta-Kanto, J.M.; Moran, M.A. Quantitative analysis of a deeply sequenced marine microbial metatranscriptome. ISME J. 2011, 5, 461–472. [Google Scholar] [CrossRef]
- Harada, M.; Yoshida, T.; Kuwahara, H.; Shimamura, S.; Takaki, Y.; Kato, C.; Miwa, T.; Miyake, H.; Maruyama, T. Expression of genes for sulfur oxidation in the intracellular chemoautotrophic symbiont of the deep-sea bivalve Calyptogena okutanii. Extremophiles 2009, 13, 895–903. [Google Scholar] [CrossRef]
- Stewart, F.J.; Dmytrenko, O.; DeLong, E.F.; Cavanaugh, C.M. Metatranscriptomic analysis of sulfur oxidation genes in the endosymbiont of solemya velum. Front Microbiol. 2011, 2. [Google Scholar]
- Bettencourt, R.; Pinheiro, M.; Egas, C.; Gomes, P.; Afonso, M.; Shank, T.; Santos, R.S. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics 2010, 11. [Google Scholar]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinforma 2008, 9. [Google Scholar]
- Poretsky, R.S.; Hewson, I.; Sun, S.; Allen, A.E.; Zehr, J.P.; Moran, M.A. Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ. Microbiol. 2009, 11, 1358–1375. [Google Scholar] [CrossRef]
- Hewson, I.; Poretsky, R.S.; Dyhrman, S.T.; Zielinski, B.; White, A.E.; Tripp, H.J.; Montoya, J.P.; Zehr, J.P. Microbial community gene expression within colonies of the diazotroph, Trichodesmium, from the Southwest Pacific Ocean. ISME J. 2009, 3, 1286–1300. [Google Scholar] [CrossRef]
- Dam, B.; Ghosh, W.; Das Gupta, S.K. Conjugative Type 4 secretion system of a novel large plasmid from the chemoautotroph Tetrathiobacter kashmirensis and construction of shuttle vectors for Alcaligenaceae. Appl. Environ. Microbiol. 2009, 75, 4362–4373. [Google Scholar]
- Hurtado, L.A.; Mateos, M.; Lutz, R.A.; Vrijenhoek, R.C. Coupling of bacterial endosymbiont and host mitochondrial genomes in the hydrothermal vent clam Calyptogena magnifica. Appl. Environ. Microbiol. 2003, 69, 2058–2064. [Google Scholar] [CrossRef]
- Degnan, P.H.; Leonardo, T.E.; Cass, B.N.; Hurwitz, B.; Stern, D.; Gibbs, R.A.; Richards, S.; Moran, N.A. Dynamics of genome evolution in facultative symbionts of aphids. Environ. Microbiol. 2010, 12, 2060–2069. [Google Scholar]
- Newton, I.; Bordenstein, S. Correlations between bacterial ecology and mobile DNA. Curr. Microbiol. 2011, 62, 198–208. [Google Scholar]
- Zander, U.; Faust, A.; Klink, B.U.; de Sanctis, D.; Panjikar, S.; Quentmeier, A.; Bardischewsky, F.; Friedrich, C.G.; Scheidig, A.J. Structural basis for the oxidation of protein-bound sulfur by the sulfur cycle molybdohemo-enzyme sulfane dehydrogenase SoxCD. J. Biol. Chem. 2011, 286, 8349–8360. [Google Scholar]
- Frigaard, N.-U.; Dahl, C. Sulfur Metabolism in Phototrophic Sulfur Bacteria. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: New York, NY, USA, 2008; Volume 54, pp. 103–200. [Google Scholar]
- Stewart, F.J.; Young, C.R.; Cavanaugh, C.M. Evidence for homologous recombination in intracellular chemosynthetic clam symbionts. Mol. Biol. Evol. 2009, 26, 1391–1404. [Google Scholar] [CrossRef]
- Beller, H.R.; Chain, P.S.G.; Letain, T.E.; Chakicherla, A.; Larimer, F.W.; Richardson, P.M.; Coleman, M.A.; Wood, A.P.; Kelly, D.P. The genome sequence of the obligately chemolithoautotrophic, facultatively anaerobic bacterium Thiobacillus denitrificans. J. Bacteriol. 2006, 188, 1473–1488. [Google Scholar]
- Kappler, U.; Dahl, C. Enzymology and molecular biology of prokaryotic sulfite oxidation. FEMS Microbiol. Lett. 2001, 203, 1–9. [Google Scholar]
- Loy, A.; Duller, S.; Baranyi, C.; Mußmann, M.; Ott, J.; Sharon, I.; Béjà, O.; Le Paslier, D.; Dahl, C.; Wagner, M. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ. Microbiol. 2009, 11, 289–299. [Google Scholar] [CrossRef]
