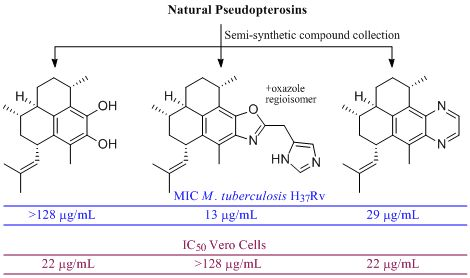
Evaluation of Pseudopteroxazole and Pseudopterosin Derivatives against Mycobacterium tuberculosis and Other Pathogens


Abstract
: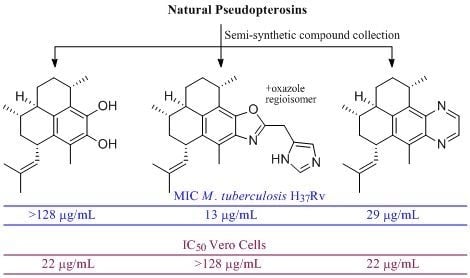
1. Introduction
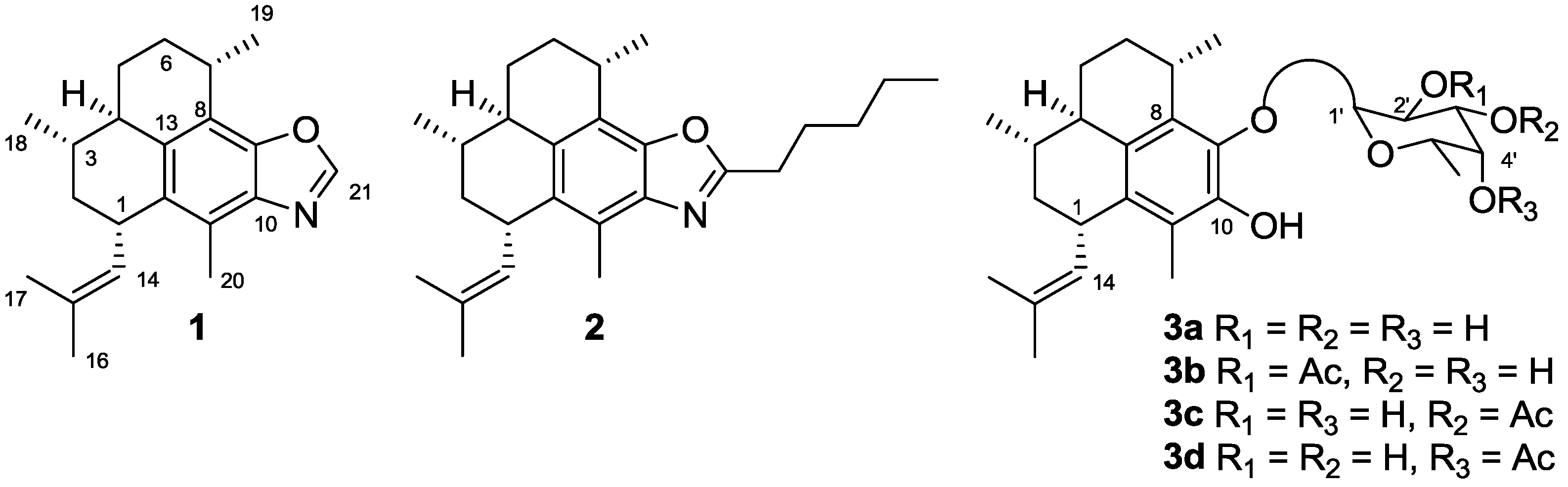
2. Results and Discussion
2.1. Chemistry
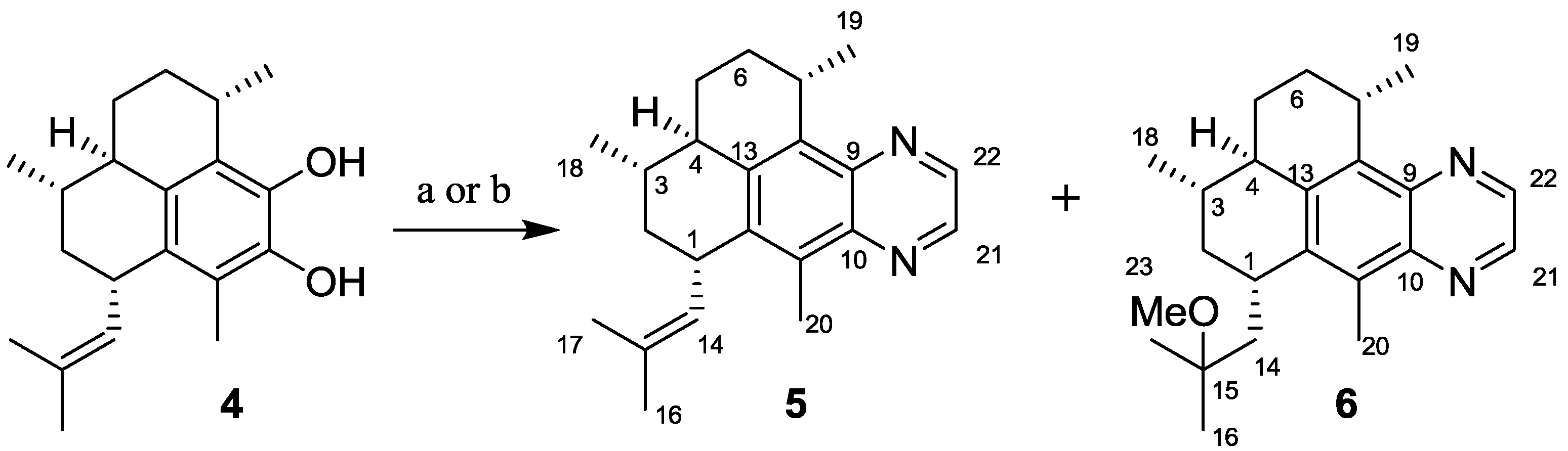
2.1.1. Synthesis of Pseudopteroquinoxalines 5 and 6
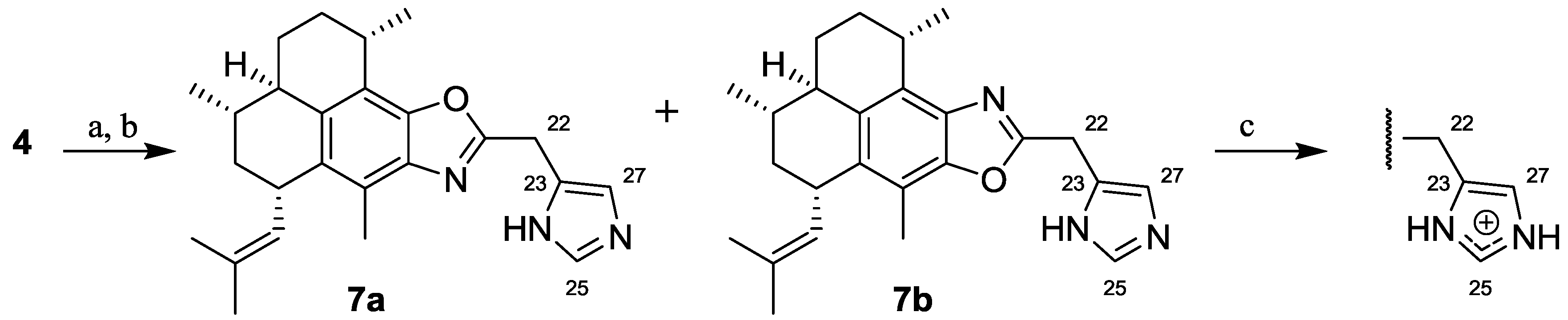
2.1.2. Synthesis of Pseudopteroxazoles 7a and 7b
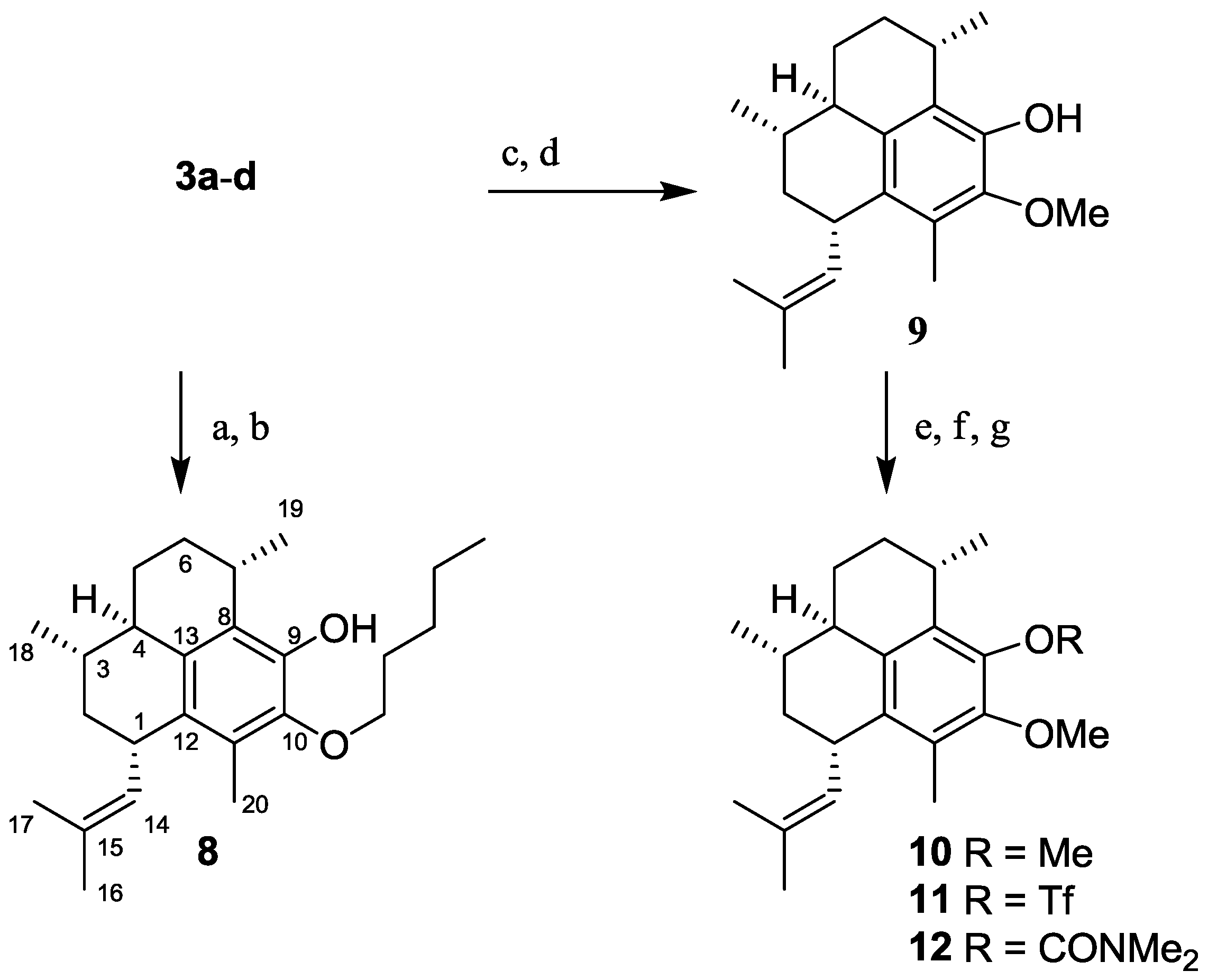
2.1.3. Synthesis of Pseudopterosin Derivatives 8–12
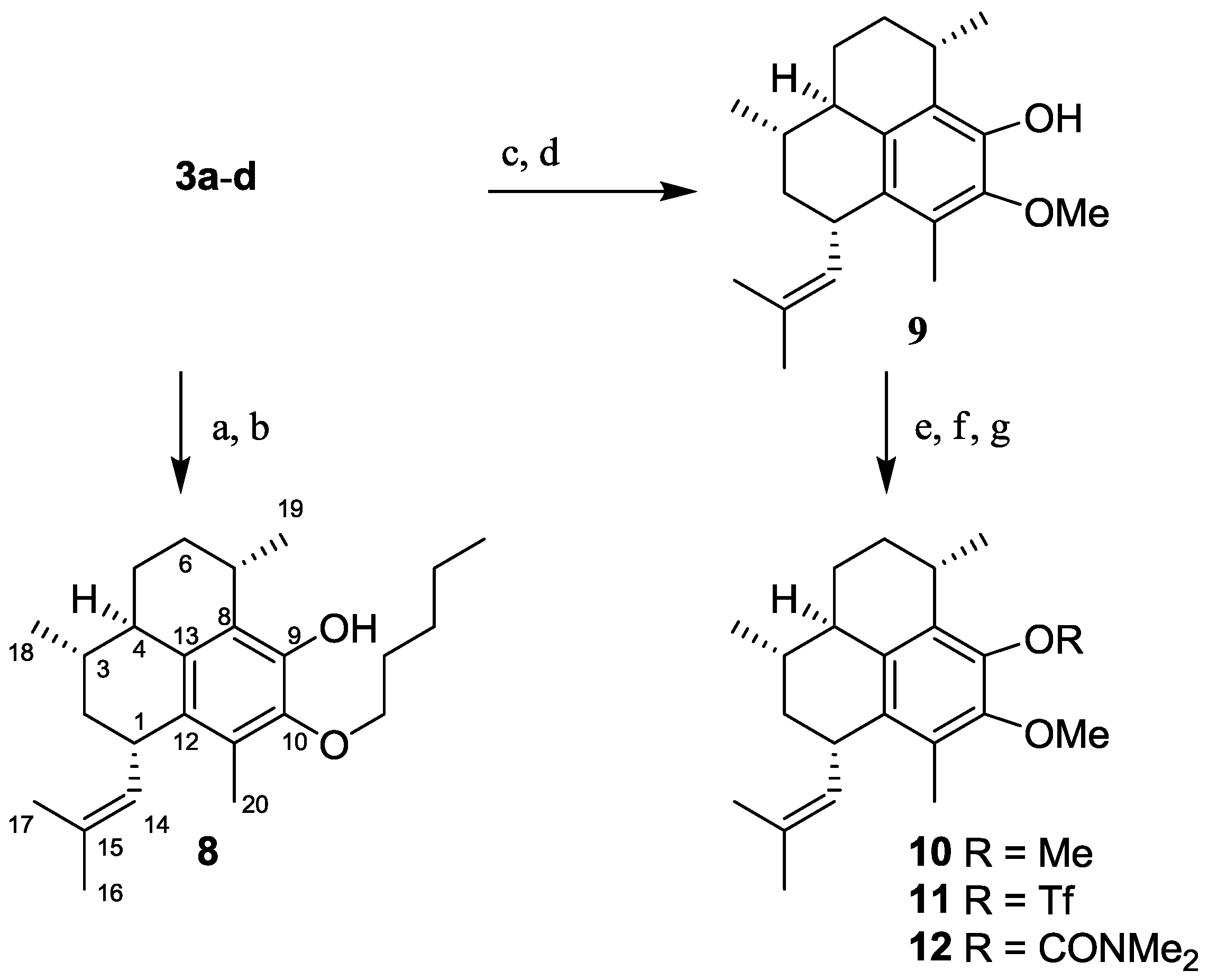
2.1.4. Synthesis of Pseudopterosin Mimics 14–20
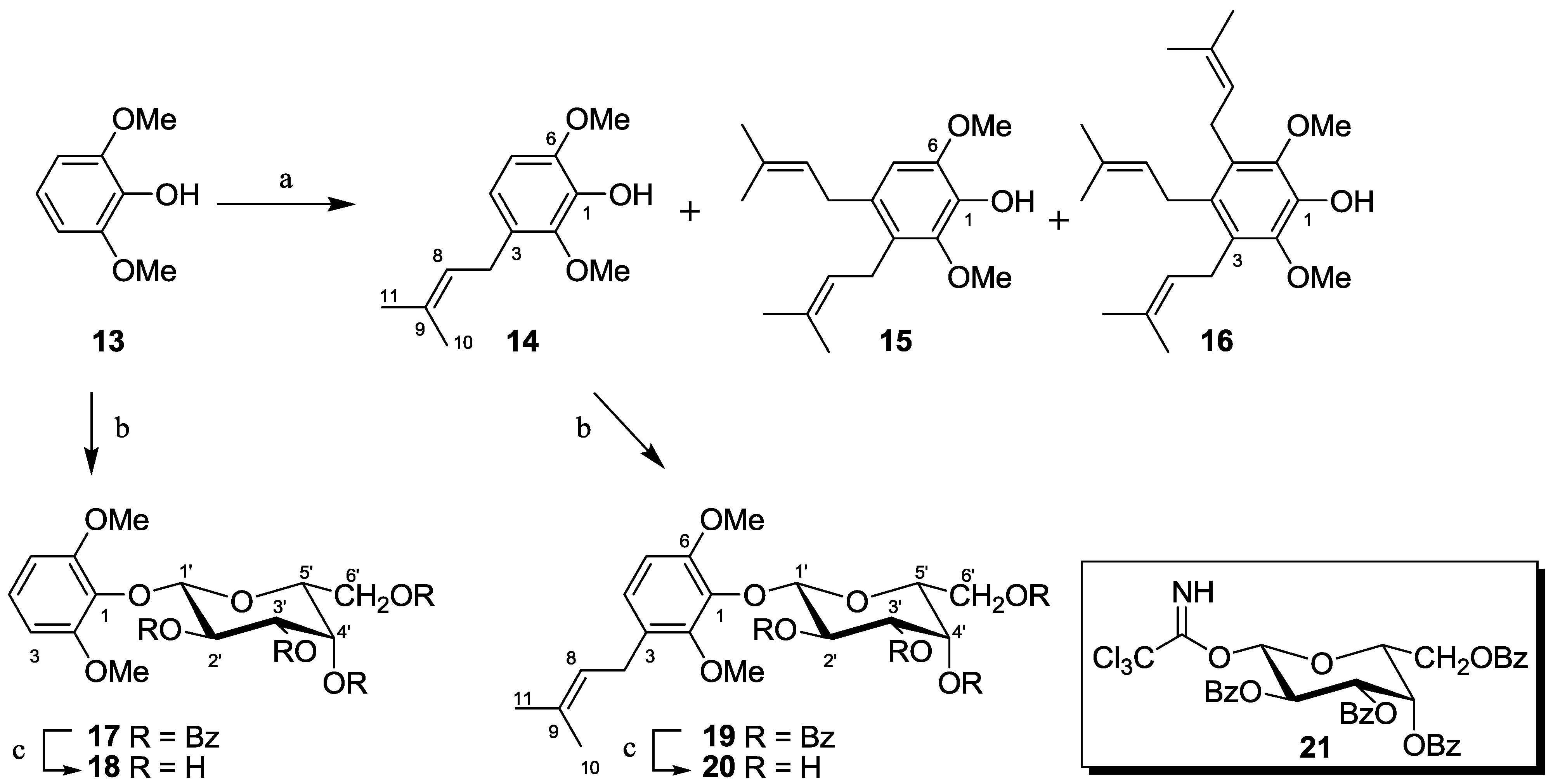
2.2. Antibacterial Activity
2.2.1. Activity of Semi-Synthetic Pseudopteroxazoles in M. tuberculosis Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MABA aMIC [μg/mL] (% inh) | LORA b % inh c | LORA b MIC [μg/mL] | Vero cell IC50 [μg/mL] (% inh) | SI d |
