Preparation and Characterization of O-Acylated Fucosylated Chondroitin Sulfate from Sea Cucumber

Abstract
:1. Introduction
2. Results and Discussion
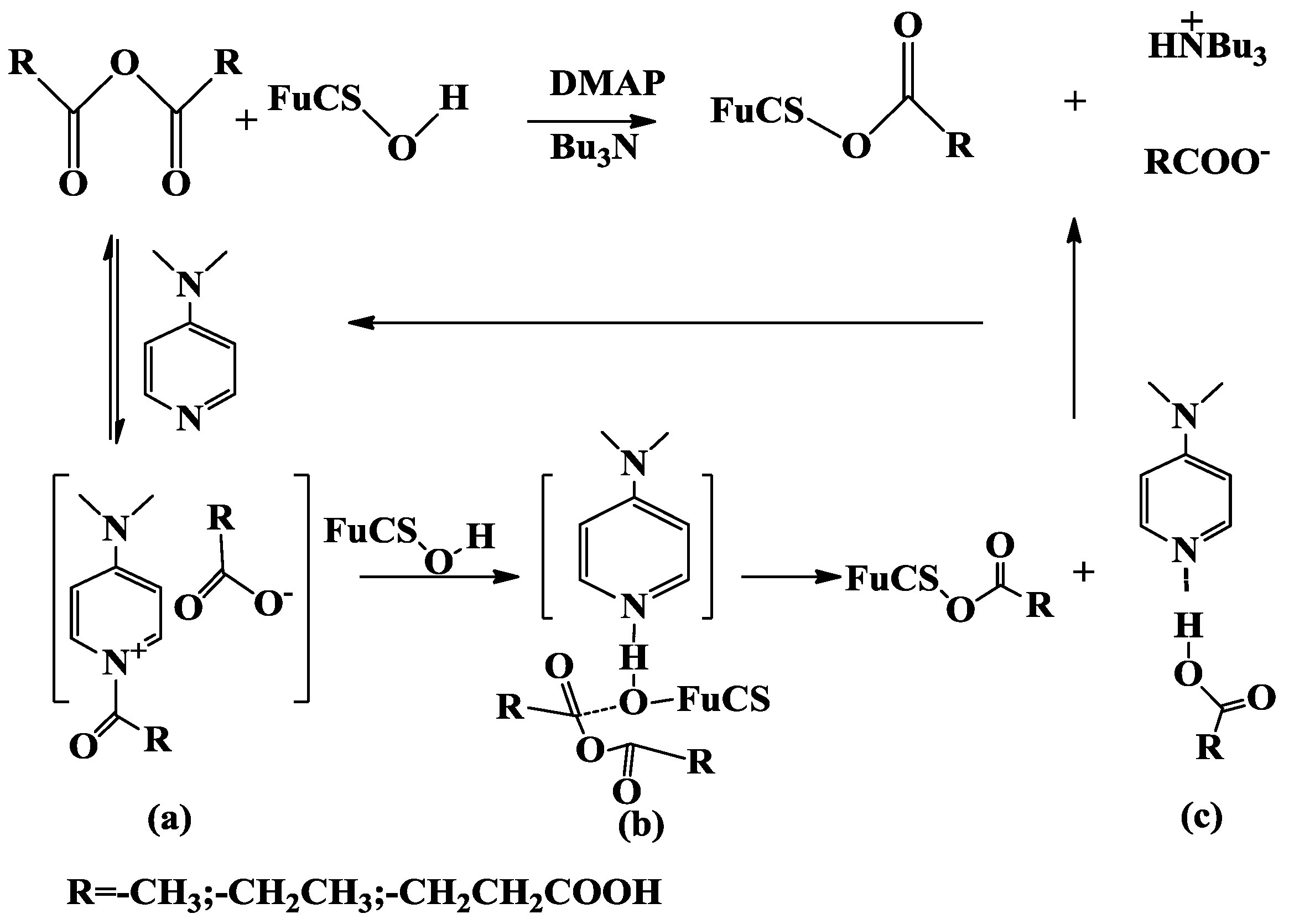
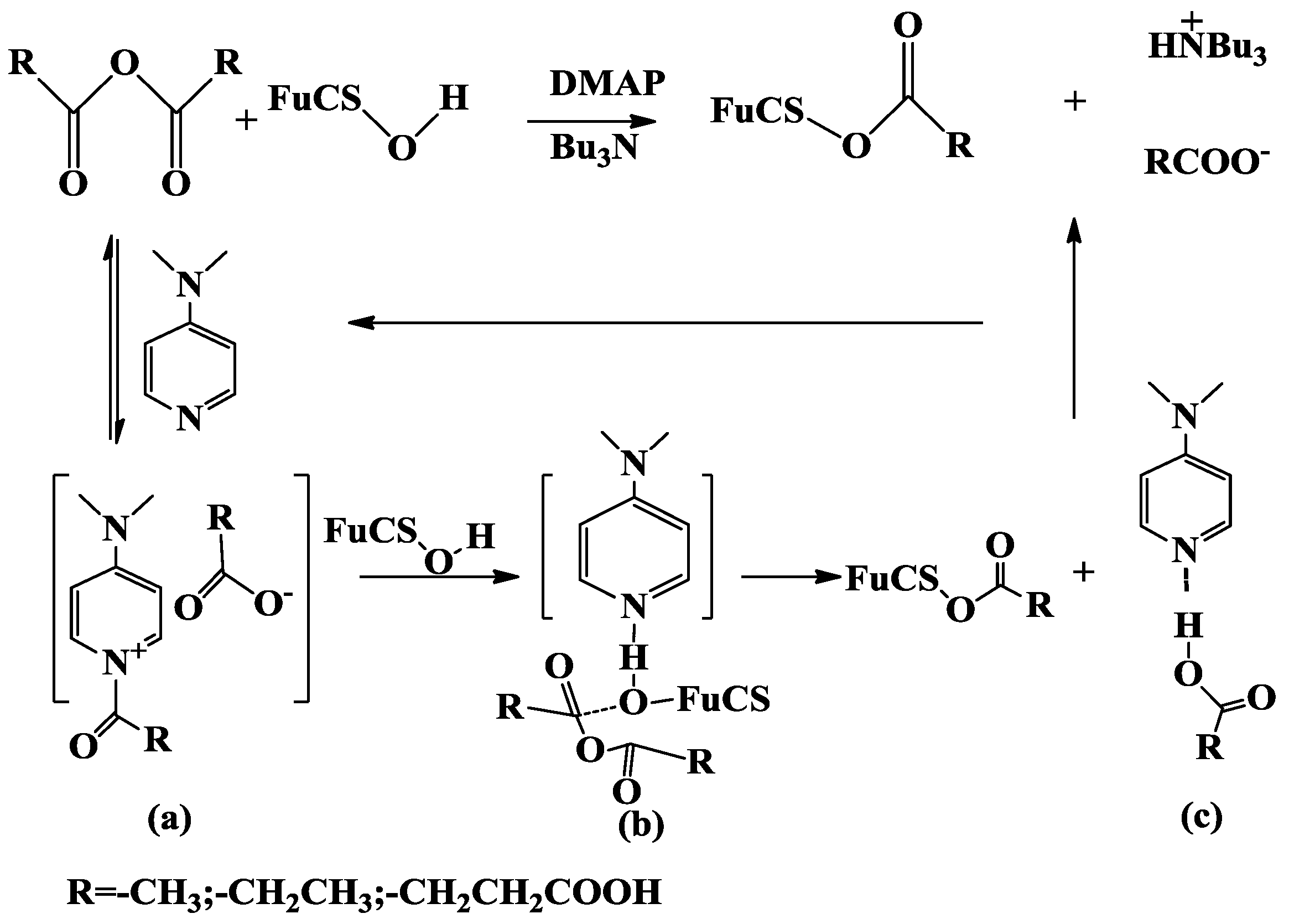
2.1. Analysis of the O-Acylation Reaction
2.1.1. Effects of Reaction Conditions on Degrees of Substitution
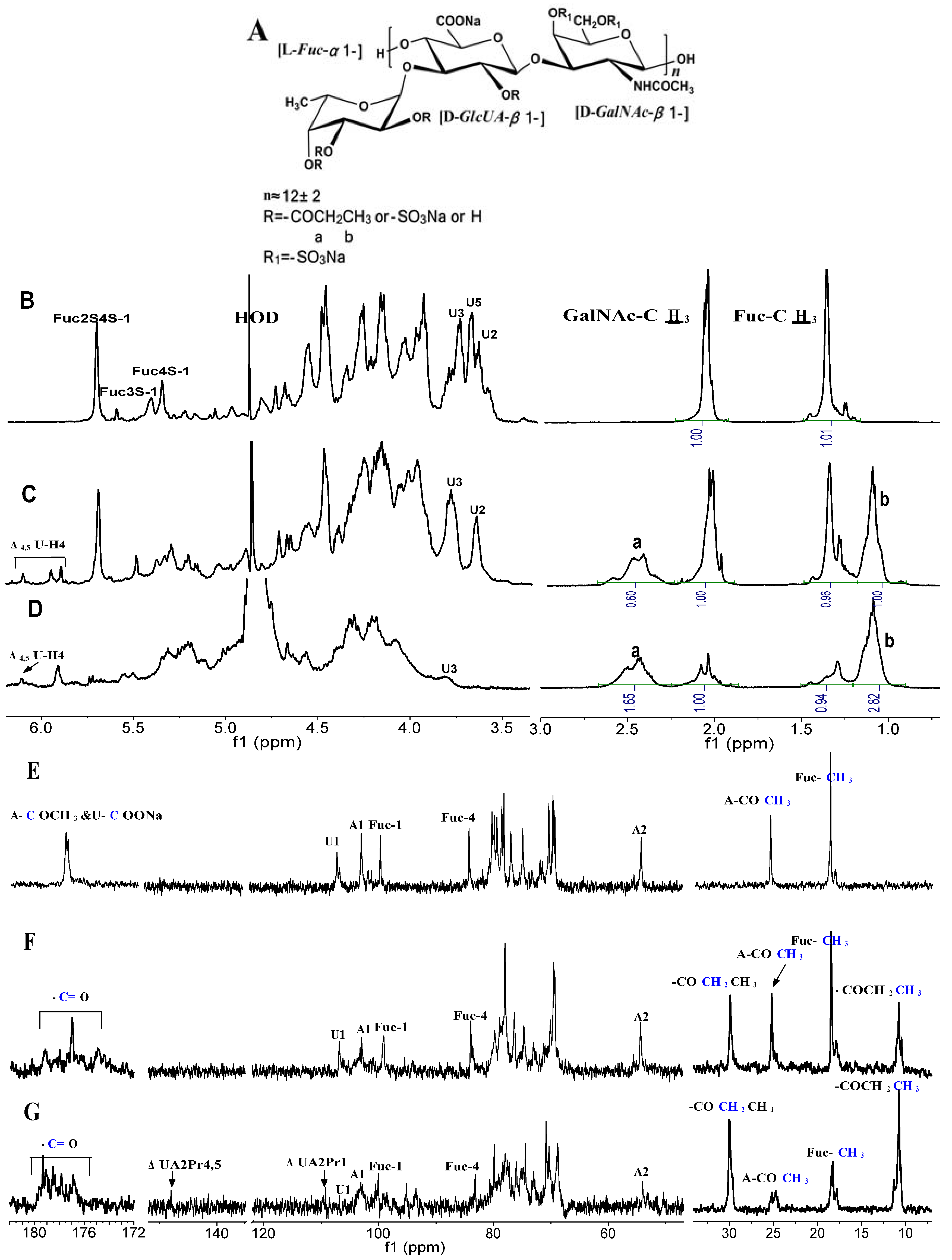

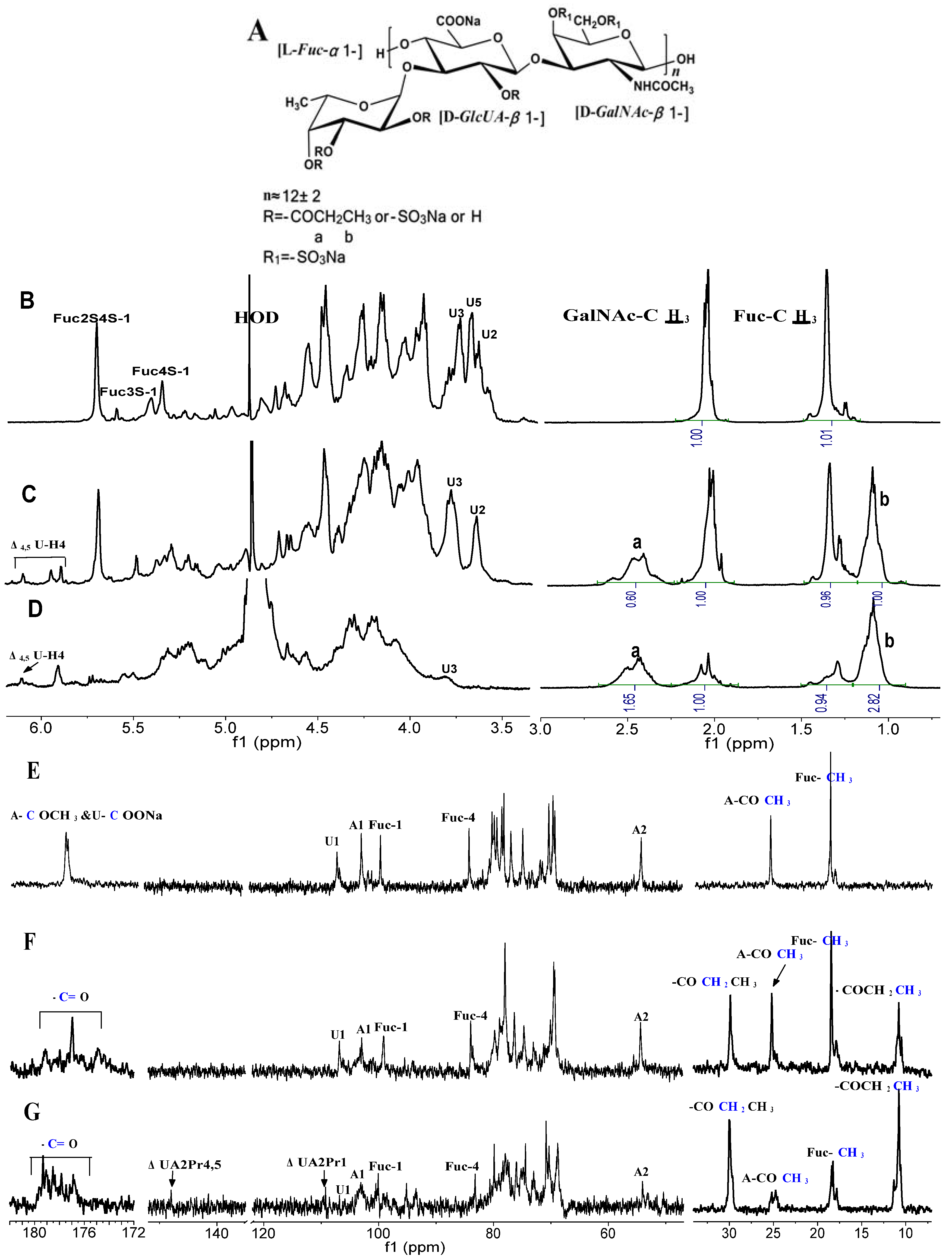{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
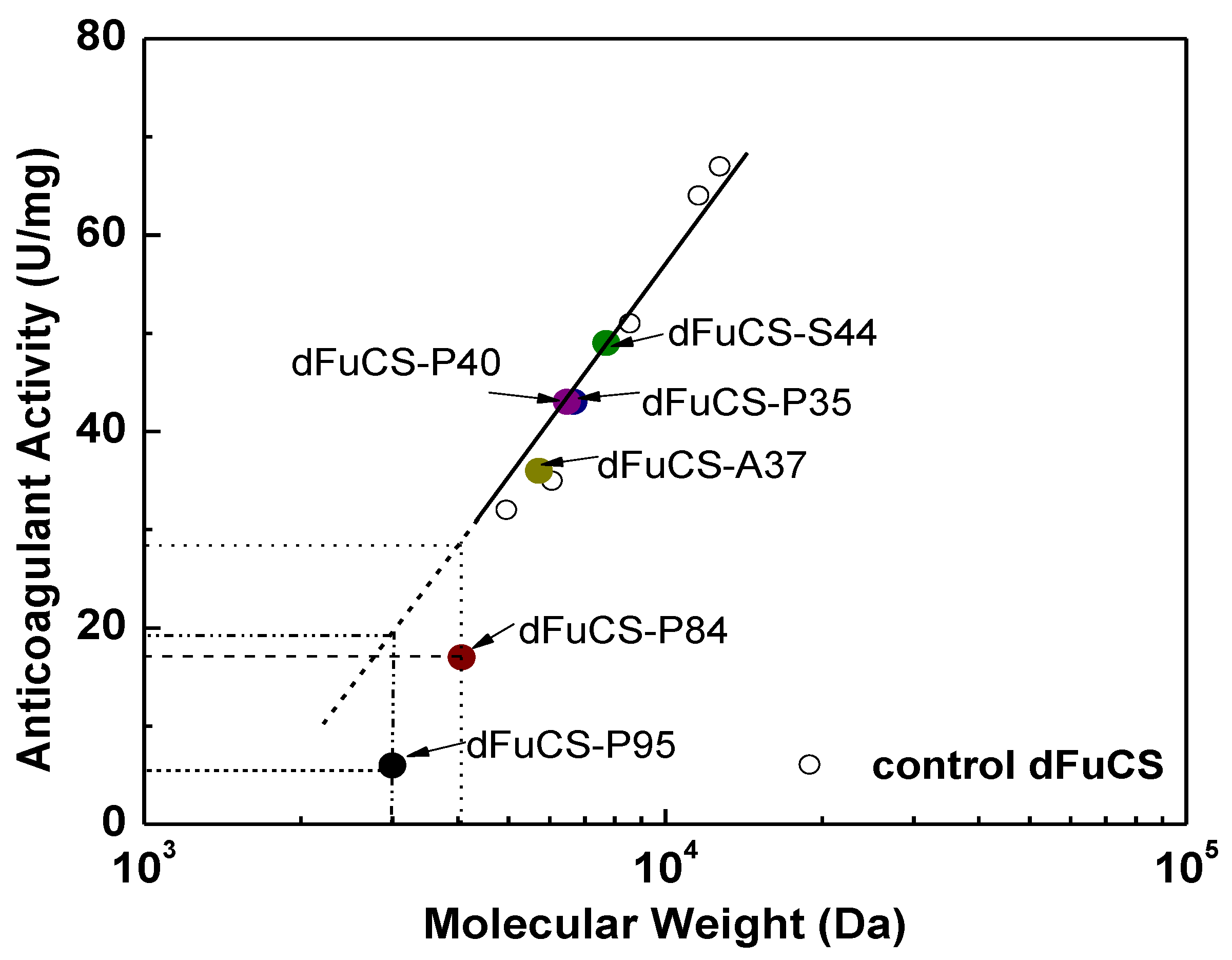
| Sample | Acylation a | DMAP (mg) | Acid Anhydride | Temperature (°C) | Time (h) | Degree of substitution b | Percentage of substitution (%) | Mw (Da) c | APTT d | |
|---|---|---|---|---|---|---|---|---|---|---|
| μg/mL | U/mg | |||||||||
| dFuCS-P35 | Propionoylation | 9.9 | 0.2 mL | 40 | 24 | 0.88 | 35 | 6970 | 5.11 | 43 |
| dFuCS-P40 | Propionoylation | 9.4 | 0.2 mL | 40 | 48 | 1.00 | 40 | 6480 | 5.04 | 43 |
| dFuCS-P84 | Propionoylation | 19.3 | 0.4 mL | 40 | 48 | 2.08 | 84 | 4070 | 12.8 | 17 |
| dFuCS-P95 | Propionoylation | 9.6 | 0.4 mL | 60 | 48 | 2.38 | 95 | 3000 | >44 | <5 |
| dFuCS-A37 | Acetylation | 10.2 | 0.3 mL | 40 | 24 | 0.92 | 37 | 5830 | 6.11 | 36 |