- Duperron, S.; Sibuet, M.; MacGregor, B.J.; Kuypers, M.M.M.; Fisher, C.R.; Dubilier, N. Diversity, relative abundance and metabolic potential of bacterial endosymbionts in three Bathymodiolus mussel species from cold seeps in the Gulf of Mexico. Environ. Microbiol. 2007, 9, 1423–1438. [Google Scholar] [CrossRef]
- Esparza, M.; Cárdenas, J.; Bowien, B.; Jedlicki, E.; Holmes, D.S. Genes and pathways for CO2 fixation in the obligate, chemolithoautotrophic acidophile, Acidithiobacillus ferrooxidans, Carbon fixation in A. ferrooxidans. BMC Microbiol. 2010, 10. [Google Scholar]
- Byrne, N.; Strous, M.; Crépeau, V.; Kartal, B.; Birrien, J.-L.; Schmid, M.; Lesongeur, F.; Schouten, S.; Jaeschke, A.; Jetten, M.; et al. Presence and activity of anaerobic ammonium-oxidizing bacteria at deep-sea hydrothermal vents. ISME J. 2009, 3, 117–123. [Google Scholar] [CrossRef]
- Xie, W.; Wang, F.; Guo, L.; Chen, Z.; Sievert, S.M.; Meng, J.; Huang, G.; Li, Y.; Yan, Q.; Wu, S.; et al. Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J. 2011, 5, 414–426. [Google Scholar] [CrossRef]
- Petersen, J.M.; Zielinski, F.U.; Pape, T.; Seifert, R.; Moraru, C.; Amann, R.; Hourdez, S.; Girguis, P.R.; Wankel, S.D.; Barbe, V.; et al. Hydrogen is an energy source for hydrothermal vent symbioses. Nature 2011, 476, 176–180. [Google Scholar] [CrossRef]
- Bettencourt, R. Department of Oceanography and Fisheries, University of Azores: Horta, Portugal, Unpublished work, 2012.
- Biddle, J.F.; Fitz-Gibbon, S.; Schuster, S.C.; Brenchley, J.E.; House, C.H. Metagenomic signatures of the peru margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. USA 2008, 105, 10583–10588. [Google Scholar]
- McCarren, J.; Becker, J.W.; Repeta, D.J.; Shi, Y.; Young, C.R.; Malmstrom, R.R.; Chisholm, S.W.; DeLong, E.F. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc. Natl. Acad. Sci. USA 2010, 107, 16420–16427. [Google Scholar]
- Newton, I.L.G.; Woyke, T.; Auchtung, T.A.; Dilly, G.F.; Dutton, R.J.; Fisher, M.C.; Fontanez, K.M.; Lau, E.; Stewart, F.J.; Richardson, P.M.; et al. The Calyptogena magnifica chemoautotrophic symbiont genome. Science 2007, 315, 998–1000. [Google Scholar]
- Nakagawa, S.; Takaki, Y.; Shimamura, S.; Reysenbach, A.-L.; Takai, K.; Horikoshi, K. Deep-sea vent epsilon-proteobacterial genomes provide insights into emergence of pathogens. Proc. Natl. Acad. Sci. USA. 2007, 104, 12146–12150. [Google Scholar]
- Kato, S.; Kobayashi, C.; Kakegawa, T.; Yamagishi, A. Microbial communities in iron-silica-rich microbial mats at deep-sea hydrothermal fields of the Southern Mariana Trough. Environ. Microbiol. 2009, 11, 2094–2111. [Google Scholar] [CrossRef]
- Sylvan, J.B.; Toner, B.M.; Edwards, K.J. Life and death of deep-sea vents: Bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. mBio 2012, 3. [Google Scholar]
- Stingl, U.; Desiderio, R.A.; Cho, J.-C.; Vergin, K.L.; Giovannoni, S.J. The SAR92 clade: An abundant coastal clade of culturable marine bacteria possessing proteorhodopsin. Appl. Environ. Microbiol. 2007, 73, 2290–2296. [Google Scholar]
- Buchan, A.; González, J.M.; Moran, M.A. Overview of the marine Roseobacter lineage. Appl. Environ. Microbiol. 2005, 71, 5665–5677. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life v2: Online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011, 39, W475–W478. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar]
- Xu, Y.; Nogi, Y.; Kato, C.; Liang, Z.; Rüger, H.-J.; de Kegel, D.; Glansdorff, N. Psychromonas profunda sp. nov., a psychropiezophilic bacterium from deep Atlantic sediments. Int. J. Syst. Evol. Microbiol. 2003, 53, 527–532. [Google Scholar] [CrossRef]