|---|---|---|---|---|---|
| 1 (Ptx-H) | 15 | 99.7 | 50 | >128 (0%) | >8.6 |
| 2 (Ptx-(CH2)4CH3) | >128 (6.3%) | 22.6 | NT | >128 (0%) | NA |
| 7a/7b (Ptx-CH2-(1H-imidazol-5-yl)) e,f | 13 | 92.5 | 12 | >128 (4%) | >9.7 |
| 22 (iso-Ptx-H) | 14 | 100.0 | 44 | 52 | 3.6 |
| 23 (Ptx-(2-CH3O-Ph)) | >82 (48%) | 83.3 | NT g | 34 | NA h |
| 24 (Ptx-(4-F-Ph)) | >31 (20%) | −19.0 | NT | >31 (0%) | NA |
| 25 (Ptx-CH3) | 15 | 99.0 | NT | 12 | 0.8 |
| 26 (Ptx-CH(CH3)CH2CH3) | >103 (0%) | 59.1 | NT | 73 | NA |
| 27 (Ptx-(CH2)2SCH3) | 106.8 | 90.0 | NT | >128 (0%) | >1.2 |
| 28 (Ptx-CH2Ph) | >128 (28%) | 13.7 | NT | 82 | NA |
| 29 (Ptx-CHOHCH3) | 53 | 99.8 | NT | 24 | 0.5 |
| 30 (Ptx-(CH2)2CO2CH3) | >128 (81%) | 99.1 | 62 | 31 | NA |
| 31 (Ptx-(CH2)2CO2H) | 95 | 99.1 | NT | 102 | 1.1 |
| 32 (Ptx-(CH2)2CONH2) | 29 | 99.0 | NT | 54 | 1.9 |
| 33 (Ptx-CH2CONH2) | 59 | 97.1 | NT | 45 | 0.8 |