| dFuCS-S44 | Succininoylation | 9.8 | 300 mg | 40 | 24 | 1.09 | 44 | 7700 | 4.47 | 49 |
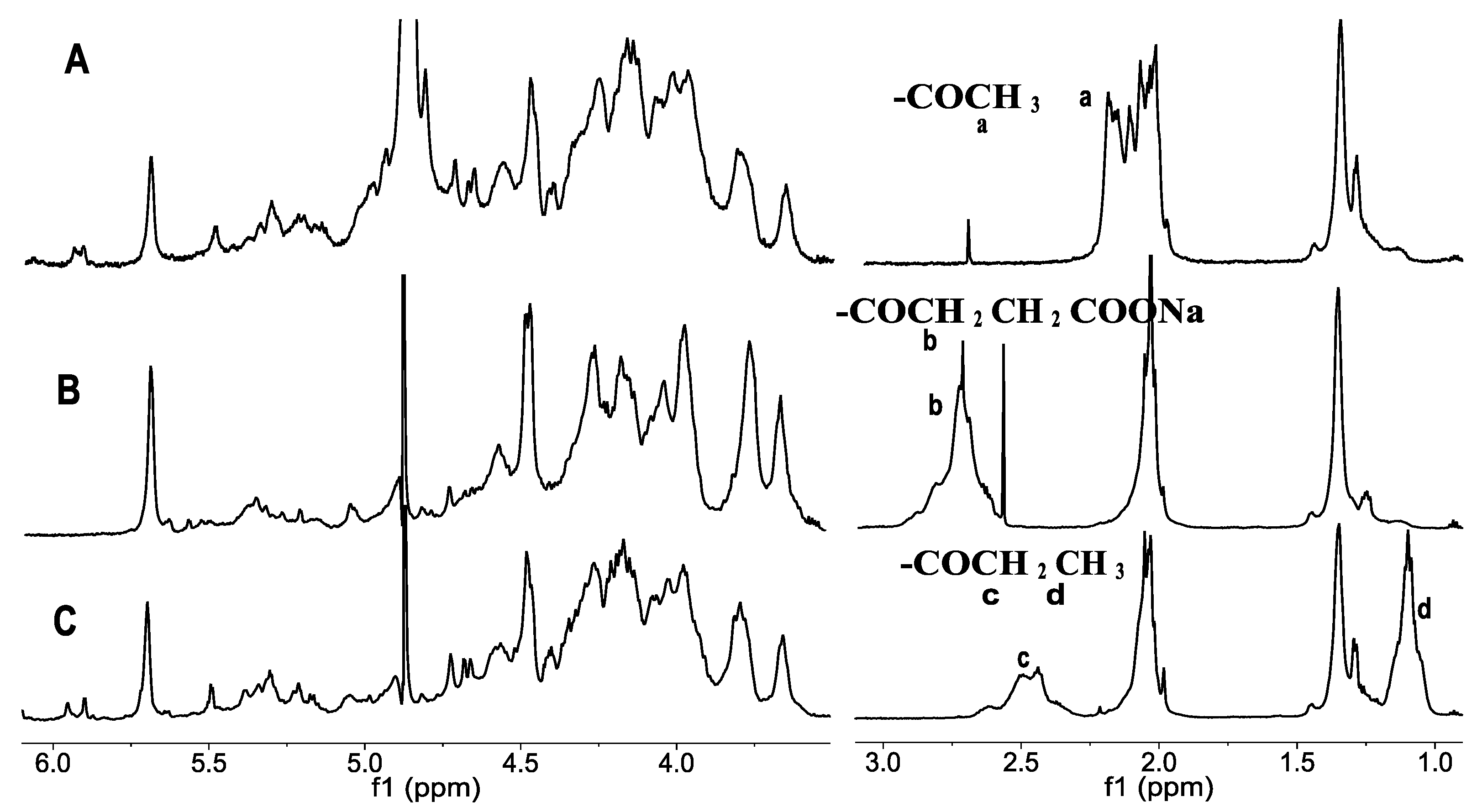
2.1.2. Effects Reaction Conditions on the Glycosidic Bond Cleavage
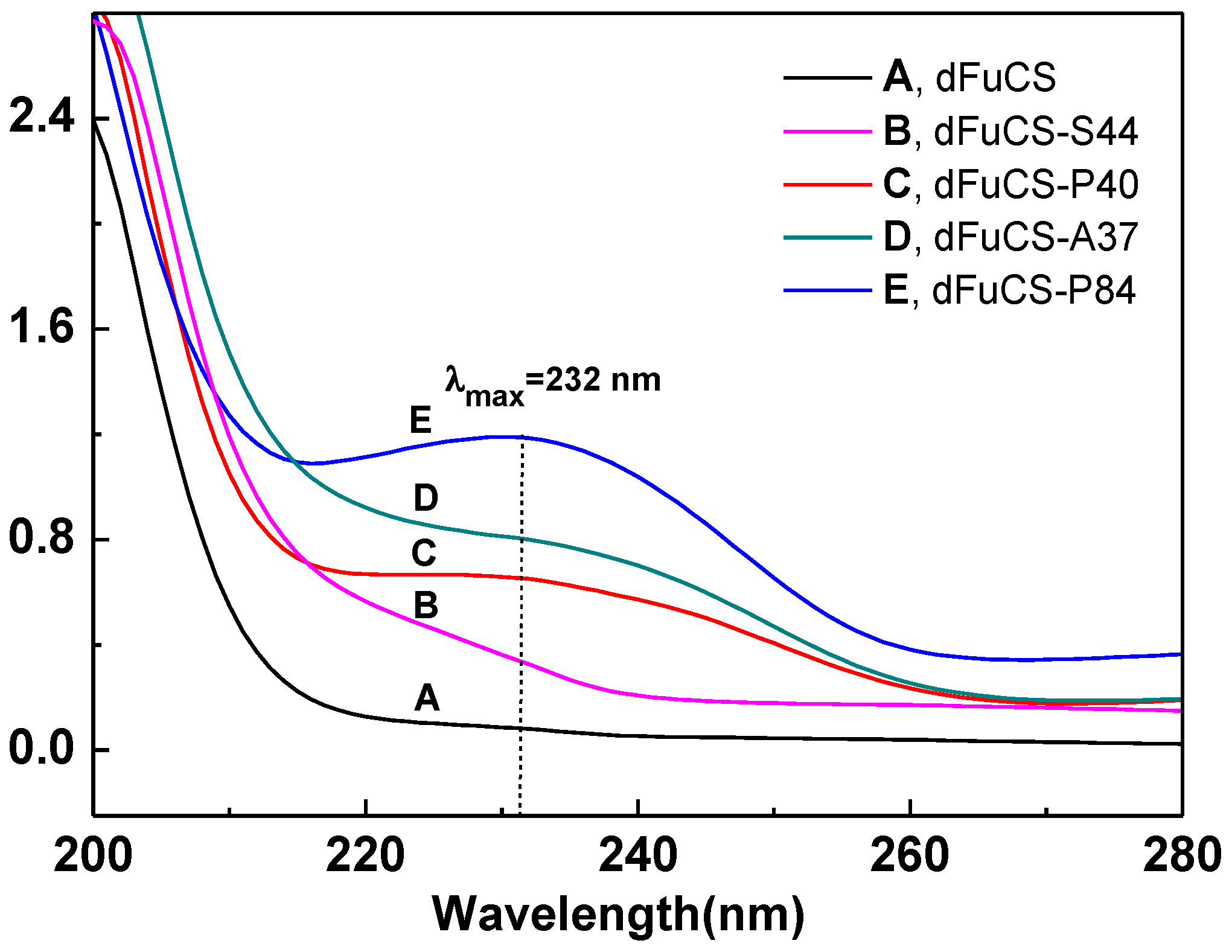
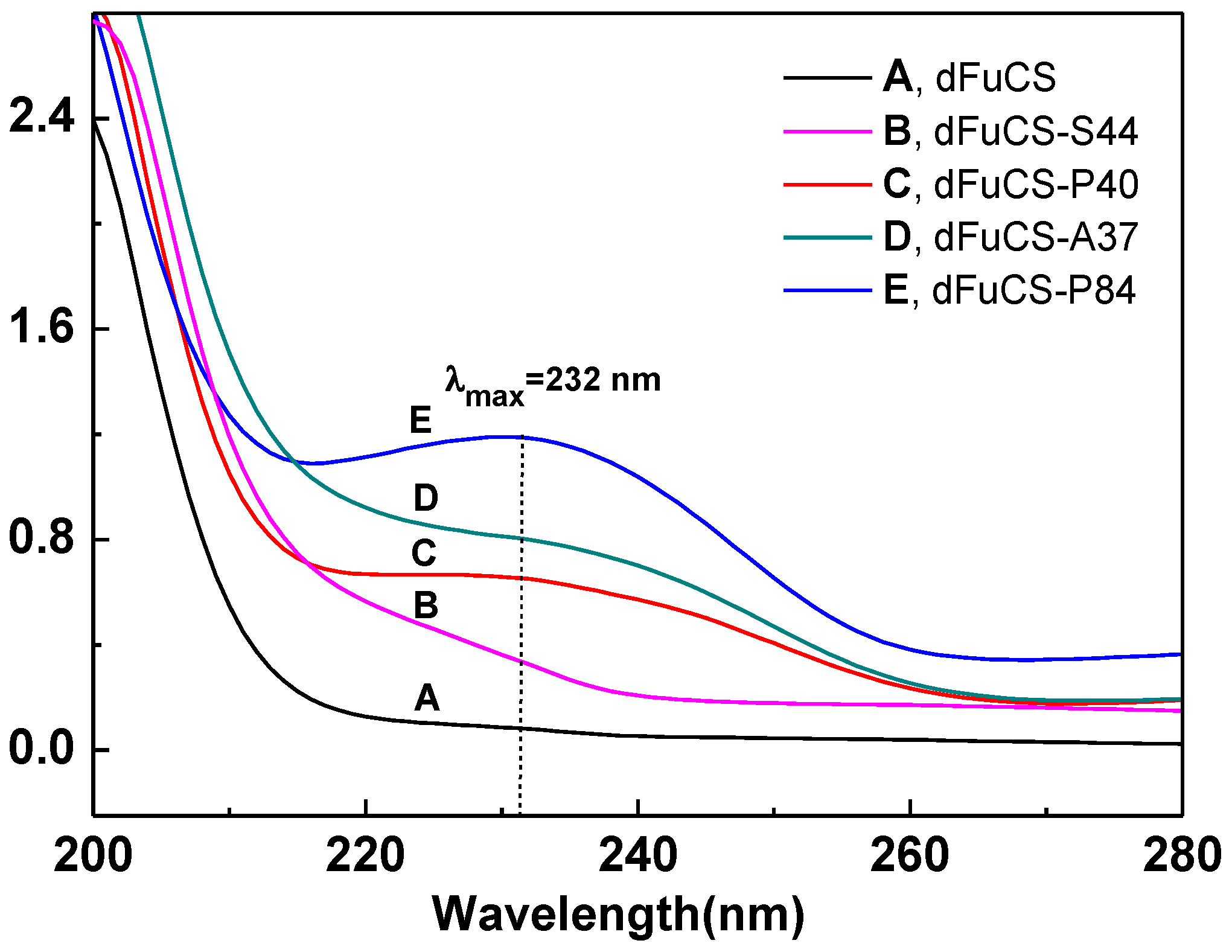
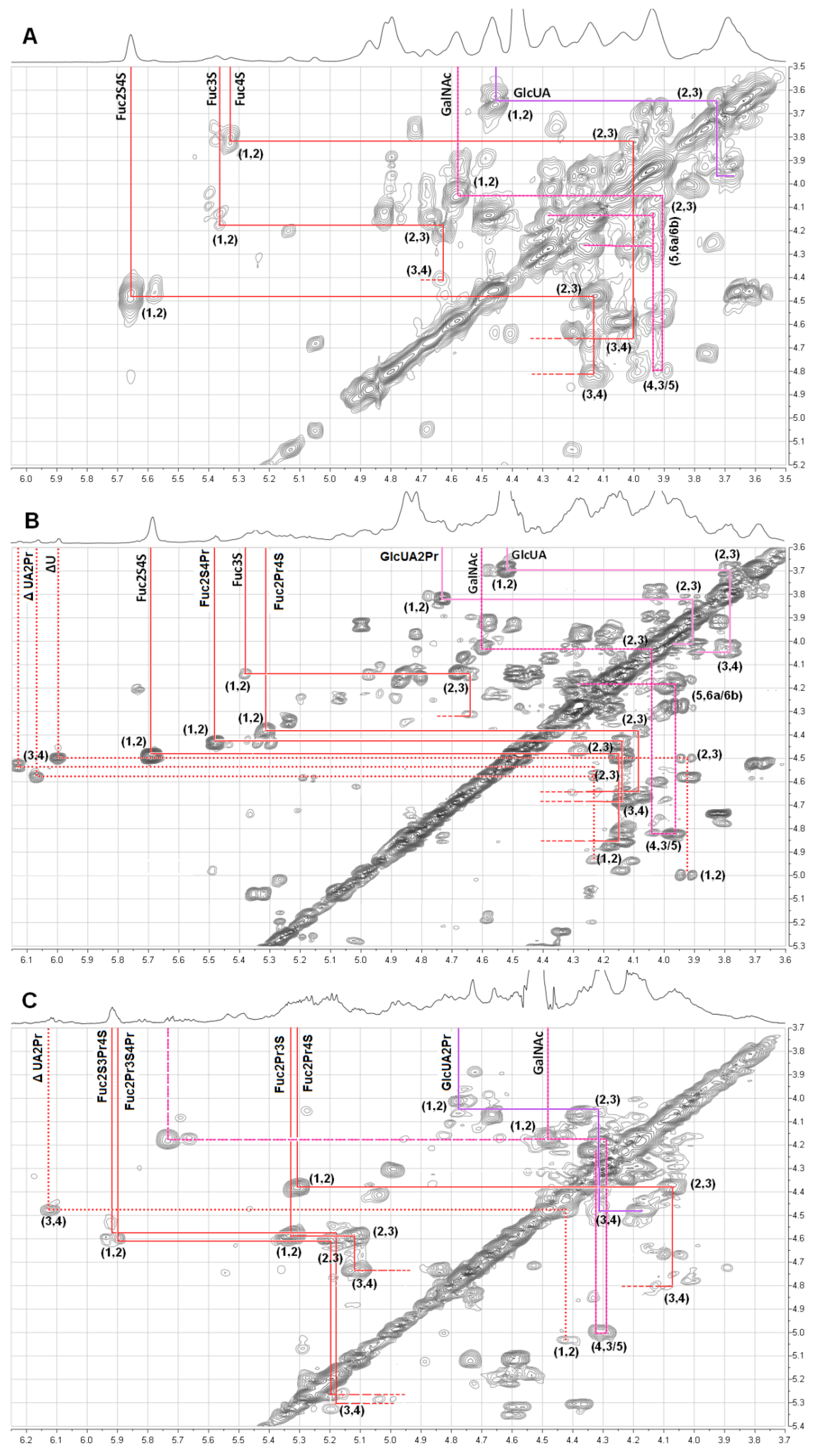
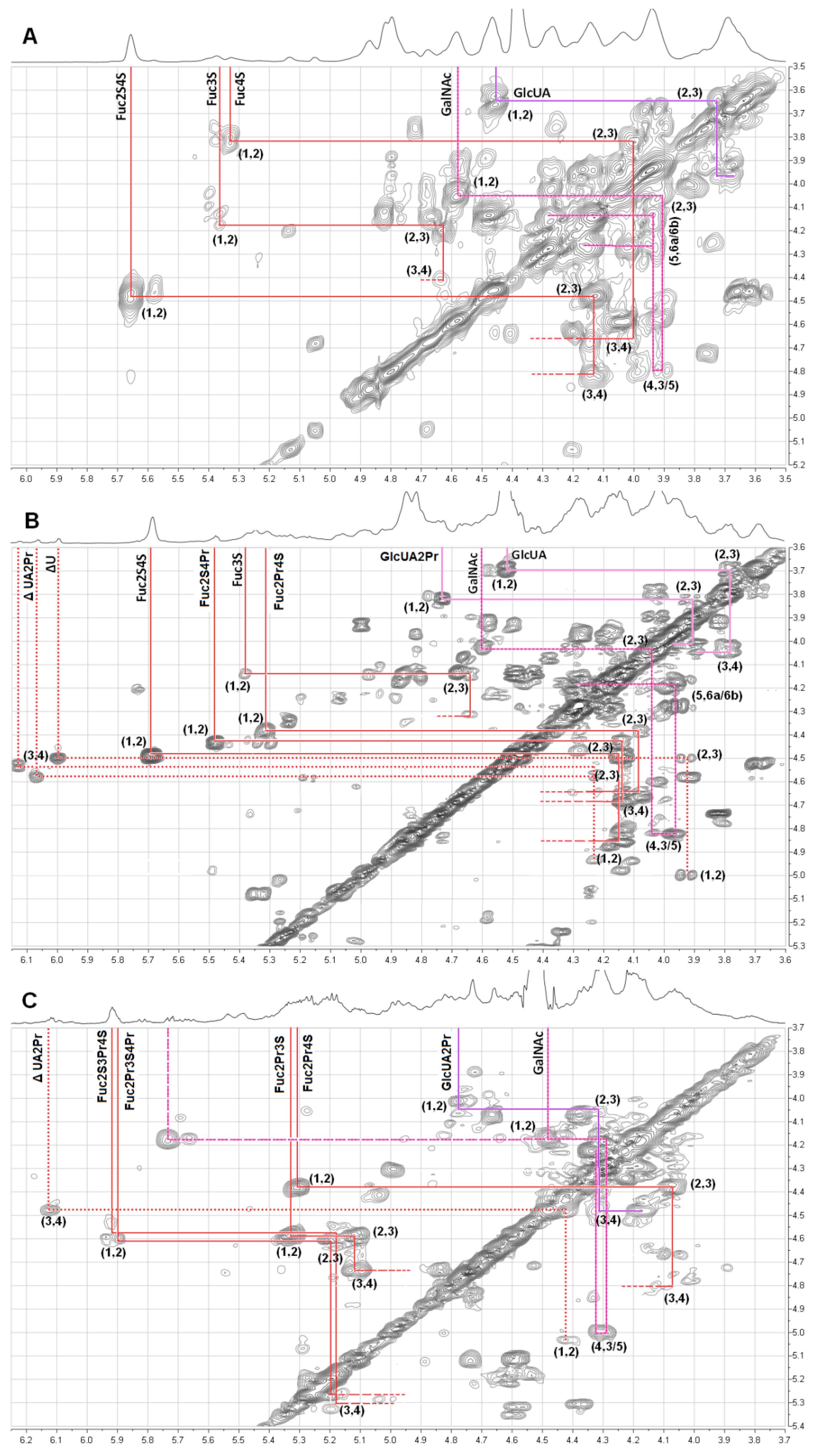
2.2. Structural Analysis of Products by NMR
| Compounds | Unit | Chemical Shift ( ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
| H-1 | H-2 | H-3 | H-4 | H-5 | H-6 | Ac | ||
| dFuCS | β- D-GalNAc4S6S | 4.58 | 4.05 | 3.93 | 4.82 | 3.97 | 4.16, 4.25 | 2.05 |
| β- D-GlcUA | 4.46 | 3.64 | 3.73 | 3.93 | 3.67 | / | / | |
| α- L-Fuc2S4S | 5.66 | 4.47 | 4.13 | 4.82 | 4.87 | 1.35 | / | |
| α- L-Fuc4S | 5.33 | 3.82 | 4.00 | 4.80 | 4.86 | 1.35 | / | |
| α- L-Fuc3S | 5.36 | 4.14 | 4.63 | 4.40 | 4.51 | 1.25 | / | |
| dFuCS-P40 | GalNAc4S6S | 4.67 | 4.13 | 4.05 | 4.83 | 3.96 | 4.17, 4.28 | 2.03 |
| GlcUA | 4.52 | 3.68 | 3.79 | 4.03 | 3.88 | / | / | |
| GlcUA2Pr | 4.73 | 3.82 | 3.91 | 4.02 | 3.95 | / | / | |
| ΔUA | 4.99 | 3.92 | 4.50 | 5.99 | / | / | / | |
| ΔUA2Pr | 4.94 | 4.23 | 4.52 | 6.12 | / | / | / | |
