
Hitting the Bull’s-Eye in Metastatic Cancers—NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Evidence for Amplified Mitochondrial Metabolism and Increased Oxphos in Metastatic Cancer
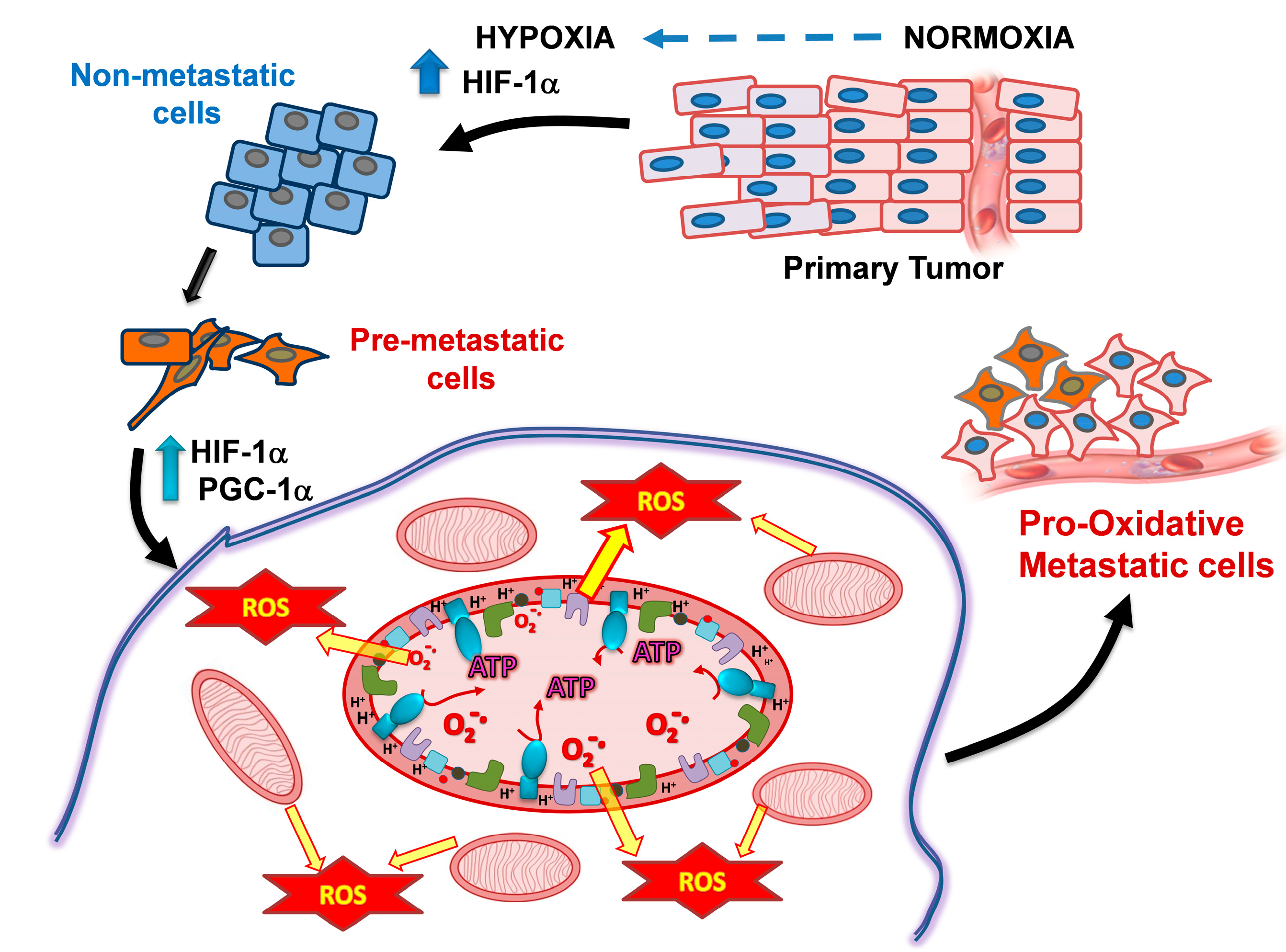
2.1. PGC1-α Increases Metastatic Cancer Cell Metabolism
3. Mechanism of Elevated ROS Production in Metastatic Cancer and the Role of Succinate
4. The Role of Mitochondrial DNA (MtDNA) Mutation in Metastatic Cancers, Increasing ROS Production and Multidrug Resistance
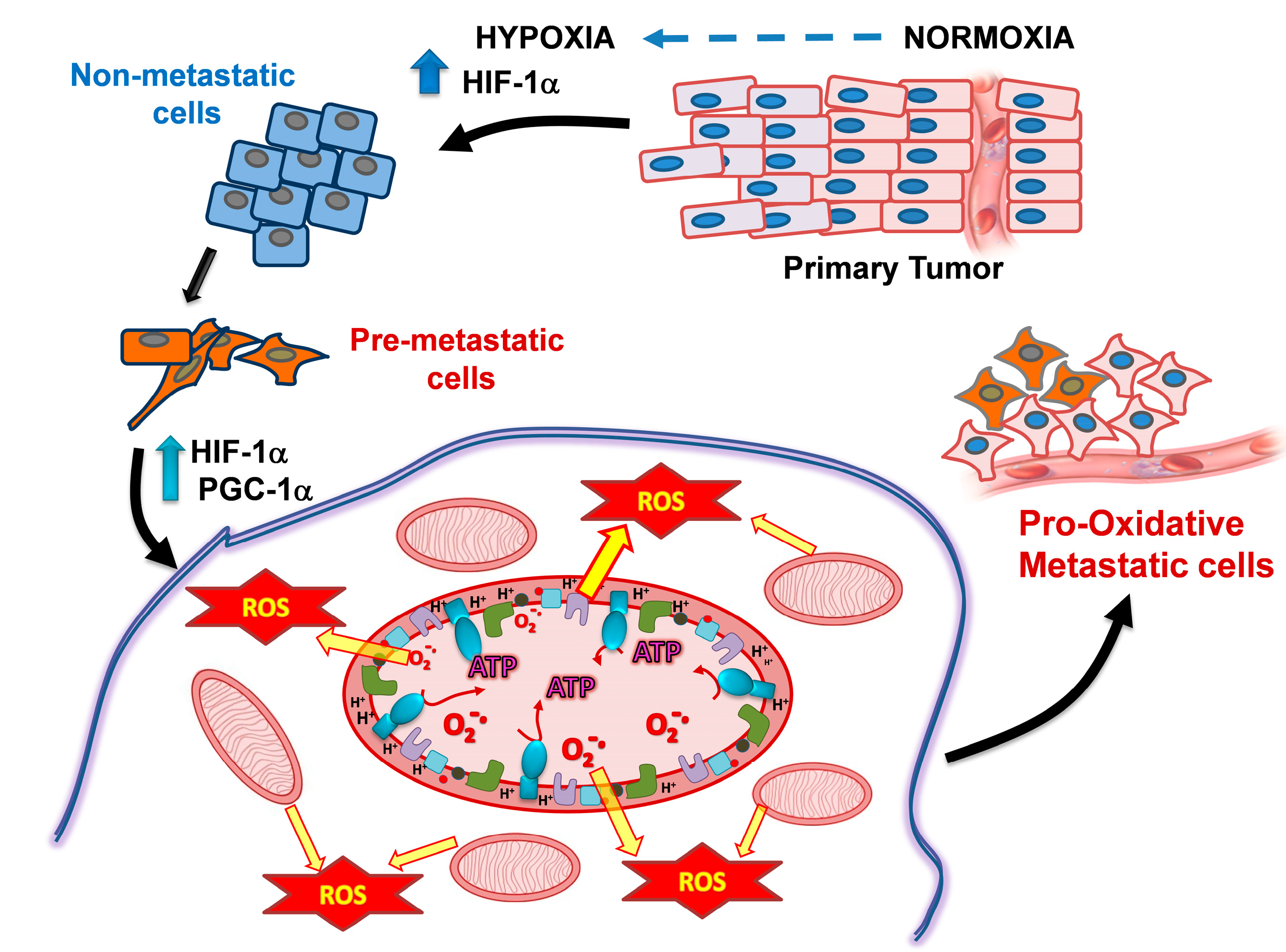
5. Hypoxia and Changes in Mitochondrial Metabolism of Metastatic Cells as Targets for Their Preferential Killing
6. Advances in Mitocans with a Focus on Metastasis
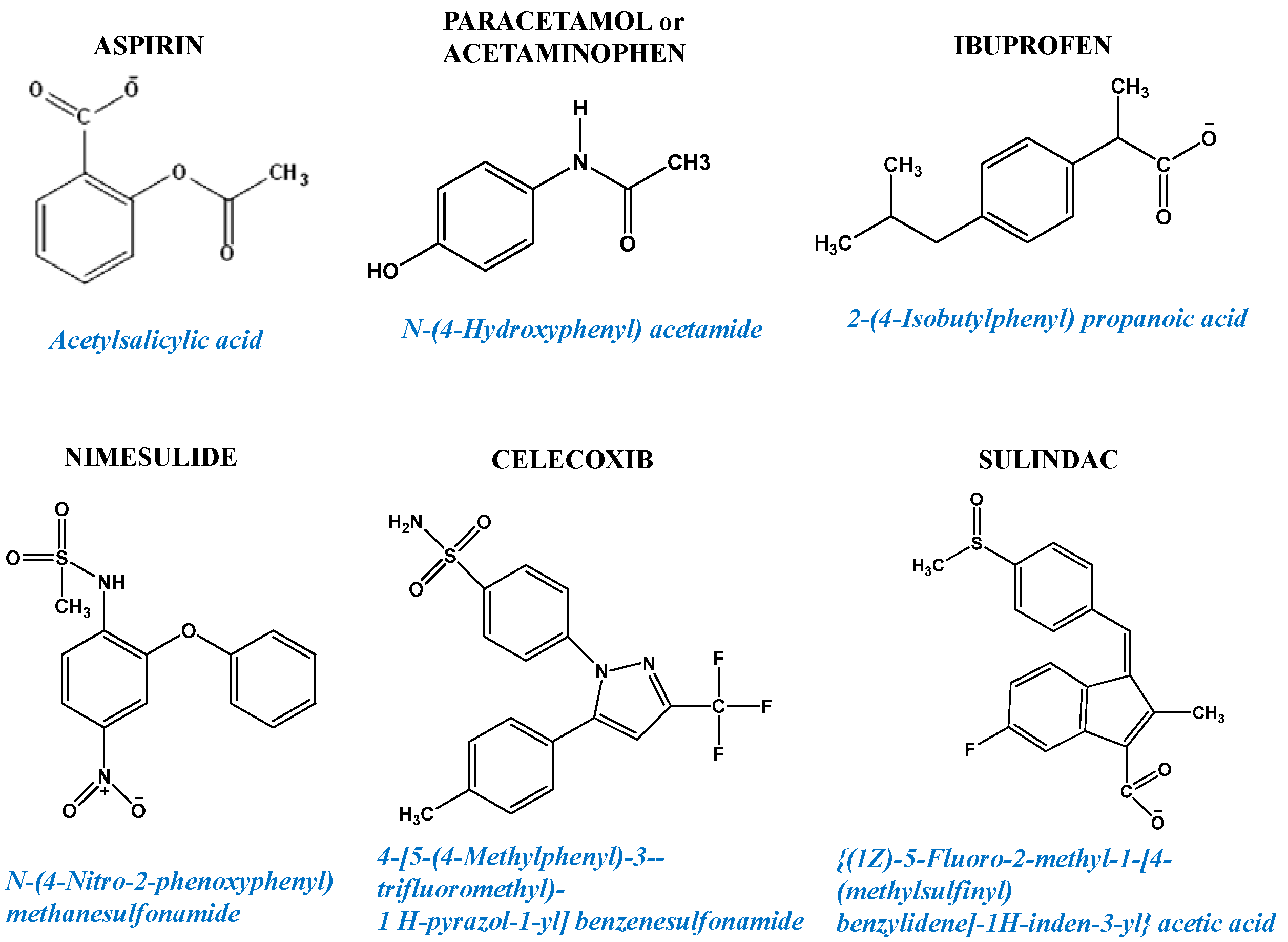
6.1. NSAIDs: Loading the Mitochondrial Gun in Highly Malignant Cells
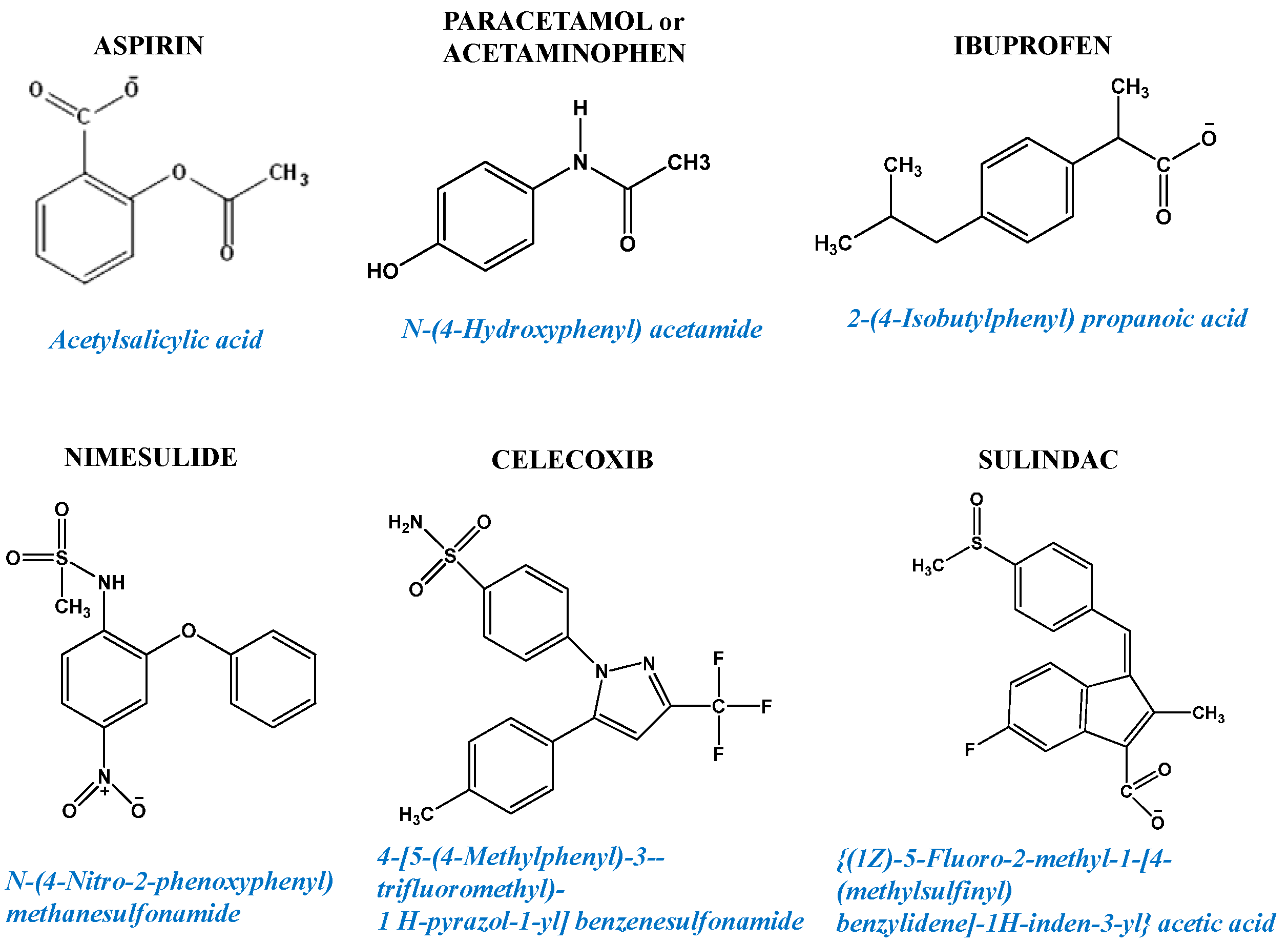
6.1.1. In Vitro Studies of NSAIDs as Anticancer Agents Inducing Apoptosis and Cancer Cell Death
6.1.2. In Vivo Studies of NSAIDs as Inhibitors of Xenografted Human Cancers
6.1.3. NSAID Action on Nag-1 and p75(NTR) Affecting Cancer Cell Survival. Pre-Clinical Studies Where NSAIDs Were Used to Treat Xenografted Tumors
6.1.4. NSAIDs in Clinical Trials of Human Cancer
7. Selective Anticancer Action of NSAIDs
7.1. The First Firing Mechanism–Destabilizing Essential Thioredoxin/Glutaredoxin Redox Systems
7.2. Mitochondrial Redox Protein Modification by Glutathionylation
8. The Pro-Oxidative Trigger Induced By NSAIDs
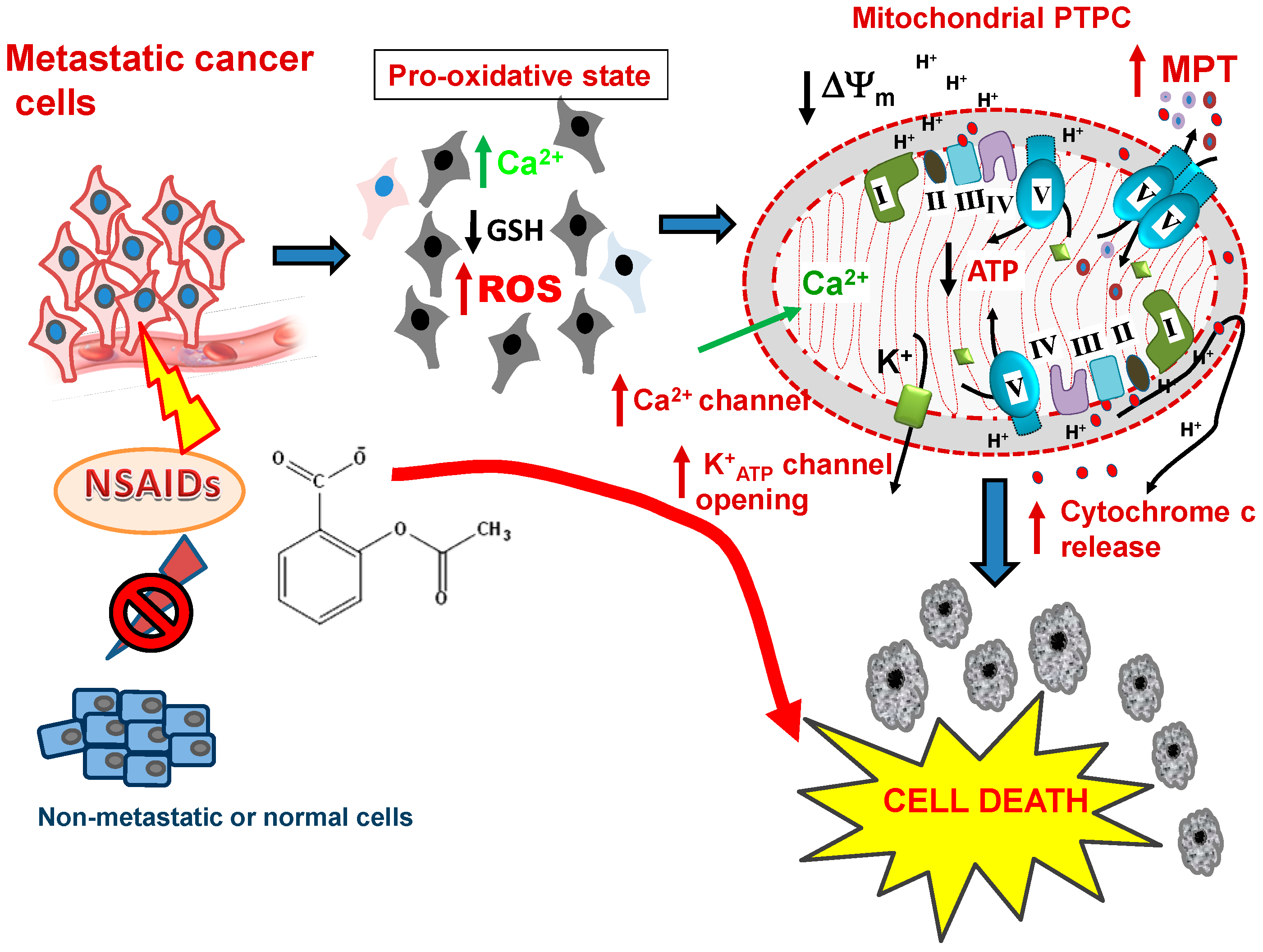
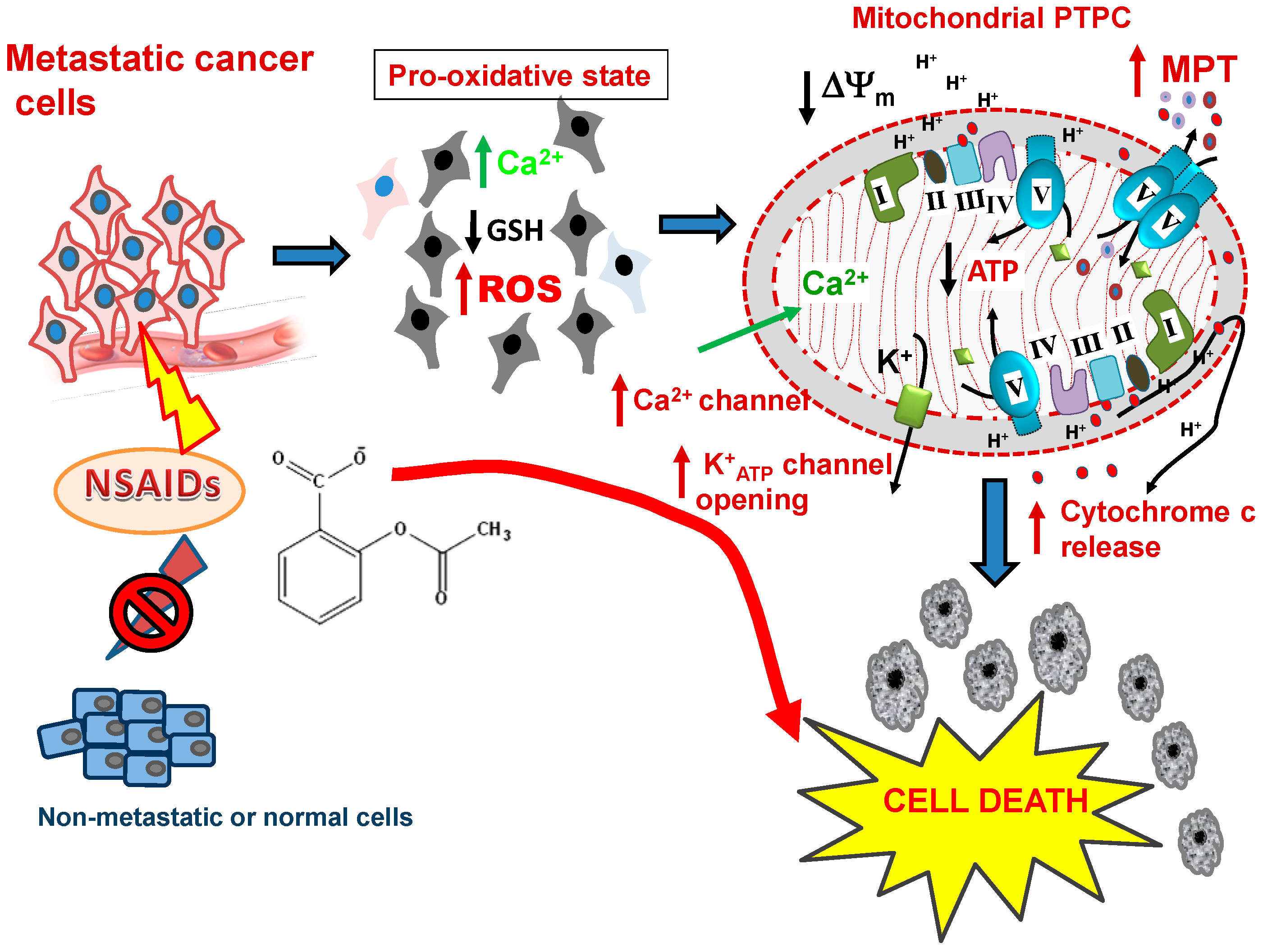
9. Selectivity of NSAIDs for Preferentially Killing Malignant but not Normal Cells
10. The Firing Mechanisms for NSAID-Induced Cancer Cell Death: The Mitochondrial ROS Burst
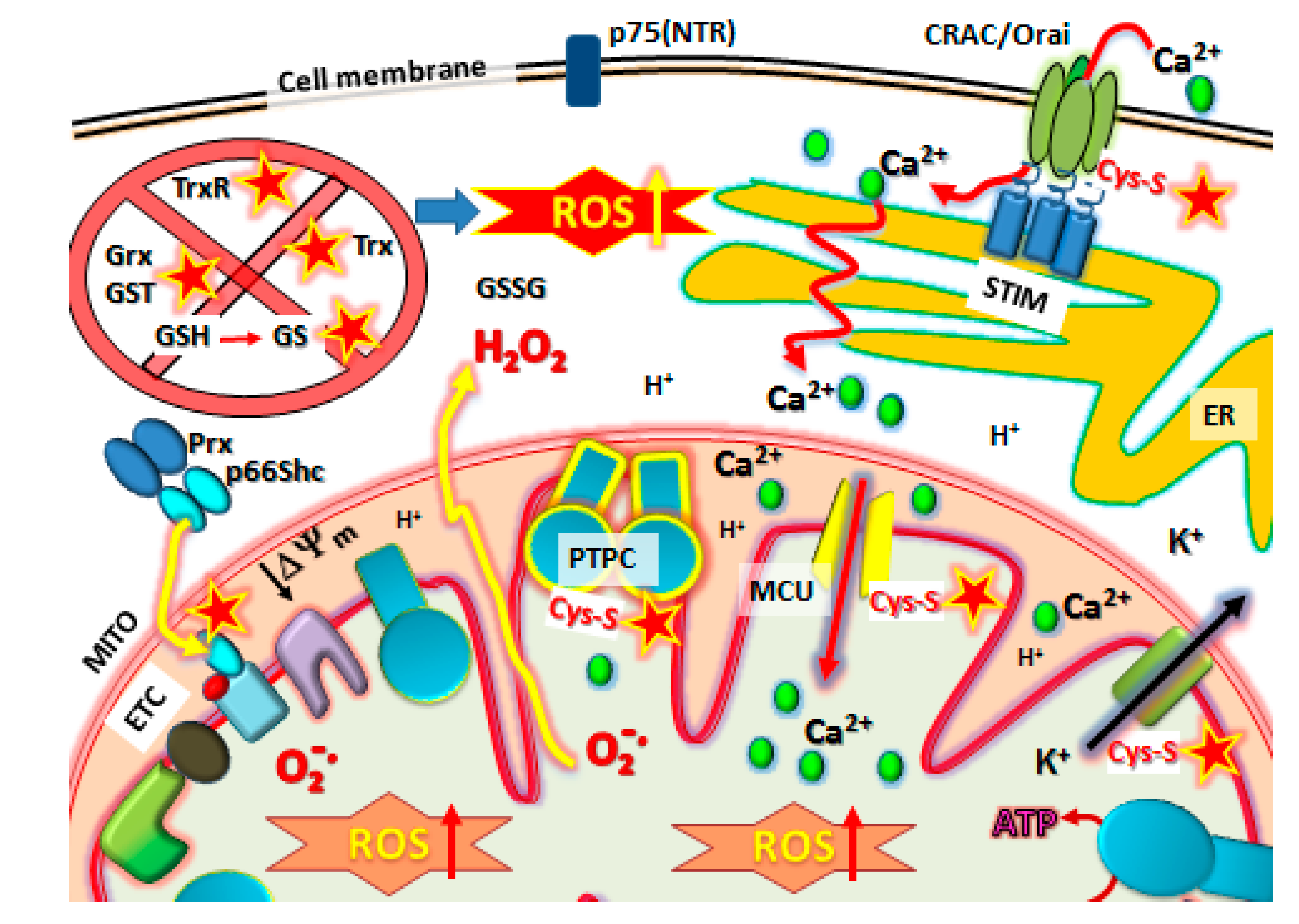
 ) protein targets such as Orai1 to block SOCE, Prx/p66shc complexes to release p66shc which then translocates into the mitochondrial intramembraneous space binding to cytochrome C to promote ROS, modification of Kv ATP channels and mitochondrial Calcium (Ca2+) uniporter (MCU) regulators to cause mCa2+ matrix influx, increasing ROS production and promoting mPTP complex formation (PTPC) leading to cell death. Abbreviations: MITO, mitochondria. ER, endoplasmic reticulum. SOCE, Store-Operated Calcium Entry. STIM, Stromal Interaction Molecules.
) protein targets such as Orai1 to block SOCE, Prx/p66shc complexes to release p66shc which then translocates into the mitochondrial intramembraneous space binding to cytochrome C to promote ROS, modification of Kv ATP channels and mitochondrial Calcium (Ca2+) uniporter (MCU) regulators to cause mCa2+ matrix influx, increasing ROS production and promoting mPTP complex formation (PTPC) leading to cell death. Abbreviations: MITO, mitochondria. ER, endoplasmic reticulum. SOCE, Store-Operated Calcium Entry. STIM, Stromal Interaction Molecules.
) protein targets such as Orai1 to block SOCE, Prx/p66shc complexes to release p66shc which then translocates into the mitochondrial intramembraneous space binding to cytochrome C to promote ROS, modification of Kv ATP channels and mitochondrial Calcium (Ca2+) uniporter (MCU) regulators to cause mCa2+ matrix influx, increasing ROS production and promoting mPTP complex formation (PTPC) leading to cell death. Abbreviations: MITO, mitochondria. ER, endoplasmic reticulum. SOCE, Store-Operated Calcium Entry. STIM, Stromal Interaction Molecules.
) protein targets such as Orai1 to block SOCE, Prx/p66shc complexes to release p66shc which then translocates into the mitochondrial intramembraneous space binding to cytochrome C to promote ROS, modification of Kv ATP channels and mitochondrial Calcium (Ca2+) uniporter (MCU) regulators to cause mCa2+ matrix influx, increasing ROS production and promoting mPTP complex formation (PTPC) leading to cell death. Abbreviations: MITO, mitochondria. ER, endoplasmic reticulum. SOCE, Store-Operated Calcium Entry. STIM, Stromal Interaction Molecules.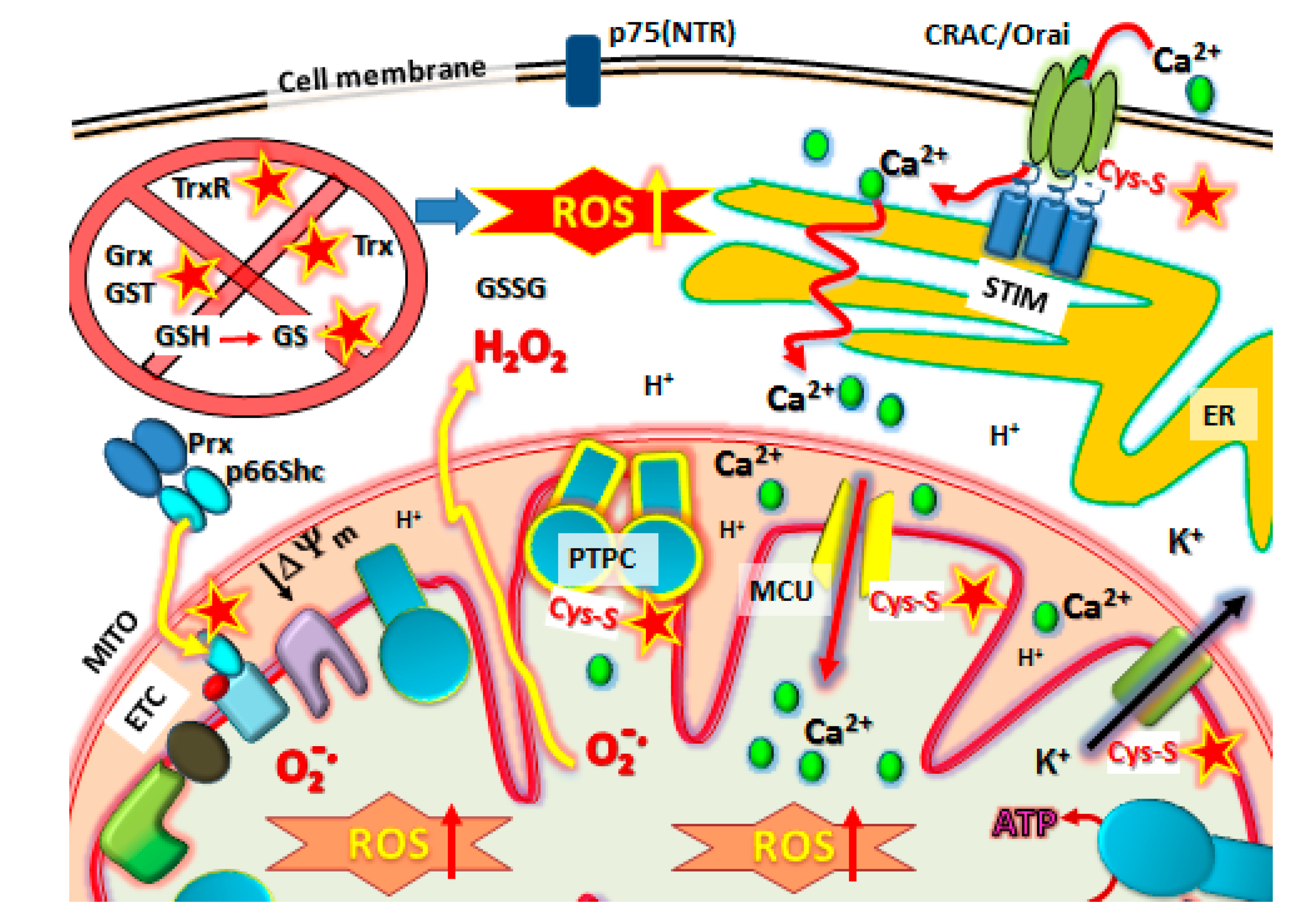
10.1. The p66Shc/PRX Couple
10.2. Kv-ATP Channels and Mitochondrial Membrane Depolarization
11. The Final Stage: Activation of Metastatic Cancer Cell Death by PTPC Opening
NSAID Effects on Calcium (Ca2+) Channels, Role in MPT Activation and Increased ROS Production
12. How the NSAIDs Fire the PTPC. Targeting Critical Cys-Thiol Reactivity to Initiate the PTPC Opening and the Cell Death Cascade in Metastatic Cancer Cells
13. Pro-Drugs with Critical Cys-Thiol Reactivity—A Common Modality for Activating the PTPC in Metastatic Cancer Cells
14. Conclusions
Acknowledgements
Author Contributions
Abbreviations
| CSC | cancer stem cell |
| EMT | epithelial-mesenchymal transition |
| MPT | mitochondrial membrane permeability transition forming the megachannel |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| OxPhos | oxidative phosphorylation |
| PGC-1α | peroxisome proliferator-activated receptor Gamma Co-activator 1-alpha |
| PTPC | inner mitochondrial membrane channel or mitochondrial permeability transition pore (mPTP) complex |
| ROS | reactive oxygen species |
| TCA cycle | tricarboxylic acid or Krebs cycle |
| Δψm | difference in electrical potential between the bulk water phases separated by the inner mitochondrial membrane |
Conflicts of Interest
References
- Menzies, A.M.; Long, G.V. Recent advances in melanoma systemic therapy. Braf inhibitors, ctla4 antibodies and beyond. Eur. J. Cancer 2013, 49, 3229–3241. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Chester, J.D. Personalised cancer medicine. Int. J. Cancer 2014. [Google Scholar] [CrossRef]
- Spano, D.; Heck, C.; de Antonellis, P.; Christofori, G.; Zollo, M. Molecular networks that regulate cancer metastasis. Semin. Cancer Biol. 2012, 22, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Rodriguez-Enriquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sanchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ros-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- Kim, Y.; Lin, Q.; Glazer, P.M.; Yun, Z. Hypoxic tumor microenvironment and cancer cell differentiation. Curr. Mol. Med. 2009, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Dewhirst, M.W.; Agarwal, N.; Sorg, B.S. Novel imaging provides new insights into mechanisms of oxygen transport in tumors. Curr. Mol. Med. 2009, 9, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.M.; Suo, Z. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. Hif induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rich, J.N. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr. Top. Microbiol. Immunol. 2010, 345, 21–30. [Google Scholar] [PubMed]
- Fabian, A.; Vereb, G.; Szollosi, J. The hitchhikers guide to cancer stem cell theory: Markers, pathways and therapy. Cytom. Part A J. Int. Soc. Anal. Cytol. 2013, 83, 62–71. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, J.; Rigas, B. Chemopreventive agents induce oxidative stress in cancer cells leading to cox-2 overexpression and cox-2-independent cell death. Carcinogenesis 2009, 30, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Rodriguez-Enriquez, S.; Neuzil, J.; Moreno-Sanchez, R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ros-induced apoptotic trigger. Mol. Asp. Med. 2010, 31, 29–59. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Gallardo-Perez, J.C.; Marin-Hernandez, A.; Aguilar-Ponce, J.L.; Mandujano-Tinoco, E.A.; Meneses, A.; Moreno-Sanchez, R. Oxidative phosphorylation as a target to arrest malignant neoplasias. Curr. Med. Chem. 2011, 18, 3156–3167. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Marin-Hernandez, A.; Saavedra, E.; Pardo, J.P.; Ralph, S.J.; Rodriguez-Enriquez, S. Who controls the atp supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 2014, 50, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major atp source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Harris, A.L. Hypoxia and metabolism in cancer. Adv. Exp. Med. Biol. 2014, 772, 1–39. [Google Scholar] [PubMed]