| Rifampin | 0.04 | NT | 0.93 | NT | NA |
| Isoniazid | 0.03 | NT | >128 (65%) | NT | NA |
| PA824 | 0.15 | NT | NT | NT | NA |
 | |||||
| MABA a MIC [μg/mL] | |||||||
|---|---|---|---|---|---|---|---|
| Compound | H37Rv | RMPr | INHr | SMr | KMr | CSr | MOXr |
| 1 | 7 | 8 | 8 | 14 | 16 | 14 | 8 |
| Rifampin | 0.03 | >3.3 | 0.02 | 0.08 | 0.02 | 0.01 | 0.02 |
| Isoniazid | 0.03 | 0.12 | >1.10 | 0.13 | 0.13 | 0.12 | 0.03 |
2.2.2. Anti-Microbial Activity of Semi-Synthetic Pseudopteroquinoxalines, Pseudopterosins, and the Prenylated Mimics
| IC50 [μg/mL] | MIC [μg/mL] (% inh) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | MRSA a | VRE a | M. smegmatisa | M. diernhoferia | M. tuberculosis b (% inh) | LORA c % inh d | LORA c MIC [μg/mL] | Vero cell IC50 [μg/mL] (% inh) | SI e |
| 3a–d f (Ps G–J mixture) | <1 | <1 | 2 | 2 | 30 | 97.9 | NT g | >128 (32%) | >4.3 |