| α- L-Fuc2S4S | 5.68 | 4.49 | 4.14 | 4.85 | 4.84 | 1.36 | / | |
| α- L-Fuc2S4Pr | 5.47 | 4.44 | 4.13 | 4.79 | 4.87 | 1.36 | / | |
| α- L-Fuc2Pr4S | 5.31 | 4.34 | 4.07 | 4.63 | 4.86 | 1.35 | / | |
| α- L-Fuc3S | 5.38 | 4.14 | 4.64 | 4.30 | 4.33 | 1.28 | / | |
| dFuCS-P95 | GalNAc4S6S | 4.67 | 4.13 | 4.05 | 4.83 | 3.96 | 4.17, 4.28 | 2.03 |
| GlcUA | 4.52 | 3.68 | 3.79 | 4.03 | 3.88 | / | / | |
| GlcUA2Pr | 4.73 | 3.82 | 3.91 | 4.02 | 3.95 | / | / | |
| ΔUA2Pr | 5.03 | 4.40 | 4.48 | 6.12 | / | / | / | |
| α- L-Fuc2S3Pr4S | 5.92 | 4.53 | 5.17 | 5.29 | 4.31–4.33 | 1.30–1.33 | / | |
| α- L-Fuc2Pr3S4Pr | 5.89 | 4.59 | 5.21 | 5.27 | / | |||
| α- L-Fuc2Pr3S | 5.33 | 4.58 | 5.12 | 4.73 | / | |||
| α- L-Fuc2Pr4S | 5.31 | 4.38 | 4.07 | 4.80 | / | |||
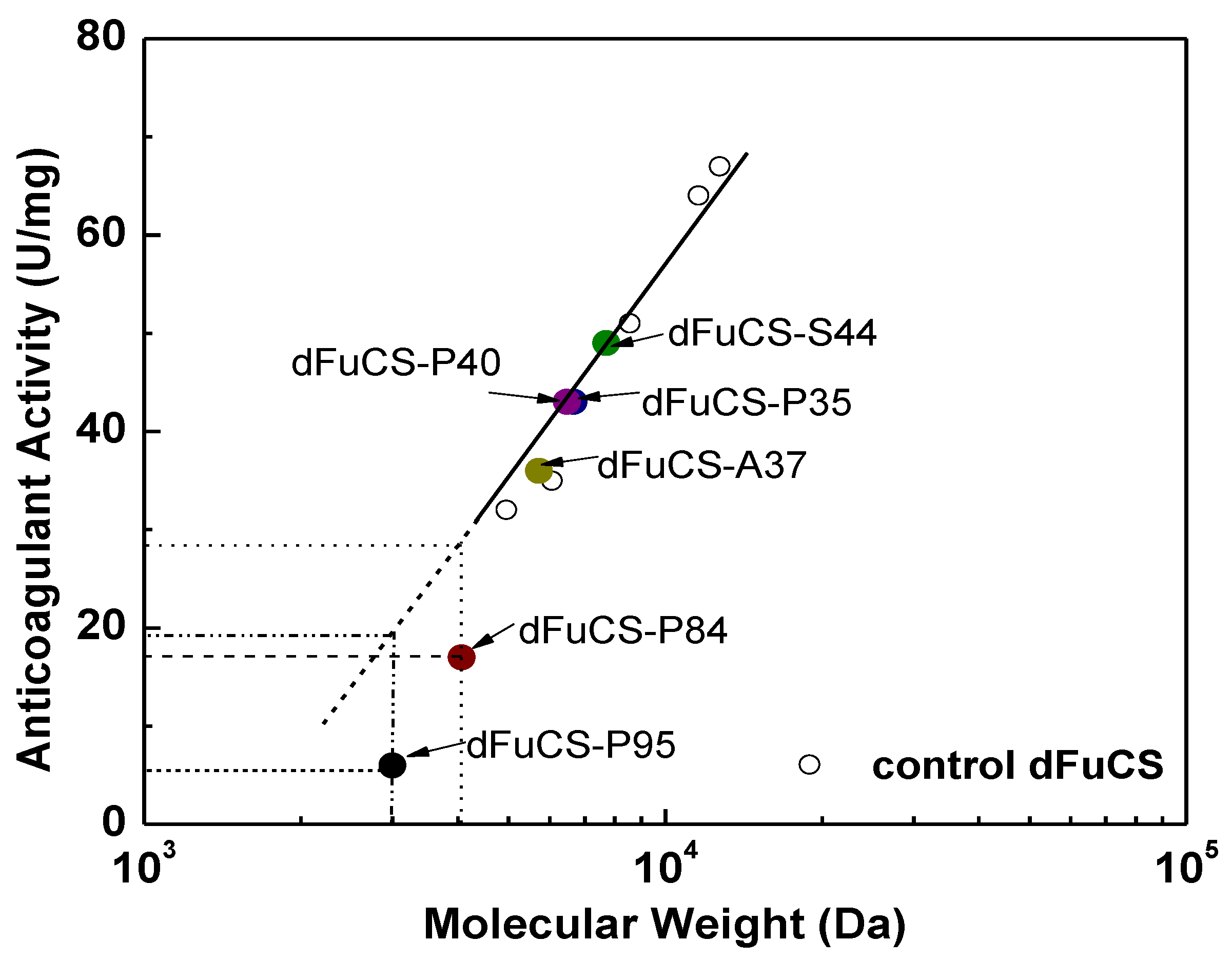
2.3. Anticoagulant Activity
3. Experimental Section
3.1. Preparation of O-Acylated Derivatives of Depolymerized FuCS
3.2. Analysis of Molecular Weight
3.3. Spectrometry Analysis
3.4. Anticoagulant Activity
4. Conclusion
Acknowledgements
References