- Tokuda, G.; Yamada, A.; Nakano, K.; Arita, N.O.; Yamasaki, H. Colonization of Sulfurovum sp. on the gill surfaces of Alvinocaris longirostris, a deep-sea hydrothermal vent shrimp. Mar. Ecol. 2007, 29, 106–114. [Google Scholar]
- Husmann, G.; Gerdts, G.; Wichels, A. Spirochetes in crystalline styles of marine bivalves: Group-specific PCR detection and 16S rRNA sequence analysis. J. Shellfish Res. 2010, 29, 1069–1075. [Google Scholar] [CrossRef]
- Takai, K.; Campbell, B.J.; Cary, S.C.; Suzuki, M.; Oida, H.; Nunoura, T.; Hirayama, H.; Nakagawa, S.; Suzuki, Y.; Inagaki, F.; et al. Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of epsilonproteobacteria. Appl. Environ. Microbiol. 2005, 71, 7310–7320. [Google Scholar] [CrossRef]
- Corre, E.; Reysenbach, A.L.; Prieur, D. Epsilon-proteobacterial diversity from a deep-sea hydrothermal vent on the Mid-Atlantic Ridge. FEMS Microbiol. Lett. 2001, 205, 329–335. [Google Scholar]
- Suzuki, Y.; Kojima, S.; Sasaki, T.; Suzuki, M.; Utsumi, T.; Watanabe, H.; Urakawa, H.; Tsuchida, S.; Nunoura, T.; Hirayama, H.; et al. Host-symbiont relationships in hydrothermal vent gastropods of the genus Alviniconcha from the Southwest Pacific. Appl. Environ. Microbiol. 2006, 72, 1388–1393. [Google Scholar] [CrossRef]
- Hügler, M.; Gärtner, A.; Imhoff, J.F. Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. Ecol. 2010, 73, 526–537. [Google Scholar]
- Petersen, J.M.; Ramette, A.; Lott, C.; Cambon-Bonavita, M.-A.; Zbinden, M.; Dubilier, N. Dual symbiosis of the vent shrimp Rimicaris exoculata with filamentous gamma- and epsilonproteobacteria at four Mid-Atlantic Ridge hydrothermal vent fields. Environ. Microbiol. 2010, 12, 2204–2218. [Google Scholar]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.G.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef]
- DeepSeaVent Database. Available online: http://transcriptomics.biocant.pt:8080/deepSeaVent (accessed on 19 June 2010).
- Stajich, J.E.; Block, D.; Boulez, K.; Brenner, S.E.; Chervitz, S.A.; Dagdigian, C.; Fuellen, G.; Gilbert, J.G.R.; Korf, I.; Lapp, H.; et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002, 12, 1611–1618. [Google Scholar] [CrossRef]
- MG-RAST Metagenomics Analysis Server. Available online: http://metagenomics.anl.gov (accessed on 5 February 2012).
- Wang, Y.; Qian, P.-Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 2009, 4. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP: Phylogenetic Inference Package, Version 3.5c; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Jukes, T.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: New York, NY, USA, 1969; Volume 3, pp. 121–132. [Google Scholar]
- Felsenstein, J. Confidence limits on Phylogenies: An approach using bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Mid-Atlantic Ridge 2008 (MAR08) Homepage. Available online: http://www.deepseavoyage.research.pdx.edu (accessed on 5 February 2010).
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Egas, C.; Pinheiro, M.; Gomes, P.; Barroso, C.; Bettencourt, R. The Transcriptome of Bathymodiolus azoricus Gill Reveals Expression of Genes from Endosymbionts and Free-Living Deep-Sea Bacteria. Mar. Drugs 2012, 10, 1765-1783. https://doi.org/10.3390/md10081765
Egas C, Pinheiro M, Gomes P, Barroso C, Bettencourt R. The Transcriptome of Bathymodiolus azoricus Gill Reveals Expression of Genes from Endosymbionts and Free-Living Deep-Sea Bacteria. Marine Drugs. 2012; 10(8):1765-1783. https://doi.org/10.3390/md10081765
Chicago/Turabian StyleEgas, Conceição, Miguel Pinheiro, Paula Gomes, Cristina Barroso, and Raul Bettencourt. 2012. "The Transcriptome of Bathymodiolus azoricus Gill Reveals Expression of Genes from Endosymbionts and Free-Living Deep-Sea Bacteria" Marine Drugs 10, no. 8: 1765-1783. https://doi.org/10.3390/md10081765