- Neuzil, J.; Dong, L.F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Exploiting mitochondrial apoptosis for the treatment of cancer. Mitochondrion 2010, 10, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Targeting oxidative phosphorylation: Why, when, and how. Cancer Cell 2013, 23, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Heerdt, B.G.; Houston, M.A.; Augenlicht, L.H. The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res. 2005, 65, 9861–9867. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.A.; Augenlicht, L.H.; Heerdt, B.G. Stable differences in intrinsic mitochondrial membrane potential of tumor cell subpopulations reflect phenotypic heterogeneity. Int. J. Cell Biol. 2011, 2011, 978583. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Philp, N.J.; Pestell, R.G.; Lisanti, M.P. Mitochondrial metabolism in cancer metastasis: Visualizing tumor cell mitochondria and the “reverse warburg effect” in positive lymph node tissue. Cell Cycle 2012, 11, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Lisanti, M.P. Mitochondrial oxidative stress drives tumor progression and metastasis: Should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. Pgc1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. Pgc-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of pgc-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Diaz-Delfin, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; St-Pierre, J.; Giguere, V. The pgc-1/err signaling axis in cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ros) and ros-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.L.; Brookes, P.S. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J. Biol. Chem. 2009, 284, 16236–16245. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.M.; Parish, G.; Ho, Y.S. Effects of hypoxia on expression of superoxide dismutases in cultured atii cells and lung fibroblasts. Am. J. Physiol. 1996, 271, L955–962. [Google Scholar] [PubMed]
- Fan, J.; Cai, H.; Yang, S.; Yan, L.; Tan, W. Comparison between the effects of normoxia and hypoxia on antioxidant enzymes and glutathione redox state in ex vivo culture of cd34(+) cells. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2008, 151, 153–158. [Google Scholar] [CrossRef]
- Xi, H.; Gao, Y.H.; Han, D.Y.; Li, Q.Y.; Feng, L.J.; Zhang, W.; Ji, G.; Xiao, J.C.; Zhang, H.Z.; Wei, Q. Hypoxia inducible factor-1alpha suppresses peroxiredoxin 3 expression to promote proliferation of ccrcc cells. FEBS Lett. 2014, 588, 3390–3394. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Wu, C.P.; Lee, H.T.; Liang, J.A.; Yu, C.Y.; Lin, Y.J. Nadph oxidase subunit 4 mediates cycling hypoxia-promoted radiation resistance in glioblastoma multiforme. Free Radic. Biol. Med. 2012, 53, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Scannell, G.; Waxman, K.; Kaml, G.J.; Ioli, G.; Gatanaga, T.; Yamamoto, R.; Granger, G.A. Hypoxia induces a human macrophage cell line to release tumor necrosis factor-alpha and its soluble receptors in vitro. J. Surg. Res. 1993, 54, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Hernandez-Esquivel, L.; Rivero-Segura, N.A.; Marin-Hernandez, A.; Neuzil, J.; Ralph, S.J.; Rodriguez-Enriquez, S. Reactive oxygen species are generated by the respiratory complex ii—Evidence for lack of contribution of the reverse electron flow in complex i. FEBS J. 2013, 280, 927–938. [Google Scholar] [PubMed]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, L.; Yu, C.A. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem. 1998, 273, 33972–33976. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Covian, R.; Trumpower, B.L. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J. Biol. Chem. 2009, 284, 19203–19210. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997, 17, 3–8. [Google Scholar] [CrossRef] [PubMed]
- McLennan, H.R.; Degli Esposti, M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr. 2000, 32, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; D’Aurelio, M.; Merlo Pich, M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta 2000, 1459, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex ii of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L710–L719. [Google Scholar] [PubMed]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 1099–1106. [Google Scholar]
- Brookes, P.S. Mitochondrial h(+) leak and ROS generation: An odd couple. Free Radic. Biol. Med. 2005, 38, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; O’Rourke, B.; Aon, M.A. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ros. Biochim. Biophys. Acta 2014, 1837, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Biagioni Angeli, E.; Ardestani, M.R.; Goracci, G.; Adam-Vizi, V. Reversible inhibition of hydrogen peroxide elimination by calcium in brain mitochondria. J. Neurosci. Res. 2011, 89, 1965–1972. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Mari, M.; Colell, A.; Morales, A.; Basanez, G.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim. Biophys. Acta 2010, 1797, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex iii of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Paranagama, M.P.; Sakamoto, K.; Amino, H.; Awano, M.; Miyoshi, H.; Kita, K. Contribution of the fad and quinone binding sites to the production of reactive oxygen species from ascaris suum mitochondrial complex ii. Mitochondrion 2010, 10, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [PubMed]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhou, A.; Lu, H.; Chen, Y.; Huang, G.; Yue, X.; Zhao, P.; Wu, Y. Suppression of mitochondrial complex i influences cell metastatic properties. PLoS One 2013, 8, e61677. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; de Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. Ros-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Hayashi, J. A novel function of mtdna: Its involvement in metastasis. Ann. N. Y. Acad. Sci. 2010, 1201, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Matsui, H.; Tomita, T.; Sadakata, H.; Indo, H.P.; Majima, H.J.; Kaneko, T.; Hyodo, I. Mitochondrial reactive oxygen species accelerate gastric cancer cell invasion. J. Clin. Biochem. Nutr. 2014, 54, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jones, A.W.; Fassone, E.; Sweeney, M.G.; Lebiedzinska, M.; Suski, J.M.; Wieckowski, M.R.; Tajeddine, N.; Hargreaves, I.P.; Yasukawa, T.; et al. Pgc-1beta mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene 2013, 32, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, K.A.; Cocchiglia, S.; Zhdanov, A.V.; Tambuwala, M.M.; Cummins, E.P.; Monfared, M.; Agbor, T.A.; Garvey, J.F.; Papkovsky, D.B.; Taylor, C.T.; et al. Pgc-1alpha is coupled to hif-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2188–2193. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (mdr1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Xia, S.; Yu, S.Y.; Yuan, X.L.; Xu, S.P. Effects of hypoxia on expression of p-glycoprotein and multidrug resistance protein in human lung adenocarcinoma a549 cell line. Zhonghua yi xue za zhi 2004, 84, 663–666. [Google Scholar] [PubMed]