| 4 f (Ps G–J aglycone) | 88 | >128 | 8 | 8 | >128 (86%) | 53.1 | NT g | 50 | NA h |
| 5 (pseudopteroquinoxaline) f | >128 | >128 | 16 | 64 | 29 | 99.9 | NT | 22 | 0.8 |
| 6 (Me ether of pseudopteroquinoxaline) | NT | NT | NT | NT | 84 | 99.9 | NT | 15 | 0.2 |
| 8 f (Ps G–J mono-pentyl ether) | 47 | 22 | >128 | >128 | >128 (85%) | 35.1 | NT | 49 | NA |
| 9 f (Ps G–J mono-methyl ether) | 9 | 12 | 4 | 4 | 30 | 99.7 | 52 | >128 (26%) | >4.3 |
| 10 f (Ps G–J di-methyl ether) | >128 | 25 | >64 | >64 | >128 (63%) | 77.5 | NT | 51 | NA |
| 11 f (triflate) | >128 | >128 | >64 | >64 | >128 (44%) | 37.5 | NT | 51 | NA |
| 12 f (carbamate) | >128 | 70 | 64 | >64 | >128 (77%) | 80.0 | NT | >128 (0%) | NA |
| 14 f (mono-prenylated mimic) | >128 | >128 | 128 | 64 | >128 (24%) | −4.7 | NT | 44 | NA |
| 15 f (di-prenylated mimic) | 20 | 3 | 8 | 8 | 59 | 99.7 | 58 | 64 | 1.1 |
| 16 f (tri-prenylated mimic) | >128 | 3 | >128 | 8 | 56 | 79.7 | NT | >128 (29%) | >2.3 |