- Mourão, P.A.S.; Pereira, M.S.; Pavão, M.S.G.; Mulloy, B.; Tollefsen, D.M.; Mowinckel, M.C.; Abildgaard, U. Structure and anticoagulant activity of a fucosylated chondroitin sulfate from echinoderm: Sulfated fucose branches on the polysaccharide account for its high anticoagulant action. J. Biol. Chem. 1996, 271, 23973–23984. [Google Scholar]
- Wu, M.Y.; Huang, R.; Wen, D.D.; Gao, N.; He, J.B.; Li, Z.; Zhao, J.H. Structure and effect of sulfated fucose branches on anticoagulant activity of the fucosylated chondroitin sulfate from sea cucumber Thelenota ananas. Carbohydr. Polym. 2012, 87, 862–868. [Google Scholar]
- Wu, M.Y.; Xu, S.M.; Zhao, J.H.; Kang, H.; Ding, H. Physicochemical characteristics and anticoagulant activities of low molecular weight fractions by free radical depolymerization of a fucosylated chondroitin sulfate from sea cucumber Thelenota ananas. Food Chem. 2010, 122, 716–723. [Google Scholar]
- Wu, M.Y.; Xu, S.M.; Zhao, J.H.; Kang, H.; Ding, H. Free-radical depolymerization of glycosaminoglycan from sea cucumber Thelenota ananas by hydrogen peroxide and copper ions. Carbohydr. Polym. 2010, 80, 1116–1124. [Google Scholar]
- Yoshida, K.; Minami, Y.; Nemoto, H.; Numata, K.; Yamanaka, E. Structure of DHG, a depolymerized glycosaminoglycan from sea cucumber Stichopus japonicus. Tetrahedron Lett. 1992, 33, 4959–4962. [Google Scholar]