- Milane, L.; Duan, Z.; Amiji, M. Role of hypoxia and glycolysis in the development of multi-drug resistance in human tumor cells and the establishment of an orthotopic multi-drug resistant tumor model in nude mice using hypoxic pre-conditioning. Cancer Cell Int. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Chen, Q.; He, S.; Liu, S.; Ma, Y. Hypoxia-induced increases in a549/cddp cell drug resistance are reversed by rna interference of hif-1alpha expression. Mol. Med. Rep. 2012, 5, 228–232. [Google Scholar] [PubMed]
- Zhan, M.; Yu, D.; Lang, A.; Li, L.; Pollock, R.E. Wild type p53 sensitizes soft tissue sarcoma cells to doxorubicin by down-regulating multidrug resistance-1 expression. Cancer 2001, 92, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.F.; Sillence, D.; Ackerley, C.; Lingwood, C. Role of multiple drug resistance protein 1 in neutral but not acidic glycosphingolipid biosynthesis. J. Biol. Chem. 2004, 279, 7867–7876. [Google Scholar]
- Baran, Y.; Gunduz, U.; Ural, A.U. Expression of multidrug resistance (mdr-1) gene in human promyelocytic leukemia cell line selected with vincristine. Turk. J. Cancer 2005, 35, 88–92. [Google Scholar]
- Loe, D.W.; Deeley, R.G.; Cole, S.P. Biology of the multidrug resistance-associated protein, mrp. Eur. J. Cancer 1996, 32A, 945–957. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- Kruspig, B.; Zhivotovsky, B.; Gogvadze, V. Mitochondrial substrates in cancer: Drivers or passengers? Mitochondrion 2014, 19, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Neuzil, J. Mitochondria as targets for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Low, P.; Dong, L.; Lawen, A.; Neuzil, J. Mitocans: Mitochondrial targeted anti-cancer drugs as improved therapies and related patent documents. Recent Pat. Anti-Cancer Drug Discov. 2006, 1, 327–346. [Google Scholar] [CrossRef]
- Neuzil, J.; Dyason, J.C.; Freeman, R.; Dong, L.F.; Prochazka, L.; Wang, X.F.; Scheffler, I.; Ralph, S.J. Mitocans as anti-cancer agents targeting mitochondria: Lessons from studies with vitamin e analogues, inhibitors of complex ii. J. Bioenerg. Biomembr. 2007, 39, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Wang, X.F.; Dong, L.F.; Low, P.; Ralph, S.J. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and bcl-2 family proteins. FEBS Lett. 2006, 580, 5125–5129. [Google Scholar] [CrossRef] [PubMed]
- Makris, U.E.; Abrams, R.C.; Gurland, B.; Reid, M.C. Management of persistent pain in the older patient: A clinical review. JAMA 2014, 312, 825–836. [Google Scholar] [CrossRef] [PubMed]
- McCarberg, B.H.; Cryer, B. Evolving therapeutic strategies to improve nonsteroidal anti-inflammatory drug safety. Am. J. Ther. 2014. [Google Scholar] [CrossRef]
- Wehling, M. Non-steroidal anti-inflammatory drug use in chronic pain conditions with special emphasis on the elderly and patients with relevant comorbidities: Management and mitigation of risks and adverse effects. Eur. J. Clin. Pharmacol. 2014, 70, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Unzueta, A.; Vargas, H.E. Nonsteroidal anti-inflammatory drug-induced hepatoxicity. Clin. Liver Dis. 2013, 17, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Sostres, C.; Gargallo, C.J.; Lanas, A. Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res. Ther. 2013, 15 (Suppl 3), S3. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Aleo, M.D.; Strock, C.J.; Stedman, D.B.; Wang, H.; Will, Y. Toxicity assessments of nonsteroidal anti-inflammatory drugs in isolated mitochondria, rat hepatocytes, and zebrafish show good concordance across chemical classes. Toxicol. Appl. Pharmacol. 2013, 272, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Ness, R.B.; Lo-Ciganic, W.H.; Murphy, M.A.; Goode, E.L.; Poole, E.M.; Brinton, L.A.; Webb, P.M.; Nagle, C.M.; Jordan, S.J.; et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: A pooled analysis in the ovarian cancer association consortium. J. Natl. Cancer Inst. 2014, 106, djt431. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef]
- Cuzick, J.; Thorat, M.A.; Andriole, G.; Brawley, O.W.; Brown, P.H.; Culig, Z.; Eeles, R.A.; Ford, L.G.; Hamdy, F.C.; Holmberg, L.; et al. Prevention and early detection of prostate cancer. Lancet. Oncol. 2014, 15, e484–e492. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Squires, H.; Carroll, C.; Papaioannou, D.; Booth, A.; Logan, R.F.; Maguire, C.; Hind, D.; Tappenden, P. Chemoprevention of colorectal cancer: Systematic review and economic evaluation. Health Technol. Assess. 2010, 14, 1–206. [Google Scholar] [CrossRef] [PubMed]
- Guadagni, F.; Ferroni, P.; Palmirotta, R.; Del Monte, G.; Formica, V.; Roselli, M. Non-steroidal anti-inflammatory drugs in cancer prevention and therapy. Anticancer Res. 2007, 27, 3147–3162. [Google Scholar] [PubMed]
- Liggett, J.L.; Zhang, X.; Eling, T.E.; Baek, S.J. Anti-tumor activity of non-steroidal anti-inflammatory drugs: Cyclooxygenase-independent targets. Cancer Lett. 2014, 346, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Waskewich, C.; Blumenthal, R.D.; Li, H.; Stein, R.; Goldenberg, D.M.; Burton, J. Celecoxib exhibits the greatest potency amongst cyclooxygenase (cox) inhibitors for growth inhibition of cox-2-negative hematopoietic and epithelial cell lines. Cancer Res. 2002, 62, 2029–2033. [Google Scholar] [PubMed]
- Song, X.; Lin, H.P.; Johnson, A.J.; Tseng, P.H.; Yang, Y.T.; Kulp, S.K.; Chen, C.S. Cyclooxygenase-2, player or spectator in cyclooxygenase-2 inhibitor-induced apoptosis in prostate cancer cells. J. Natl. Cancer Inst. 2002, 94, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Kan, M.; Qiao, P.; Pan, Y.; Wang, Z.; Xiao, X.; Li, J.; Chen, L. Celecoxib induces apoptosis via a mitochondriadependent pathway in the h22 mouse hepatoma cell line. Mol. Med. Rep. 2014, 10, 2093–2098. [Google Scholar] [PubMed]
- Hashitani, S.; Urade, M.; Nishimura, N.; Maeda, T.; Takaoka, K.; Noguchi, K.; Sakurai, K. Apoptosis induction and enhancement of cytotoxicity of anticancer drugs by celecoxib, a selective cyclooxygenase-2 inhibitor, in human head and neck carcinoma cell lines. Int. J. Oncol. 2003, 23, 665–672. [Google Scholar] [PubMed]
- Duncan, K.; Uwimpuhwe, H.; Czibere, A.; Sarkar, D.; Libermann, T.A.; Fisher, P.B.; Zerbini, L.F. NSAIDs induce apoptosis in nonproliferating ovarian cancer cells and inhibit tumor growth in vivo. IUBMB Life 2012, 64, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yim, G.W.; Nam, E.J.; Kim, Y.T. Synergistic effect of cox-2 inhibitor on paclitaxel-induced apoptosis in the human ovarian cancer cell line ovcar-3. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2014, 46, 81–92. [Google Scholar]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, G.; Balzan, R. The proapoptotic effect of traditional and novel nonsteroidal anti-inflammatory drugs in mammalian and yeast cells. Oxidative Med. Cell. Longev. 2013, 2013, 504230. [Google Scholar] [CrossRef]
- Khwaja, F.; Allen, J.; Lynch, J.; Andrews, P.; Djakiew, D. Ibuprofen inhibits survival of bladder cancer cells by induced expression of the p75ntr tumor suppressor protein. Cancer Res. 2004, 64, 6207–6213. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.; Djakiew, D.; Krygier, S.; Andrews, P. Superior effectiveness of ibuprofen compared with other NSAIDs for reducing the survival of human prostate cancer cells. Cancer Chemother. Pharmacol. 2002, 50, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.; Zhao, X.; Allen, J.; Li, F.; Chang, M. A comparison of the effectiveness of selected non-steroidal anti-inflammatory drugs and their derivatives against cancer cells in vitro. Cancer Chemother. Pharmacol. 2008, 61, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, B.A.; Narayanan, N.K.; Pittman, B.; Reddy, B.S. Regression of mouse prostatic intraepithelial neoplasia by nonsteroidal anti-inflammatory drugs in the transgenic adenocarcinoma mouse prostate model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7727–7737. [Google Scholar] [CrossRef]