| 20 (galactoside of 14) | >128 | >128 | >128 | >128 | >128 (1.4%) | −24.8 | NT | 82 | NA |
| Vancomycin | 1.23 | NT | NT | NT | NT | NT | NT | NT | NA |
| Rifampin | NT | 0.88 | 4 | 4 | 0.04 | NT | 0.93 | NT | NA |
| Isoniazid | NT | NT | NT | NT | 0.03 | NT | >128 (65%) | NT | NA |
| PA824 | NT | NT | NT | NT | 0.15 | NT | NT | NT | NA |
2.2.3. Relevance of the Use of Model Mycobacteria
3. Experimental Section
3.1. General Experimental Procedures
3.2. Synthesis
3.2.1. Synthesis of Pseudopteroquinoxaline (5)
3.2.2. Synthesis of 14,15-Dihydro-15-methoxy-pseudopteroquinoxaline (6)
3.2.3. Synthesis of 21-((1H-Imidazol-5-yl)methyl)-pseudopteroxazole (7a) and 21-((1H-Imidazol-5-yl)methyl)-isopseudopteroxazole (7b)
3.2.4. Synthesis of 10-Pentoxy-pseudopterosin G–J Aglycone (8)
3.2.5. Synthesis of 9,10-Dimethoxy-pseudopterosin G–J Aglycone (10)