- Buyue, Y.; Sheehan, J.P. Fucosylated chondroitin sulfate inhibits plasma thrombin generation via targeting of the factor IXa heparin-binding exosite. Blood 2009, 114, 3092–3100. [Google Scholar]
- Glauser, B.F.; Pereira, M.S.; Monteiro, R.Q.; Mourão, P.A.S. Serpin-independent anticoagulant activity of a fucosylated chondroitin sulfate. Thromb. Haemost. 2008, 100, 420–428. [Google Scholar]
- Sheehan, J.P.; Walke, E.N. Depolymerized holothurian glycosaminoglycan and heparin inhibit the intrinsic tenase complex by a common antithrombin independent mechanism. Blood 2006, 107, 3876–3882. [Google Scholar] [CrossRef]
- Hoshino, H.; Heiwamachi, M. Anti-HIV Drug. European Patent EP 0410002A1, 30 January 1991. [Google Scholar]
- Melo-Filho, N.M.; Belmir, C.L.; Gonçalves, R.G.; Takiya, C.M.; Leite, M., Jr.; Pavão, M.S.G.; Mourão, P.A.S. Fucosylated chondroitin sulfate attenuates renal fibrosis in animals submitted to unilateral ureteral obstruction: A P-selectin-mediated event. Am. J. Physiol. Renal Physiol. 2010, 299, F1299–F1307. [Google Scholar] [CrossRef]