- Ishiguro, H.; Kawahara, T. Nonsteroidal anti-inflammatory drugs and prostatic diseases. BioMed Res. Int. 2014, 2014, 436123. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.; Jutooru, I.; Chadalapaka, G.; Nair, V.; Lee, S.O.; Safe, S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (sp) transcription factors. PLoS One 2012, 7, e48208. [Google Scholar] [CrossRef] [PubMed]
- Valle, B.L.; D’Souza, T.; Becker, K.G.; Wood, W.H., 3rd; Zhang, Y.; Wersto, R.P.; Morin, P.J. Non-steroidal anti-inflammatory drugs decrease e2f1 expression and inhibit cell growth in ovarian cancer cells. PLoS One 2013, 8, e61836. [Google Scholar] [CrossRef] [PubMed]
- Goluboff, E.T.; Shabsigh, A.; Saidi, J.A.; Weinstein, I.B.; Mitra, N.; Heitjan, D.; Piazza, G.A.; Pamukcu, R.; Buttyan, R.; Olsson, C.A. Exisulind (sulindac sulfone) suppresses growth of human prostate cancer in a nude mouse xenograft model by increasing apoptosis. Urology 1999, 53, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Cui, X.X.; Avila, G.E.; Huang, M.T.; Liu, Y.; Patel, J.; Kong, A.N.; Paulino, R.; Shih, W.J.; Lin, Y.; et al. Atorvastatin and celecoxib inhibit prostate pc-3 tumors in immunodeficient mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5480–5487. [Google Scholar] [CrossRef]
- Zheng, X.; Cui, X.X.; Gao, Z.; Zhao, Y.; Lin, Y.; Shih, W.J.; Huang, M.T.; Liu, Y.; Rabson, A.; Reddy, B.; et al. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent lncap xenograft prostate tumors to androgen independence. Cancer Prev. Res. 2010, 3, 114–124. [Google Scholar] [CrossRef]
- Wang, H.; Cui, X.X.; Goodin, S.; Ding, N.; van Doren, J.; Du, Z.; Huang, M.T.; Liu, Y.; Cheng, X.; Dipaola, R.S.; et al. Inhibition of il-6 expression in lncap prostate cancer cells by a combination of atorvastatin and celecoxib. Oncol. Rep. 2014, 31, 835–841. [Google Scholar] [PubMed]
- Huang, H.; Cui, X.X.; Chen, S.; Goodin, S.; Liu, Y.; He, Y.; Li, D.; Wang, H.; van Doren, J.; Dipaola, R.S.; et al. Combination of lipitor and celebrex inhibits prostate cancer vcap cells in vitro and in vivo. Anticancer Res. 2014, 34, 3357–3363. [Google Scholar] [PubMed]
- Lonnroth, C.; Andersson, M.; Asting, A.G.; Nordgren, S.; Lundholm, K. Preoperative low dose nsaid treatment influences the genes for stemness, growth, invasion and metastasis in colorectal cancer. Int. J. Oncol. 2014, 45, 2208–2220. [Google Scholar] [PubMed]
- Baggott, J.E.; Morgan, S.L.; Ha, T.; Vaughn, W.H.; Hine, R.J. Inhibition of folate-dependent enzymes by non-steroidal anti-inflammatory drugs. Biochem. J. 1992, 282 (Pt 1), 197–202. [Google Scholar] [PubMed]
- Reti, A.; Pap, E.; Adleff, V.; Jeney, A.; Kralovanszky, J.; Budai, B. Enhanced 5-fluorouracil cytotoxicity in high cyclooxygenase-2 expressing colorectal cancer cells and xenografts induced by non-steroidal anti-inflammatory drugs via downregulation of dihydropyrimidine dehydrogenase. Cancer Chemother. Pharmacol. 2010, 66, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Reti, A.; Pap, E.; Zalatnai, A.; Jeney, A.; Kralovanszky, J.; Budai, B. Co-inhibition of cyclooxygenase-2 and dihydropyrimidine dehydrogenase by non-steroidal anti-inflammatory drugs in tumor cells and xenografts. Anticancer Res. 2009, 29, 3095–3101. [Google Scholar] [PubMed]
- Reti, A.; Barna, G.; Pap, E.; Adleff, V.; L Komlósi, V.; Jeney, A.; Kralovanszky, J.; Budai, B. Enhancement of 5-fluorouracil efficacy on high cox-2 expressing hca-7 cells by low dose indomethacin and ns-398 but not on low cox-2 expressing ht-29 cells. Pathol. Oncol. Res. POR 2009, 15, 335–344. [Google Scholar] [CrossRef]
- Mingatto, F.E.; Santos, A.C.; Uyemura, S.A.; Jordani, M.C.; Curti, C. In vitro interaction of nonsteroidal anti-inflammatory drugs on oxidative phosphorylation of rat kidney mitochondria: Respiration and atp synthesis. Arch. Biochem. Biophys. 1996, 334, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Bravo, C.; Vasquez, C.; Ayala, G.; Silveira, L.H.; Martinez-Lavin, M. Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs: Study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem. Pharmacol. 1999, 57, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, S.; Rafi, S.; Hayllar, J.; Sigthorsson, G.; Jacob, M.; Price, A.B.; Macpherson, A.; Mahmod, T.; Scott, D.; Wrigglesworth, J.M.; et al. Mitochondrial damage: A possible mechanism of the “topical” phase of nsaid induced injury to the rat intestine. Gut 1997, 41, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, T.G.; Weis-Fogh, U.S.; Nielsen, H.H.; Olesen, H.P. Salicylate- and aspirin-induced uncoupling of oxidative phosphorylation in mitochondria isolated from the mucosal membrane of the stomach. Scand. J. Clin. Lab. Investig. 1976, 36, 649–654. [Google Scholar] [CrossRef]
- Petrescu, I.; Tarba, C. Uncoupling effects of diclofenac and aspirin in the perfused liver and isolated hepatic mitochondria of rat. Biochim. Biophys. Acta 1997, 1318, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, V.; Salvi, M.; Toninello, A. Oxidative stress is responsible for mitochondrial permeability transition induction by salicylate in liver mitochondria. J. Biol. Chem. 2005, 280, 33864–33872. [Google Scholar] [CrossRef] [PubMed]
- Pigoso, A.A.; Uyemura, S.A.; Santos, A.C.; Rodrigues, T.; Mingatto, F.E.; Curti, C. Influence of nonsteroidal anti-inflammatory drugs on calcium efflux in isolated rat renal cortex mitochondria and aspects of the mechanisms involved. Int. J. Biochem. Cell Biol. 1998, 30, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Uyemura, S.A.; Santos, A.C.; Mingatto, F.E.; Jordani, M.C.; Curti, C. Diclofenac sodium and mefenamic acid: Potent inducers of the membrane permeability transition in renal cortex mitochondria. Arch. Biochem. Biophys. 1997, 342, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Daouphars, M.; Koufany, M.; Benani, A.; Marchal, S.; Merlin, J.L.; Netter, P.; Jouzeau, J.Y. Uncoupling of oxidative phosphorylation and smac/diablo release are not sufficient to account for induction of apoptosis by sulindac sulfide in human colorectal cancer cells. Int. J. Oncol. 2005, 26, 1069–1077. [Google Scholar] [PubMed]
- Tomellini, E.; Touil, Y.; Lagadec, C.; Julien, S.; Ostyn, P.; Ziental-Gelus, N.; Meignan, S.; Lengrand, J.; Adriaenssens, E.; Polakowska, R.; et al. NGF and proNGF simultaneously promote symmetric self-renewal, quiescence and emt to enlarge the breast cancer stem cell compartment. Stem Cells 2014, 33, 342–353. [Google Scholar] [CrossRef]
- Tomellini, E.; Lagadec, C.; Polakowska, R.; le Bourhis, X. Role of p75 neurotrophin receptor in stem cell biology: More than just a marker. Cell. Mol. Life Sci.: CMLS 2014, 71, 2467–2481. [Google Scholar] [CrossRef] [PubMed]
- Wynne, S.; Djakiew, D. Nsaid inhibition of prostate cancer cell migration is mediated by nag-1 induction via the p38 mapk-p75(ntr) pathway. Mol. Cancer Res.: MCR 2010, 8, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Kim, J.; Khan, I.A.; Walker, L.A.; Khan, S.I. Nonsteroidal anti-inflammatory drug activated gene-1 (nag-1) modulators from natural products as anti-cancer agents. Life Sci. 2014, 100, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chrysovergis, K.; Kosak, J.; Kissling, G.; Streicker, M.; Moser, G.; Li, R.; Eling, T.E. Hnag-1 increases lifespan by regulating energy metabolism and insulin/IGF-1/mTOR signaling. Aging 2014, 6, 690–704. [Google Scholar] [PubMed]