3.2.6. Synthesis of 9-Trifluoromethylsulfonyloxy-10-methoxy-pseudopterosin G–J Aglycone (11)
3.2.7. Synthesis of 9-Dimethylcarbamoyloxy-10-methoxy-pseudopterosin G–J Aglycone (12)
3.2.8. Prenylation of 2,6-Dimethoxyphenol: Synthesis of 14, 15 & 16
3.2.9. Synthesis of the Glycosyl Donor 2,3,4,6-Tetra-O-benzoyl-β-D-galactopyranosyl Trichloroacetimidate (21)
3.2.10. Synthesis of 2,6-Dimethoxyphenol-2,3,4,6-tetra-O-benzoyl-β-D-galactopyranoside (17)
3.2.11. Synthesis of 2,6-Dimethoxyphenol-β-D-galactopyranoside (18)
3.2.12. Synthesis of 2,6-Dimethoxy-3-(3-methylbut-2-enyl)phenol-2,3,4,6-tetra-O-benzoyl-β-D-galactopyranoside (19)
3.2.13. Synthesis of 2,6-Dimethoxy-3-(3-methylbut-2-enyl)phenol-β-D-galactopyranoside (20)
4. Conclusions
Acknowledgments
References and Notes
- Stop TB Partnership, The Global Plan to Stop TB 2011–2015: Transforming the Fight towards Elimination of Tuberculosis; World Health Organization: Geneva, Switzerland, 2011.