- Li, J.; Lian, E. Aggregation of human platelets by acidic mucopolysaccharide extracted from Stichopus japonicas Selenka. Thromb. Haemost. 1998, 59, 435–439. [Google Scholar]
- Petitou, M.; Coudert, C.; Level, M.; Lormeau, J.-C.; Zuber, M.; Simenel, C.; Fournier, J.-P.; Choay, J. Selectively O-acylated glycosaminoglycan derivatives. Carbohydr. Res. 1992, 236, 107–119. [Google Scholar] [CrossRef]
- Bârzu, T.; Level, M.; Petitou, M.; Lormeau, J.C.; Choay, J.; Schos, D.; Baba, M.; Pauwels, R.; Witvrouw, M.; Clercq, D.E. Preparation and anti-HIV activity of O-acylated heparin and dermatan sulfate derivatives with low anticoagulant effect. J. Med. Chem. 1993, 36, 3546–3555. [Google Scholar]
- Yamada, T.; Ogmo, A.; Saito, T.; Uchiyama, H.; Nakagawa, Y. Preparation of O-acylated low-molecular-weight carrageenans with potent anti-HIV activity and low anticoagulant effect. Carbohydr. Polym. 2000, 41, 115–120. [Google Scholar]
- Jiang, Y.P.; Guo, X.K.; Tian, X.F. Synthesis and NMR structural analysis of O-succinyl derivative of low-molecular-weight κ-carrageenan. Carbohydr. Polym. 2005, 61, 399–406. [Google Scholar] [CrossRef]