- Chrysovergis, K.; Wang, X.; Kosak, J.; Lee, S.H.; Kim, J.S.; Foley, J.F.; Travlos, G.; Singh, S.; Baek, S.J.; Eling, T.E. Nag-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int. J. Obes. 2014, 38, 1555–1564. [Google Scholar] [CrossRef]
- Vaish, V.; Piplani, H.; Rana, C.; Vaiphei, K.; Sanyal, S.N. NSAIDs may regulate egr-1-mediated induction of reactive oxygen species and non-steroidal anti-inflammatory drug-induced gene (nag)-1 to initiate intrinsic pathway of apoptosis for the chemoprevention of colorectal cancer. Mol. Cell. Biochem. 2013, 378, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Legge, F.; Paglia, A.; D’Asta, M.; Fuoco, G.; Scambia, G.; Ferrandina, G. Phase ii study of the combination carboplatin plus celecoxib in heavily pre-treated recurrent ovarian cancer patients. BMC Cancer 2011, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Reyners, A.K.; de Munck, L.; Erdkamp, F.L.; Smit, W.M.; Hoekman, K.; Lalisang, R.I.; de Graaf, H.; Wymenga, A.N.; Polee, M.; Hollema, H.; et al. A randomized phase ii study investigating the addition of the specific cox-2 inhibitor celecoxib to docetaxel plus carboplatin as first-line chemotherapy for stage ic to iv epithelial ovarian cancer, fallopian tube or primary peritoneal carcinomas: The docacel study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO 2012, 23, 2896–2902. [Google Scholar] [CrossRef]
- Chung, Y.M.; Bae, Y.S.; Lee, S.Y. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic. Biol. Med. 2003, 34, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, L.; Mackenzie, G.G.; Rigas, B. Oxidative stress mediates through apoptosis the anticancer effect of phospho-nonsteroidal anti-inflammatory drugs: Implications for the role of oxidative stress in the action of anticancer agents. J. Pharmacol. Exp. Ther. 2011, 338, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B.; Sun, Y. Induction of oxidative stress as a mechanism of action of chemopreventive agents against cancer. Br. J. Cancer 2008, 98, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Niwa, K.; Takemura, M.; Sun, W.; Onogi, K.; Wu, Y.; Seishima, M.; Mori, H.; Tamaya, T. Significant anti-proliferation of human endometrial cancer cells by combined treatment with a selective cox-2 inhibitor ns398 and specific mek inhibitor u0126. Int. J. Oncol. 2005, 26, 737–744. [Google Scholar] [PubMed]
- Bank, A.; Yu, J.; Zhang, L. NSAIDs downregulate bcl-x(l) and dissociate bax and bcl-x(l) to induce apoptosis in colon cancer cells. Nutr. Cancer 2008, 60 (Suppl. 1), 98–103. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Fairbairn, D.W.; Bradshaw, W.S.; O’Neill, K.L.; Ewert, D.L.; Simmons, D.L. Nsaid-induced apoptosis in rous sarcoma virus-transformed chicken embryo fibroblasts is dependent on v-src and c-myc and is inhibited by bcl-2. Prostaglandins 1997, 54, 549–568. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in hepg2 cells. PLoS One 2012, 7, e36325. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma hepg2 cells. Eur. J. Pharmacol. 2011, 668, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A.; Howarth, F.C. Alterations in glutathione redox metabolism, oxidative stress, and mitochondrial function in the left ventricle of elderly zucker diabetic fatty rat heart. Int. J. Mol. Sci. 2012, 13, 16241–16254. [Google Scholar] [CrossRef] [PubMed]
- Rousar, T.; Parik, P.; Kucera, O.; Bartos, M.; Cervinkova, Z. Glutathione reductase is inhibited by acetaminophen-glutathione conjugate in vitro. Physiol. Res./Acad. Sci. Bohemoslov. 2010, 59, 225–232. [Google Scholar]
- Sun, Y.; Rigas, B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: Implications for the mechanism of action of anticancer agents. Cancer Res. 2008, 68, 8269–8277. [Google Scholar] [CrossRef] [PubMed]
- Uwakwe, A.A.; Monanu, M.O. In vitro effects of aspirin and paracetamol on human erythrocyte glutathione-s-transferase activity. Glob. J. Med. Sci. 2004, 3, 33–36. [Google Scholar]
- Duffy, C.P.; Elliott, C.J.; O’Connor, R.A.; Heenan, M.M.; Coyle, S.; Cleary, I.M.; Kavanagh, K.; Verhaegen, S.; O’Loughlin, C.M.; NicAmhlaoibh, R.; et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti-inflammatory drugs (NSAIDs). Eur. J. Cancer 1998, 34, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Costa, N.J.; Dahm, C.C.; Beer, S.M.; Brown, S.E.; Filipovska, A.; Murphy, M.P. Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal. 2005, 7, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Zhang, J.; Grek, C.; Ye, Z.W.; Manevich, Y.; Tew, K.D.; Townsend, D.M. Pleiotropic functions of glutathione s-transferase p. Adv. Cancer Res. 2014, 122, 143–175. [Google Scholar] [PubMed]
- Singh, S. Cytoprotective and regulatory functions of glutathione s-transferases in cancer cell proliferation and cell death. Cancer Chemother. Pharmacol. 2014, 75, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant defense. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.J.; Reed, D.J. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J. Biol. Chem. 1982, 257, 3747–3753. [Google Scholar] [PubMed]
- Costa, N.J.; Dahm, C.C.; Hurrell, F.; Taylor, E.R.; Murphy, M.P. Interactions of mitochondrial thiols with nitric oxide. Antioxid. Redox Signal. 2003, 5, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.J. Glutathione: Toxicological implications. Annu. Rev. Pharmacol. Toxicol. 1990, 30, 603–631. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex i increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.B.; Kurten, R.C.; McCullough, S.S.; Brock, R.W.; Hinson, J.A. Mechanisms of acetaminophen-induced hepatotoxicity: Role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J. Pharmacol. Exp. Ther. 2005, 312, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Van Bladeren, P.J. Glutathione conjugation as a bioactivation reaction. Chem.-Biol. Interact. 2000, 129, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Resnick, L.; Gamliel, E.; Kesaraju, S.; Weissbach, H.; Binninger, D. Sulindac enhances the killing of cancer cells exposed to oxidative stress. PLoS One 2009, 4, e5804. [Google Scholar] [CrossRef] [PubMed]
- Al-Belooshi, T.; John, A.; Tariq, S.; Al-Otaiba, A.; Raza, H. Increased mitochondrial stress and modulation of mitochondrial respiratory enzyme activities in acetaminophen-induced toxicity in mouse macrophage cells. Food Chem. Toxicol. 2010, 48, 2624–2632. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.S.; Chae, H.J. Sodium salicylate inhibits expression of cox-2 through suppression of erk and subsequent nf-kappab activation in rat ventricular cardiomyocytes. Arch. Pharm. Res. 2003, 26, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Carrett-Dias, M.; Votto, A.P.; Filgueira Dde, M.; Almeida, D.V.; Vallochi, A.L.; D’Oca, M.G.; Marins, L.F.; Trindade, G.S. Anti-mdr and antitumoral action of acetylsalicylic acid on leukaemic cells. Biosci. Rep. 2011, 31, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P. Nonsteroidal anti-inflammatory drug-induced hepatotoxicity. Clin. Liver Dis. 2007, 11, 563–575, vi-vii. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Leipelt, M.; Wolters, D.; Steegborn, C. Identification of peroxiredoxin 1 as a novel interaction partner for the lifespan regulator protein p66shc. Aging 2009, 1, 254–265. [Google Scholar] [PubMed]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; Snider, G.W.; Fredette, N.; Stumpff, J.; Hondal, R.J.; Heintz, N.H. Resolution of oxidative stress by thioredoxin reductase: Cysteine versus selenocysteine. Redox Biol. 2014, 2, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Soethoudt, M.; Peskin, A.V.; Dickerhof, N.; Paton, L.N.; Pace, P.E.; Winterbourn, C.C. Interaction of adenanthin with glutathione and thiol enzymes: Selectivity for thioredoxin reductase and inhibition of peroxiredoxin recycling. Free Radic. Biol. Med. 2014, 77, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Zhang, B.; Yao, J.; Liu, Y.; Fang, J. Shikonin targets cytosolic thioredoxin reductase to induce ros-mediated apoptosis in human promyelocytic leukemia hl-60 cells. Free Radic. Biol. Med. 2014, 70, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Zhang, B.; Yao, J.; Liu, Y.; Sun, J.; Ge, C.; Peng, S.; Fang, J. Gambogic acid induces apoptosis in hepatocellular carcinoma smmc-7721 cells by targeting cytosolic thioredoxin reductase. Free Radic. Biol. Med. 2014, 69, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhang, H.; Zhang, X.; Lu, J.; Holmgren, A. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J. Biol. Chem. 2013, 288, 32241–32247. [Google Scholar] [CrossRef] [PubMed]