- Kaneko, T.; Cooper, C.; Mdluli, K. Challenges and opportunities in developing novel drugs for TB. Future Med. Chem. 2011, 3, 1373–1400. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The Challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar]
- Dover, L.G.; Coxon, G.D. Current status and research strategies in tuberculosis drug development. J. Med. Chem. 2011, 54, 6157–6165. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural product scaffolds as leads to drugs. Future Med. Chem. 2009, 1, 1415–1427. [Google Scholar] [CrossRef]
- Rodriguez, A.D.; Ramirez, C.; Rodriguez, I.I.; Gonzalez, E. Novel antimycobacterial benzoxazole alkaloids, from the west Indian Sea whip Pseudopterogorgia elisabethae. Org. Lett. 1999, 1, 527–530. [Google Scholar] [CrossRef]
- Rodriguez, I.I.; Rodriguez, A.D. Homopseudopteroxazole, a new antimycobacterial diterpene alkaloid from Pseudopterogorgia elisabethae. J. Nat. Prod. 2003, 66, 855–857. [Google Scholar] [CrossRef]
- Harmata, M.; Hong, X. Benzothiazines in synthesis. A total synthesis of pseudopteroxazole. Org. Lett. 2005, 7, 3581–3583. [Google Scholar] [CrossRef]
- Harmata, M.; Cai, Z.; Chen, Y. Benzothiazines in synthesis. A formal total synthesis of pseudopteroxazole. J. Org. Chem. 2009, 74, 5559–5561. [Google Scholar] [CrossRef]
- Davidson, J.P.; Corey, E.J. First enantiospecific total synthesis of the antitubercular marine natural product pseudopteroxazole. Revision of assigned stereochemistry. J. Am. Chem. Soc. 2003, 125, 13486–13489. [Google Scholar] [CrossRef]
- McCulloch, M.W.; Berrue, F.; Haltli, B.; Kerr, R.G. One-pot syntheses of pseudopteroxazoles from pseudopterosins: A rapid route to non-natural congeners with improved antimicrobial activity. J. Nat. Prod. 2011, 74, 2250–2256. [Google Scholar] [CrossRef]
- More than 26 different pseudopterosins have been identified in nature, these can possess different configurations about the aglycone and/or different sugars.
- Berrue, F.; McCulloch, M.W.B.; Kerr, R.G. Marine diterpene glycosides. Bioorg. Med. Chem. 2011, 19, 6702–6719. [Google Scholar] [CrossRef]
- Look, S.A.; Fenical, W.; Jacobs, R.S.; Clardy, J. The pseudopterosins: Anti-inflammatory and analgesic natural products from the sea whip Pseudopterogorgia elisabethae. Proc. Natl. Acad. Sci. USA 1986, 83, 6238–6240. [Google Scholar] [CrossRef]
- Rodriguez, I.I.; Shi, Y.P.; Garcia, O.J.; Rodriguez, A.D.; Mayer, A.M.; Sanchez, J.A.; Ortega-Barria, E.; Gonzalez, J. New pseudopterosin and seco-pseudopterosin diterpene glycosides from two Colombian isolates of Pseudopterogorgia elisabethae and their diverse biological activities. J. Nat. Prod. 2004, 67, 1672–1680. [Google Scholar] [CrossRef]
- Correa, H.; Aristizabal, F.; Duque, C.; Kerr, R. Cytotoxic and antimicrobial activity of pseudopterosins and seco-pseudopterosins isolated from the octocoral Pseudopterogorgia elisabethae of San Andres and Providencia Islands (Southwest Caribbean Sea). Mar. Drugs 2011, 9, 334–343. [Google Scholar] [CrossRef]
- Ata, A.; Win, H.Y.; Holt, D.; Holloway, P.; Segstro, E.P.; Jayatilake, G.S. New antibacterial diterpenes from Pseudopterogorgia elisabethae. Helv. Chim. Acta 2004, 87, 1090–1098. [Google Scholar] [CrossRef]