- Monfregola, L.; Leone, M.; Vittoria, V.; Amodeoc, P.; Luca, S.D. Chemical modification of pectin: Environmental friendly process for new potential material development. Polym. Chem. 2011, 2, 800–804. [Google Scholar]
- Edward, A. Conformations in polysaccharides and complex carbohydrates. J. Biosci. 1985, 8, 375–387. [Google Scholar]
- Sakakura, A.; Kawajiri, K.; Ohkubo, T.; Kosugi, Y.; Ishihara, K. Widely useful DMAP-catalyzed esterification under auxiliary base- and solvent-free conditions. J. Am. Chem. Soc. 2007, 129, 14775–14779. [Google Scholar]
- Xu, S.; Held, I.; Kempf, B.; Mayr, H.; Steglich, W.; Zipse, H. The DMAP-catalyzed acetylation of alcohols—A mechanistic study (DMAP = 4-(Dimethylamino) pyridine). Chem. Eur. J. 2005, 11, 4751–4757. [Google Scholar]
- Kiss, J. β-Eliminative degradation of carbohydrates containing uronic acid residues. Adv. Carbohydr. Chem. Biochem. 1974, 29, 229–303. [Google Scholar] [CrossRef]
- Vieira, R.P.; Mulloy, B.; Mourão, P.A.S. Structure of a fucose-branched chondroitin sulfate from sea cucumber: Evidence for the presence of 3-O-sulfo-β-D-glucuronosyl residues. J. Biol. Chem. 1991, 266, 13530–13536. [Google Scholar]