- Galimov, E.R. The role of p66shc in oxidative stress and apoptosis. Acta Nat. 2010, 2, 44–51. [Google Scholar]
- Galimov, E.R.; Chernyak, B.V.; Sidorenko, A.S.; Tereshkova, A.V.; Chumakov, P.M. Prooxidant properties of p66shc are mediated by mitochondria in human cells. PLoS One 2014, 9, e86521. [Google Scholar] [CrossRef] [PubMed]
- Savino, C.; Pelicci, P.; Giorgio, M. The p66shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxidative Med. Cell. Longev. 2013, 2013, 719407. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Trinei, M.; Giorgio, M.; Cicalese, A.; Barozzi, S.; Ventura, A.; Migliaccio, E.; Milia, E.; Padura, I.M.; Raker, V.A.; Maccarana, M.; et al. A p53-p66shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Y.G.; Chen, C.L. Anti-apoptotic role of peroxiredoxin iii in cervical cancer cells. FEBS Open Bio 2013, 3, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, H.C.; Patel, D.; Howat, W.J.; Warren, A.Y.; Kay, J.D.; Sangan, T.; Marioni, J.C.; Mitchell, J.; Aldridge, S.; Luxton, H.J.; et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 2013, 109, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabo, I. Mitochondrial ion channels as oncological targets. Oncogene 2014, 33, 5569–5581. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-k+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial kv1.3 channels induce bax/bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Schennach, R.; Obermeier, M.; Seemuller, F.; Jager, M.; Schmauss, M.; Laux, G.; Pfeiffer, H.; Naber, D.; Schmidt, L.G.; Gaebel, W.; et al. Evaluating depressive symptoms in schizophrenia: A psychometric comparison of the calgary depression scale for schizophrenia and the hamilton depression rating scale. Psychopathology 2012, 45, 276–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ros-hif-1alpha-kv1.5 o2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–578. [Google Scholar] [CrossRef] [PubMed]
- Caouette, D.; Dongmo, C.; Berube, J.; Fournier, D.; Daleau, P. Hydrogen peroxide modulates the kv1.5 channel expressed in a mammalian cell line. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 479–486. [Google Scholar] [CrossRef]
- Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Yu, Y.; Remillard, C.V.; Yuan, J.X. Acute hypoxia selectively inhibits kcna5 channels in pulmonary artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2006, 290, C907–916. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Ma, W.L.; Yang, M.L.; Liu, S.Y.; Wang, D.X. Effect of chronic cigarette smoking on large-conductance calcium-activated potassium channel and kv1.5 expression in bronchial smooth muscle cells of rats. Sheng li xue bao : [Acta Physiol. Sin.] 2004, 56, 573–578. [Google Scholar]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel kv1.3 mediates bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Leanza, L.; Gulbins, E.; Zoratti, M. Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Archiv.: Eur. J. Physiol. 2012, 463, 231–246. [Google Scholar] [CrossRef]
- Szabo, I.; Soddemann, M.; Leanza, L.; Zoratti, M.; Gulbins, E. Single-point mutations of a lysine residue change function of bax and bcl-xl expressed in bax- and bak-less mouse embryonic fibroblasts: Novel insights into the molecular mechanisms of bax-induced apoptosis. Cell Death Differ. 2011, 18, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Annis, M.G.; Soucie, E.L.; Dlugosz, P.J.; Cruz-Aguado, J.A.; Penn, L.Z.; Leber, B.; Andrews, D.W. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005, 24, 2096–2103. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Lin, J.C. A set of homology models of pore loop domain of six eukaryotic voltage-gated potassium channels kv1.1-kv1.6. Proteins 2004, 55, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Ruppersberg, J.P.; Stocker, M.; Pongs, O.; Heinemann, S.H.; Frank, R.; Koenen, M. Regulation of fast inactivation of cloned mammalian Ik(A) channels by cysteine oxidation. Nature 1991, 352, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Weng, J.; Levin, E.J.; Zhou, M. Oxidation of nadph on kvbeta1 inhibits ball-and-chain type inactivation by restraining the chain. Proc. Natl. Acad. Sci. USA 2011, 108, 5885–5890. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T. Inhibition of the activity of glutathione peroxidase by tertiary-butylhydroperoxide in cultured chinese hamster cells and the role of cellular glutathione in the recovery of the activity. Toxicology 1992, 71, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Kushnareva, Y.E.; Sokolove, P.M. Prooxidants open both the mitochondrial permeability transition pore and a low-conductance channel in the inner mitochondrial membrane. Arch. Biochem. Biophys. 2000, 376, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.M.; Lemasters, J.J.; Nieminen, A.L. Contribution of increased mitochondrial free ca2+ to the mitochondrial permeability transition induced by tert-butylhydroperoxide in rat hepatocytes. Hepatology 1999, 29, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.; Nutt, L.; Orrenius, S.; Gogvadze, V. Arsenic stimulates release of cytochrome c from isolated mitochondria via induction of mitochondrial permeability transition. Toxicol. Appl. Pharmacol. 2005, 207, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V.; Bernardi, P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur. J. Biochem./FEBS 1996, 238, 623–630. [Google Scholar] [CrossRef]
- Connern, C.P.; Halestrap, A.P. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem. J. 1994, 302 (Pt 2), 321–324. [Google Scholar] [PubMed]
- Costantini, P.; Belzacq, A.S.; Vieira, H.L.; Larochette, N.; de Pablo, M.A.; Zamzami, N.; Susin, S.A.; Brenner, C.; Kroemer, G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000, 19, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Rettig, J.; Heinemann, S.H.; Wunder, F.; Lorra, C.; Parcej, D.N.; Dolly, J.O.; Pongs, O. Inactivation properties of voltage-gated k+ channels altered by presence of beta-subunit. Nature 1994, 369, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Lee, J.E.; Park, J.M.; Suh, C.K. T-butyl hydrogen peroxide increases the activities of the maxi-k channels of rat brain. Life Sci. 2000, 67, 2485–2491. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.K.; Schyvens, C.G.; Wyse, K.R.; Bursill, J.A.; Owe-Young, R.A.; Macdonald, P.S.; Campbell, T.J. Role of non-steroidal anti-inflammatory drugs (NSAIDs) in modulating vascular smooth muscle cells by activating large-conductance potassium ion channels. In Patch Clamp Technique; InTech: Bratislava, Slovakia, 2012; pp. 283–300. [Google Scholar]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Giorgio, V.; Fogolari, F.; Glick, G.D.; Bernardi, P.; Lippe, G. The oligomycin-sensitivity conferring protein of mitochondrial atp synthase: Emerging new roles in mitochondrial pathophysiology. Int. J. Mol. Sci. 2014, 15, 7513–7536. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Watanabe, K.; Uechi, Y.; Yoshioka, H.; Ohta, Y. Repetitive transient depolarizations of the inner mitochondrial membrane induced by proton pumping. Biophys. J. 2005, 88, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Barsukova, A.; Komarov, A.; Hajnoczky, G.; Bernardi, P.; Bourdette, D.; Forte, M. Activation of the mitochondrial permeability transition pore modulates ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci. 2011, 33, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Dworetzky, S.I.; Bonanni, L.; Zhang, P.; Sacchetti, S.; Li, H.; Signore, A.P.; Smith, P.J.; Gribkoff, V.K.; Jonas, E.A. The mitochondrial complex v-associated large-conductance inner membrane current is regulated by cyclosporine and dexpramipexole. Mol. Pharmacol. 2014, 87, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial atp synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faccenda, D.; Campanella, M. Molecular regulation of the mitochondrial f(1)f(o)-atpsynthase: Physiological and pathological significance of the inhibitory factor 1 (if(1)). Int. J. Cell Biol. 2012, 2012, 367934. [Google Scholar] [CrossRef] [PubMed]
- Formentini, L.; Pereira, M.P.; Sanchez-Cenizo, L.; Santacatterina, F.; Lucas, J.J.; Navarro, C.; Martinez-Serrano, A.; Cuezva, J.M. In vivo inhibition of the mitochondrial h+-atp synthase in neurons promotes metabolic preconditioning. EMBO J. 2014, 33, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Formentini, L.; Sanchez-Arago, M.; Sanchez-Cenizo, L.; Cuezva, J.M. The mitochondrial atpase inhibitory factor 1 triggers a ros-mediated retrograde prosurvival and proliferative response. Mol. Cell 2012, 45, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Faccenda, D.; Tan, C.H.; Duchen, M.R.; Campanella, M. Mitochondrial if(1) preserves cristae structure to limit apoptotic cell death signaling. Cell Cycle 2013, 12, 2530–2532. [Google Scholar] [CrossRef] [PubMed]
- Faccenda, D.; Tan, C.H.; Seraphim, A.; Duchen, M.R.; Campanella, M. If1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ. 2013, 20, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Arago, M.; Formentini, L.; Garcia-Bermudez, J.; Cuezva, J.M. If1 reprograms energy metabolism and signals the oncogenic phenotype in cancer. Cell Cycle 2012, 11, 2963–2964. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Arago, M.; Formentini, L.; Martinez-Reyes, I.; Garcia-Bermudez, J.; Santacatterina, F.; Sanchez-Cenizo, L.; Willers, I.M.; Aldea, M.; Najera, L.; Juarranz, A.; et al. Expression, regulation and clinical relevance of the atpase inhibitory factor 1 in human cancers. Oncogenesis 2013, 2, e46. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Arago, M.; Formentini, L.; Cuezva, J.M. Mitochondria-mediated energy adaption in cancer: The h(+)-atp synthase-geared switch of metabolism in human tumors. Antioxid. Redox Signal. 2013, 19, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 2014, 56, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Chantome, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. Kca and Ca2+ channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.; Lindemann, O.; Schwab, A. Trp channels and stim/orai proteins: Sensors and effectors of cancer and stroma cell migration. Br. J. Pharmacol. 2014, 171, 5524–5540. [Google Scholar] [CrossRef] [PubMed]
- Nunez, L.; Valero, R.A.; Senovilla, L.; Sanz-Blasco, S.; Garcia-Sancho, J.; Villalobos, C. Cell proliferation depends on mitochondrial ca2+ uptake: Inhibition by salicylate. J. Physiol. 2006, 571, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Munoz, E.; Valero, R.A.; Quintana, A.; Hoth, M.; Nunez, L.; Villalobos, C. Nonsteroidal anti-inflammatory drugs inhibit vascular smooth muscle cell proliferation by enabling the ca2+-dependent inactivation of calcium release-activated calcium/orai channels normally prevented by mitochondria. J. Biol. Chem. 2011, 286, 16186–16196. [Google Scholar] [CrossRef]
- Brueggemann, L.I.; Mani, B.K.; Mackie, A.R.; Cribbs, L.L.; Byron, K.L. Novel actions of nonsteroidal anti-inflammatory drugs on vascular ion channels: Accounting for cardiovascular side effects and identifying new therapeutic applications. Mol. Cell. Pharmacol. 2010, 2, 15–19. [Google Scholar]
- Brueggemann, L.I.; Mackie, A.R.; Mani, B.K.; Cribbs, L.L.; Byron, K.L. Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol. Pharmacol. 2009, 76, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.S.; Lim, P.L.; Gupta, R.; Boelsterli, U.A. Critical role of free cytosolic calcium, but not uncoupling, in mitochondrial permeability transition and cell death induced by diclofenac oxidative metabolites in immortalized human hepatocytes. Toxicol. Appl. Pharmacol. 2006, 217, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Poon, G.K.; Chen, Q.; Teffera, Y.; Ngui, J.S.; Griffin, P.R.; Braun, M.P.; Doss, G.A.; Freeden, C.; Stearns, R.A.; Evans, D.C.; et al. Bioactivation of diclofenac via benzoquinone imine intermediates-identification of urinary mercapturic acid derivatives in rats and humans. Drug Metab. Dispos.: Biol. Fate Chem. 2001, 29, 1608–1613. [Google Scholar]
- Grillo, M.P.; Ma, J.; Teffera, Y.; Waldon, D.J. A novel bioactivation pathway for 2-[2-(2,6-dichlorophenyl)aminophenyl]ethanoic acid (diclofenac) initiated by cytochrome p450-mediated oxidative decarboxylation. Drug Metab. Dispos.: Biol. Fate Chem. 2008, 36, 1740–1744. [Google Scholar] [CrossRef]
- Hansson, M.J.; Mansson, R.; Morota, S.; Uchino, H.; Kallur, T.; Sumi, T.; Ishii, N.; Shimazu, M.; Keep, M.F.; Jegorov, A.; et al. Calcium-induced generation of reactive oxygen species in brain mitochondria is mediated by permeability transition. Free Radic. Biol. Med. 2008, 45, 284–294. [Google Scholar] [CrossRef] [PubMed]
- LoGuidice, A.; Ramirez-Alcantara, V.; Proli, A.; Gavillet, B.; Boelsterli, U.A. Pharmacologic targeting or genetic deletion of mitochondrial cyclophilin d protects from nsaid-induced small intestinal ulceration in mice. Toxicol. Sci.: Off. J. Soc. Toxicol. 2010, 118, 276–285. [Google Scholar] [CrossRef]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin frames a common drug-binding site in the atp synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Bononi, A.; de Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the fo atp synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Odinokova, I.; Bakunts, A.; Ternovsky, V.; Krestinina, O.; Tyynela, J.; Saris, N.E. Potential role of subunit c of f0f1-atpase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014, 55, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, G.; Martinez-Diez, M.; Fabregat, I.; Cuezva, J.M. Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial h+-atp synthase. Carcinogenesis 2006, 27, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Chen, X.; Boitano, A.; Swenson, L.; Opipari, A.W., Jr.; Glick, G.D. Identification and validation of the mitochondrial f1f0-atpase as the molecular target of the immunomodulatory benzodiazepine bz-423. Chem. Biol. 2005, 12, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Cleary, J.; Fierke, C.A.; Opipari, A.W., Jr.; Glick, G.D. Mechanistic basis for therapeutic targeting of the mitochondrial f1f0-atpase. ACS Chem. Biol. 2006, 1, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid. Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Cuezva, J.M. The h(+)-atp synthase: A gate to ros-mediated cell death or cell survival. Biochim. Biophys. Acta 2014, 1837, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xl regulates metabolic efficiency of neurons through interaction with the mitochondrial f1fo atp synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xl regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Costantini, P.; Colonna, R.; Bernardi, P. Induction of the mitochondrial permeability transition by n-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochim. Biophys. Acta 1998, 1365, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J. Arsenic-based antineoplastic drugs and their mechanisms of action. Met.-Based Drugs 2008, 2008, 260146. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Costantini, P.; Scorrano, L.; Colonna, R.; Passamonti, S.; Bernardi, P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J. Biol. Chem. 1994, 269, 16638–16642. [Google Scholar] [PubMed]