- Lazerwith, S.E.; Johnson, T.W.; Corey, E.J. Syntheses and stereochemical revision of pseudopterosin G–J aglycon and helioporin E. Org. Lett. 2000, 2, 2389–2392. [Google Scholar] [CrossRef]
- Roussis, V.; Wu, Z.; Fenical, W.; Strobel, S.A.; Van Duyne, G.D.; Clardy, J. New anti-inflammatory pseudopterosins from the marine octocoral Pseudopterogorgia elisabethae. J. Org. Chem. 1990, 55, 4916–4922. [Google Scholar]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar]
- Falzari, K.; Zhu, Z.; Pan, D.; Liu, H.; Hongmanee, P.; Franzblau, S.G. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 1447–1454. [Google Scholar] [CrossRef]
- Cho, S.H.; Warit, S.; Wan, B.; Hwang, C.H.; Pauli, G.F.; Franzblau, S.G. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [Google Scholar] [CrossRef]
- Collins, L.; Franzblau, S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar]
- Molina-Salinas, G.M.; Rivas-Galindo, V.M.; Said-Fernandez, S.; Lankin, D.C.; Munoz, M.A.; Joseph-Nathan, P.; Pauli, G.F.; Waksman, N. Stereochemical analysis of leubethanol, an anti-TB-active serrulatane, from Leucophyllum frutescens. J. Nat. Prod. 2011, 74, 1842–1850. [Google Scholar]
- Arbex, M.A.; Varella Mde, C.; Siqueira, H.R.; Mello, F.A. Antituberculosis drugs: Drug interactions, adverse effects, and use in special situations. Part 2: Second line drugs. J. Bras. Pneumol. 2010, 36, 641–656. [Google Scholar] [CrossRef]
- Shi, R.; Itagaki, N.; Sugawara, I. Overview of anti-tuberculosis (TB) drugs and their resistance mechanisms. Mini Rev. Med. Chem. 2007, 7, 1177–1185. [Google Scholar] [CrossRef]
- Altaf, M.; Miller, C.H.; Bellows, D.S.; O’Toole, R. Evaluation of the Mycobacterium smegmatis and BCG models for the discovery of Mycobacterium tuberculosis inhibitors. Tuberculosis (Edinb.) 2010, 90, 333–337. [Google Scholar] [CrossRef]
- Marchbank, D.H.; Kerr, R.G. Semisynthesis of fuscoside B analogues and eunicosides, and analysis of anti-inflammatory activity. Tetrahedron 2011, 67, 3053–3061. [Google Scholar] [CrossRef]
- Mbadugha, B.N.; Menger, F.M. Sugar/steroid/sugar conjugates: Sensitivity of lipid binding to sugar structure. Org. Lett. 2003, 5, 4041–4044. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McCulloch, M.W.B.; Haltli, B.; Marchbank, D.H.; Kerr, R.G. Evaluation of Pseudopteroxazole and Pseudopterosin Derivatives against Mycobacterium tuberculosis and Other Pathogens. Mar. Drugs 2012, 10, 1711-1728. https://doi.org/10.3390/md10081711
McCulloch MWB, Haltli B, Marchbank DH, Kerr RG. Evaluation of Pseudopteroxazole and Pseudopterosin Derivatives against Mycobacterium tuberculosis and Other Pathogens. Marine Drugs. 2012; 10(8):1711-1728. https://doi.org/10.3390/md10081711
Chicago/Turabian StyleMcCulloch, Malcolm W. B., Brad Haltli, Douglas H. Marchbank, and Russell G. Kerr. 2012. "Evaluation of Pseudopteroxazole and Pseudopterosin Derivatives against Mycobacterium tuberculosis and Other Pathogens" Marine Drugs 10, no. 8: 1711-1728. https://doi.org/10.3390/md10081711
APA StyleMcCulloch, M. W. B., Haltli, B., Marchbank, D. H., & Kerr, R. G. (2012). Evaluation of Pseudopteroxazole and Pseudopterosin Derivatives against Mycobacterium tuberculosis and Other Pathogens. Marine Drugs, 10(8), 1711-1728. https://doi.org/10.3390/md10081711