- Vilela-Silva, A.-C.E.S.; Castro, M.O.; Valente, A.-P.; Biermann, C.H.; Mourão, P.A.S. Sulfated fucans from the egg jellies of the closely related sea urchins Strongylocentrotus droebachiensis and Strongylocentrotus pallidus ensure species-specific fertilization. J. Biol. Chem. 2002, 277, 379–387. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gao, N.; Wu, M.; Liu, S.; Lian, W.; Li, Z.; Zhao, J. Preparation and Characterization of O-Acylated Fucosylated Chondroitin Sulfate from Sea Cucumber. Mar. Drugs 2012, 10, 1647-1661. https://doi.org/10.3390/md10081647
Gao N, Wu M, Liu S, Lian W, Li Z, Zhao J. Preparation and Characterization of O-Acylated Fucosylated Chondroitin Sulfate from Sea Cucumber. Marine Drugs. 2012; 10(8):1647-1661. https://doi.org/10.3390/md10081647
Chicago/Turabian StyleGao, Na, Mingyi Wu, Shao Liu, Wu Lian, Zi Li, and Jinhua Zhao. 2012. "Preparation and Characterization of O-Acylated Fucosylated Chondroitin Sulfate from Sea Cucumber" Marine Drugs 10, no. 8: 1647-1661. https://doi.org/10.3390/md10081647
APA StyleGao, N., Wu, M., Liu, S., Lian, W., Li, Z., & Zhao, J. (2012). Preparation and Characterization of O-Acylated Fucosylated Chondroitin Sulfate from Sea Cucumber. Marine Drugs, 10(8), 1647-1661. https://doi.org/10.3390/md10081647