- Costantini, P.; Chernyak, B.V.; Petronilli, V.; Bernardi, P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 1996, 271, 6746–6751. [Google Scholar] [CrossRef] [PubMed]
- Skonberg, C.; Olsen, J.; Madsen, K.G.; Hansen, S.H.; Grillo, M.P. Metabolic activation of carboxylic acids. Expert Opin. Drug Metab. Toxicol. 2008, 4, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Nam, J.H.; Lee, E.S.; Zhang, Y.; Kim, S.J. Inhibition of Ca2+ release-activated Ca2+ channel (CRAC) by curcumin and caffeic acid phenethyl ester (CAPE) via electrophilic addition to a cysteine residue of orai1. Biochem. Biophys. Res. Commun. 2012, 428, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Callister, M.E.; Pinhu, L.; Catley, M.C.; Westwell, A.D.; Newton, R.; Leaver, S.K.; Quinlan, G.J.; Evans, T.W.; Griffiths, M.J.; Burke-Gaffney, A. Pmx464, a thiol-reactive quinol and putative thioredoxin inhibitor, inhibits nf-kappab-dependent proinflammatory activation of alveolar epithelial cells. Br. J. Pharmacol. 2008, 155, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kufe, D. Triterpenoid cddo-methyl ester inhibits the janus-activated kinase-1 (jak1)-->signal transducer and activator of transcription-3 (stat3) pathway by direct inhibition of jak1 and stat3. Cancer Res. 2008, 68, 2920–2926. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid cddo-me blocks the nf-kappab pathway by direct inhibition of ikkbeta on cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Qi, C.; Liu, J.; Sun, X.; Zhang, Q.; Sima, Z.; Liu, J.; Li, W.; Yu, Q. 2-methoxystypandrone inhibits signal transducer and activator of transcription 3 and nuclear factor-kappab signaling by inhibiting janus kinase 2 and ikappab kinase. Cancer Sci. 2014, 105, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, D.H.; Kim, J.H.; Hong, S.; Choi, D.; Kim, Y.J.; Kwak, M.K.; Jung, Y. Caffeic acid phenethyl ester-mediated nrf2 activation and ikappab kinase inhibition are involved in nfkappab inhibitory effect: Structural analysis for nfkappab inhibition. Eur. J. Pharmacol. 2010, 643, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Jeong, J.Y.; Jue, D.M. Thiol-reactive metal compounds inhibit nf-kappa b activation by blocking i kappa b kinase. J. Immunol. 2000, 164, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T. Chemical biology of inflammatory cytokine signaling. J. Antibiot. 2009, 62, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of i(kappa)b kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Kudugunti, S.K.; Thorsheim, H.; Yousef, M.S.; Guan, L.; Moridani, M.Y. The metabolic bioactivation of caffeic acid phenethyl ester (cape) mediated by tyrosinase selectively inhibits glutathione s-transferase. Chem.-Biol. Interact. 2011, 192, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Bioreductive activation of quinones: Redox properties and thiol reactivity. Free Radic. Res. Commun. 1990, 8, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Heinze, J.; Haehnel, W. Site-specific binding of quinones to proteins through thiol addition and addition-elimination reactions. J. Am. Chem. Soc. 2005, 127, 6140–6141. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.; Bellomo, G.; Thor, H.; Nicotera, P.; Orrenius, S. Menadione-induced cytotoxicity is associated with protein thiol oxidation and alteration in intracellular ca2+ homeostasis. Arch. Biochem. Biophys. 1984, 235, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.; Ross, D.; Bellomo, G.; Eklow, L.; Orrenius, S. Alterations in intracellular thiol homeostasis during the metabolism of menadione by isolated rat hepatocytes. Arch. Biochem. Biophys. 1984, 235, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, N.; Aniya, Y. The role of a membrane-bound glutathione transferase in the peroxynitrite-induced mitochondrial permeability transition pore: Formation of a disulfide-linked protein complex. Arch. Biochem. Biophys. 2011, 516, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Aniya, Y.; Imaizumi, N. Mitochondrial glutathione transferases involving a new function for membrane permeability transition pore regulation. Drug Metab. Rev. 2011, 43, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Shimoji, M.; Imaizumi, N.; Aniya, Y. Modulation of membrane-bound glutathione transferase activity by phospholipids including cardiolipin. Biol. Pharm. Bull. 2011, 34, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Ulziikhishig, E.; Lee, K.K.; Hossain, Q.S.; Higa, Y.; Imaizumi, N.; Aniya, Y. Inhibition of mitochondrial membrane bound-glutathione transferase by mitochondrial permeability transition inhibitors including cyclosporin a. Life Sci. 2010, 86, 726–732. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Pestana, C.R.; Silva, C.H.; Uyemura, S.A.; Santos, A.C.; Curti, C. Impact of adenosine nucleotide translocase (ant) proline isomerization on ca2+-induced cysteine relative mobility/mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2010, 42, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Pestana, C.R.; Silva, C.H.; Pardo-Andreu, G.L.; Rodrigues, F.P.; Santos, A.C.; Uyemura, S.A.; Curti, C. Ca2+ binding to c-state of adenine nucleotide translocase (ant)-surrounding cardiolipins enhances (ant)-cys(56) relative mobility: A computational-based mitochondrial permeability transition study. Biochim. Biophys. Acta 2009, 1787, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front. Oncol. 2013, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Global Cancer Facts & Figures, 2nd. edition; Atlanta, GA, USA, 2011; pp. 1–57. [Google Scholar]
- World Cancer Report; Stewart, B.W.; Kleihues, P. (Eds.) IARC Press: Lyon, France, 2003.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ralph, S.J.; Pritchard, R.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R.; Ralph, R.K. Hitting the Bull’s-Eye in Metastatic Cancers—NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death. Pharmaceuticals 2015, 8, 62-106. https://doi.org/10.3390/ph8010062
Ralph SJ, Pritchard R, Rodríguez-Enríquez S, Moreno-Sánchez R, Ralph RK. Hitting the Bull’s-Eye in Metastatic Cancers—NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death. Pharmaceuticals. 2015; 8(1):62-106. https://doi.org/10.3390/ph8010062
Chicago/Turabian StyleRalph, Stephen John, Rhys Pritchard, Sara Rodríguez-Enríquez, Rafael Moreno-Sánchez, and Raymond Keith Ralph. 2015. "Hitting the Bull’s-Eye in Metastatic Cancers—NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death" Pharmaceuticals 8, no. 1: 62-106. https://doi.org/10.3390/ph8010062
APA StyleRalph, S. J., Pritchard, R., Rodríguez-Enríquez, S., Moreno-Sánchez, R., & Ralph, R. K. (2015). Hitting the Bull’s-Eye in Metastatic Cancers—NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death. Pharmaceuticals, 8(1), 62-106. https://doi.org/10.3390/ph8010062